Back to Journals » OncoTargets and Therapy » Volume 13
MiR-138 Suppresses the PDK1 Expression to Decrease the Oxaliplatin Resistance of Colorectal Cancer
Authors Wang Y, Zhang D, Li Y, Fang F
Received 19 December 2019
Accepted for publication 29 March 2020
Published 29 April 2020 Volume 2020:13 Pages 3607—3618
DOI https://doi.org/10.2147/OTT.S242929
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Yao Wang, Duo Zhang, Yao Li, Fang Fang
Inspection Institute, Jilin Medical University, Jilin City, Jilin Province 132013, People’s Republic of China
Correspondence: Fang Fang No. 5, Jilin Street, Jilin City, Jilin Province 132013, People’s Republic of China
Email [email protected]
Background: Oxaliplatin is one kind of platinum-based drug. It is effective and commonly used in the treatment of colorectal cancer (CRC). However, development of acquired drug resistance is still a big obstacle during the oxaliplatin therapy. It is urgent to take strategies to decrease the oxaliplatin resistance of CRC.
Materials and Methods: Oxaliplatin-resistant HT29 and SW480 (HT29/R and SW480/R) cells were acquired through long-term exposure to oxaliplatin by using the routine HT29 and SW480 cells. Relative glucose consumption, lactate generation and LDH activity were tested to evaluate the glycolysis of CRC cell lines. MTT assays were conducted to evaluate the differences of oxaliplatin sensitivity between HT29/R (SW480/R) cells and their parental HT29 (SW480) cells. Regulation of miR-138 on PDK1 was confirmed through qRT-PCR, Western blot and dual-luciferase reporter assays. Reactive oxygen species (ROS) levels were measured by flow cytometry.
Results: HT29/R and SW480/R cells exhibited higher glucose consumption, lactate production and LDH activity compared to their parental HT29 and SW480 cells. However, oxygen consumption rate (OCR) in HT29/R and SW480/R cells is lower than that in HT29 and SW480 cells, respectively. Results of MTT assays showed that treatment with miR-138 can increase the cytotoxicity of oxaliplatin to HT29/R and SW480/R cells. Research on mechanisms showed that PDK1 was the target of miR-138. Overexpression of miR-138 can inhibit the expression of PDK1, and thus increase the OCR of HT29/R and SW480/R cells. Under the treatment of oxaliplatin, the miR-138-overexpressed HT29/R and SW480/R cells generated more amount of ROS to get into the apoptosis process.
Conclusion: Overexpression of miR-138 suppressed the PDK1 expression to decrease the oxaliplatin resistance of CRC.
Keywords: miR-138, oxaliplatin, colorectal cancer, PDK1
Introduction
Colorectal cancer (CRC) is one of the most common types of malignant cancers in the world. As CRC cells are very aggressive with high metastatic potential, many CRC patients are diagnosed as advanced cancer at their first interview of doctor. To make matters worse, overall 5-year survival rate of CRC is very low.1,2 CRC has been one of the most serious diseases that threaten human health. Despite surgery is the most effective treatment for cancers, advanced CRC patients have lost the chance of surgery. Nevertheless, platinum-based chemotherapy is still a standard treatment for these advanced CRC patients.3–5
MicroRNAs (miRNAs) are a class of small, conservative and non-coding RNAs that negatively regulate more than 30% of coding genes. MiRNAs regulate genes through directly binding to the 3ʹ-untranslated region (3ʹ-UTR) of the targeted mRNAs.6–8 Previous research has proved that miRNAs modulate proliferation, migration and apoptosis of various cancers including CRC.9–11 Recently, studies indicate that aberrant expression of miRNAs is closely associated with drug resistance.12,13 Furthermore, miRNAs can be used to sensitize tumors to chemotherapy.14,15 Therefore, altering the expression of miRNAs may be a potential and promising strategy for overcoming the acquired drug resistance of CRC.
It is recognized that oxaliplatin is an effective chemotherapeutic drug in the treatment of CRC. Thus, oxaliplatin is commonly used in CRC therapy. However, development of acquired drug resistance against oxaliplatin usually occurs during the chemotherapy treatment course.16,17 Novel approaches are urgently required to reduce the oxaliplatin resistance of CRC.
Previous studies have reported that miR-138 acts as a tumor suppressor. It inhibits growth and metastasis of various cancers including lung cancer, gastric cancer and hepatocellular carcinoma. Furthermore, it has been reported that decrease of miR-138 predicts poor prognosis in some cancer patients.18–21 More importantly, studies indicate that overexpression of miR-138 can increase the chemosensitivity of some cancers.22,23 However, the role of miR-138 in affecting the acquired drug resistance of CRC is still unclear. In the present study, we tried to explore the effect of miR-138 on reducing the oxaliplatin resistance of CRC.
Materials and Methods
Cell Culture
HT29/R and SW480/R cells were obtained by stepwise exposure of HT29 and SW480 cells to increasing concentrations of oxaliplatin from 0.2 μM to 2 μM for 6 months. HT29 and SW480 cell lines were obtained from the Cell Bank of the Institute of Biochemistry and Cell Biology (Shanghai, China) and cultured in RPMI-1640 (Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific, Inc.) at 37°C in a humidified 5% CO2 incubator.
Cell Transfection
Human miR-138 mimics, negative control oligonucleotides (NCO), PDK1 siRNA (siRNA-PDK1), control siRNA (siRNA-control) and pcDNA3.1 vector were purchased from Genechem Co., Ltd. (Shanghai, China). The pcDNA3.1-PDK1 plasmid (plasmid-PDK1) was constructed by inserting the PDK1 open reading frame into the pcDNA3.1 vector. For transfection, cells were seeded into six-well plates, with the density of 4×105 cells/well. Subsequently, the above RNA oligonucleotides or plasmid were transfected into cells with Lipofectamine®2000 (Thermo Fisher Scientific, Inc.) following the manufacturer’s instruction.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Cellular total RNAs were extracted by using Trizol reagent (Thermo Fisher Scientific, Inc.). To obtain the cDNA, extracted RNAs were reversely transcribed by One Step PrimeScript miRNA cDNA Synthesis Kit (TaKaRa, Dalian, China) according to the manufacturer’s instruction. qPCR was performed by using a standard protocol from the SYBR Green PCR kit (Toyobo, Japan). The qPCR primer for miR-138 is as follows: 5ʹ-AGCTGGTGTTGTGAATCAGGCCG-3ʹ. U6 small nuclear RNA (snRNA U6) was used as the internal reference to determine the relative expression of miR-138 through the 2−ΔΔCT method.
MTT Assay
Cells with the density of 5×103/well were seeded on a 96-well plate with 100 μL culture medium. Cells were incubated at 37°C in a humid atmosphere with 5% CO2 for 24 h before treatment with different concentrations of oxaliplatin. At the end of 48 h treatment with oxaliplatin, 20 μL MTT reagent (5 mg/mL; Sigma-Aldrich, St. Louis, MO, USA) was added to the culture medium for 4 h incubation. The cells were then suspended in 150 μL dimethyl sulfoxide followed by detection of the absorbance at 570 nm on an enzyme-linked immunosorbent assay microplate reader (Sunrise Microplate Reader; TECAN, Männedorf, Switzerland). Then, we calculated the IC50 of oxaliplatin to CRC cells according to the cell viability curve.
