Back to Journals » OncoTargets and Therapy » Volume 13
miR-137-3p Modulates the Progression of Prostate Cancer by Regulating the JNK3/EZH2 Axis
Authors Zang Y, Zhu J, Li Q, Tu J, Li X, Hu R ![]() , Yang D
, Yang D
Received 30 March 2020
Accepted for publication 22 July 2020
Published 10 August 2020 Volume 2020:13 Pages 7921—7932
DOI https://doi.org/10.2147/OTT.S256161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Yachen Zang, 1,* Jin Zhu, 1,* Qin Li, 2, 3,* Jian Tu, 4 Xiaoqing Li, 5 Rongkuan Hu, 3 Dongrong Yang 1
1Department of Urology, The Second Affiliated Hospital of Soochow University, Suzhou 215004, People’s Republic of China; 2School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, People’s Republic of China; 3GenePharma-Deakin University Joint Laboratory of Aptamer Medicine, Suzhou 215123, People’s Republic of China; 4Department of Pathology, The Second Affiliated Hospital of Soochow University, Suzhou 215006, People’s Republic of China; 5Suzhou Cancer Center Core Laboratory, Nanjing Medical University Affiliated Suzhou Hospital, Suzhou 215001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dongrong Yang
Department of Urology, The Second Affiliated Hospital of Soochow University, 1055 Sanxiang Road, Suzhou 215004, People’s Republic of China
Email [email protected]
Rongkuan Hu
GenePharma-Deakin University Joint Laboratory of Aptamer Medicine, 199 Dongping Street, Suzhou 215123, People’s Republic of China
Email [email protected]
Background: Prostate cancer (PCa) is one of the most common cancers in men worldwide. Early detection of prostate cancer by prostate-specific antigen (PSA) screening still has limitations. The discovery of new candidates is urgent and can provide insights into the mechanism involved in prostate cancer tumorigenesis.
Methods: We conducted a cross-sectional study involving prostate cancer cell lines and clinical samples. qPCR and IHC were used to evaluate the expression of miR-137-3p/JNK3/EZH2. Furthermore, cell growth, migration, invasion, cell cycle and apoptosis were analyzed to describe the function of this axis. Moreover, xenograft models, pathology platforms and TCGA data were generated to confirm the role of the miR-137-3p/JNK3/EZH2 axis.
Results: In this study, we determined that miR-137-3p was significantly reduced in prostate cancer, and low expression of miR-137-3p was correlated with tumor stage . The overexpression of miR-137-3p suppressed cell proliferation, migration and invasion in prostate cancer by enhancing cell apoptosis. We also validated JNK3 (MAPK10) as a direct target gene of miR-137-3p. Down-regulation of JNK3 in prostate cancer also inhibited cell proliferation and invasion and promoted apoptosis. Moreover, JNK3 expression was up-regulated and negatively correlated with miR-137-3p in prostate cancer tissues. Furthermore, JNK3 modulated EZH2 expression, which is a key oncogene in prostate cancer. Survival data indicated that patients with high levels of JNK3 and EZH2 had a worse prognosis.
Conclusion: Collectively, the identification of miR-137-3p and the JNK3/EZH2 pathway might facilitate the development of biomarkers and therapeutic targets for prostate cancer.
Keywords: microRNA-137-3p, prostate cancer, biomarker, MAPK10, EZH2
Background
Prostate cancer is one of the most common cancers, and its incidence is still increasing in many countries. Early diagnosis and treatment of early prostate cancer is one of the most challenging and hot topics.1 To increase the detection accuracy of early-stage prostate cancer, there have been many reports about early effective biomarkers and the management of prostate cancer.2 Currently, serum PSA-based screening for prostate cancer is the most common tool in the clinic. Although the PSA level is considered to be the most useful biomarker correlated with prostate cancer risk, aggressiveness and outcome, it still has limitations, including sensitivity and specificity.3 Moreover, PSA testing is often associated with over diagnosis. Thus, interest is growing in developing additional biomarkers that have the potential to minimize over diagnosis and can serve as alternative biomarkers, such as microRNAs (miRNAs).
miRNAs are 22-nucleotide-long, noncoding RNAs that regulate several cellular processes at the posttranscriptional level.4 Compelling evidence demonstrates that miRNAs are widely altered in prostate cancer, suggesting that miRNA dysfunction is deeply involved in tumorigenesis, especially in cell proliferation and metastasis. Several studies have indicated that miRNAs are associated with stage, grade, biochemical recurrence and metastasis in prostate cancer.5 Among these, many miRNAs have been commonly reported to be up-regulated in prostate cancer, including miR-106a, miR-17-5p, miR-375 and miR-93.6 These miRNAs are invariably identified as oncogenes that promote cell proliferation, migration, invasion and drug resistance. On the other hand, several miRNAs are commonly reported to be down-regulated in prostate cancer, including miR-125, miR-130a and miR-221.7 These miRNAs may contribute to suppressing the initiation and progression of prostate cancer and can serve as potential diagnostic and prognostic biomarkers, as well as treatment targets.
Very recently, Pashaei E conducted a meta-analysis of 6 miRNA expression datasets to identify a panel of down-regulated miRNAs in recurrent prostate cancer samples in comparison to non-recurrent prostate cancer samples. Among these miRNAs, miR-137 was significantly down-regulated in recurrent prostate cancer patients, suggesting a role for miR-137 in the initiation and progression of prostate cancer.8 miR-137 is one of the most commonly down-regulated tumor suppressors in many types of cancer, including ovarian carcinoma, gastric cancer, colorectal cancer and lung cancer.9–13 It has also been reported that miR-137 inhibits proliferation and metastasis in hepatocellular carcinoma by targeting the EZH2-STAT3 pathway.12 Overexpression of miR-137 inhibits EZH2 and regulates cisplatin resistance in ovarian cancer.14 Furthermore, miR-137 mediates tumorigenesis in triple-negative breast cancer by perturbing the BCL11A/DNMT1 interaction.11 In particular, miR-137 targets P160 and down-regulates the expression of SRC1 and SRC2, which in turn inhibits cell proliferation in breast cancer, melanoma and prostate cancer.15 However, the specific subtype of miR-137 regulating prostate cancer is largely unknown.
The complex molecular events that occur at the level of miR-137-3p and oncogene families during prostate cancer tumorigenesis are still unclear. Herein, we reveal a central role for miR-137-3p down-regulation in the progression of prostate cancer. Additionally, we clarify how miR-137-3p regulates the JNK3/EZH2 signaling pathway to inhibit prostate cancer progression. Finally, the clinical relevance of miR-137-3p/JNK3/EZH2 was observed, which might facilitate the development of early detection and treatment for prostate cancer.
