")
Back to Journals » International Journal of Nanomedicine » Volume 17
miR-186-5p Dysregulation in Serum Exosomes from Patients with AMI Aggravates Atherosclerosis via Targeting LOX-1
Authors Ding J, Li H, Liu W, Wang X, Feng Y, Guan H, Chen Z
Received 27 July 2022
Accepted for publication 23 November 2022
Published 13 December 2022 Volume 2022:17 Pages 6301—6316
DOI https://doi.org/10.2147/IJN.S383904
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Jiaxing Ding,1,2,* Huili Li,1,* Wei Liu,1,* Xuehua Wang,1 Yu Feng,1 Hongquan Guan,1 Zhijian Chen1
1Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Department of Cardiology, Henan Provincial Key Lab for Control of Coronary Heart Disease, Henan Provincial People’s Hospital Heart Center, Central China Fuwai Hospital of Zhengzhou University, Zhengzhou, Henan Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhijian Chen; Hongquan Guan, Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China, Tel + 86 27 85726011, Fax +86 27 85727340, Email [email protected]; [email protected]
Purpose: The formation of macrophage-derived foam cells via the uptake of modified lipoproteins is a pivotal development event in atherosclerosis. It has been reported that clinical and experimental myocardial infarction could accelerate atherosclerosis. Several studies have suggested the critical role of exosomes in cardiovascular diseases. However, the role of exosomes from patients with acute myocardial infarction (AMI) patients in atherogenesis remains unclear.
Patients and Methods: Serum exosomes from AMI patients (AMI-Exo) and control individuals (Con-Exo) were isolated and characterized. These exosomes were studied in vitro and in vivo to determine their impact on macrophage foaming and atherogenesis.
Results: Our results showed that AMI-Exo promoted foam cell formation in oxidized low-density lipoprotein (ox-LDL)-treated macrophages and progression of atherosclerosis in high-fat/cholesterol diet-fed ApoE−/− mice together with a significantly upregulated levels of lectin-like ox-LDL receptor-1 (LOX-1). The miR-186-5p was found to be downregulated in AMI-Exo and macrophages administered with AMI-Exo. Moreover, serum exosomal miR-186-5p achieved high diagnostic performance for AMI. Luciferase reporter assay indicated that miR-186-5p directly inhibited LOX-1. The endogenous or exogenous miR-186-5p deficiency enhanced lipid accumulation by upregulating LOX-1, whereas miR-186-5p mimics had a reverse effect.
Conclusion: In conclusion, the current findings suggest that dysregulated miR-186-5p in AMI-Exo may explain the contribution of acute ischemia events to the advancement of atherosclerosis by enhancing macrophage foaming via its target, LOX-1.
Keywords: atherosclerosis, exosomes, acute myocardial infarction, LOX-1, macrophages
Introduction
Atherosclerosis is a progressive disease characterized by excessive lipid accumulation in the intima of arteries.1 Within the atherosclerotic plaque, macrophages ingest modified lipoproteins and transform into foam cells, which are recognized as the initial and critical steps of atherosclerosis.2,3 The formation of macrophage-derived foam cells is regulated by the balance of cholesterol uptake, cholesterol esterification, and free cholesterol efflux.4–6 Lectin-like ox-LDL receptor 1 (LOX-1), a critical receptor for binding and internalizing lipoproteins in macrophages, is known to play a vital role in modulating the intracellular cholesterol and consequently influencing foam cell formation.6,7
Following acute ischemia incidents, accelerated atherosclerosis has been seen. Studies have reported that secondary plaque rupture occurs more commonly in individuals with acute myocardial infarction (AMI) than in those with stable angina.8 Moreover, a more severe progression of non-culprit lesions (lesions not responsible for the acute events) was found in patients presenting with AMI when compared to patients with stable angina.9–12 AMI has been demonstrated as an independent risk factor for the progression of the non-culprit coronary lesion.8 Interestingly, this assumption has also been supported by recent experimental model system research. A model of acute experimental myocardial infarction in ApoE-deficient mice accelerated atherogenesis, as evidenced by increased lipid deposits and the growing instability of plaque.13,14
Exosomes are nanosized membrane vesicles that transfer bioactive molecules for intercellular communication.15 Increasing evidence has indicated that miRNAs cargo in exosomes takes an essential part in atherosclerosis.16 For instance, exosomal miR-21-3p from nicotine-stimulated macrophages was found to aggravate atherosclerosis by targeting phosphatase and tension homologue (PTEN).17 Exosomal miRNAs, including miR-30e and miR-92a, have also been suggested as potential biomarkers to diagnose atherosclerosis.18 In addition, miRNAs in AMI patients’ circulating exosomes have been associated with pathological processes like angiogenesis.19,20 We hypothesized a potential role of exosomal miRNAs isolated from AMI patients in accelerated atherosclerosis.
Therefore, this study aimed to determine whether exosomes isolated from AMI patients accelerated atherosclerosis and the underlying mechanism involving miRNAs. Serum exosomes from patients with AMI (AMI-Exo) and control individuals (Con-Exo) were isolated to evaluate their effects on atherogenesis. AMI-Exo treatment caused lipid accumulation in macrophages in vitro and accelerated atherosclerosis in vivo, according to our findings. As a result, our study revealed that miR-186-5p expression was lower in AMI-Exo than in Con-Exo, and exosomal miR-186-5p achieved high diagnostic performance against AMI. Furthermore, miR-186-5p dysregulation in AMI-Exo was demonstrated to regulate macrophage foam cell formation via targeting LOX-1, indicating a critical biological role of exosomal miRNAs in the pathogenesis of atherosclerosis.
Materials and Methods
Ethics Statement
The study conformed to the principles outlined in the Declaration of Helsinki. Informed consents were obtained from all participants and institutional review board approval (IEC Approval Letter No. 0266) was granted by the ethics committee of Wuhan Union Hospital of Huazhong University of Science and Technology. Animal studies were conducted following the Guidelines for the Care and Use of Laboratory Animals and with the approval (IACUC No. 2741) by the Animal Care and Use Committee of Huazhong University of Science and Technology.