Luciferase Reporter Assay
PDK1 3ʹ-UTR fragment containing predicted miR-138 binding site was amplified from cDNA of CRC cells. Subsequently, the fragment was inserted into pGL3 luciferase reporter vector (Promega, Madison, WI, USA). The pGL3 reporter with mutant PDK1 3ʹ UTR was conducted by using QuikChange Site-Directed Mutagenesis kit (Stratagene, Missouri, TEX, USA). For luciferase reporter assay, CRC cells were co-transfected with pGL3 reporter with wild-type (wt) or mutant-type (mt) PDK1 3ʹ UTR together with either NCO or miR-138 mimics via Lipofectamine®2000. 24 h after transfection, cells were collected and lysed. Then, the luciferase activities were confirmed by using dual-luciferase reporter assay system (Promega) according to the manufacturer’s instruction.
Western Blot Analysis
Treated cells were harvested and incubated in RIPA lysis buffer (Cell Signaling Technology Inc., Danvers, MA, USA) for 30 min at 4°C to isolate the total proteins. The extracted proteins were then separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride membrane (Roche, Basel, Switzerland). Subsequently, membranes were incubated with primary antibodies (Santa Cruze Biotechnology, Santa Cruz, CA, USA) at 4°C overnight and then incubated with secondary antibody (Santa Cruze Biotechnology) at room temperature for 2 h. Protein bands were visualized by using ECL Western Blot Substrate (Thermo Fisher Scientific, Inc.).
Analysis of Glycolysis
Treated cells were harvested and washed with PBS twice. To evaluate the glycolysis of cells, relative glucose consumption, lactate generation and LDH activity were detected by using Amplex Red Glucose/Glucose Oxidase assay kit (Molecular Probes; Thermo Fisher Scientific, Inc. USA), Lactate Assay Kit (BioVision, Milpitas, CA, USA) and LDH Quantification Kit (Biovision), respectively.
Flow Cytometry Analysis
OCR, ROS and cell apoptosis were detected by flow cytometry analysis. For evaluation of OCR, oxygen-sensitive fluorescent probe Mito-Xpress (Luxcel Bioscience, Cork, Ireland) was used to analyze the oxygen consumption of cells. For measurement of apoptotic rate, Annexin V-FITC and propidium iodide (BD Pharmigen, San Diego, CA, USA) were used to calculate the Annexin V-positive cells. For the detection of cellular ROS, cells were stained with dihydroethidium (Molecular Probes) according to the manufacturer’s instruction.
Statistical Analysis
SPSS 16.0 statistical software (IBM Corporation, Armonk, NY, USA) was used to analyze the data. Non-paired t test was used to estimate the statistical differences between two groups. One-way analysis of variance (ANOVA) was applied to verify differences among three or more groups. P value <0.05 was considered to indicate a statistically significant difference.
Results
Oxaliplatin Resistance of SW480/R and HT29/R
To study the oxaliplatin resistance in CRC, we continuously treated the HT29 and SW480 cell lines with oxaliplatin to establish the oxaliplatin-resistant CRC models. Next, we evaluated the sensitivity of oxaliplatin to these oxaliplatin-resistant HT29 and SW480 (HT29/R and SW480/R) cells. We showed that cell viability of HT29/R was higher than their parental HT29 cells when they were under the equal concentration of oxaliplatin (Figure 1A). Specifically, IC50 of oxaliplatin to HT29/R was 8.6 fold higher than that to HT29 cells, and the IC50 of oxaliplatin to SW480/R was 11.5 fold higher than that to SW480 cells (Figure 1B). We demonstrated that long-term exposure to oxaliplatin can induce obvious oxaliplatin resistance in CRC cells.
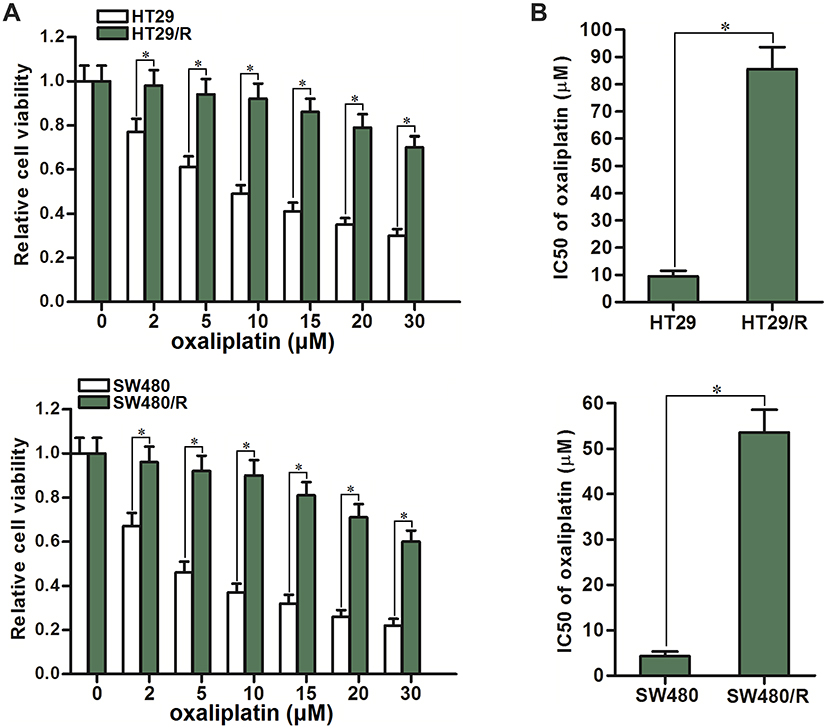 |
Figure 1 Resistance of HT29/R and SW480/R to oxaliplatin. (A) Differences of oxaliplatin sensitivity between HT29/R and SW480 cells and their parental HT29 and SW480 cells. (B) Differences of oxaliplatin IC50 between HT29/R and SW480 cells and their parental HT29 and SW480 cells. Notes: Data were expressed as mean±SD. *P<0.05. Abbreviation: IC50, half-maximal inhibitory concentration. |
HT29/R and SW480/R Cells Exhibit High Level of Glycolysis and Low Level of Oxygen Consumption Rate (OCR)
We next evaluated the difference of glycolysis between HT29/R (SW480/R) cells and their parental HT29 (SW480) cells, because some studies have reported that glycolysis is essential for chemoresistance in some cancers.24,25 As shown in Figure 2A, HT29/R and SW480/R cells consumed more amount of glucose compared to the HT29 and SW480 cells. Furthermore, we observed that HT29/R and SW480/R cells produced more amount of lactate compared to the HT29 and SW480 cells (Figure 2B). Consistent with this, HT29/R and SW480/R cells showed higher activity of lactic dehydrogenase (LDH) rather than the HT29 and SW480 cells (Figure 2C). These data indicated that HT29/R and SW480/R cells exhibited higher level of glycolysis compared to the routine HT29 and SW480 cells. We next evaluated the difference of OCR between HT29/R (SW480/R) cells and their parental HT29 (SW480) cells. By contrast to the high level of glycolysis rate in HT29/R and SW480/R, OCR level in HT29/R and SW480/R was significantly lower than that in HT29 and SW480 cells (Figure 2D). Taken together, we demonstrated that oxaliplatin-resistant CRC cells exhibited higher rate of glycolysis and lower level of OCR compared to the routine CRC cells.