Methods
Materials
Lipofectamine 2000, FBS, DMEM, RPMI-1640, and Keratinocyte Serum Free Medium (K-SFM) kits were purchased from Thermo Fisher (MA, USA). Antibodies against JNK3 (MAPK10), EZH2, and GAPDH were purchased from Proteintech (Wuhan, China). Goat anti-rabbit and goat anti-mouse antibodies were obtained from Sigma-Aldrich (St. Louis, USA). The following oligos were used in this study: miR-137-3p mimics, sense: 5ʹ-UUAUUGCUUAAGAAUACGCGUAG-3ʹ; antisense: 5ʹ- ACGCGUAUUCUUAAGCAAUAAUU-3ʹ; negative control, sense: 5ʹ- UUCUCCGAACGUGUCACGUTT-3ʹ; antisense: 5ʹ- ACGUGACACGUUCGGAGAATT-3ʹ; and siRNA targeting JNK3, sense1: 5ʹ- GCUUCAUGAUGACUCCAUATT-3ʹ; antisense1: 5ʹ- UAUGGAGUCAUCAUGAAGCTT-3ʹ; sense2: 5ʹ-CCAGGGACUUGUUGUCAAATT-3ʹ; antisense2: 5ʹ- UUUGACAACAAGUCCCUGGTT-3ʹ. All oligos were obtained got from GenePharma Co., Ltd. (Suzhou, China).
Cell Culture
All prostate cancer cell lines used in this study were obtained from the Cell Bank of the Chinese Academy of Sciences (http://www.cellbank.com.cn/index.asp, Shanghai, China). DU145 (RRID:CVCL_0105) cells were cultured in DMEM supplemented with 10% FBS. RWPE-1 (RRID:CVCL_3791) cells were cultured in K-SFM with BPE and EGF, and LNCAP (RRID:CVCL_0395) and PC-3 (RRID:CVCL_0035) cells were cultured in RPMI-1640 with 10% FBS at 37 °C in 5% CO2. All human cell lines had been authenticated using STR profiling within the last three years. All experiments were performed with mycoplasma-free cells.
Western Blotting
Western blotting analysis was performed as described previously.16 Briefly, 20 µg of cell lysate was loaded onto 10% SDS-PAGE and transferred to nitrocellulose blotting membranes (GE Healthcare, Chicago, IL). The membranes were blotted with primary antibodies against JNK3, Bcl-2, EZH2 and GAPDH separately (1:1000 dilution, 4 °C, overnight). After that, membranes were incubated with secondary antibodies (1:5000) for 1 h at room temperature. Signals were developed using enhanced chemiluminescence (ECL), and the protein bands were visualized using a Tannon Western blotting detection system (Shanghai, China).
Reverse Transcription and Quantitative PCR (RT-qPCR)
Total RNA was extracted by TRIzol (Thermo Fisher, Waltham, MA). Then, 1 µg RNA was reverse transcribed using the Super-Script II reverse Transcriptase kit (Thermo Fisher, Waltham, MA). qRT-PCR was performed using the Power SYBR Green Master Mix (Thermo Fisher, Waltham, MA) for 40 cycles. The relative expression of mRNA was calculated as the inverse log of the delta/delta CT; microRNA expression was normalized to U6 expression, while mRNA was normalized to GAPDH. The following primers were used: miR-137 FP (forward primer): 5ʹ-CCATTCATTCGTTATTGCTTAAGA-3ʹ, miR-137 RP (reverse primer): 5ʹ-TATGCTTGTTCTCGTCTCTGTGTC-3ʹ; JNK3 FP: 5ʹ-ACAGAGCCCCTGAGGTCATCCT-3ʹ; JNK3 RP: 5ʹ-GGCGAACCATTTCTCCCATAA-3ʹ; Actin FP: 5ʹ-CGTGGACATCCGCAAAGA-3ʹ; Actin RP: 5ʹ-GAAGGTGGACAGCGAGGC-3ʹ; U6 FP: 5ʹ-ATTGGAACGATACAGAGAAGATT-3ʹ; U6 RP: 5ʹ-GGAACGCTTCACGAATTTG-3ʹ; And GAPDH sense, 5ʹ-ACAACTTTGGTATCGTGGAAGG-3ʹ; GAPDH anti-sense, 5ʹ-GCCATCACGCCACAGTTTC-3ʹ.
Dual-Luciferase Reporter Assay
For the dual-luciferase reporter assay, DU-145 and PC-3 cells were seeded in 24-well plates and cotransfected 24 h later with PGL-3 control plasmid, wild-type or mutant JNK3 3ʹUTR vector, and miR-137 mimics or control (GenePharma Co., Ltd, Suzhou, China). The cells were harvested 24 h later, and firefly and Renilla luciferase activities were analyzed as previously reported.17
Cell Proliferation and Colony Formation
Cell proliferation was detected using Cell Counting Kit-8 (CCK8, Dojindo, Japan) reagent. The absorbance of each sample after transfection with or without miR-137 was measured at 450 nm. All experiments included at least three tests, and average values were calculated. The colony formation assay protocol was reported previously.4 Briefly, 300 prostate cancer cells were seeded per well. Approximately two weeks later, colonies were fixed with 4% formaldehyde and stained with crystal violet (0.5% w/v).
Cell Migration and Invasion
Cell migration was detected by the wound healing assay, and cells transfected with miR-137-3p or negative control were seeded in 6-well plates. After 24 h, the cell monolayer was gently scratched with a pipette tip across the center of the well and imaged by a Nikon Eclipse Ti system (Tokyo, Japan) after 48 h. Cell invasion assays were performed using cell culture insert membranes coated with diluted Matrigel (BD Biosciences, USA). Briefly, after transfection with miR-137-3p or negative control, 106 cells were seeded in serum-free medium. After 48 h, the cells were fixed with prechilled methanol and stained using 0.5% crystal violet.4
Cell Apoptosis
Cells were harvested and suspended in PI and FITC staining solution following the manufacturer’s instructions for the Annexin V-FITC Apoptosis Detection Kit (Thermo Fisher). Cell apoptosis was measured on a flow cytometer (BD Biosciences, San Jose, USA).
Fluorescence in situ Hybridization (FISH)
The probe for miR-137 (5ʹ-CTACGCGTATTCTTAAGCAATAA-3ʹ, underlined bases were modified with locked nucleic acid (LNA) modification) was synthesized and labeled with 6-carboxyfluorescein (FAM) and purified via HPLC (GenePharma, Suzhou, China). Tissue sections were deparaffinized and hybridized overnight at 50 °C with the probe at a concentration of 25 nM as previously described.18
Xenograft Experiments
Tumors were generated by subcutaneous injection of DU-145 cells (107) into the right flanks of 6-week-old male BALB/c nude mice (n=6, Vital River Laboratory Animal Technology Co., Ltd, Beijing, China). The experiment was approved by the animal ethics committee of Soochow University, and procedures were conducted in accordance with the guidelines of the Soochow University Animal Care and Use Committee (IACUC). The mice were monitored for tumor growth twice a week. Tumor volume was calculated as (width2 x length)/2 at each time point. When tumors reached 3000 mm3, mice were sacrificed, and tumors were collected for molecular analyses and hematoxylin and eosin (H&E) staining as described previously.4
Patient Sample Collection
Twenty pairs of prostate cancer samples were collected from 2013 to 2018 at the Second Affiliated Hospital of Soochow University (Table 1). Pathological analysis confirmed that all the specimens were human prostate cancer samples and correlated adjacent normal control samples were also analyzed. All of the samples were obtained with informed consent from patients and approval from the Ethics Committee at the authors’ institution.
