Back to Journals » OncoTargets and Therapy » Volume 9
MiR-126 regulates proliferation and invasion in the bladder cancer BLS cell line by targeting the PIK3R2-mediated PI3K/Akt signaling pathway
Authors Xiao J, Lin H, Zhu Y, Zhu Y, Chen L ![]()
Received 27 January 2016
Accepted for publication 5 May 2016
Published 22 August 2016 Volume 2016:9 Pages 5181—5193
DOI https://doi.org/10.2147/OTT.S105198
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr William C. Cho
This paper has been retracted.
Jun Xiao, 1 Huan-Yi Lin, 2 Yuan-Yuan Zhu, 3 Yu-Ping Zhu, 1 Ling-Wu Chen 2
1Department of Urology, Anhui Provincial Hospital, Anhui Medical University, Hefei, 2Department of Urology, First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, 3Clinical Laboratory, Anhui Provincial Hospital, Anhui Medical University, Hefei, People’s Republic of China
Objective: To assess whether microRNA-126 (miR-126) targets phosphatidylinositol 3-kinase regulatory subunit beta (PIK3R2) and to determine the potential roles of miR-126 in regulating proliferation and invasion via the PIK3R2-mediated phosphatidylinositol 3 kinase (PI3K)-protein kinase B (Akt) signaling pathway in the human bladder BLS cell line.
Materials and methods: A recombinant lentivirus (Lv) vector expressing miR-216 (Lv-miR-126) was successfully constructed, and Lv-miR-126 and Lv vector were transfected into the BLS cell line. A direct regulatory relationship between miR-126 and the PIK3R 2 gene was demonstrated by luciferase reporter assays. To determine whether PIK3R2 directly participates in the miR-126-induced effects in BLS cells, anti-miR-126 and a PIK3R2 small interfering RNA (siRNA) were transfected into the BLS cells. Quantitative real-time polymerase chain reaction was used to measure miR-126 and PIK3R2 expressions. 5-Ethynyl-2'-deoxyuridine and colony formation assays to assess cell proliferation, flow cytometry for cell apoptosis and cell cycle analysis, Transwell assays for cell migration and invasion, and Western blots for PIK3R2, PI3K, phosphorylated PI3K (p-PI3K), Akt, and phosphorylated Akt (p-Akt) protein expressions were performed.
Results: Lv-miR-126 significantly enhanced the relative expression of miR-126 in the BLS cells after infection (P< 0.0001). MiR-126 overexpression inhibited the proliferation, cloning, migration, and invasion of BLS cells, promoted cell apoptosis, and induced S phase arrest (all P< 0.05). PIK3R2, p-PI3K, and p-Akt protein expressions were significantly decreased in the BLS cells infected with Lv-miR-126. Luciferase assays showed that miR-126 significantly inhibited the PIK3R2 3' untranslated region (3'UTR) luciferase reporter activity (P< 0.05). The anti-miR-126 + PIK3R2 siRNA group had significantly decreased PIK3R2, p-PI3K, and p-Akt expressions compared with those of anti-miR-126 alone, as well as significantly decreased proliferation, invasion, and metastasis and increased apoptosis compared with the anti-miR-126 group (all P< 0.05). Additionally, proliferation, invasion, and metastasis were significantly increased, and cell apoptosis was decreased compared with the PIK3R2 siRNA group (all P< 0.05).
Conclusion: Overexpression of miR-126 negatively regulated the target gene PIK3R2 and further inhibited the PI3K/Akt signaling pathway, thereby inhibiting proliferation, migration, and invasion and promoting apoptosis in BLS cells.