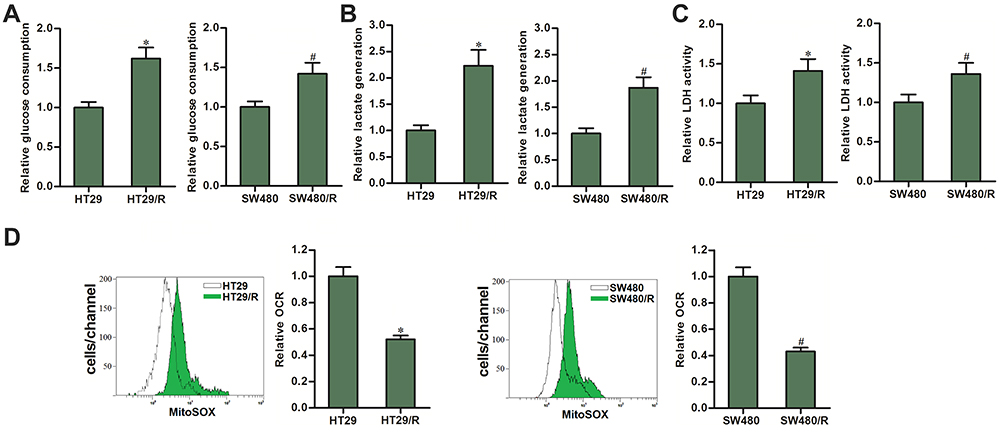 |
Figure 2 Differences of OCR between HT29/R and SW480/R cells and their parental HT29 and SW480 cells. (A) Differences of glucose consumption between HT29/R and SW480/R cells and their parental HT29 and SW480 cells. (B) Differences of lactate production between HT29/R and SW480/R cells and their parental HT29 and SW480 cells. (C) Differences of LDH activity between HT29/R and SW480/R cells and their parental HT29 and SW480 cells. (D) Differences of OCR between HT29/R and SW480/R cells and their parental HT29 and SW480 cells. Notes: Data were expressed as mean±SD. *P<0.05 vs HT29, #P<0.05 vs SW480. Abbreviations: OCR, glycolysis and oxygen consumption rate; LDH, dehydrogenase. |
Expression of PDK1 Partially Determines the Oxaliplatin Sensitivity of CRC
To explore the mechanism by which HT29/R and SW480/R cells showed low level of OCR, we compared the expression of PDK1 between HT29/R (SW480/R) cells and their parental HT29 (SW480) cells, because PDK1 is a key enzyme that reduces available pyruvate for mitochondrial metabolism.26 As shown in Figure 3A, expression of PDK1 was obviously overexpressed in HT29/R and SW480/R cells. To explore the association between oxaliplatin resistance and PDK1 expression, we overexpressed the PDK1 in the routine HT29 and SW480 cells (Figure 3B). We then found that overexpression of PDK1 significantly decreased the sensitivity of HT29 and SW480 cells to oxaliplatin (Figure 3C). Specifically, transfection with plasmid-PDK1 increased the oxaliplatin IC50 by 4.4 fold to HT29/R and 5.3 fold to SW480/R (Figure 3D). On the other hand, we knockdown the PDK1 gene in HT29/R and SW480/R cells by transfection with siRNA-PDK1 (Figure 3E). We then found that direct inhibition of PDK1 increased the sensitivity of HT29/R and SW480/R cells to oxaliplatin treatment (Figure 3F). Specifically, siRNA-PDK1 decreased the oxaliplatin IC50 by 68.0% to HT29/R and 75.7% to SW480/R (Figure 3G). These data indicated that expression profile of PDK1 partially determined the oxaliplatin sensitivity of CRC. Overexpression of PDK1 is responsible for oxaliplatin resistance in HT29/R and SW480/R.
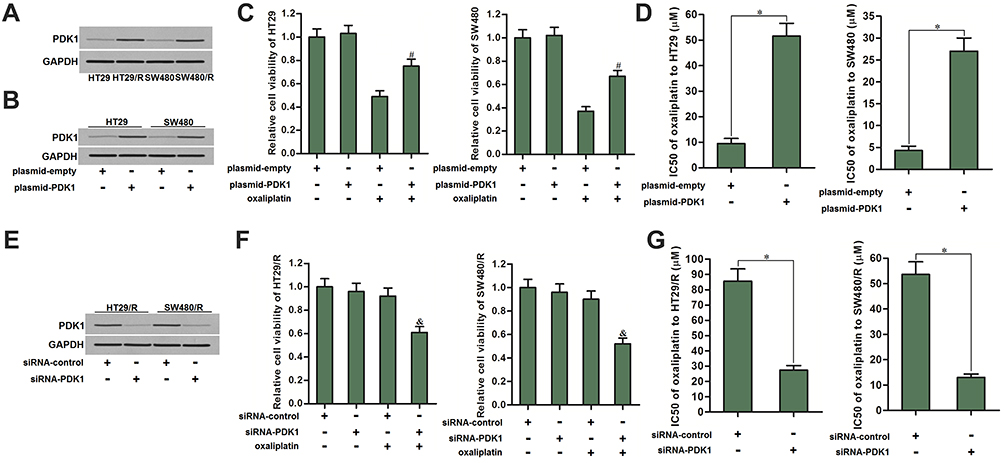 |
Figure 3 PDK1 partially determined the oxaliplatin sensitivity of CRC. (A) HT29/R and SW480/R cells expressed higher level of PDK1 compared to the HT29 and SW480 cells. (B) Transfection with plasmid-PDK1 increased the expression of PDK1 in HT29 and SW480 cells. (C) Transfection with plasmid-PDK1 decreased the cytotoxicity of oxaliplatin (10 μM) against HT29 and SW480 cells. (D) Transfection with plasmid-PDK1 increased the IC50 of oxaliplatin to HT29 and SW480 cells. (E) Transfection with siRNA-PDK1 decreased the expression of PDK1 in HT29/R and SW480/R cells. (F) Transfection with siRNA-PDK1 increased the cytotoxicity of oxaliplatin (10 μM) against HT29/R and SW480/R cells. (G) Transfection with siRNA-PDK1 decreased the IC50 of oxaliplatin to HT29/R and SW480/R cells. Notes: Data were expressed as mean±SD. #P<0.05 vs oxaliplatin+plasmid-empty, &P<0.05 vs oxaliplatin+siRNA-control, *P<0.05. Abbreviations: CRC, colorectal cancer; LDH, dehydrogenase; IC50, half maximal inhibitory concentration; siRNA, small interfering RNA; PDK1, pyruvate dehydrogenase kinase 1. |
PDK1 Is Targeted by miR-138 in HT29/R and SW480/R
Since expression level of numerous genes was regulated by miRNAs, we searched the potential upstream regulator of PDK1 through the public miRNA prediction databases of TargetScan, miRanda, and PicTar. Among the candidate miRNAs, miR-138 was commonly predicted by all of these databases. The potential complementary site paired with miR-138 and PDK1 was shown in Figure 4A. Additionally, expression of miR-138 was found to be downregulated in HT29/R and SW480/R cells compared to the HT29 and SW480 cells (Figure 4B). These results suggested the potential negative correlation between PDK1 expression and miR-138 level in HT29/R and SW480/R. Next, we observed significant inhibition of PDK1 expression after transfection with miR-138 in HT29/R and SW480/R cells (Figure 4C). Furthermore, results of luciferase reporter assays showed that co-transfection with miR-138 significantly inhibited the luciferase activities of the reporters contained wild type PDK1 3ʹ-UTR but not the mutant one (Figure 4D). These data indicated that miR-138 targeted PDK1 in CRC. Overexpression of PDK1 was induced by decrease of miR-138 expression in HT29/R and SW480/R.