Participants
The study population included patients with AMI and control individuals. The inclusion criteria of AMI patients were as follows: (1) diagnosed with AMI based on the expert consensus of the Fourth General Definition of Myocardial Infarction (2018);21 (2) within 72 hours of the acute myocardial infarction onset; (3) did not receive the reperfusion therapy; (4) age older than 18 years old. Controls were the non-AMI patients hospitalized over the same period of time. Population with comorbidities such as chronic or acute infections, malignant tumors, severe kidney or liver injuries, and hematological disorders were excluded. Demographic and clinical details of the participants were collected from the electronic medical records. In this study, patients with AMI and age-matched controls (n = 19 each) were enrolled. Clinical information about all the subjects was listed in Table 1.
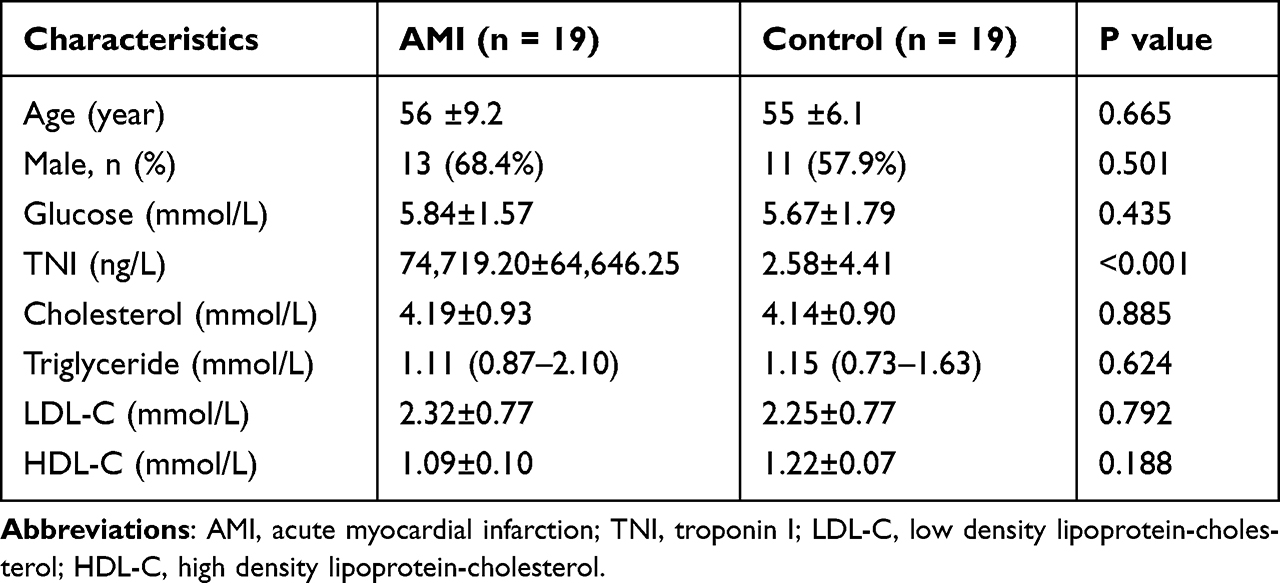 |
Table 1 The Baseline Characteristics of Enrolled Participants |
Serum Collection and Exosome Isolation
The serum was separated from whole blood by centrifugation at 3000 g for 15 min at room temperature and then diluted with PBS to lower viscosity for exosome extraction. Exosomes were isolated from diluted serum through an ultracentrifugation procedure as described in our previous study.22 Briefly, the diluted serum was centrifuged at 3000 g for 15 min at 4°C to eliminate cellular debris. Supernatant was then collected and spun at 12,000 g for 30 min at 4°C and filtered using 0.22 μm Millipore filters (MA, USA) for removing the large vesicles. The filtrate was collected and ultracentrifuged at 100,000 g for 2 hours at 4°C to pellet the exosomes. The final exosome pellets were carefully resuspended in pre-cooled PBS and stored in liquid nitrogen for further analysis.
Transmission Electron Microscopy
The morphology of the separated exosomes was identified using a transmission electron microscope. After fixation with 2.5% glutaraldehyde, the samples were incubated overnight at 4°C. Next, the exosomes were spotted onto carbon-coated copper grids and stained with the phosphotungstic acid solution. The droplets of exosomes were air-dried at room temperature and analyzed using a JEM-1400 transmission electron microscope (JEOL, Tokyo, Japan) at 80 kV.
Nanoparticle Tracking Analysis
Exosomes were diluted to achieve between 140–200 particles per frame. ZetaView inspection instrument (Particle Metrix, Meerbusch, Germany) was used to determine the size and number of exosome particles. After loading exosome samples into the sample chamber, the manufacturer’s settings for nanospheres were set and the data were captured and calculated by the Nanoparticle Tracking Analysis (NTA) software (ZetaView 8.04.02).
Western Blot
Western blot was done following previously described protocols.23 Total proteins of exosomes, cells, and arterial tissues were extracted using the RIPA lysis buffer (Beyotime, Shanghai, China). After electroblotting, the membranes were blocked with 3% BSA (Sigma-Aldrich, USA) and then incubated with primary antibodies against CD63 (1:1000, Abcam, Cambridgeshire, UK), CD81 (1:1000, Abcam), tumor susceptibility gene 101 (Tsg101, 1:500, Proteintech, Rosemont, USA), LOX-1 (1:1000, Abcam), and β-actin (1:1000, Proteintech) overnight at 4°C. The membranes were then washed and incubated with secondary antibodies (1:3000, Ant Gene, Wuhan, China). The protein bands were visualized and analyzed with a Bio-Rad Chemidoc system (Bio-Rad Laboratories, CA, USA).
Cell Culture and Treatment
RAW264.7 macrophage cells were purchased from American Type Culture Collection (VA, USA), sub-cultured in DMEM medium (Gibco, CA, USA) containing 10% FBS (Gibco, CA, USA) and 1% penicillin/streptomycin (Thermo Fisher Scientific, MA, USA) in a humidified incubator at 37°C under 5% CO2. We used cells between passages 3–7 in this study. To explore the effects of exosomes on macrophages, the macrophages were incubated with AMI-Exo or Con-Exo and exposed to ox-LDL. Foam cells derived from macrophages are known to play a crucial role in the initiation and progression of atherosclerosis. For atherosclerosis model construct in vitro, macrophages were treated with 50 μg/mL of ox-LDL (Yiyuan biotech, Guangzhou, China) for 48 h. In the exosome treatment experiments, macrophages cultured in the ox-LDL medium were assigned to the following three groups: (a) PBS (b) Con-Exo (50ug/mL) and (c) AMI-Exo (50ug/mL).