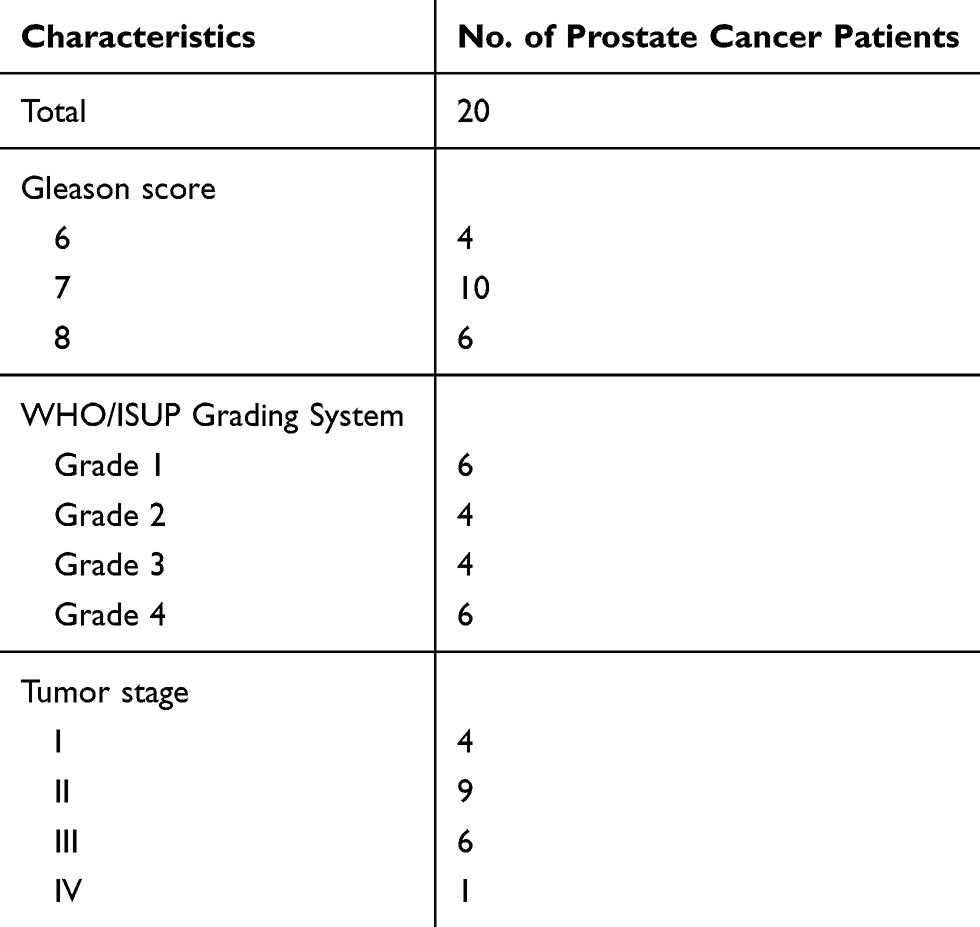 |
Table 1 Patient Clinical Information |
Statistical Analysis
All data described are shown as the mean ± SD. Significance tests were performed using GraphPad Prism 6.0 (GraphPad Inc.). Statistical significance of the differences between groups was calculated using ANOVA or two-tailed Student’s t-test. For correlation analysis, gene expression was converted to log10, and linear regression was performed by GraphPad Prism 6.0 (GraphPad Inc.). A p value of <0.05 was considered significant.
Data Availability Statement
Information for the 18 cohorts of approximately 4850 prostate cancer samples can be found on the website (http://www.cbioportal.org/). Other data will be made available upon reasonable request.
Results
miR-137-3p is Decreased in Prostate Cancer
To identify the role of miR-137-3p in prostate cancer development, we first detected its abundance in 3 prostate cancer cell lines, LNCAP, DU-145 and PC-3. RWPE-1 is a normal epithelial cell line that was used as a negative control. miR-137-3p was found to be significantly down-regulated in PC-3, LNCAP and DU-145 cells compared to RWPE-1 cells (Figure 1A). Subsequently, we measured the levels of miR-137-3p in 20 pairs of tumor tissues and corresponding adjacent controls using qRT-PCR and FISH. In accordance with the cell line data, miR-137-3p was reduced in tumor samples, suggesting a gradual loss of miR-137-3p during prostate tumorigenesis (Figure 1B and C). Among those tumor specimens, miR-137-3p was lower in late stage patient samples (III/IV) than in early stage (I/II) samples (Figure 1D).
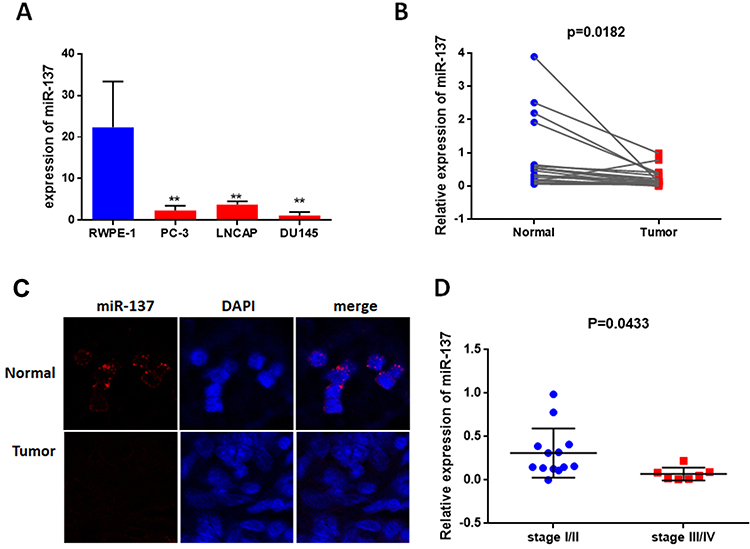 |
Figure 1 miR-137-3p is downregulated in prostate cancer. (A) Expression of miR-137-3p was analyzed by qRT-PCR in 3 prostate cancer cell lines, and RWPE-1 was used as a normal control. (B) miR-137-3p levels were shown to be downregulated in 20 prostate cancer tissues compared to paired normal samples using qRT-PCR. (C) The expression of miR-137-3p in normal and prostate cancer tissues was detected by FISH (Gleason score 7). (D) miR-137 expression in different clinical stages of prostate cancer (n=20). **p < 0.01. |
miR-137-3p Suppresses Tumor Growth Both in vitro and in vivo
As mentioned above, our hypothesis was that miR-137-3p may act as a tumor suppressor in prostate cancer. To confirm this hypothesis, miR-137-3p mimics were introduced into two prostate cancer cell lines with low expression of miR-137 (PC-3 and DU-145) (Figure S1A). The proliferation rate of the two prostate cancer cell lines was decreased after transfection with miR-137-3p (Figure 2A and B). Furthermore, the colony number of prostate cancer cells transfected with miR-137-3p was also decreased, confirming the suppressor role of miR-137-3p in tumor growth (Figures 2C and S1B). In addition, to evaluate the in vivo function, xenograft models were constructed in male BALB/c nude mice. The results showed that treatment with miR-137-3p dramatically decreased the tumor volume and tumor weight (Figure 2D–F). These results both indicate that miR-137-3p may function as a tumor suppressor in human prostate cancer.