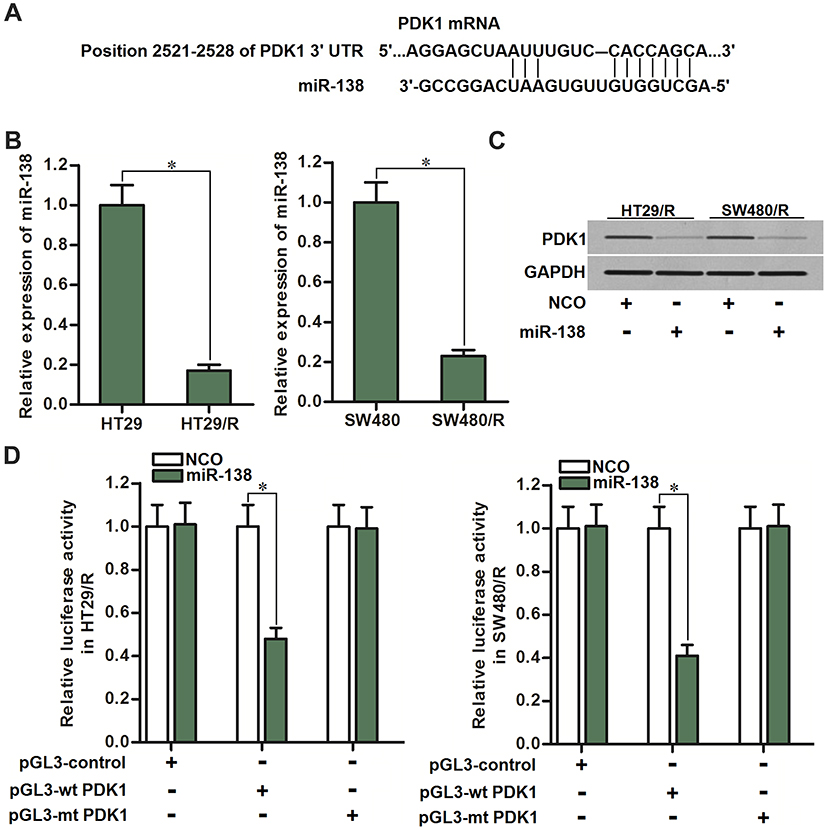 |
Figure 4 PDK1 was the target of miR-138 in HT29/R and SW480/R. (A) PDK1 mRNA 3ʹ UTR exits potential binding site paired with miR-138. (B) Significant downregulation of miR-138 in HT29/R and SW480/R compared to their parental cells. (C) Effect of miR-138 on inhibiting the expression of PDK1 in HT29/R and SW480/R cells. (D) Co-transfection with miR-138 decreased the luciferase activities of the pGL3 reporters contained wild type PDK1 3ʹ-UTR but not the mutant one. Notes: Data were expressed as mean±SD. *P<0.05. Abbreviation: PDK1, pyruvate dehydrogenase kinase 1. |
MiR-138/PDK1 Axis Fails to Change the Glycolysis of HT29/R and SW480/R
To explore the role of PDK1 in regulating the glycolysis in HT29/R and SW480/R, we transfected the HT29/R and SW480/R cells with siRNA-PDK1 or miR-138. As shown in Figure 5A, neither siRNA-PDK1 nor miR-138 obviously changed the consumption of glucose in HT29/R and SW480/R cells. Similarly, we can not observe significant differences of lactate production (Figure 5B) and LDH activity (Figure 5C) in HT29/R and SW480/R cells when they were transfected with siRNA-PDK1 or miR-138. These data showed that knockdown of PDK1 expression can not influence the glycolysis in HT29/R and SW480/R cells.