Exosome Labeling and Uptake by Cells
The isolated exosomes were incubated with PKH26 (Sigma-Aldrich, MO, USA) at room temperature for 10 min. The labeled exosomes were washed with PBS and re-purified via ultracentrifugation. The macrophages grown on coverslips (NEST Biotechnology, Wuxi, China) were cultured with PKH26-labeled exosome for 6 h at 37°C. DAPI (Beyotime, Shanghai, China) was then used to stain the nuclei at room temperature for 10 min. Images were captured by a NikonA1R confocal microscope (Nikon Instruments, NY, USA).
Observation of Adipocytes
The distribution of adipose cells in RAW264.7 macrophage-derived foam cells was determined using oil red O (Solarbio, Beijing, China) staining. Macrophages were fixed with 4% paraformaldehyde for 15 min and stained with oil red O working solution at room temperature for 60 min. After washing with 60% isopropanol and PBS, cells were imaged using an optical microscope (Olympus, Tokyo, Japan).
Visualized Cellular Uptake of Cholesterol
The fluorescence labeled oxidative LDL (Dil-oxLDL, Yiyuan biotech, Guangzhou, China) was used for the uptake analysis. Cells were co-incubated with Dil-ox-LDL (20 μg/mL) at 37°C for 4 h following serum starvation in the presence or absence of exosomes. After staining the nuclei with DAPI (Beyotime, Shanghai, China), Dil-ox-LDL uptake was visualized by a confocal microscope (Nikon Instruments, NY, USA). The images were analyzed using ImagePro Plus 6.0 software (Media Cybernetics, Silver Spring, USA).
Cell Transfection
The miR-186-5p mimic (chemically synthesized oligonucleotides according to sequences of miR-186-5p), miR-186-5p inhibitor (chemically modified single-stranded RNA molecules that inhibits the function of miR-186-5p), and their negative controls (NC-mimic, NC-inhibitor) were purchased from RiboBio (Guangzhou, China). The transfection of miRNAs into macrophages was conducted with the TransIT-X2 Dynamic Delivery System (Mirus Bio, WI, USA), following the manufacturer’s protocols.
Quantitative Reverse Transcriptional Polymerase Chain Reaction Analysis
Total RNA was extracted using RNAiso Plus (Vazyme, Nanjing, China). After quantification, total RNA was transcribed into cDNA with a PrimeScript RT reagent kit (Vazyme, Nanjing, China). The real-time reactions were performed with SYBR Green master mix (Vazyme, Nanjing, China) with a CFX96 real-time system thermocycler (Bio-Rad, Hercules, CA, USA). The relative miRNA or mRNA expression level was quantified using the 2−∆∆Ct method and normalized against U6 or β-actin. The primer sequences are presented in Table 2.
 |
Table 2 The Primer Sequence for qRT-PCR |
Luciferase Reporter Assay
The 3′-UTR of the LOX-1 sequence containing the predicted miR-186-5p binding sites and its mutant were cloned into the psiCHECK-2 vector (Promega, WI, USA) to generate psiCHECK-2-LOX-1-Wt and psiCHECK-2-LOX-1-Mut. Next, RAW264.7 macrophage cells were co-transfected with psiCHECK-2 empty vector, psiCHECK-2-LOX-1-Wt, or psiCHECK-2-LOX-1-Mut with either the miR-186-5p mimics or control. A dual-luciferase reporter assay system (Promega, WI, USA) was used to detect luciferase activity after 48 h incubation.
Animal Experiments
Male ApoE−/− mice (8-week-old) were purchased from Vital River Laboratories (Beijing, China). Mice were maintained in a specific pathogen-free laboratory with a 12 h light-dark cycle. Mice were fed with a high-fat diet (MD12015, Medicine Ltd, Jiangsu, China) for 10 weeks to develop an atherosclerosis mouse model. During administration, the model mice received an injection with PBS or exosomes (200 μg each time) once a week via tail vein for 4 weeks. At the termination of the experimental period, the aortas of mice were removed under a stereo microscope and processed for further analysis.
Atherosclerotic Lesion Assessment
The heart and the entire length of the aorta were carefully removed and fixed in 4% paraformaldehyde. The aortas were unfolded longitudinally and stained with oil red O (Solarbio, Beijing, China) to analyze the en face lesions. The images of aortas were obtained using a digital camera (Canon, Tokyo, Japan). The hearts were immersed in optimal cutting temperature compound (OCT) and cryosectioned into 7 μm slices. Oil red O and hematoxylin staining of cryostat sections were performed to detect the atherosclerotic lesions in the aortic root. The images were obtained using a microscope (Olympus, Tokyo, Japan) and analyzed with ImagePro Plus 6.0 software (Media Cybernetics, Silver Spring, USA).
Histology and Immunofluorescence Staining of Aortic Sections
For the necrotic core analysis, aortic sinus sections were stained with hematoxylin and eosin (H&E) (Solarbio, Beijing, China). Then, the slices were observed using an optical microscope. For detecting macrophage-derived foam cells in atherosclerotic lesions, cryostat sections were immunostained with anti-CD68 antibodies (1:250, Abcam) at 4°C overnight and then incubated with the corresponding fluorescein-conjugated secondary antibodies (1:200, Life Technologies, NY, USA) for 1 h. After immunostaining with CD68 antibody, the slides were stained with Bodipy 493/503 (Invitrogen, Karlsruhe, Germany) working solution for 1 h at room temperature. Nuclei were stained with DAPI (Beyotime, Shanghai, China) for 10 min. Confocal imaging was recorded in a laser-scanning microscope (Nikon Instruments, NY, USA). The captured images above were analyzed with ImagePro Plus 6.0 software (Media Cybernetics, Silver Spring, USA).
Statistical Analyses
All statistical analyses were performed using SPSS version 23.0 (SPSS Inc., Chicago, IL, USA). Each experimental group had at least three replicates. Data has been presented as numbers and percentages for categorical variables and as mean ± standard error of the mean (SEM) for continuous variables. Categorical variables were evaluated by the Chi-square test or Fisher’s exact test. The Kolmogorov–Smirnov test was used to assess data normalization. Unpaired t-test or non-parametric Mann–Whitney U-test was used to compare the two groups. Continuous variable data between multiple groups were tested using one-way ANOVA. Two-sided P values < 0.05 were considered statistically significant.