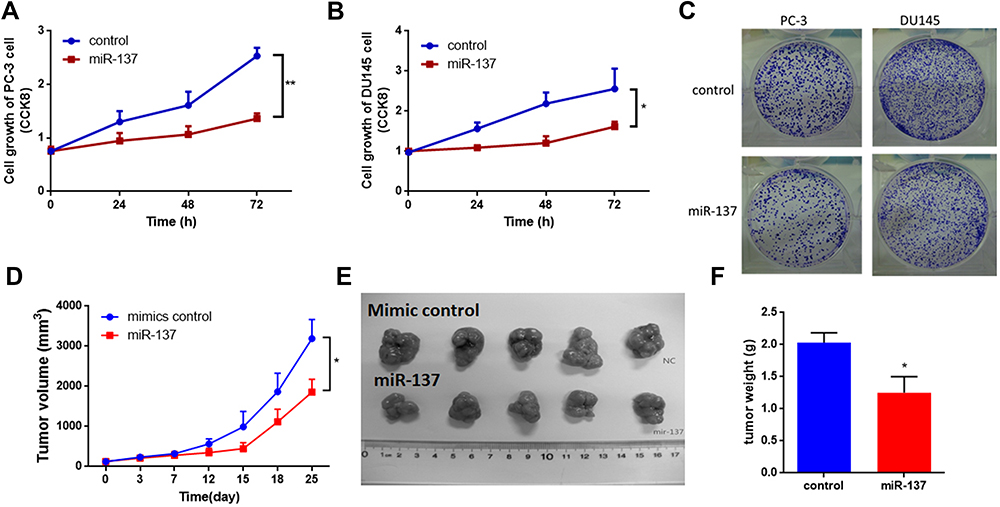 |
Figure 2 miR-137-3p suppresses prostate cancer tumor growth both in vitro and in vivo. After transfection of miR-137-3p mimics into two prostate cancer cell lines (A) and DU-145 cells (B), cell proliferation was detected by CCK8 assay at different time points. (C) Colony formation assays were performed after transfection with miR-137-3p (48 h). (D) Xenograft experiments were performed to confirm the effects of miR-137-3p on tumor growth in vivo. Each group (5 mice) was injected with DU145 (107) cells overexpressing miR-137-3p or scramble control. Tumors were photographed (E) and weighed (F). *p < 0.05, **p < 0.01. |
miR-137-3p Inhibits Tumor Metastasis by Promoting Cell Apoptosis
To investigate whether miR-137-3p regulates tumor metastasis, cell migration and invasion were measured. Briefly, after transfection of miR-137-3p, the cell monolayer was scratched across the center of the well. The gaps were imaged and indicated that miR-137-3p mimics decreased the migration of the two prostate cancer cell lines (Figure 3A). Consistent with the migration results, reduced invasion activity after incubation with miR-137-3p was also observed in the Transwell assay (Figure 3B). These results demonstrate that miR-137-3p inhibits migration and invasion in prostate cancer metastasis.
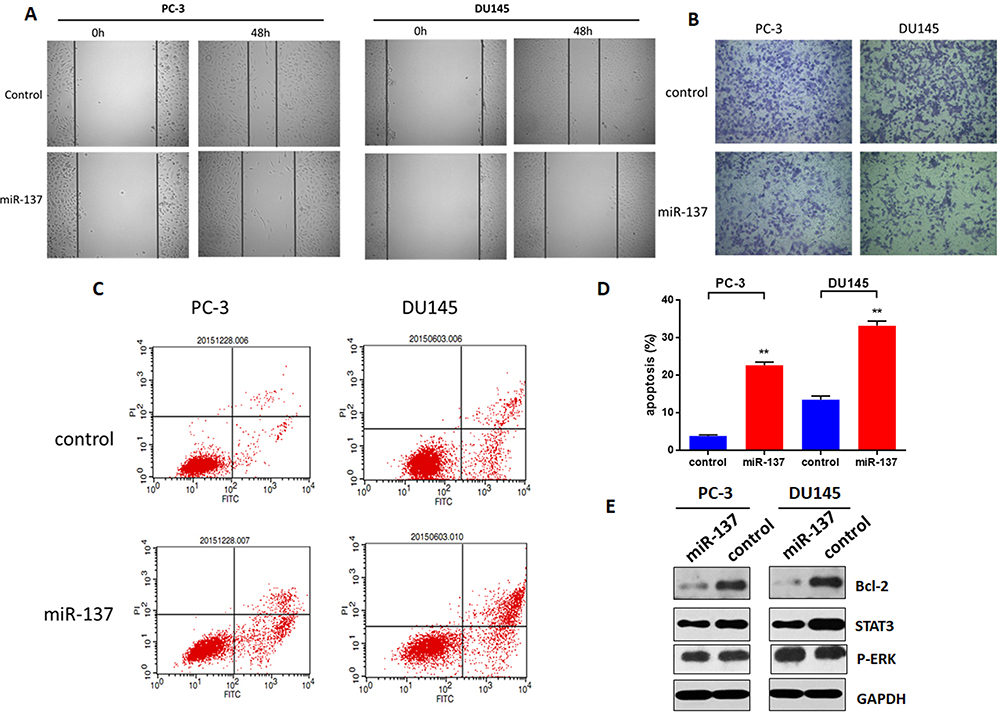 |
Figure 3 miR-137-3p inhibits prostate cancer metastasis and modulates cell apoptosis. After transfection of miR-137-3p mimics or negative control into two prostate cancer cell lines (48 h), (A) cell migration was measured by wound healing assay. (B) Cell invasion was measured by Transwell assay, and the number of cells in the lower chamber was counted. (C) Cell apoptosis was measured after staining with PI & Annexin V. (D) The percentage of apoptotic cells was counted. (E) Cells were harvested, and Bcl-2 was detected by Western blot. **p<0.01. Experiments were performed at least in triplicate. |
In general, many physiological processes, including cell growth and metastasis, require a balance between cell growth and apoptosis. To determine whether the antitumor function of miR-137-3p is based on apoptosis, a PI and Annexin V staining kit (Thermo Fisher) was applied, and the cells were measured by FACS. As shown in Figure 3C and D, the percentage of cell apoptosis in the miR-137 mimic group was markedly increased in both cell lines. Furthermore, miR-137-3p downregulated the protein expression of bcl-2, a key factor in cell apoptosis (Figure 3E). This specific evidence supports the hypothesis that miR-137-3p regulates tumor metastasis by promoting cell apoptosis.