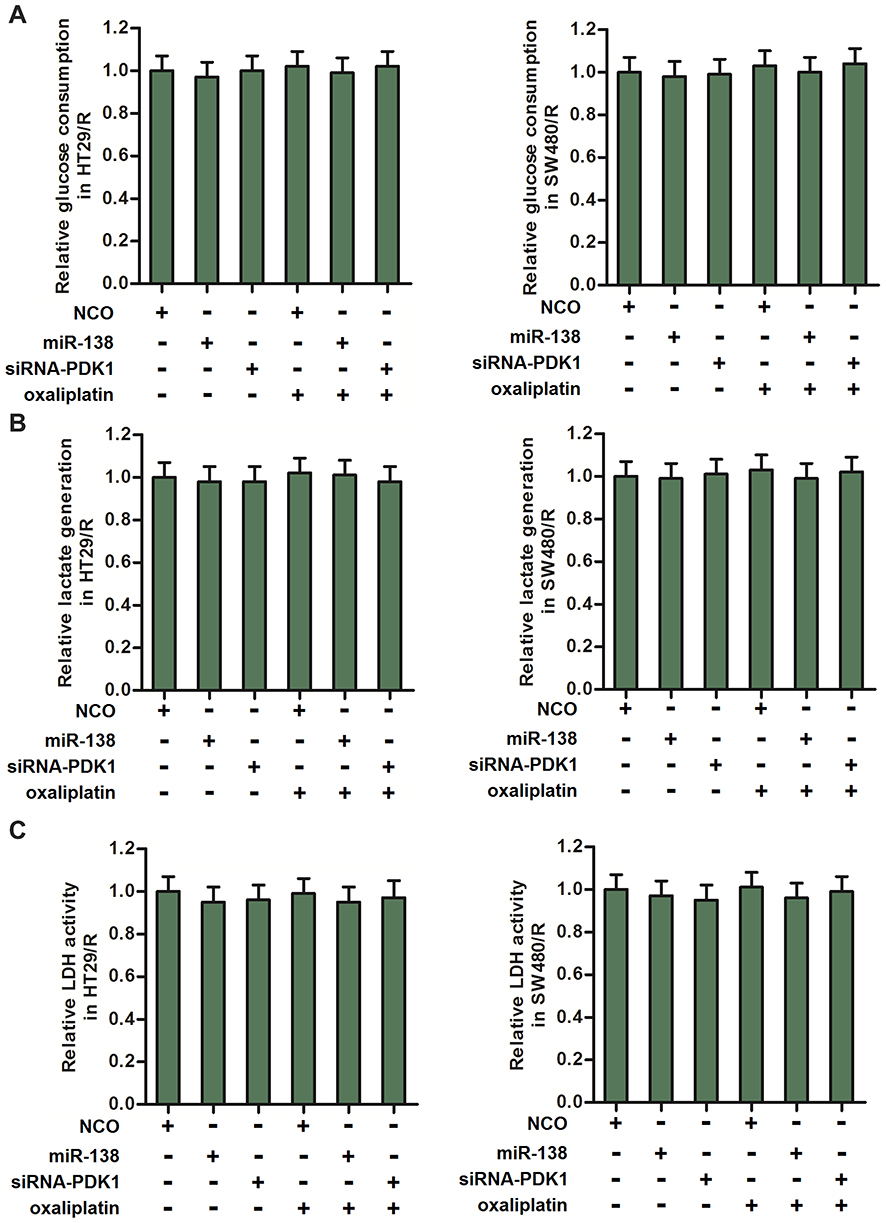 |
Figure 5 MiR-138 and siRNA-PDK1 failed to change the glycolysis level of HT29/R and SW480/R. (A) MiR-138 and siRNA-PDK1 exhibited no activity on glucose consumption of HT29/R and SW480/R cells. (B) MiR-138 and siRNA-PDK1 failed to change the lactate generation in HT29/R and SW480/R cells. (C) MiR-138 and siRNA-PDK1 failed to change the LDH activity in HT29/R and SW480/R cells. Note: Data were expressed as mean±SD. Abbreviations: LDH, dehydrogenase; siRNA, small interfering RNA; PDK1, pyruvate dehydrogenase kinase 1. |
MiR-138 Targets PDK1 to Increase the OCR and Decrease the Oxaliplatin Resistance of HT29/R and SW480/R
Despite miR-138 exhibited no activity on glycolysis, we found that either siRNA-PDK1 or miR-138 significantly increased the OCR of HT29/R and SW480/R cells (Figure 6A). Next, we co-transfected the HT29/R and SW480/R cells with miR-138 and plasmid-PDK1 before measurement of OCR. As shown in Figure 6B, despite miR-138 increased the OCR of HT29/R and SW480/R, enforced expression of PDK1 abolished the activity of miR-138 on OCR. We thus confirmed that the miR-138-induced enhancement of OCR was dependent on the inhibition of PDK1. Furthermore, results of MTT assays showed that treatment with miR-138 increased the sensitivity of HT29/R and SW480/R cells to oxaliplatin. However, co-transfection with plasmid-PDK1 increased the cell viability of HT29/R and SW480/R cells which were co-treated with oxaliplatin and miR-138 (Figure 6C). Specifically, miR-138 decreased the IC50 of oxaliplatin by 76.2% to HT29/R and 83.2% to SW480/R (Figure 6D). These data indicated that overexpression of miR-138 can increase the OCR and partially reverse the oxaliplatin resistance of HT29/R and SW480/R cells through suppression of PDK1.
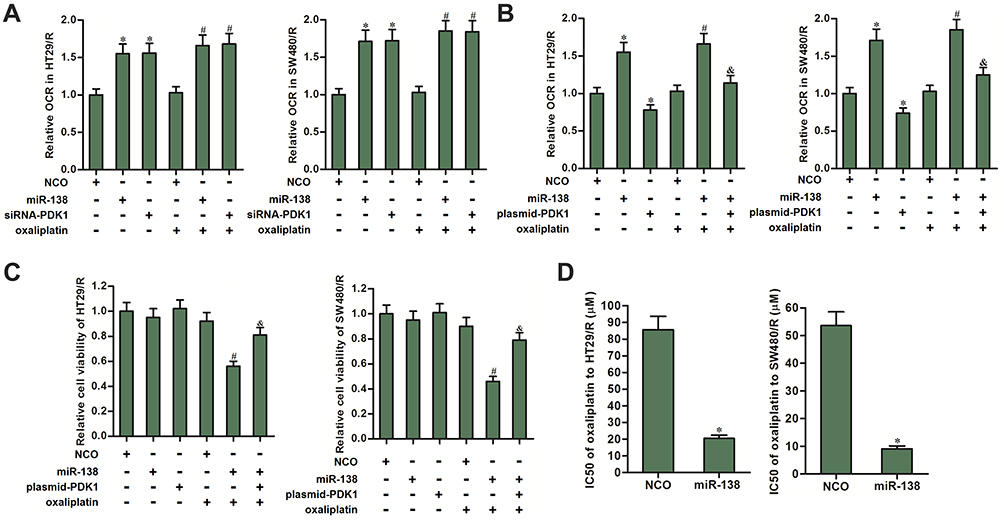 |
Figure 6 MiR-138 increased the OCR and decreased the oxaliplatin resistance of HT29/R and SW480/R through suppression of PDK1. (A) MiR-138 and siRNA-PDK1 significantly increased the OCR of HT29/R and SW480/R cells. (B) Transfection with plasmid-PDK1 inhibited the effect of miR-138 on increasing the OCR in oxaliplatin-treated (10 μM) HT29/R and SW480/R cells. (C) Transfection with plasmid-PDK1 decreased the sensitization of miR-138 on oxaliplatin-induced (10 μM) cytotoxicity against HT29/R and SW480/R cells. (D) Transfection with plasmid-PDK1 decreased the IC50 of oxaliplatin to HT29/R and SW480/R cells. Notes: Data were expressed as mean±SD. *P<0.05 vs NCO group, #P<0.05 vs oxaliplatin+NCO group, &P<0.05 vs oxaliplatin+miR-138 group. Abbreviations: OCR, glycolysis and oxygen consumption rate; IC50, half-maximal inhibitory concentration; siRNA, small interfering RNA; PDK1, pyruvate dehydrogenase kinase 1; NCO, negative control oligonucleotides. |
MiR-138 Promotes the Generation of ROS in HT29/R and SW480/R Through Suppression of PDK1
To explore the mechanism by which miR-138/PDK1 axis changed the oxaliplatin sensitivity of HT29/R and SW480/R, we measured the level of ROS which is mainly generated from mitochondria.27 As shown in Figure 7A, despite miR-138 single treatment cannot induce the generation of ROS, it obviously promoted production of ROS in HT29/R and SW480/R cells. On the other hand, overexpression of PDK1 decreased the ROS level in the oxaliplatin and miR-138 co-treated HT29/R and SW480/R cells. Our results showed that miR-138 can enhance the effect of oxaliplatin on generating the ROS in HT29/R and SW480/R through suppression of PDK1. To evaluate the role of ROS in HT29/R and SW480/R, N-acetylcysteine (NAC) was used to remove the ROS.28 Despite miR-138 obviously promoted the oxaliplatin-induced cell death in HT29/R and SW480/R, addition with NAC significantly reduced the cytotoxicity of them against HT29/R and SW480/R cells (Figure 7B). These results indicated that miR-138 targeted PDK1 to promote the generation of ROS. Increase of ROS level was a key mechanism for miR-138 to partially reverse the oxaliplatin-resistance of HT29/R and SW480/R.