Keywords: human bladder BLS cell line, microRNA-126, PIK3R2, PI3K/Akt signaling pathway, proliferation, migration, apoptosis
Introduction
Bladder cancer, the most common malignancy of the urinary system, accounted for 74,690 new cases and 15,580 deaths in the United States in 2014.1 The incidence of bladder cancer is higher in developed countries, and the burden of urinary bladder cancer will also strongly increase in developing countries due to the aging population, the progression of the tobacco epidemic, and increasing exposure to occupational chemicals.2,3 Although 5-year survival among all cases is high at 81%, survival among patients with regional and distant spread of the disease drops substantially to 36% and 6%, respectively.4 Thus, effective intervention is imperative for cancer prevention and control, and significant efforts have been undertaken to investigate the molecular mechanisms of carcinogenesis and develop potential therapeutic targets for bladder cancer.5,6 Currently, the elucidation of signaling pathways has led to new advances in the understanding of the pathogenic mechanisms of bladder cancer, including the phosphatidylinositol 3 kinase (PI3K)-protein kinase B (Akt) pathway.7–9
As demonstrated by a growing body of evidence, microRNAs (miRs) are aberrantly expressed in bladder cancer and may be functionally implicated in the molecular pathogenesis of bladder cancer by regulating related molecular pathways.10,11 miRs were frequently altered in most urologic cancers, playing significant roles as oncogenes or tumor suppressors, and miRs also target common pathways involved in the regulation of cell growth, proliferation, invasion, and apoptosis in bladder carcinogenesis.10,12,13 It has been suggested that miR-125b targets E2F transcription factor 3 (E2F3) and that miR-125b may be involved in the regulation of the G1/S transition via the E2F3-cyclin A2 signaling pathway in bladder cancer.14 Down-regulated miR-143 and miR-145 have been found in human bladder cancer, and miR-143 and miR-145 were shown to play a role in cell growth via the regulation of the PI3K/Akt and mitogen-activated protein kinase signaling pathways.11 As shown in a previous study, most miRs were down-regulated in bladder cancer, and the target genes of the miRs were identified to further assess their biological functions in bladder cancer.15 MiR-126 is located in the seventh intron of the EGFL7 gene, which resides on human chromosome 9.16 More specifically, down-regulated miR-126 was observed and may act as a tumor suppressor in bladder cancer. The invasive potential of bladder cancer cells can be attenuated with increased miR-126 levels by mechanistically targeting disintegrin and metalloproteinase domain-containing protein 9.17,18 By synergistically targeting oncogenes (PI3KR2 and adaptor protein Crk) and tumor suppressors (polo-like kinase 2), miR-126 was shown to have an inhibitory effect on the growth of gastric cancer cell lines.19 Mechanistically, silencing miR-126 may alter the activation of the PI3K/Akt pathway, suggesting an important regulatory role for miR-126 in PI3K/Akt pathway transduction, which is considered to play a major role in bladder carcinogenesis.20,21 Therefore, we aimed to evaluate the potential role of miR-126 in targeting the phosphatidylinositol 3-kinase regulatory subunit beta (PIK3R2)-mediated PI3K/Akt signaling pathway and to elucidate the mechanisms of miR-126 in bladder cancer progression.
Materials and methods
Cell culture
Stable passaging of the human bladder transitional cell carcinoma cell line BLS was established by our research laboratory. The cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 0.1 mg/mL streptomycin and were incubated in a 5% CO2 incubator at 37°C. Cells were maintained with digestion and passage. This study was approved by the Ethics Committee of Anhui Provincial Hospital, Anhui Medical University, all study participants provided written informed consent before the experiments.
Lentivirus construction, package, and titer determination
DNA of normal mucosa tissues was used as a template, and polymerase chain reaction (PCR)-amplified miR-126 and the target gene fragments were recovered. The double-stranded DNA fragments were ligated to the linearized lentivirus (Lv) vector digested by EcoRI and BamHI (Thermo Fisher Scientific, Waltham, MA, USA) with T4 DNA ligase at 16°C overnight. The ligated products were transformed into DH5α competent cells (System Biosciences, Mountain View, CA, USA), and positive clones were selected for sequencing verification. The upstream and downstream primers for miR-126 were as follows: 5′-TGTCTAGATGTGGCTGTTAGGCATGG (EcoRI)-3′ and 5′-ATAGGTACCAAGACTCAGGCCCAGGC (BamHI)-3′, respectively. The upstream and downstream primers contained EcoRI and BamHI sites, respectively, as well as additional base pairs. The primers were synthesized by Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, People’s Republic of China).