Results
Identification and Uptake of Serum Exosomes
Serum exosomes from AMI patients (AMI-Exo) and controls (Con-Exo) both exhibited a spherical or cup-shaped structure under TEM (Figure 1A). Specific exosomal markers (CD63, CD81, and Tsg101) were expressed in both two types of exosomes (Figure 1B). Meanwhile, NTA measurements showed that the average size and main peak of the two exosome groups was approximately 100 nm (Figure 1C). After PKH26-labeled AMI-Exo and Con-Exo were incubated with DAPI-labeled RAW264.7 macrophages for 6 h; fluorescence images demonstrated that red exosomes were localized on the cytoplasm of cells, indicating that AMI-Exo and Con-Exo could be internalized by RAW264.7 macrophages (Figure 1D).
 |
Figure 1 Identification and uptake of AMI-Exo and Con-Exo. (A) Morphology of exosomes by transmission electron microscope, scale bar = 100 nm. (B) The expression of exosomal markers CD63, CD81, and Tsg101. (C) The diameter distribution of exosomes by nanoparticle tracking analysis. (D) Uptake of PKH26-labelled exosomes (red) in DAPI-labeled macrophages (blue). Scale bar = 10 μm. |
AMI-Exo Promotes Lipid Accumulation in Macrophages by Upregulating LOX-1
As shown in Figures 2A and B, amounts of oil-red O stained lipid droplets were observed more in macrophages treated with AMI-Exo, indicating an enhanced capacity of AMI-Exo to induce foam cell formation. Lipid metabolism in macrophages involves three processes: cholesterol uptake, esterification, and efflux. The mRNA levels of cholesterol metabolism were analyzed in RAW264.7 cells, including CD36, SR-AI, LOX-1, ACAT1, NECH1, ABCA1, and ABCG1. Compared with Con-Exo treated macrophages, we found that AMI-Exo significantly enhanced the expression of LOX-1 mRNA (Figure 2C). To support the role of AMI-Exo in LOX-1 regulation, we compared the protein expression of LOX-1 following exosome intervention, and observed an upregulation of LOX-1 in AMI-Exo treated macrophages (Figure 2D and E). LOX-1 is critical for the internalization of ox-LDL in macrophages. For obtaining further evidences for the involvement of LOX-1, we visualized the uptake of Dil-ox-LDL in macrophages by confocal fluorescence microscopy. Figure 2F and G show that the intracellular fluorescence intensity of Dil-ox-LDL markedly increased after treatment with AMI-Exo. Thus, these results suggested that AMI-Exo promoted lipid accumulation through upregulating LOX-1 in macrophages.
 |
Figure 2 AMI-Exo promotes lipid accumulation in macrophages by upregulating LOX-1. (A) Representative images and quantification of oil-red O staining of macrophages treated with AMI-Exo or Con-Exo, scale bar = 20 µm. (C) qRT-PCR estimating lipid catabolism gene expression relative to β-actin in macrophages, respectively. (D and E) Western blotting estimating LOX-1 protein levels relative to β-actin. (F and G) Representative fluorescence images from macrophages incubated with Dil-ox-LDL following serum starvation in the presence or absence of exosomes. (D) Quantification of Dil-ox-LDL fluorescence intensity, scale bar = 10 μm. **P < 0.01, ***P < 0.001. |
Downregulated Expression of miR-186-5p, and Its Diagnostic Value for AMI
We utilized both miRNA-target interaction databases (miRDB, miRmap, TargetScan) and the exosomal cargos database (Exocarta) for analysis to identify the miRNAs upstream of LOX-1 mRNA (Figure 3A). The overlapping and highly conserved miRNAs (miR-17-5p, miR-98-5p, miR-9-5p, miR-141-3p, miR-186-5p, miR-93-5p, miR-185-5p, miR-377-3p, and miR-200a-3p) were presented using Cytoscape software (Figure 3B). Based on the action mechanism of miRNAs in regulation of target mRNA, we expected a negative correlation between upstream miRNA and LOX-1 in AMI-Exo treated macrophages. Thus, we performed qRT-PCR for validation. It was found that the expression of miR-186-5p was markedly downregulated after AMI-Exo administration (Figure 3C). Subsequently, the differential expression of miR-186-5p in the serum exosomes of ten AMI patients and ten control individuals was validated. Based on the qRT-PCR analysis, we identified exosomal miR-186-5p downregulated in AMI-Exo (Figure 3D). Recent studies have indicated that miR-186-5p was involved in regulating ischemic injury.24,25 Since the miR-186-5p expression in exosomes might be related to myocardial ischemia, a correlation analysis was performed. We found that the level of exosomal miR-186-5p in patients with AMI was negatively linked to the TNI levels (Figure 3E). In addition, ROC analysis was conducted to evaluate the predictive power of exosomal miR-186-5p toward AMI. The results suggested that exosomal miR-186-5p achieved high diagnostic performance with an area under the curve (AUC) of 0.910 for AMI (Figure 3F). A low exosomal miR-186-5p expression might serve as a reliable potential biomarker for AMI. We hypothesized that low miR-186-5p levels meditated by AMI-Exo could be related to its role in macrophage foaming.
 |
Figure 3 Downregulated expression of miR-186-5p, and its diagnostic value for AMI. (A) Bioinformatics tools (miRDB, miRmap, TargetScan, and Exocarta) were utilized to identify upstream miRNAs of LOX-1. (B) The overlapping and highly conserved miRNAs were listed. (C) miR-186-5p expression in ox-LDL treated macrophages was measured using qRT-PCR after incubation with PBS, Con-Exo, or AMI-Exo. (D) qRT-PCR detected the exosomal miR-186-5p level in serum from AMI patients and control individuals, respectively. (E) Pearson’s test analyzed the correlation between miR-186-5p expression level and serum TNI level. (F) ROC plot showing the diagnostic value of exosomal miR-186-5p to distinguish AMI patients. *P < 0.05, **P < 0.01. Abbreviation: AUC, area under the curve. |
miR-186-5p Dysregulation Promotes Lipid Accumulation by Directly Targeting LOX-1
Based on the effect of miR-186-5p dysregulation on regulating LOX-1, we predicted the relevant binding sites using miRBD (https://mirdb.org/). It was found that two binding sites existed between miR-186-5p and the 3′-untranslated regions (UTR) of LOX-1, which were located in two base fragments, including 65–71 (target sites 1) and 1523–1529 (target sites 2) (Figure 4A). Next, plasmids with wild-type (WT) or mutant targets were constructed for dual luciferase assay. As shown in Figure 4B, miR-186-5p inhibited the luciferase activity of plasmids containing WT sequences, and the mutation of target site 1 abolished this repressive effect. The results indicated that LOX-1 was the direct downstream target of miR-186-5p.