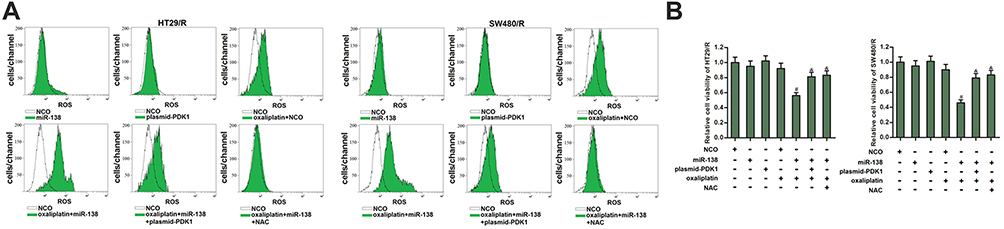 |
Figure 7 MiR-138 promoted generation of ROS in oxaliplatin-treated HT29/R and SW480/R cells. (A) MiR-138 enhanced the effect of oxaliplatin (10 μM) on producing the ROS in HT29/R and SW480/R cells. (B) NAC (2 mM) decreased the sensitization of miR-138 on oxaliplatin-induced (10 μM) cytotoxicity against HT29/R and SW480/R cells. Notes: Data were expressed as mean±SD. #P<0.05 vs oxaliplatin+NCO group, &P<0.05 vs oxaliplatin+miR-138 group. Abbreviations: NCO, negative control oligonucleotides; ROS, reactive oxygen species; NAC, N-acetylcysteine. |
MiR-138 Promotes Oxaliplatin-Dependent Apoptosis in HT29/R and SW480/R Through the PDK1/ROS/ASK1/JNK/Bcl-2 Pathway
The preceding results have proved that miR-138 promoted oxaliplatin-induced cell death of HT29/R and SW480/R through the ROS pathway. We thus investigated the potential role of cellular ROS in activation of apoptosis signal-regulating kinase 1 (ASK1). As shown in Figure 8A, miR-138 promoted phosphorylation of ASK1 in the oxaliplatin-treated HT29/R and SW480/R cells. However, either plasmid-PDK1 or NAC inhibited the phosphorylation of ASK1. Furthermore, as the downstream of ASK1, c-Jun N-terminal kinase (JNK) was significantly phosphorylated in the miR-138 and oxaliplatin co-treated HT29/R and SW480/R cells (Figure 8B). Previous research has proved that activation of JNK induces phosphorylation of Bcl-2, and thus suppressing the anti-apoptotic function of Bcl-2.29 As shown in Figure 8C, combination with oxaliplatin and miR-138 induced significant phosphorylation of Bcl-2 in HT29/R and SW480/R cells. However, either plasmid-PDK1 or NAC inhibited the phosphorylation of it. As the results of Bcl-2 inhibition, we finally observed significant apoptotic cell death of HT29/R and SW480/R when they were co-treated with oxaliplatin and miR-138 (Figure 8D). Taken together, we demonstrated that miR-138 promoted oxaliplatin-dependent apoptosis in HT29/R and SW480/R through the PDK1/ROS/ASK1/JNK/Bcl-2 pathway.
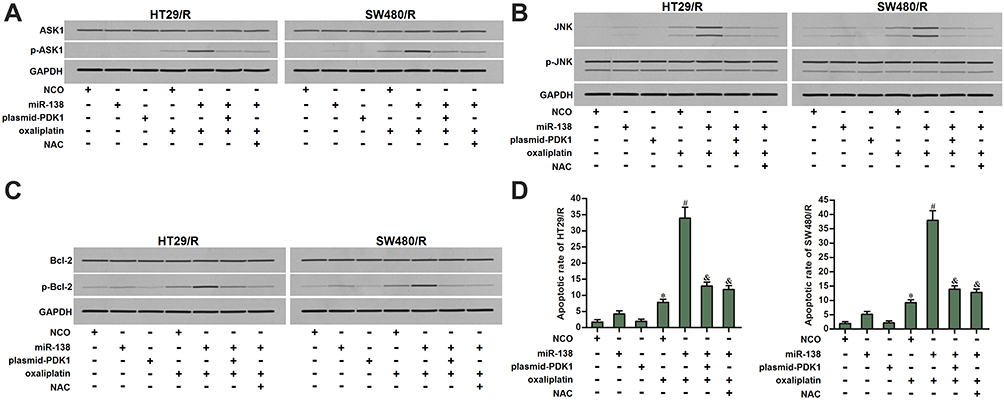 |
Figure 8 ASK1/JNK/Bcl-2 pathway in miR-138 and oxaliplatin co-treated HT29/R and SW480/R cells. (A) MiR-138 enhanced the phosphorylation of ASK1 in oxaliplatin-treated (10 μM) HT29/R and SW480/R cells, whereas plasmid-PDK1 or NAC (2 mM) inhibited the ASK1 phosphorylation. (B) Effect of miR-138, plasmid-PDK1, oxaliplatin (10 μM) and NAC (2 mM) on changing the phosphorylation of JNK in HT29/R and SW480/R cells. (C) Effect of miR-138, plasmid-PDK1, oxaliplatin (10 μM) and NAC (2 mM) on changing the phosphorylation of Bcl-2 in HT29/R and SW480/R cells. (D) miR-138 enhanced oxaliplatin-induced (10 μM) apoptosis of HT29/R and SW480/R cells, whereas plasmid-PDK1 or NAC (2 mM) decreased the apoptotic rate of them. Notes: Data were expressed as mean±SD. *P<0.05 vs NCO group, #P<0.05 vs oxaliplatin+NCO group, &P<0.05 vs oxaliplatin+miR-138 group. Abbreviations: NCO, negative control oligonucleotides; NAC, N-acetylcysteine; PDK1, pyruvate dehydrogenase kinase 1; ASK1, apoptosis signal-regulating kinase 1; JNK, c-Jun N-terminal kinase; Bcl-2, B-cell lymphoma-2. |
Discussion
Oxaliplatin is one of the first-line chemotherapeutic drugs in the treatment of CRC.30 As a platinum-based drug, oxaliplatin cross-links with DNAs and thus induces apoptotic cell death of cancer cells.31,32 However, under the long-term use of oxaliplatin, cancer cells including CRC cells usually show poor response to oxaliplatin-induced apoptosis.33–35 It is urgent to explore the underlying mechanism of the apoptosis-resistance and develop novel strategies to reverse the drug resistance.