The 293T human embryonic kidney cells in logarithmic phase growth were collected and then seeded in a 10 cm culture dish (2×106 cells per culture dish) using dulbecco’s modified eagle medium (DMDM) culture medium containing 10% FBS and cultivated in an incubator (37°C, 5% CO2) for 24 hours. The expression vector and Lv packaging mixture were cotransfected into 293T cells with Lipofectamine™ 2000 (Thermo Fisher Scientific). The primary culture medium was replaced with a DMDM culture medium containing 1% FBS 24 hours after transfection, and the supernatant was collected 48 hours after transfection. The collected supernatant was centrifuged at 5,000 rpm/min for 10 minutes to remove cell sedimentation and then filter-sterilized with a 0.45 μm filter membrane for packaging preservation. The expression of green fluorescent protein (GFP) was observed under a fluorescence microscope 72 hours after transfection, the number of GFP-expressing cells was counted in 100 cells, and virus packaging efficiency was monitored. Virus packaging efficiency (%) = the number of GFP-expressing cells/cell count.
The 293T cells (100 μL) were inoculated in 96-well culture plates (4×104 cells per well) and cultured at 37°C with 5% CO2 and 100% humidity, and viral titer was determined after 36 hours of culturing. Eight sterile eppendorf (EP) tubes were numbered from 1 to 8, and Opti-MEM (90 μL) was added to each EP tube. Viral particles (10 μL) were added to the first tube and evenly mixed; 10 μL of the mixture containing viral particles was then added to the second tube and successively diluted with a double-dilution method to the eighth tube. The dilution gradient ratio was 10−1–10−8, and each dilution gradient was assessed in three wells. Culture medium (90 μL) taken from the corresponding culture wells was added to the corresponding diluted viral particles, and the virus solution was placed in the incubator. After 24 hours, an additional incubation was performed after replacing the virus solution with complete culture medium (100 μL) for 72 hours. Then, GFP expression was observed under an inverted fluorescence microscope, and viral titers were calculated based on the number of GFP-positive cells with the maximum dilution. Viral titer = the number of GFP-positive cells × dilution.
Cell grouping and transfection
1) Lv infection: BLS cells in the logarithmic phase were digested by trypsin to prepare cell suspensions and seeded in eight-well cell culture plates (4×105 cells per well). BLS cells were divided into the BLS-Lv-miR126 group, the BLS-Lv-vector group, and the noninfected group. According to the FuGENE instructions, BLS cells were infected with the viral supernatant, and the infection efficiency was 70%–80%.
2) Cell transfection: BLS cells were seeded in a 50 mL culture flask and grown to 30%–50% confluence in complete culture medium. Then, 5×104 cells/well were inoculated in 24-well plates and cultured for 24 hours until the cell density reached to 70%–80% confluence. The miR-126 mimics, anti-miRNA-126, and PIK3R2 small interfering RNA (siRNA) were transfected into BLS cells using Lipofectamine™ 2000. All the reagents were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, People’s Republic of China). Specific steps were performed according to the manufacturer’s instructions. The BLS cells transfected with miRNA-126 mimics were used for luciferase activity assays, and BLS cells transfected with anti-miRNA-126 and PIK3R2 siRNA were used to determine whether PIK3R2 was directly involved in the effects of miR-126 in BLS cells. Then, the BLS cells were divided into four groups: the anti-miRNA-126 group (transfected with anti-miRNA-126), the PIK3R2 siRNA group (transfected with PIK3R2 siRNA), the anti-miRNA-126 + PIK3R2 siRNA group (transfected with anti-miRNA-126 and PIK3R2 siRNA), and the negative control (NC) group.