JNK3 is a Downstream Target of miR-137-3p
To explore the potential mechanism of miR-137-3p, miRDB, TargetScan and miRbase were used to identify the targets of miR-137-3p (Table S1). Among the predicted genes, JNK3 (MAPK10), which may regulate tumor growth, metastasis and apoptosis, was further confirmed by the dual-luciferase reporter assay. Reporter vectors that had either a wild-type or mutant 3ʹUTR of JNK3 were constructed (Figure 4A). Luciferase activity was reduced in the miR-137-3p plus wild-type JNK3 3ʹUTR group in both PC-3 and DU-145 cells. However, the effect was abolished in the mutant group (Figure 4B and C). To further confirm the posttranscriptional regulation of JNK3 by miR-137-3p, JNK3 protein levels were measured after transfection. The results indicated that miR-137-3p significantly reduced JNK3 protein levels (Figure 4D). Finally, we measured the protein level of JNK3 in xenograft samples transfected with mimics or miR-137-3p. As shown in Figure 4E JNK3 was also reduced in the miR-137-3p treatment group compared with the mimic control group.
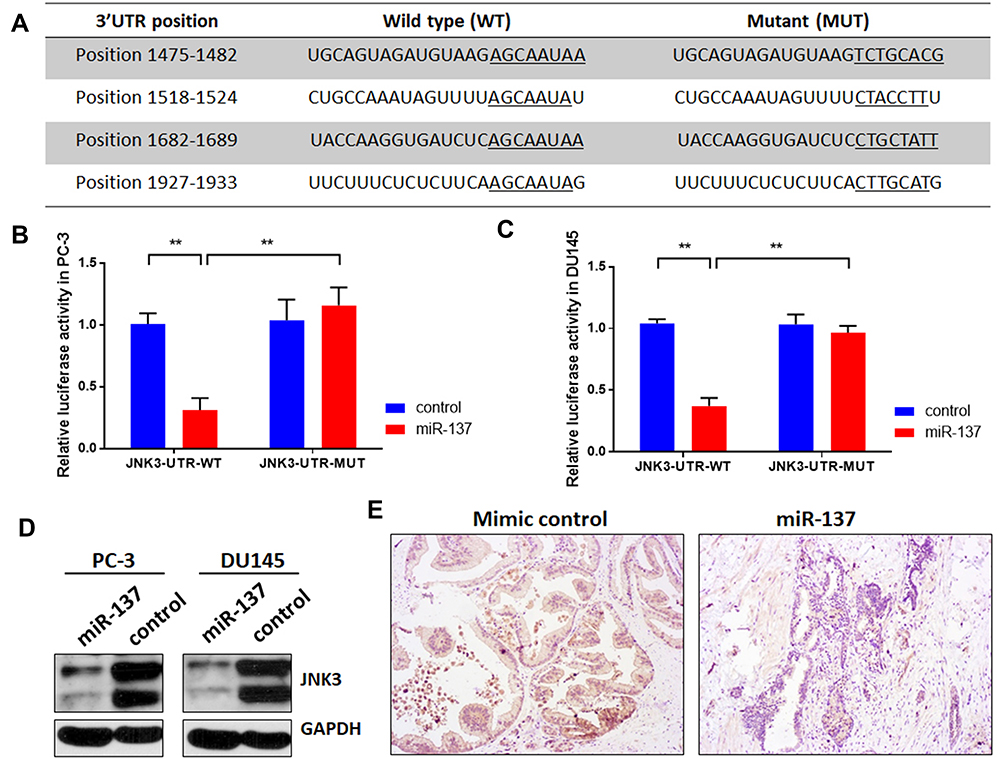 |
Figure 4 miR-137-3p directly targets JNK3. (A) miR-137-3p-binding sites located on the 3ʹUTR of JNK3 mRNA and the wild-type or mutant site of JNK3 3ʹUTR were cloned into the pmirGLO plasmid. (B) and (C) miR-137-3p mimics or scramble control and wild-type or mutant JNK3 3ʹUTR were cotransfected into PC-3 or DU145 cells. miR-137-3p reduced luciferase activities in prostate cancer cells transfected with the wild-type 3ʹUTR but did not alter luciferase activities in cells transfected with the mutant JNK3 3ʹUTR. (D) Western blotting was performed after transfecting cells with miR-137-3p mimics or control, and (E) JNK3 protein levels were detected in xenograft samples. **p<0.01 compared to the corresponding controls. |
JNK3 is Overexpressed in Prostate Cancer Cells and Tissues
To further explore the expression of JNK3 in prostate cancer and the relationship between JNK3 and miR-137-3p, we measured the expression of JNK3 in prostate cancer cell lines and 20 pairs of patient samples. The mRNA and protein levels of JNK3 in prostate cancer cells were higher than those in RWPE-1 cells (Figure 5A and B), which was negatively correlated with miR-137-3p. Similarly, we also measured JNK3 mRNA and protein levels in clinical samples by qPCR and IHC separately. As predicted, JNK3 mRNA and protein were both up-regulated in tumor tissues compared to normal controls (Figure 5C and D). Interestingly, JNK3 was found to be expressed in prostate cancer tissues but not in normal tissues, indicating that JNK3 can serve as a tissue-specific biomarker. In addition, JNK3 mRNA was negatively correlated with miR-137-3p, confirming that miR-137-3p negatively regulates JNK3 (Figure 5E).
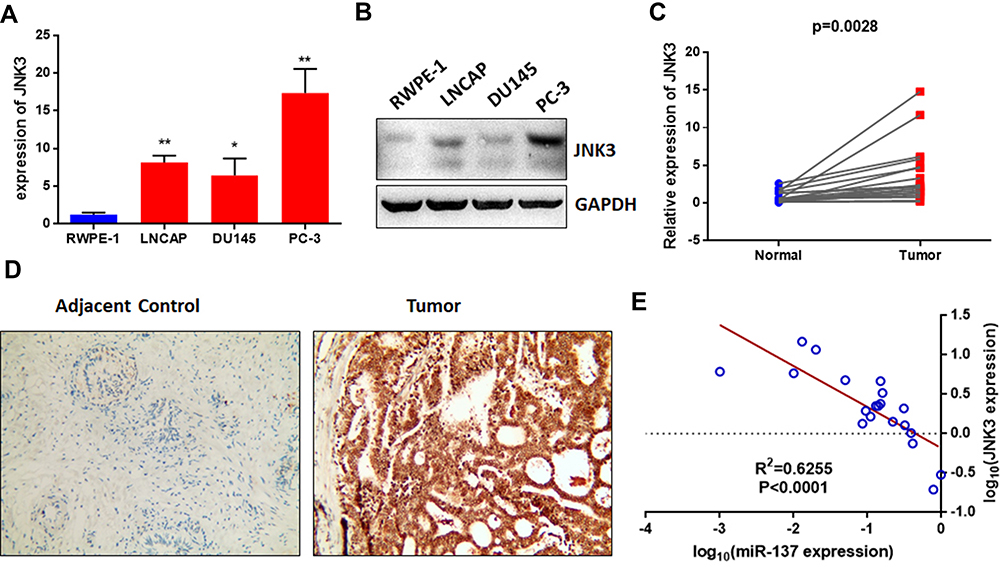 |
Figure 5 The JNK3 level is highly expressed and negatively correlated with miR-137-3p in both prostate cancer cell lines and tissues. The mRNA and protein levels of JNK3 in prostate cancer cell lines were detected by qRT-PCR (A) and Western blotting (B). qRT-PCR was used to analyze the expression of JNK3 in 20 paired prostate cancer tumors and adjacent controls (C). (D) JNK3 protein levels were measured by IHC. (E) The expression of JNK3 was negatively correlated with miR-137-3p in prostate cancer samples. *p<0.05, **p<0.01. |
JNK3 Modulates Tumorigenesis of Prostate Cancer
Next, we measured the effects of JNK3 knockdown by siRNAs on biological processes in both PC-3 and DU-145 cells. JNK3 expression was detected via qRT-PCR and Western blot (Figure 6A and B). As we can see, depletion of JNK3 reduced JNK3 levels along with suppressed cell growth (Figure 6C and D) and metastasis (Figure 6E and F) in the two prostate cancer cell lines. Moreover, we further evaluated the function of JNK3 in apoptosis. As shown in Figure 6G, the percentage of apoptotic JNK3 knockdown cells was dramatically inhibited compared to that in the negative control. These results indicate that JNK3 is an oncogene and a potential treatment target in human prostate cancer.