 |
Figure 4 miR-186-5p dysregulation promotes lipid accumulation by directly targeting LOX-1. (A) The binding sites of miR-186-5p and LOX-1 predicted by online website. (B) Relative luciferase activity in RAW264.7 transfected with miR-186-5p mimic or mimic-negative control (NC) and reporter plasmids carrying wild-type or mutant binding site of miR-186-5p in 3ʹ-UTR of LOX-1. (C) qRT-PCR estimating miR-186-5p expression after administered with miR-186-5p mimic, mimic NC, miR-186-5p inhibitor, or inhibitor NC in RAW264.7 macrophage cells. (D) mRNA expression of LOX-1 in macrophages transfected with miR-186-5p mimic or inhibitor. (E and F) LOX-1 protein expression after transfection with miR-186-5p mimic or inhibitor was detected by Western blot. (G and H) Foam cell formation in macrophages measured by oil-red O staining after administration with miR-186-5p mimic or inhibitor, scale bar = 20 μm. (I and J) Evaluation of Dil-ox-LDL uptake for macrophages treated with miR-186-5p mimic or inhibitor, scale bar = 10 μm. *P < 0.05, **P < 0.01, ***P < 0.001. |
In order to further address whether the regulation of LOX-1 by miR-186-5p affects lipid metabolism, macrophages were transfected with miR-186-5p mimic, mimic negative control (NC), miR-186-5p inhibitor, or inhibitor NC, respectively. The results of qRT-PCR revealed that the miR-186-5p mimic group exhibited a significantly augmented level of miR-186-5p, and the miR-186-5p inhibitor group presented a significantly downregulated expression of miR-186-5p (Figure 4C). The miR-186-5p mimic significantly decreased the mRNA and protein levels of LOX-1 induced by ox-LDL in macrophages (Figure 4D–F). Marked increases in LOX-1 mRNA and protein were also observed in the miR-186-5p inhibitor group (Figure 4D–F). Next, lipid accumulation was evaluated. In the foam cell formation assay, the overexpression of miR-186-5p significantly inhibited macrophage foaming, while the knockdown of miR-186-5p had an inverse effect (Figure 4G and H). Consistently, the upregulation of miR-186-5p suppressed the uptake of Dil-ox-LDL, whereas it increased in the miR-186-5p inhibitor group (Figure 4I and J). These findings suggested that miR-186-5p inhibited atherosclerosis-related phenotypes of macrophages by regulating its downstream molecule LOX-1.
AMI-Exo Aggravates Atherosclerosis in ApoE−/− Mice
High-fat/cholesterol diet-fed ApoE−/− mice were treated weekly with PBS, Con-Exo, or AMI-Exo for 4 weeks (Figure 5A) to determine whether AMI-Exo had a role in the development of atherosclerosis in vivo. The atherosclerotic lesion area was evaluated by an en-face analysis of the entire aorta and the cross-sections of the aortic sinus (Figure 5B–D). The en face analysis suggested that atherosclerotic plaque areas in the whole aortas in ApoE−/− mice were markedly increased in the AMI-Exo group compared with the PBS and Con-Exo administered mice (Figure 5C). On aortic cross-sections, severe lipid deposition and aortic root lesion burden were observed in AMI-Exo treated mice (Figure 5E). The aortic root’s necrotic core was examined in the H&E stained cross-sections (Figure 5F). We found that AMI-Exo administration significantly increased the necrotic core area within the lesions (Figure 5G). The above results indicated that AMI-Exo treatment accelerated atherosclerosis progression in vivo.
 |
Figure 5 AMI-Exo aggravates atherosclerosis in ApoE−/− mice. (A) Scheme describing the experimental workflow of animal study. (B) Representative en face oil-red O stained images of ApoE−/− mice aortic atherosclerotic plaques. (C) Lesion quantification of atherosclerotic plaques from the aortic arch to the iliac bifurcation. (D) Representative oil-red O stained images of aortic sinus cross-sections, scale bar = 200 μm. (E) Quantification of aortic sinus lesion. (F) Representative hematoxylin and eosin staining of aortic sinus cross-sections, scale bar = 100 μm. (G) Quantification of necrotic core area as a percent of total lesion area in ApoE−/− mice. **P < 0.01, ***P < 0.001. |
miR-186-5p Mediated Macrophage-Derived Foam Cell Formation via LOX-1 in ApoE−/− Mice
To further investigate whether the poorly expressed exosomal miR-186-5p could impact macrophage-derived foam cell formation in lesion areas, we used immunofluorescent staining on aortic root cross-sections with Bodipy 493/503 (green) and CD68 (red) (Figure 6A). Compared with Con-Exo treated mice, there was a high degree of co-localization of neutral lipids and macrophages in the AMI-Exo group, suggesting a marked increase of macrophage-derived foam cell formation by the AMI-Exo treatment (Figure 6B). In addition, we investigated the effect of miR-186-5p on LOX-1 expression in vivo. The aorta from each group was used for the assessment. The expression of LOX-1 and miR-186-5p detected by qRT-PCR showed that LOX-1 was significantly increased, while miR-186-5p expression was markedly decreased in the mice treated with AMI-Exo (Figure 6C and D). Western blotting results also displayed LOX-1 protein expression pattern similar to that of qRT-PCR (Figure 6E and F). Thus, these results indicate that miR-186-5p dysregulation aggravates the development of aortic atherosclerosis, probably through upregulation of LOX-1.