Construction of the luciferase reporter vector and determination of luciferase activity
Based on the 3′ untranslated region (3′UTR) of the PIK3R2 gene, the sequence was designed and synthesized. The restriction enzyme XhoI and NotI recognition sites were introduced into the upstream and downstream primers. The primer sequences for the 3′UTR of PIK3R2 were as follows: upstream 5′-GCACCACGAGGAACGCACTT-3′, downstream 5′-CGTCCACTACCACGGAGCAG-3′. The binding site of the wild-type PIK3R2 3′UTR for miR-126 was ACGGTACG, and the binding site of the mutant PIK3R2 3′UTR for miR-126 was TGGCTTCC. DNA from healthy human peripheral blood was used as a template for PCR with a total reaction volume of 25 μL. The PCR amplification conditions were as follows: pre-denaturation (5 minutes, 94°C), followed by a total of 35 amplification cycles of 94°C for 1 minute, 60°C for 30 seconds, and 72°C for 1 minute, and an extension step (72°C, 7 minutes). Then, the PCR products were detected by 1% agarose gel electrophoresis, purified, and recovered. The recovered PCR products and the pGL4 vector were digested by restriction enzymes XhoI and NotI, and the enzyme-digested products were recovered. Finally, the linearized dual luciferase reporter vector (pGL4-Ctrl) was ligated with the 3′UTR of the PIK3R2 gene fragment using T4 DNA ligase with the following protocol: PCR products were mixed with the luciferase reporter vector at a ratio of 3:1, and the ligated reaction products (4 μL) were transformed into competent DH5a cells, followed by the selection of single colonies and growth and extraction to obtain isolated plasmid. Using a XhoI and NotI double digestion, the correct wild-type (Wt-miR-126/PIK3R2) and mutant (Mut-miR-126/PIK3R2) recombinant plasmids were obtained for sequencing. BLS cells were seeded in 12-well plates (1×105 cells per well) and cotransfected into the corresponding groups with recombinant plasmids and miR-126 mimics. After 48 hours, the primary cell culture solution was discarded, and cells were washed with phosphate-buffered saline (PBS) three times. BLS cells in each well were treated with cell lysate solution (100 μL) from the luciferase reporter gene assay kit for 30 minutes, and Luciferase Assay Reagent II (LAR II) (100 μL) was added to the cell lysates (20 μL) to measure luciferase activity (A). Stop & Glo reagent (Promega Corporation, Beijing, People’s Republic of China) (100 μL) was used to measure luciferase activity (B), and the firefly fluorescence (A) was used as a reference to calculate the luciferase activity value (C = B/A).
Quantitative real-time polymerase chain reaction
After 72 hours of virus infection, the medium was removed, and cells were washed using precooled Dulbecco’s Phosphate-Buffered Saline (4°C) twice. Then, 1 mL of TRIzol (Thermo Fisher Scientific) was added to each well to lyse the cells, and total cell RNA was extracted by the phenol chloroform method; 1 μg of RNA was subjected to reverse transcription for cDNA preparation, and specific reverse transcription primers were used. The primers were 5′-GTCGTATCCAGTGTCGTGGAGTCGGCAATTGCACTGGATACGACCGCGTA-3′ for miR-126, and 5′-AAAATATGGAACGCTTCACGAATTTG-3′ for U6. Homo sapiens U6 snRNA was used as a reference gene, and 1 μL of reverse transcription product was subjected to PCR. The primer sequences include the U6-forward primer, 5′-GTGCTCGCTTCGGCAGCACAT-3′; the U6-reverse primer, 5′-TACCTTGCGAAGTGCTTAAAC-3′; the miR126-forward primer, 5′-GCCGGCGCCCGAGCTCTGGCTC-3′; and the miR-126 reverse primer, 5′-CATTATTACTTTTGGTACGCG -3′. For the analysis of mRNA expression, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal control, and oligo-(dT) primer was used for reverse transcription. Then, quantitative real-time PCR (qRT-PCR) was carried out with the following primers: PIK3R2 forward primer: 5′-GCACCACGAGGAACGCACTT-3′; PIK3R2 reverse primer: 5′-CGTCCACTACCACGGAGCAG-3′; GAPDH forward primer: 5′-TGGGTGTGAACCATGAGAAGT-3′; GAPDH reverse primer: 5′-TGAGTCCTTCCACGATACCAA-3′. The PCR conditions were as follows: 95°C for 5 minutes, 60°C for 20 seconds (40 cycles), and 72°C for 20 seconds. PCR results were analyzed using Bio-Rad CFX96 software for the real-time fluorescence quantitative PCR instrument to obtain threshold cycle (Ct) values. Data were analyzed using the 2−ΔΔCt method.22 The 2−ΔΔCt demonstrates the ratios of the target gene relative expression in the case group to that of the control group (ΔΔCt = ΔCtcase group − ΔCtcontrol group ΔCt = Cttarget gene − Ctinternal reference gene). Ct is the number of amplification cycles when the real-time fluorescence intensity of the reaction reaches the threshold values. The amplification is performed during a period of logarithmic growth. The experiment was performed in triplicate.