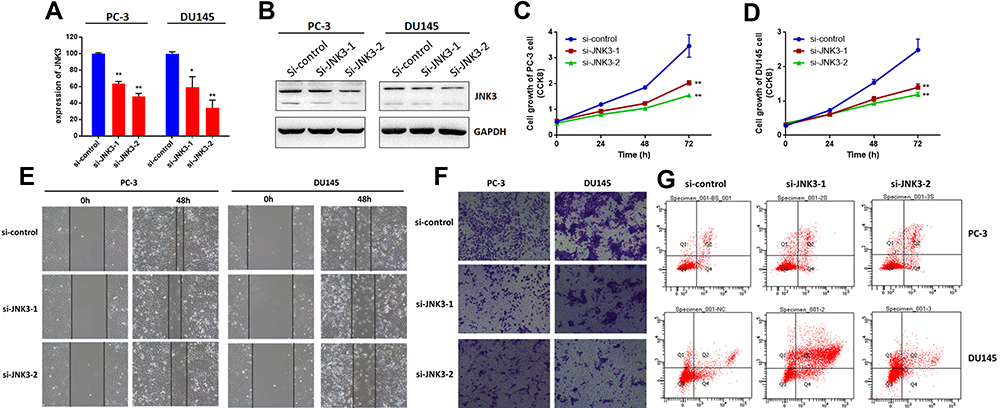 |
Figure 6 JNK3 regulates the tumorigenesis of prostate cancer cells. JNK3 was knocked down by two separate siRNAs in two prostate cancer cell lines. After 48 h, the expression of JNK3 was detected by qRT-PCR (A) and Western blot (B). Cell growth was measured by CCK8 assay in PC-3 (C) and DU145 cells (D). Cell migration was detected by wound healing assay (E). Cell invasion was detected by Transwell assay (F). Cell apoptosis was measured by FACS (G). *p<0.05, **p<0.01 compared to the corresponding controls. |
JNK3 Regulates EZH2 and is a Potential Prognostic Factor in Prostate Cancer
As previously reported, miR-137-3p was found to regulate cell apoptosis and suppress EZH2 expression.12,19 EZH2 is overexpressed in metastatic prostate cancer and acts as an epigenetic-driven gene.20 Additionally, a relationship between MAPK and EZH2 was further validated.21 Therefore, we aimed to test the hypothesis that JNK3 regulates EZH2. To this end, we detected the expression of EZH2 after depletion of JNK3 in prostate cancer (Figure 7A). The results indicated that EZH2 expression but not phosphorylation of EZH2 (Thr 311) was down-regulated after knockdown of JNK3. Furthermore, we overexpressed JNK3 in LNCAP cells, and as shown in Figure 7B, overexpression of JNK3 promoted EZH2 expression. Moreover, EZH2 was positively correlated with JNK3 in prostate cancer samples (Figure 7C). Consistent with the expression of JNK3, the EZH2 protein level was also higher in cancer tissues than in adjacent controls (Figure 7D). We further analyzed TCGA data for EZH2 expression in 550 prostate cancer patients (https://tcga.xenahubs.net/download/TCGA.PRAD.sampleMap/HiSeqV2.gz). These results indicate that JNK3 modulates tumorigenesis by regulating EZH2 in prostate cancer (Figure 7E).
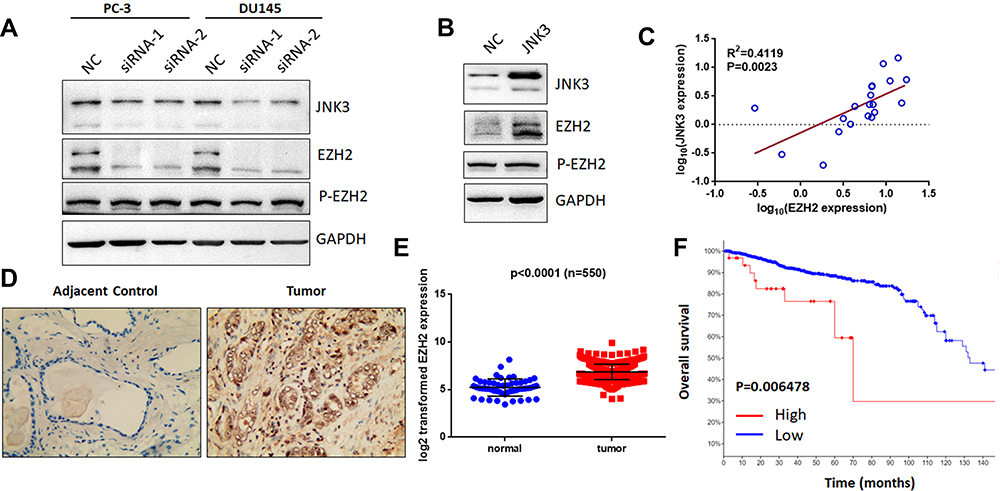 |
Figure 7 JNK3 modulates EZH2 expression and can serve as a prognostic biomarker. (A) After knockdown of JNK3 for 48 h in two prostate cancer cell lines, EZH2 and p-EZH2 (Thr 311) were detected by Western blot. (B) JNK3 was overexpressed in LNCAP cells, which in turn promoted EZH2 expression. (C) EZH2 positively correlated with JNK3 in prostate cancer clinical samples (n=20). (D) EZH2 protein expression was higher in prostate cancer tumor samples than in adjacent controls (Gleason score 7). (E) TCGA data for EZH2 in prostate cancer samples were analyzed (n =550). (F) Patients with high JNK3 and EZH2 expression had worse outcomes (high expression: 30, low expression: 1445). Each group was compared to the corresponding controls (one-way ANOVA). |
We subsequently evaluated the role of the JNK3/EZH2 pathway in prostate cancer using 18 validation cohorts consisting of 4850 samples (http://www.cbioportal.org/). We found that the frequency of alterations in JNK3 and EZH2 was significantly increased in most of the categories (10/18 and 12/18, respectively). Among these alterations, JNK3 and EZH2 were overexpressed in prostate cancers (Figure S2A and S2B). Furthermore, we analyzed the survival of patients with JNK3/EZH2 alterations. The survival rate of the JNK3/EZH2 alteration (high expression) group was significantly worse than the rate observed in the non-alteration (low expression) group (Figure 7F), suggesting that JNK3/EZH2 amplification predicts a worse outcome of prostate cancer patients.