Recently, studies report that chemoresistant cancer show high level of glycolysis.24,25 However, the potential mechanism for illustrating the association between glycolysis and chemoresistance is still unclear. In the present study, we found that the oxaliplatin-resistant CRC cells consumed more amount of glucose and exhibited higher level of glycolysis compared to the routine CRC cells. However, we observed a significantly lower level of OCR in oxaliplatin-resistant CRC cells. These results suggested that oxaliplatin-resistant CRC cells were more dependent on glycolysis than sensitive ones.
PDK1 reduces available pyruvate for mitochondrial oxidative phosphorylation through inactivating the pyruvate dehydrogenase.26 Therefore, PDK1 is a key enzyme that connects glycolysis and OCR. Previous studies have demonstrated that PDK1 acts as a tumor promoter and exhibits anti-apoptotic activity in some cancers.36–38 Furthermore, overexpression of PDK1 is reported to be associated with development of chemoresistance in different types of malignancy.39 In this study, we observed significantly higher level of PDK1 in oxaliplatin-resistant CRC cells. We then proved that overexpression of PDK1 induced low OCR and is responsible for oxaliplatin resistance in oxaliplatin-resistant CRC cells. Knockdown of PDK1 can increase the OCR level and partially reversed the oxaliplatin resistance of oxaliplatin-resistant CRC cells. Furthermore, we found that overexpression of PDK1 was induced by downregulation of miR-138 in oxaliplatin-resistant CRC cells. Restore of miR-138 in oxaliplatin-resistant CRC cells can resensitize them to oxaliplatin treatment through inhibition of PDK1.
ROS is a cellular product under stress conditions. Previous research have indicated that the anti-tumor effect of oxaliplatin is closely related to the ROS accumulation.40,41 ASK1 is a serine/threonine protein kinase that acts as downstream molecule of ROS.42 As a key intermediate-bridge of ROS-dependent apoptotic pathway, activated ASK1 induces apoptotic cell death through phosphorylating the JNK which is one member of MAPK family.43–45 Moreover, activation of JNK sensitizes mitochondrial apoptosis by inactivating the Bcl-2 through phosphorylation modification of it.29,46
In our further research, we demonstrated that miR-138-inhibited expression of PDK1 through binding to the 3ʹ UTR of PDK1 mRNA. Therefore, miR-138 enhanced OCR to facilitate the generation of ROS caused by oxaliplatin treatment. Cellular high level of ROS induced phosphorylation of ASK1 and subsequent phosphorylation of JNK and Bcl-2. Despite dephosphorylated Bcl-2 exhibited powerful anti-apoptotic activity, Bcl-2 with phosphorylation modification lost its function. As a result, apoptotic cell death occurred in the oxaliplatin and miR-138 co-treated HT29/R and SW480/R cells. In summary, miR-138 promoted oxaliplatin-dependent apoptosis in HT29/R and SW480/R through the PDK1/ROS/ASK1/JNK/Bcl-2 pathway.
Acknowledgments
This study is supported by the Natural Scientific Foundation of China (grant no. 81202031); the Scientific Research Project of Jilin Province Science and Technology Department (grant no. 20160204033YY); the Technical Innovation Project of Jilin Province Health Department (grant no. 2016J100).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi:10.3322/caac.21254
2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.20107
3. Manfredi S, Lepage C, Hatem C, Coatmeur O, Faivre J, Bouvier AM. Epidemiology and management of liver metastases from colorectal cancer. Ann Surg. 2006;244(2):254–259. doi:10.1097/01.sla.0000217629.94941.cf
4. Yao Y, Rao C, Zheng G, Wang S. Luteolin suppresses colorectal cancer cell metastasis via regulation of the miR-384/pleiotrophin axis. Oncol Rep. 2019;42(1):131–141. doi:10.3892/or.2019.7136
5. Ciombor KK, Wu C, Goldberg RM. Recent therapeutic advances in the treatment of colorectal cancer. Annu Rev Med. 2015;66(1):83–95. doi:10.1146/annurev-med-051513-102539
6. Esquela-Kerscher A, Slack F. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–269. doi:10.1038/nrc1840
7. Filipowicz W, Bhattacharyya S, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9(2):102–114. doi:10.1038/nrg2290
8. Wang D, Qiu C, Zhang H, Wang J, Cui Q, Yin Y. Human microRNA oncogenes and tumor suppressors show significantly different biological patterns: from functions to targets. PLoS One. 2010;5(9):e13067. doi:10.1371/journal.pone.0013067
9. Li L, Luo J, Wang B, et al. MicroRNA-124 targets flotillin-1 to regulate proliferation and migration in breast cancer. Mol Cancer. 2013;12(1):163. doi:10.1186/1476-4598-12-163
10. Zhu E-D, Na L, Bo-Sheng L, et al. miR-30b, down-regulated in gastric cancer, promotes apoptosis and suppresses tumor growth by targeting plasminogen activator inhibitor-1. PLoS One. 2014;9(8):e106049. doi:10.1371/journal.pone.0106049
11. Balacescu O, Sur D, Cainap C, et al. The impact of miRNA in colorectal cancer progression and its liver metastases. Int J Mol Sci. 2018;19(12):E3711. doi:10.3390/ijms19123711
12. Hsu HH, Kuo WW, Shih HN, et al. FOXC1 regulation of miR-31-5p confers oxaliplatin resistance by targeting LATS2 in colorectal cancer. Cancers (Basel). 2019;11(10):E1576. doi:10.3390/cancers11101576
13. Jia G, Tang Y, Deng G, et al. miR-590-5p promotes liver cancer growth and chemotherapy resistance through directly targeting FOXO1. Am J Transl Res. 2019;11(4):2181–2193.
14. Zhang L, He L, Zhang H, Chen Y. Knockdown of MiR-20a enhances sensitivity of colorectal cancer Cells to cisplatin by increasing ASK1 expression. Cell Physiol Biochem. 2018;47(4):1432–1441. doi:10.1159/000490834
15. Wang CQ. MiR-195 reverses 5-FU resistance through targeting HMGA1 in gastric cancer cells. Eur Rev Med Pharmacol Sci. 2019;23(9):3771–3778. doi:10.26355/eurrev_201905_17803
16. Lu WQ, Hu YY, Lin XP, Fan W. Knockdown of PKM2 and GLS1 expression can significantly reverse oxaliplatin-resistance in colorectal cancer cells. Oncotarget. 2017;8(27):44171–44185. doi:10.18632/oncotarget.17396
17. Qin Y, Li L, Wang F, et al. Knockdown of Mir-135b sensitizes colorectal cancer cells to oxaliplatin-induced apoptosis through increase of FOXO1. Cell Physiol Biochem. 2018;48(4):1628–1637. doi:10.1159/000492284
18. Zhu D, Gu L, Li Z, Jin W, Lu Q, Ren T. MiR-138-5p suppresses lung adenocarcinoma cell epithelial-mesenchymal transition, proliferation and metastasis by targeting ZEB2. Pathol Res Pract. 2019;215(5):861–872. doi:10.1016/j.prp.2019.01.029
19. Pang L, Li B, Zheng B, Niu L, Ge L. miR-138 inhibits gastric cancer growth by suppressing SOX4. Oncol Rep. 2017;38(2):1295–1302. doi:10.3892/or.2017.5745
20. Liu Y, Zhang W, Liu K, Liu S, Ji B, Wang Y. miR-138 suppresses cell proliferation and invasion by inhibiting SOX9 in hepatocellular carcinoma. Am J Transl Res. 2016;8(5):2159–2168.