5-Ethynyl-2′-deoxyuridine cell proliferation assay
After 72 hours of virus infection, BLS cells were seeded in 96-well plates. A Click-iT 5-ethynyl-2′-deoxyuridine (EdU) kit (Molecular Probes, Carlsbad, CA, USA) was used to measure cell proliferation according to the manufacturer’s procedures. Cells were labeled with EdU. Culture medium (100 μL) containing EdU (5 mol/L) was added to each well, followed by a 2-hour incubation. After fixation (4% paraformaldehyde [Shanghai biotechwell Co Ltd, Shanghai, People’s Republic of China], 30 minutes) and transparency (0.5% Triton X-l00 [Sigma-Aldrich Co., St Louis, MO, USA], 10 minutes) treatment, l00 μL × Hoechst 33342 reaction solution (Sigma-Aldrich Co) was added to each well, followed by 4′,6-diamidino-2-phenylindole nuclear staining. After rinsing three times, cells were observed under an inverted fluorescence microscope with three random fields of view. All images were obtained and processed with ImagePro software (Media Cybernetics, Rockville, MD, USA).
Plate colony formation assay
After a 72-hour infection, cells in logarithmic growth phase were inoculated in six-well culture plates (200 cells/well). Three parallel wells were arranged, and cells were subjected to stationary culture for 2 weeks; the culturing was complete when the white clone spots were visible to the naked eye. Methanol (2 mL) was added to fix the cells at room temperature for 15 minutes, and cells were stained with Giemsa staining solution for 15 minutes at room temperature, slowly rinsed with running water, and air-dried. Cell clones with cell numbers >50 were counted under an optical microscope to calculate the rate of colony formation. The colony formation rate = (number of clones/number of inoculated cells) ×100%. The experiment was repeated three times.
Cell cycle detected by flow cytometry
The cells were seeded in six-well culture plates (1×106 per well). After cell attachment, the cells were subjected to synchronization culture for 12 hours, and the primary culture was discarded. Cells were digested, centrifuged, collected, and washed with PBS twice, resuspended with precooled 75% ethanol, fixed at −20°C overnight, centrifuged to remove the supernatant, and washed with PBS two times. Each sample was suspended in 450 μL PBS, and propidium iodide (PI, 0.5 mg/mL) was added to the cell suspension, mixed, and incubated in a water bath at 37°C for 30 minutes. The mixture was centrifuged to remove the supernatant, and cells were resuspended in PBS and assessed by flow cytometry (model: FACSCalibur; BD Company, USA) to analyze the cell cycle distribution.
Cell apoptosis detected by flow cytometry
After a 72-hour virus infection, cells were collected and washed with PBS three times, and then, precooled in 1× binding buffer (500 μL), 5 μL Annexin-V-fluorescein isothiocyanate (FITC) and 2.5 μL PI were added to the cell suspension, gently mixed, and detected by flow cytometry (FACSAria I cell sorter; BD). Based on the results in the scatter diagram, the left lower quadrant (Q4) indicates healthy living cells (FITC−/PI−), the right lower quadrant (Q3) indicates early apoptotic cells (FITC+/PI–), the right upper quadrant (Q2) indicates late necrotic and apoptotic cells (FITC+/PI+), and the apoptosis rate = early apoptosis percentage (Q3) + late apoptosis percentage (Q2).