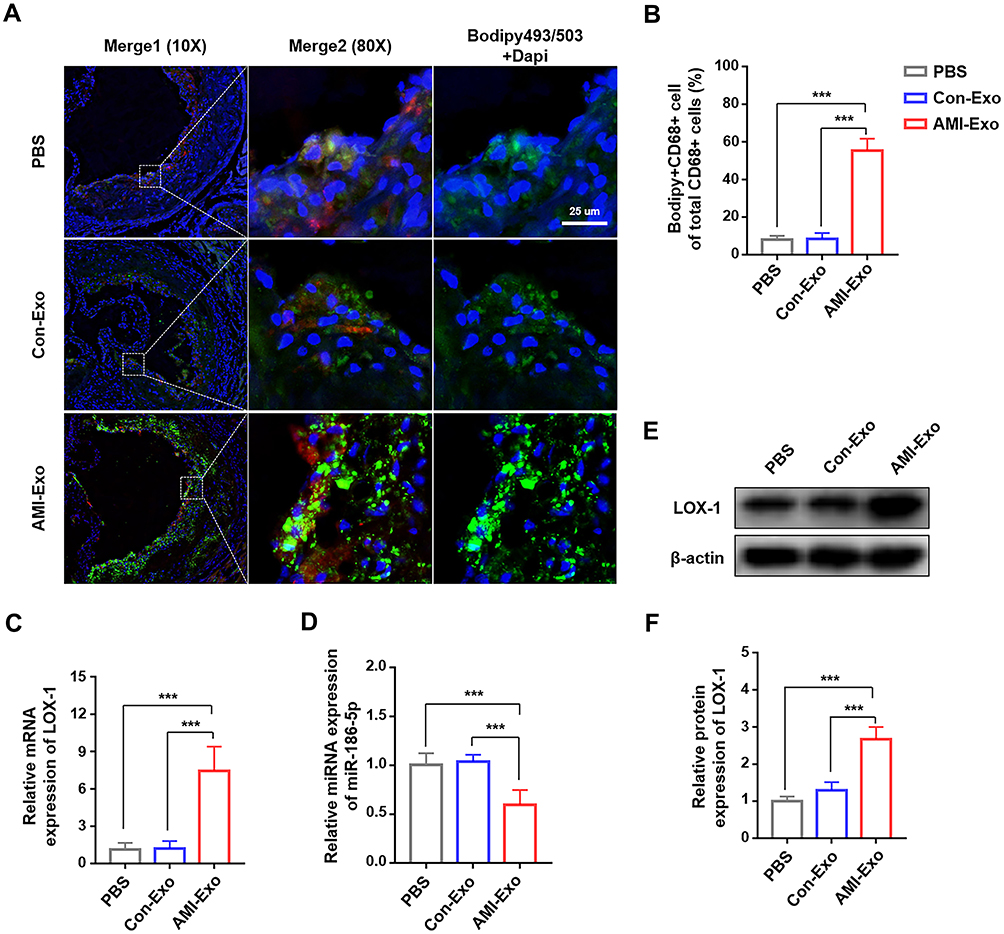 |
Figure 6 miR-186-5p mediated macrophage-derived foam cell formation via LOX-1 in ApoE−/− mice. (A) Representative immunofluorescence microscopy images of neutral lipid of the lesional macrophages, by staining Bodipy 493/503 (green) and CD68 (red)), scale bar = 25 μm. (B) Quantification of the co-localization of Bodipy 493/503 and CD68 fluorescence intensity. (C) Expression of LOX-1 mRNA after AMI-Exo administration was determined by qRT-PCR. (D) miR-186-5p expression in vivo measured by RT-qPCR. (E and F) The protein expression of LOX-1 in ApoE−/− mice treated with AMI-Exo detected by Western blot analysis. ***P < 0.001. |
Discussion
Atherosclerosis is a chronic degenerative disease that is associated with a high incidence of morbidity and mortality of cardiovascular events.26 Previous studies have suggested that AMI accelerates the atherosclerotic progression in non-culprit vessels and is an independent risk factor for the rupture of atherosclerotic plaque.8–10 However, further analysis is required to understand the specific mechanism. Exosomes from myocardial infarction patients have emerged as essential players in various pathophysiological processes. Recent studies have shown that circulating exosomes from patients with AMI participate in angiogenesis in experimental models of left anterior descending coronary artery ligation.19,20 Another study reported that exosomes isolated from AMI patients significantly promoted cell injury when incubated with HUVECs.27 This study is the first to show the proatherogenic effect of exosomes from patients with AMI, which might partly account for the observations that clinical and experimental myocardial infarction facilitated atherosclerosis progression.
The accumulation of lipids is essential for the formation of macrophages-derived foam cells that deposit in the artery wall.2 The recognition and uptake of modified lipid is mainly mediated by macrophage scavenger receptors, such as CD36, SR-AI and LOX-1.28 Our findings suggested that in the aortic root plaques of mice of the AMI-Exo group, lipid accumulation in macrophages was aggravated and there was a significant increase in the levels of scavenger receptor LOX-1. Evidence has increasingly revealed the importance of LOX-1 in atherogenesis. Previously, LOX-1 upregulation was discovered in atherosclerotic plaques of atherosclerosis patients.29 Inoue et al reported that mice overexpressing LOX-1 exhibited an acceleration of atherosclerotic lesions.30 Besides, compared with other scavenger receptors, LOX-1 could recognize and bind to moderately oxidatively modified or incompletely ox-LDL,31 suggesting a more significant potential contribution of LOX-1 in atherogenesis.
In this study, we conducted in vitro study in macrophages to explore the role of LOX-1 in lipid-laden foam cells. The administration of AMI-Exo enhanced lipid uptake and promoted foam cell formation with the upregulation of LOX-1. A previous study had revealed that LOX-1 increased the internalization of ox-LDL by more than 40% in macrophages.32 The inflammatory cytokines upregulated the LOX-1 expression in macrophages while other scavenger receptors were downregulated.33 Thus, LOX-1 mediated ox-LDL uptake in macrophages might play a central role in the chronic inflammatory environment of atherosclerosis.