21. Zheng S, Zhang X, Wang X, Li J. Downregulation of miR-138 predicts poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Biomark. 2017;20(1):49–54. doi:10.3233/CBM-170079
22. Gao Y, Fan X, Li W, Ping W, Deng Y, Fu X. miR-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124. Biochem Biophys Res Commun. 2014;446(1):179–186. doi:10.1016/j.bbrc.2014.02.073
23. Golubovskaya VM, Sumbler B, Ho B, Yemma M, Cance WG. MiR-138 and MiR-135 directly target focal adhesion kinase, inhibit cell invasion, and increase sensitivity to chemotherapy in cancer cells. Anticancer Agents Med Chem. 2014;14(1):18–28. doi:10.2174/187152061401140108113435
24. Song YD, Li DD, Guan Y, Wang YL, Zheng J. miR-214 modulates oxaliplatin sensitivity of osteosarcoma cells through regulation of anaerobic glycolysis. Cell Mol Biol (Noisy-Le-Grand). 2017;63(9):75–79. doi:10.14715/cmb/2017.63.9.14
25. Wang T, Ning K, Sun X, Zhang C, Jin LF, Hua D. Glycolysis is essential for chemoresistance induced by transient receptor potential channel C5 in colorectal cancer. BMC Cancer. 2018;18(1):207. doi:10.1186/s12885-018-4123-1
26. Chambers KT, Leone TC, Sambandam N, et al. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. J Biol Chem. 2011;286(13):11155–11162. doi:10.1074/jbc.M110.217349
27. Yang Y, Karakhanova HW, D’Haese JG, Philippov PP, Werner J, Bazhin AV. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. 2016;231(12):2570–2581. doi:10.1002/jcp.25349
28. Halasi M, Wang M, Chavan TS, Gaponenko V, Hay N, Gartel AL. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem J. 2013;454(2):201–208. doi:10.1042/BJ20130282
29. Stander XX, Stander BA, Joubert AM. Synergistic anticancer potential of dichloroacetate and estradiol analogue exerting their effect via ROS-JNK-Bcl-2-mediated signalling pathways. Cell Physiol Biochem. 2015;35(4):1499–1526. doi:10.1159/000369710
30. Liu Z, Kong J, Kong Y, et al. Association of XPD Asp312Asn polymorphism and response to oxaliplatin-based first-line chemotherapy and survival in patients with metastatic colorectal cancer. Adv Clin Exp Med. 2019;28(11):1459–1468. doi:10.17219/acem/108552
31. Zhang Y, Xie C, Li A, et al. PKI-587 enhances chemosensitivity of oxaliplatin in hepatocellular carcinoma through suppressing DNA damage repair pathway (NHEJ and HR) and PI3K/AKT/mTOR pathway. Am J Transl Res. 2019;11(8):5134–5149.
32. Seo SU, Min KJ, Woo SM, Kwon TK. Z-FL-COCHO, a cathepsin S inhibitor, enhances oxaliplatin-mediated apoptosis through the induction of endoplasmic reticulum stress. Exp Mol Med. 2018;50(8):107. doi:10.1038/s12276-018-0138-6
33. Virag P, Brie I, Fischer-Fodor E, et al. Assessment of cytotoxicity, apoptosis and DNA damages in Colo320 colorectal cancer cells selected for oxaliplatin resistance. Cell Biochem Funct. 2011;29(5):351–355. doi:10.1002/cbf.1754
34. Hsu HH, Chen MC, Baskaran R, et al. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J Cell Physiol. 2018;233(7):5458–5467. doi:10.1002/jcp.26406
35. Zhang Y, Xu Z, Sun Y, Chi P, Lu X. Knockdown of KLK11 reverses oxaliplatin resistance by inhibiting proliferation and activating apoptosis via suppressing the PI3K/AKT signal pathway in colorectal cancer cell. Onco Targets Ther. 2018;11:809–821. doi:10.2147/OTT.S151867
36. Peng F, Wang JH, Fan WJ, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene. 2018;37(8):1062–1074. doi:10.1038/onc.2017.368
37. Ruggieri V, Agriesti F, Scrima R, et al. Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment. Oncotarget. 2015;6(2):1217–1230. doi:10.18632/oncotarget.2721
38. Raimondi C, Falasca M. Targeting PDK1 in cancer. Curr Med Chem. 2011;18(18):2763–2769. doi:10.2174/092986711796011238
39. Emmanouilidi A, Falasca M. Targeting PDK1 for chemosensitization of cancer cells. Cancers (Basel). 2017;9(12):E140. doi:10.3390/cancers9100140
40. Cao P, Xia Y, He W, et al. Enhancement of oxaliplatin-induced colon cancer cell apoptosis by alantolactone, a natural product inducer of ROS. Int J Biol Sci. 2019;15(8):1676–1684. doi:10.7150/ijbs.35265
41. Zhang P, Shi L, Zhang T, et al. Piperlongumine potentiates the antitumor efficacy of oxaliplatin through ROS induction in gastric cancer cells. Cell Oncol (Dordr). 2019;42(6):847–860. doi:10.1007/s13402-019-00471-x
42. Hong SW, Shin JS, Lee YM, et al. p34 SEI-1 Inhibits ROS-induced cell death through suppression of ASK1. Cancer Biol Ther. 2011;12(5):421–426. doi:10.4161/cbt.12.5.15972
43. Zheng R, You Z, Jia J, et al. Curcumin enhances the antitumor effect of ABT-737 via activation of the ROS-ASK1-JNK pathway in hepatocellular carcinoma cells. Mol Med Rep. 2016;13(2):1570–1576. doi:10.3892/mmr.2015.4715
44. Palit S, Kar S, Sharma G, Das PK. Hesperetin induces apoptosis in breast carcinoma by triggering accumulation of ROS and activation of ASK1/JNK pathway. J Cell Physiol. 2015;230(8):1729–1739. doi:10.1002/jcp.24818
45. Liang T, Zhang X, Xue W, Zhao S, Zhang X, Pei J. Curcumin induced human gastric cancer BGC-823 cells apoptosis by ROS-mediated ASK1-MKK4-JNK stress signaling pathway. Int J Mol Sci. 2014;15(9):15754–15765. doi:10.3390/ijms150915754
46. Low SY, Tan BS, Choo HL, Tiong KH, Khoo AS, Leong CO. Suppression of BCL-2 synergizes oxaliplatin sensitivity in nasopharyngeal carcinoma cells. Cancer Lett. 2012;314(2):166–175. doi:10.1016/j.canlet.2011.09.025
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.