Cell migration and invasion detected by Transwell assays
After 72 hours of virus infection, BLS cells were collected and cultured in serum-free medium for 24 hours and digested. Cell suspension (100 μL) with an adjusted concentration of 1×105 cells was added to the upper chamber, and medium (500 μL) containing 10% FBS was added to the lower chamber and incubated in 5% CO2 under 37°C. The culture medium in the upper chamber was removed and moved to a 24-well plate containing 4% formaldehyde (600 μL). Giemsa reagent 1 was added to the upper chamber, incubated for 1 minute and then Giemsa reagent 2 was added to the upper chamber, with incubation for 5 minutes. The upper chamber was washed with PBS (30 minutes, three times), and results were observed and photographed under an inverted microscope. Five fields were randomly selected for further analysis with a microscope image acquisition system. In the invasion test, the number of cells in each group passing through the Matrigel was used as an index to evaluate invasion; the migration test was conducted without adding the Matrigel but shared the same steps as the invasion test.
Western blot
The cultured cells were washed with precooled PBS three times, lysed with 100 μL/50 mL protein extraction lysis solution (Radio-Immunoprecipitation Assay buffer; Pierce, Rockford, IL, USA), placed on ice for 30 minutes, and centrifuged at 12,000 rpm for 10 minutes at 4°C. The supernatant was separated in a 0.5 mL centrifuge tube and stored in −20°C. Total protein content was measured by the bicinchoninic acid method (BCA kit; Pierce, Inc.) The samples were run on electrophoresis for 1–2 hours at 4°C, and the proteins were transferred to the Polyvinylidene Fluoride (PVDF) membrane (EMD Millipore, Billerica, MA, USA) by electrotransfer for 2 hours at 4°C; the PVDF membrane was removed and blocked in 5% skim milk-Tris Buffered saline Tween (TBST), incubated for 1–2 hours at room temperature, and incubated with antibodies, including rabbit anti-human PIK3R2 (1:1,000, sc-131324; Santa Cruz Biotechnology Inc., Dallas, TX, USA), rabbit anti-human PI3K (1:1,000 sc-7177; Santa Cruz Biotechnology Inc.), rabbit anti-human phospho-PI3K (1:1,000, cs-4228; Cell Signaling, Boston, MA, USA), rabbit anti-human total Akt (1:1,000, cs-9271; Cell Signaling), and rabbit anti-human phospho-Akt (1:1,000, cs-9272; Cell Signaling) at 4°C overnight. The membranes were washed three times with TBST every 10 minutes and incubated with secondary antibodies (horseradish peroxidase [HRP]-labeled sheep anti-rabbit [l:5,000], A0208; Beyotime, Shanghai, People’s Republic of China) at room temperature for 1 hour and washed three times with TBST every 10 minutes. Beta-actin was used as a reference. The relative expression of the target protein was calculated according to the gray level of target protein and reference protein (beta-actin).
Statistical analyses
Data are presented as the mean ± standard deviation, and multiple group comparisons were performed with a one-way analysis of variance after homogeneity test of variance, and a least significant difference t-test was used for pairwise comparisons. SPSS Inc., (Chicago, IL, USA) for Versions 18 was used for statistical analysis. A P-value of <0.05 indicates statistically significant.
Results
Lentivirus construction and the expression of miR-126
Using human genomic DNA as a template, miR-126 precursor sequences were successfully amplified with a size of ~786 bp. Then, 5 μL of the PCR products was analyzed by agarose gel electrophoresis. Compared to the marker, the band size was identical to the theoretical value (786 bp), as shown in Figure 1A (1–4 bands). Using the Lv packaging technique, 293T cells were cotransfected with the Lv vector expressing miR-126 and the auxiliary packaging vector. After 72 hours of transfection, the viral supernatant was harvested, and the GFP expression of 293T cells was observed under a fluorescence microscope to determine the packaging efficiency. The number of fluorescence-positive cells and the total number of cells were determined in the same field of view, and the positive rate of 293T cells was calculated. The results showed that the transfection rate of 293T cells was >90%, as shown in Figure 1B. BLS cells were cultured in a culture bottle, and when the confluence of the cells was approximately 15% after 20 hours of culture, BLS cells were infected with Lv supernatant carrying miR-126 or no-load virus. After 72 hours of infection, the BLS cells were observed under the fluorescence microscope to assess cell infection. The infection rate reached 70%, as shown in Figure 1C. After 72 hours of recombinant Lv infections, BLS cells were harvested for total RNA extraction, and reverse transcription qRT-PCR assays were performed to measure relative miR-126 expression in the BLS cells. The relative expression of miR-126 in the Lv-infected BLS cells increased significantly, ~15 times greater than that of the BLS-Lv-vector group (P<0.0001) (Figure 1D), while no significant difference in the relative expression of miR-126 was found between the BLS-Lv-vector group and the noninfected group (P>0.05), indicating that the virus infection had no impact on the relative expression of miR-126.