We further investigated the miRNA mechanism underlying the exosome regulation of LOX-1. Several studies have reported on the role of miRNA-mediated regulation of LOX-1 expression. miR-30c-1-3p is known to be implicated in repressing macrophage foaming by binding directly to LOX-1 and inhibiting its expression.34 However, the regulation of LOX-1 by exosome mediated miRNA has not been explored. miRNAs are the most abundant cargo in human blood-derived exosomes, accounting for more than 50% of exosomal non-coding RNA.35 Also, specific exosomal miRNAs may act as potential circulating biomarkers.36 For instance, exosomal miR-214 was confirmed to be a promising diagnostic biomarker for patients with AMI.37 In this study, we identified that the level of miR-186-5p was markedly decreased in AMI-Exo and had a high diagnostic value for AMI. miR-186-5p reportedly played a regulatory role in ischemic injury, which was consistent with our findings.24,25 Subsequently, luciferase assays were performed, and the results indicated that miR-186-5p directly targeted the 3ʹ-UTR of LOX-1 and repressed LOX-1 expression. In the current study, macrophage foam cell formation was increased when the expression of endogenous or exogenous miR-186-5p was blocked. Conversely, macrophage foaming was inhibited when elevating the miR-186-5p level. Moreover, delivery of AMI-Exo with low levels of miR-186-5p in vivo increased the level of LOX-1 and accelerated atherosclerotic lesions. These data demonstrated that miR-186-5p dysregulation mediated by AMI-Exo was involved in the pathological process of atherosclerosis via LOX-1. Tao et al previously revealed that serum levels of miR-186-5p had a negative association with the progression of atherosclerosis.38 Zhang et al reported that the addition of exogenous miR-186-5p inhibited the proliferation and migration of vascular endothelial cells in atherosclerosis.39 These data demonstrated the critical function of miR-186-5p in atherosclerosis development, in agreement with our findings.
Taken together, this study provides a new understanding to interpret accelerated atherosclerosis after acute ischemic events, by exosome-mediated miRNA communication. Exosomes have been viewed as new players in translational nanomedicine.40 Growing evidence in recent years has suggested exosome-based drug delivery systems as a promising strategy for diseases.41 Our findings support the importance of exosomal miR-186-5p in atherogenesis, therapeutic miRNA cargoed in engineered exosomes may also be an alternative approach for the treatment of atherosclerosis. Nevertheless, this study had several limitations. First, the clinical sample size in this study was relatively small. A post hoc power analysis was conducted with G*Power (version 3.1.9.7, Franz Faul, University of Kiel, Germany). Our statistical decision was made using an alpha level of 0.05. Based on the sample size of this study, the power to detect significant differences was 99% (actual power, 0.9933770; critical t, 2.0399640; non-centrality parameter, 4.5839372), with a large effect size of 1.63 (Cohen’s d). Thus, the sample size, albeit small, provided sufficient power to detect statistical differences. Second, due to the complicated exosomal contents, the findings of the study might not elucidate all functional molecules in AMI-Exo. Finally, the regulatory effect of miR-186-5p on LOX-1 was confirmed in vitro experiments, and further validation should be performed to uncover the regulation effect of miR-186-5p in vivo.
Conclusion
In summary, we reported that low levels of miR-186-5p in AMI-Exo promoted macrophages foaming through the direct binding of LOX-1, aggravating atherosclerosis (Figure 7). This study provides new evidence confirming the role of exosomal miRNA as a potent regulator in atherosclerosis. Further efforts involving exosomes based miRNA delivery strategy will offer novel perspectives for the treatment of atherosclerosis.
 |
Figure 7 Schematic diagram of the experimental hypothesis. miR-186-5p dysregulation in AMI-Exo regulates the formation of foam cells by targeting LOX-1 for enhancing the macrophage lipid uptake, thereby promoting atherosclerosis. |
Acknowledgments
This work was supported by Grants from the National Natural Science Foundation of China (NO. 81770330).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Prim. 2019;5(1):56. doi:10.1038/s41572-019-0106-z
2. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118(4):653–667. doi:10.1161/CIRCRESAHA.115.306256
3. Orekhov AN. LDL and foam cell formation as the basis of atherogenesis. Curr Opin Lipidol. 2018;29(4):279–284. doi:10.1097/MOL.0000000000000525
4. Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol. 2018;330:27–42. doi:10.1016/j.cellimm.2018.01.020
5. Tall AR, Costet P, Wang N. Regulation and mechanisms of macrophage cholesterol efflux. J Clin Invest. 2002;110(7):899–904. doi:10.1172/JCI0216391
6. Poznyak AV, Wu W-K, Melnichenko AA, et al. Signaling pathways and key genes involved in regulation of foam cell formation in atherosclerosis. Cells. 2020;9(3):584. doi:10.3390/cells9030584
7. Kattoor AJ, Goel A, Mehta JL. LOX-1: regulation, signaling and its role in atherosclerosis. Antioxidants. 2019;8(7). doi:10.3390/antiox8070218
8. Hong M-K, Mintz GS, Lee CW, et al. Comparison of coronary plaque rupture between stable angina and acute myocardial infarction: a three-vessel intravascular ultrasound study in 235 patients. Circulation. 2004;110(8):928–933. doi:10.1161/01.CIR.0000139858.69915.2E
9. Han Y, Jing J, Tu S, et al. ST elevation acute myocardial infarction accelerates non-culprit coronary lesion atherosclerosis. Int J Cardiovasc Imaging. 2014;30(2):253–261. doi:10.1007/s10554-013-0354-z
10. Asakura M, Ueda Y, Yamaguchi O, et al. Extensive development of vulnerable plaques as a pan-coronary process in patients with myocardial infarction: an angioscopic study. J Am Coll Cardiol. 2001;37(5):1284–1288. doi:10.1016/S0735-1097(01)01135-4