Discussion
Prostate cancer is a common cancer in men, and its incidence is on the rise in many countries. There are several risk factors for this disease, including genetic factors, external exposure, urinary infections, smoking, diet, weight, endogenous hormones and physical activity.1 The tumorigenesis of prostate cancer is a complex process beginning with many genetic changes through its progression to malignancy. Accumulated evidence has indicated that miRNAs play indispensable roles in prostate cancer progression.22,23 Furthermore, miRNAs can be used as clinical biomarkers and treatment targets.24–26 The identification of miRNAs is relevant to these cancer processes and will greatly facilitate the understanding of cancer biology. To this end, many studies have been carried out with the aim of fully understanding the role of miRNAs in cancer cells. As previously reported, miR-137 was confirmed to target KDM4A and functions as a key mediator inhibiting cancer growth, migration and invasion.27 In addition, miR-137 plays a key role in several cancer types, including hepatocyte carcinoma, triple-negative breast cancer and ovarian cancer.11,12,14,15 miR-137 was also demonstrated to be overexpressed in recurrent prostate cancer and regulates NOX4.8,28 However, miR-137 has two subtypes: miR-137-5p and miR-137-3p. The exact subtype and role of miR-137 in the tumor progression of prostate cancer is largely unknown. In this study, we first investigated whether miR-137-5p or miR-137-3p was altered in prostate cancer cells and tissues. By using qRT-PCR, we found that miR-137-3p was decreased in prostate cancer cells (Figure 1A), while miR-137-5p was not (data not shown). Furthermore, miR-137-3p affected the tumorigenesis of prostate cancer. Cell proliferation, colony formation, migration and invasion were all assayed after transfection with miR-137 mimics. Among these phonotypes, we demonstrated that miR-137-3p suppressed tumor cell growth both in vitro and in vivo (Figure 2). An interesting result is that miR-137-3p inhibits tumor growth in androgen receptor (AR)-negative xenografts (DU-145 cells) but not in AR-positive xenografts (LNCAP cells, data not shown). This difference may be caused by the complicated network involving miR-137 and AR.29 Furthermore, inhibited cell growth may not be a result of reduced migration according to the tumor dormancy hypothesis.30 In addition, the expression of miR-137-3p was associated with tumor stage and PSA level (data not shown), which is a good supplementary marker in the clinic.
Next, we confirmed that miR-137-3p regulates JNK3 by binding to its 3ʹUTR via a luciferase reporter assay. qRT-PCR and Western blotting also validated the direct interaction between JNK3 and miR-137 in prostate cancer cell lines. These results connect miR-137-3p to the regulatory pathway of JNK3 (Figure 3). In particular, miR-137 could regulate NOX4, P160 and TRIM24, which also play important roles in prostate cancer. These data indicate that miR-137-3p regulates several genes and suppresses tumor growth in prostate cancer.
It is known that JNK3 (MAPK10) is a member of the c-Jun N-terminal kinases, which are part of the mitogen-activated protein kinase family. JNK, p38 and ERK are signal transducers involved in a broad range of cell functions, including survival, apoptosis and cell proliferation in prostate cancer.31,32 The earliest discoveries identified three mammalian JNK genes, namely, JNK1, JNK2, and JNK3. The products of JNK1 and JNK2 are expressed in almost all cell types and tissues, whereas JNK3 is localized primarily in the brain, heart and testis. Interestingly, in our study, JNK3 was also found to be expressed in prostate cancer cells but not in normal prostate cells, indicating that it can serve as a tissue-specific biomarker. Knockdown of JNK3 in two prostate cell lines also suppressed tumor growth, migration and invasion, suggesting an oncogenic role for JNK3. Many previous reports have revealed that JNK phosphorylates the EZH2 protein and contributes to cell death and apoptosis.33,34 However, in our study, there was no significant change in EZH2 phosphorylation after JNK3 knockdown. Rather, we found that depletion of JNK3 decreased EZH2 in prostate cancer cells (Figure 7).
In addition, bioinformatics results revealed that both JNK3 and EZH2 were overexpressed in 18 clinical patient cohorts (n=4850). Among these cohorts, JNK3/EZH2 amplification predicted a worse outcome of prostate cancer patients, suggesting the prognostic role of these genes in prostate cancer. Moreover, JNK3 expression was found to be negatively correlated with miR-137-3p levels in our clinical tissues, which further confirmed the linkage between miR-137-3p and JNK3.
Conclusions
Overall, the identification of interactions between miR-137-3p and JNK3/EZH2 in this study enhances our understanding of the relationship between miRNAs in prostate cancer. miR-137-3p suppresses the tumorigenesis of prostate cancer by regulating the JNK3/EZH2 pathway. JNK3 promotes prostate cancer cell growth, migration and invasion. Furthermore, these findings indicate that the miR-137-3p/JNK3 signaling cascade plays important roles in the regulation of prostate cancer. These discoveries might also facilitate the development of biomarkers for the early detection and treatment of prostate cancer.
Abbreviations
PCa, prostate cancer; JNK, c-Jun N-terminal kinases; MAPK10, mitogen-activated protein kinases 10; EZH2, enhancer of zeste homolog 2; UTR, untranslated region; PSA, prostate-specific antigen; FISH, fluorescence in situ hybridization; IHC, immunohistochemistry; LNA, locked nucleic acid.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Ethics Approval and Consent
Study approval was obtained from the independent ethics committee at the Second Affiliated Hospital of Soochow University (Suzhou, China). The privacy of the patients involved was protected. Patients provided written informed consent. Study participants provided consent for the publication of the data and any associated images.
Acknowledgments
We also thank Linquan Bai from Shanghai Jiao Tong University and Changgui Dong, Michael Chu for advice and comments.
Author Contributions
All the authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests for this work.