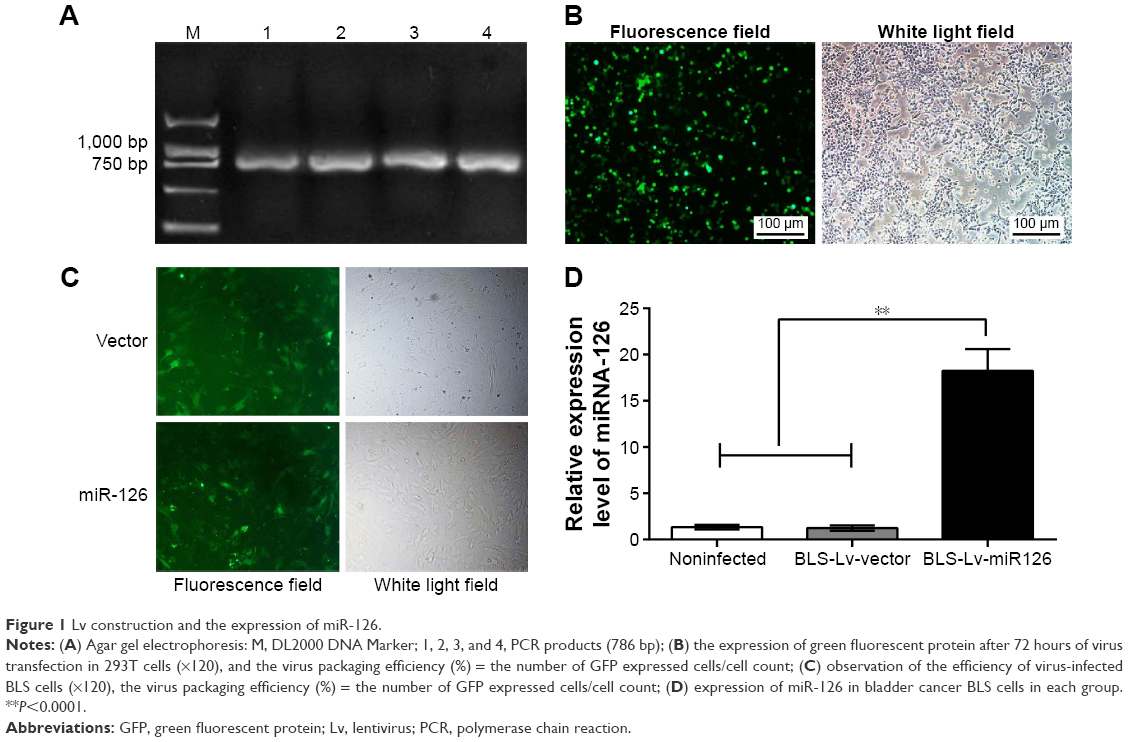 | Figure 1 Lv construction and the expression of miR-126. |
MiR-126 inhibits the proliferation of BLS cells
EdU assays showed that the proliferation rates of the BLS-Lv-miR126 group, the BLS-Lv-vector group, and the noninfected group were 6.37%±1.08%, 34.08%±4.58%, and 31.62%±5.21%, respectively. The differences in the proliferation rates between the BLS-Lv-miR126 group and the BLS-Lv-vector group or the noninfected group were significant (both P<0.001) (Figure 2A). The colony formation results further showed that the clone-forming ability of the BLS-Lv-miR126 group was significantly lower than that of the BLS-Lv-vector group and the noninfected group (both P<0.001) (Figure 2B). These experiments indicated that miR-126 could inhibit the proliferation and clone formation of BLS cells.