11. Montone RA, Meucci MC, Niccoli G. The management of non-culprit coronary lesions in patients with acute coronary syndrome. Eur Heart J Suppl. 2020;22(SupplL):L170–L175. doi:10.1093/eurheartj/suaa175
12. Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364(3):226–235. doi:10.1056/NEJMoa1002358
13. Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325–329. doi:10.1038/nature11260
14. Kyaw T, Loveland P, Kanellakis P, et al. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur Heart J. 2021;42(9):938–947. doi:10.1093/eurheartj/ehaa995
15. Kalluri R, LeBleu VS. The biology function and biomedical applications of exosomes. Science. 2020;367:6478. doi:10.1126/science.aau6977
16. Lin B, Yang J, Song Y, Dang G, Feng J. Exosomes and atherogenesis. Front Cardiovasc Med. 2021;8:738031. doi:10.3389/fcvm.2021.738031
17. Zhu J, Liu B, Wang Z, et al. Exosomes from nicotine-stimulated macrophages accelerate atherosclerosis through miR-21-3p/PTEN-mediated VSMC migration and proliferation. Theranostics. 2019;9(23):6901–6919. doi:10.7150/thno.37357
18. Wang Z, Zhang J, Zhang S, et al. MiR-30e and miR-92a are related to atherosclerosis by targeting ABCA1. Mol Med Rep. 2019;19(4):3298–3304. doi:10.3892/mmr.2019.9983
19. Geng T, Song ZY, Xing JX et al. Exosome derived from coronary serum of patients with myocardial infarction promotes angiogenesis through the miRNA-143/IGF-IR pathway. Int J Nanomedicine. 2020;15:2647–2658. doi:10.2147/IJN.S242908
20. Duan S, Wang C, Xu X, et al. Peripheral serum exosomes isolated from patients with acute myocardial infarction promote endothelial cell angiogenesis via the miR-126-3p/TSC1/mTORC1/HIF-1α pathway. Int J Nanomedicine. 2022;17:1577–1592. doi:10.2147/IJN.S338937
21. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction (2018). Circulation. 2018;138(20):e618–e651. doi:10.1161/CIR.0000000000000617
22. Liu W, Feng Y, Wang X, et al. Human umbilical vein endothelial cells-derived exosomes enhance cardiac function after acute myocardial infarction by activating the PI3K/AKT signaling pathway. Bioengineered. 2022;13(4):8850–8865. doi:10.1080/21655979.2022.2056317
23. Zheng J, Ding J, Liao M, et al. Immunotherapy against angiotensin II receptor ameliorated insulin resistance in a leptin receptor-dependent manner. FASEB J. 2021;35(1):e21157. doi:10.1096/fj.202000300R
24. Lin L, Sun J, Wu D, et al. MicroRNA-186 is associated with hypoxia-inducible factor-1α expression in chronic obstructive pulmonary disease. Mol Genet Genomic Med. 2019;7(3):e531. doi:10.1002/mgg3.531
25. Ouyang M, Lu J, Ding Q et al. Knockdown of long non-coding RNA PVT1 protects human AC16 cardiomyocytes from hypoxia/reoxygenation-induced apoptosis and autophagy by regulating miR-186/Beclin-1 axis. Gene. 2020;754:144775. doi:10.1016/j.gene.2020.144775
26. Kozarov E, Padro T, Badimon L. View of statins as antimicrobials in cardiovascular risk modification. Cardiovasc Res. 2014;102(3):362–374. doi:10.1093/cvr/cvu058
27. Gong Z, Wen M, Zhang W, et al. Plasma exosomes induce inflammatory immune response in patients with acute myocardial infarction. Arch Physiol Biochem. 2021:1–9. doi:10.1080/13813455.2021.1912102
28. Syväranta S, Alanne-Kinnunen M, Oörni K, et al. Potential pathological roles for oxidized low-density lipoprotein and scavenger receptors SR-AI, CD36, and LOX-1 in aortic valve stenosis. Atherosclerosis. 2014;235(2):398–407. doi:10.1016/j.atherosclerosis.2014.05.933
29. Kataoka H, Kume N, Miyamoto S, et al. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation. 1999;99(24):3110–3117. doi:10.1161/01.CIR.99.24.3110
30. Inoue K, Arai Y, Kurihara H, Kita T et al. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E-null mice. Circ Res. 2005;97(2):176–184. doi:10.1161/01.RES.0000174286.73200.d4
31. Levitan I, Volkov S, Subbaiah PV. Oxidized LDL: diversity, patterns of recognition, and pathophysiology. Antioxid Redox Signal. 2010;13(1):39–75. doi:10.1089/ars.2009.2733
32. Schaeffer DF, Riazy M, Parhar KS, et al. LOX-1 augments oxLDL uptake by lysoPC-stimulated murine macrophages but is not required for oxLDL clearance from plasma. J Lipid Res. 2009;50(8):1676–1684. doi:10.1194/jlr.M900167-JLR200
33. Kume N, Moriwaki H, Kataoka H, et al. Inducible expression of LOX-1, a novel receptor for oxidized LDL, in macrophages and vascular smooth muscle cells. Ann N Y Acad Sci. 2000;902:323–327. doi:10.1111/j.1749-6632.2000.tb06332.x
34. Li X, Feng S, Luo Y, et al. Expression profiles of microRNAs in oxidized low-density lipoprotein-stimulated RAW 264.7 cells. In Vitro Cell Dev Biol Anim. 2018;54(2):99–110. doi:10.1007/s11626-017-0225-3
35. Huang X, Yuan T, Tschannen M, et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics. 2013;14:319. doi:10.1186/1471-2164-14-319
36. Sahoo S, Losordo DW. Exosomes and cardiac repair after myocardial infarction. Circ Res. 2014;114(2):333–344. doi:10.1161/CIRCRESAHA.114.300639
37. Lu HQ, Liang C, He ZQ et al. Circulating miR-214 is associated with the severity of coronary artery disease. J Geriatr Cardiol. 2013;10(1):34–38. doi:10.3969/j.issn.1671-5411.2013.01.007
38. Tao Z, Cao Z, Wang X et al. Long noncoding RNA SNHG14 regulates ox-LDL-induced atherosclerosis cell proliferation and apoptosis by targeting miR-186-5p/WIPF2 axis. Hum Exp Toxicol. 2021;40(1):47–59. doi:10.1177/0960327120940363
39. Zhang S, Zhu X, Li G. E2F1/SNHG7/miR-186-5p/MMP2 axis modulates the proliferation and migration of vascular endothelial cell in atherosclerosis. Life Sci. 2020;257:118013. doi:10.1016/j.lfs.2020.118013
40. He C, Zheng S, Luo Y, et al. Exosome theranostics: biology and translational medicine. Theranostics. 2018;8(1):237–255. doi:10.7150/thno.21945
41. Antimisiaris SG, Mourtas S, Marazioti A. Exosomes and exosome-inspired vesicles for targeted drug delivery. Pharmaceutics. 2018;10(4):218. doi:10.3390/pharmaceutics10040218
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.