References
1. Cuzick J, Thorat MA, Andriole G, et al. Prevention and early detection of prostate cancer. Lancet Oncol. 2014;15(11):e484–e492. doi:10.1016/S1470-2045(14)70211-6
2. Bonci D, Coppola V, Patrizii M, et al. A microRNA code for prostate cancer metastasis. Oncogene. 2016;35(9):1180–1192. doi:10.1038/onc.2015.176
3. Kim WT, Kim WJ. MicroRNAs in prostate cancer. Prostate Int. 2013;1(1):3–9. doi:10.12954/PI.12011
4. Xue X, Fei X, Hou W, Zhang Y, Liu L, Hu R. miR-342-3p suppresses cell proliferation and migration by targeting AGR2 in non-small cell lung cancer. Cancer Lett. 2018;412:170–178. doi:10.1016/j.canlet.2017.10.024
5. Kumar B, Lupold SE. MicroRNA expression and function in prostate cancer: a review of current knowledge and opportunities for discovery. Asian J Androl. 2016;18(4):559–567. doi:10.4103/1008-682X.177839
6. Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res. 2007;67(13):6130–6135. doi:10.1158/0008-5472.CAN-07-0533
7. Ambs S, Prueitt RL, Yi M, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68(15):6162–6170. doi:10.1158/0008-5472.CAN-08-0144
8. Pashaei E, Pashaei E, Ahmady M, Ozen M, Aydin N. Meta-analysis of miRNA expression profiles for prostate cancer recurrence following radical prostatectomy. PLoS One. 2017;12(6):e0179543. doi:10.1371/journal.pone.0179543
9. Cheng Y, Li Y, Liu D, Zhang R, Zhang J. miR-137 effects on gastric carcinogenesis are mediated by targeting Cox-2-activated PI3K/AKT signaling pathway. FEBS Lett. 2014;588(17):3274–3281. doi:10.1016/j.febslet.2014.07.012
10. Liang L, Li X, Zhang X, et al. MicroRNA-137, an HMGA1 target, suppresses colorectal cancer cell invasion and metastasis in mice by directly targeting FMNL2. Gastroenterology. 2013;144(3):624–635 e624. doi:10.1053/j.gastro.2012.11.033
11. Chen F, Luo N, Hu Y, Li X, Zhang K. MiR-137 suppresses triple-negative breast cancer stemness and tumorigenesis by perturbing BCL11A-DNMT1 interaction. Cell Physiol Biochem. 2018;47(5):2147–2158. doi:10.1159/000491526
12. Huang B, Huang M, Li Q. MiR-137 suppresses migration and invasion by targeting EZH2-STAT3 signaling in human hepatocellular carcinoma. Pathol Res Pract. 2018;214(12):1980–1986. doi:10.1016/j.prp.2018.08.005
13. Zhu X, Li Y, Shen H, et al. miR-137 inhibits the proliferation of lung cancer cells by targeting Cdc42 and Cdk6. FEBS Lett. 2013;587(1):73–81. doi:10.1016/j.febslet.2012.11.004
14. Sun J, Cai X, Yung MM, et al. miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance in ovarian cancer. Oncogene. 2018.
15. Eedunuri VK, Rajapakshe K, Fiskus W, et al. miR-137 targets p160 steroid receptor coactivators SRC1, SRC2, and SRC3 and inhibits cell proliferation. Mol Endocrinol. 2015;29(8):1170–1183. doi:10.1210/me.2015-1080
16. Hu R, Huffman KE, Chu M, Zhang Y, Minna JD, Yu Y. Quantitative secretomic analysis identifies extracellular protein factors that modulate the metastatic phenotype of non-small cell lung cancer. J Proteome Res. 2016;15(2):477–486. doi:10.1021/acs.jproteome.5b00819
17. Xue X, Wang X, Zhao Y, Hu R, Qin L. Exosomal miR-93 promotes proliferation and invasion in hepatocellular carcinoma by directly inhibiting TIMP2/TP53INP1/CDKN1A. Biochem Biophys Res Commun. 2018;502(4):515–521. doi:10.1016/j.bbrc.2018.05.208
18. Shi Z, Johnson JJ, Stack MS. Fluorescence in situ hybridization for microRNA detection in archived oral cancer tissues. J Oncol. 2012;2012:903581. doi:10.1155/2012/903581
19. Sun J, Cai X, Yung MM, et al. miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance in ovarian cancer. Oncogene. 2019;38(4):564–580. doi:10.1038/s41388-018-0459-x
20. Yang YA, Yu J. EZH2, an epigenetic driver of prostate cancer. Protein Cell. 2013;4(5):331–341. doi:10.1007/s13238-013-2093-2
21. Nishioka C, Ikezoe T, Yang J, Yokoyama A. Tetraspanin family member, CD82, regulates expression of EZH2 via Inactivation of p38 MAPK signaling in leukemia cells. PLoS One. 2015;10(5):e0125017. doi:10.1371/journal.pone.0125017
22. Liu X, Chen Q, Yan J, et al. MiRNA-296-3p-ICAM-1 axis promotes metastasis of prostate cancer by possible enhancing survival of natural killer cell-resistant circulating tumour cells. Cell Death Dis. 2013;4:e928. doi:10.1038/cddis.2013.458
23. Majid S, Dar AA, Saini S, et al. miRNA-34b inhibits prostate cancer through demethylation, active chromatin modifications, and AKT pathways. Clin Cancer Res. 2013;19(1):73–84. doi:10.1158/1078-0432.CCR-12-2952
24. Bucay N, Shahryari V, Majid S, et al. miRNA expression analyses in prostate cancer clinical tissues. J Vis Exp. 2015;103. doi:10.3791/53123
25. Wagner S, Ngezahayo A, Murua Escobar H, Nolte I. Role of miRNA let-7 and its major targets in prostate cancer. Biomed Res Int. 2014;2014:376326. doi:10.1155/2014/376326
26. Osipov ID, Zaporozhchenko IA, Bondar AA, et al. Cell-free miRNA-141 and miRNA-205 as prostate cancer biomarkers. Adv Exp Med Biol. 2016;924:9–12.
27. Neault M, Mallette FA, Richard S. miR-137 modulates a tumor suppressor network-inducing senescence in pancreatic cancer cells. Cell Rep. 2016;14(8):1966–1978. doi:10.1016/j.celrep.2016.01.068
28. Wu QQ, Zheng B, Weng GB, et al. Downregulated NOX4 underlies a novel inhibitory role of microRNA-137 in prostate cancer. J Cell Biochem. 2019;120(6):10215–10227. doi:10.1002/jcb.28306
29. Nilsson EM, Laursen KB, Whitchurch J, et al. MiR137 is an androgen regulated repressor of an extended network of transcriptional coregulators. Oncotarget. 2015;6(34):35710–35725. doi:10.18632/oncotarget.5958
30. van der Toom EE, Verdone JE, Pienta KJ. Disseminated tumor cells and dormancy in prostate cancer metastasis. Curr Opin Biotechnol. 2016;40:9–15. doi:10.1016/j.copbio.2016.02.002
31. Rodriguez-Berriguete G, Fraile B, Martinez-Onsurbe P, Olmedilla G, Paniagua R, Royuela M. MAP Kinases and Prostate Cancer. J Signal Transduct. 2012;2012:169170. doi:10.1155/2012/169170
32. Ma B, Wells A. The mitogen-activated protein (MAP) kinases p38 and extracellular signal-regulated kinase (ERK) are involved in hepatocyte-mediated phenotypic switching in prostate cancer cells. J Biol Chem. 2014;289(16):11153–11161. doi:10.1074/jbc.M113.540237
33. Mazars A, Tournigand C, Mollat P, et al. Differential roles of JNK and Smad2 signaling pathways in the inhibition of c-Myc-induced cell death by TGF-beta. Oncogene. 2000;19(10):1277–1287. doi:10.1038/sj.onc.1203420
34. Yu C, Minemoto Y, Zhang J, et al. JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol Cell. 2004;13(3):329–340. doi:10.1016/S1097-2765(04)00028-0
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.