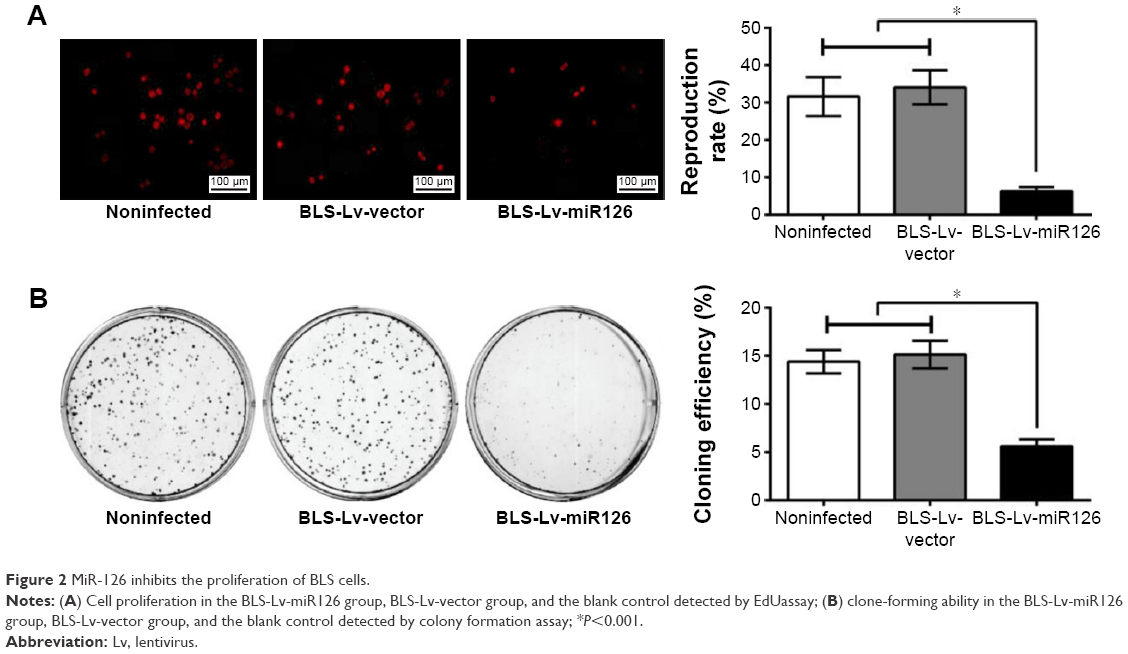 | Figure 2 MiR-126 inhibits the proliferation of BLS cells. |
MiR-126 promotes apoptosis of BLS cells
Flow cytometry analysis showed that the apoptosis rates in the BLS-Lv-miR126 group, the BLS-Lv-vector group, and the noninfected group were 31.87%±1.81%, 12.42%±0.84%, and 10.95%±0.73%, respectively. The differences in the apoptosis rates between the BLS-Lv-miR126 group and the BLS-Lv-vector group or the noninfected group were statistically significant (all P<0.01; Figure 3A). Cell cycle analysis showed that compared with the BLS-Lv-vector group and the noninfected group, an obviously decrease in S phase cells (P<0.01), an increase in G0/G1 cells (P<0.01), and a slight increase in G2/M cells (P>0.01) were found in the BLS-Lv-miR126 group. The percentages of G0/G1 phase cells in the BLS-Lv-miR126 group, the BLS-Lv-vector group, and the noninfected group were 82.70%±5.31%, 62.75%±5.94%, and 62.74%±4.96%, respectively; percentages in the S phase were 14.12%±2.64%, 33.55%±4.37%, and 33.93%±5.80%, respectively; and percentages in the G2/M phase were 3.18%±2.67%, 3.71%±1.61%, and 3.33%±0.84%, respectively (Figure 3B). These results indicated that miR-126 promotes apoptosis in BLS cells, and S phase arrest occurs.
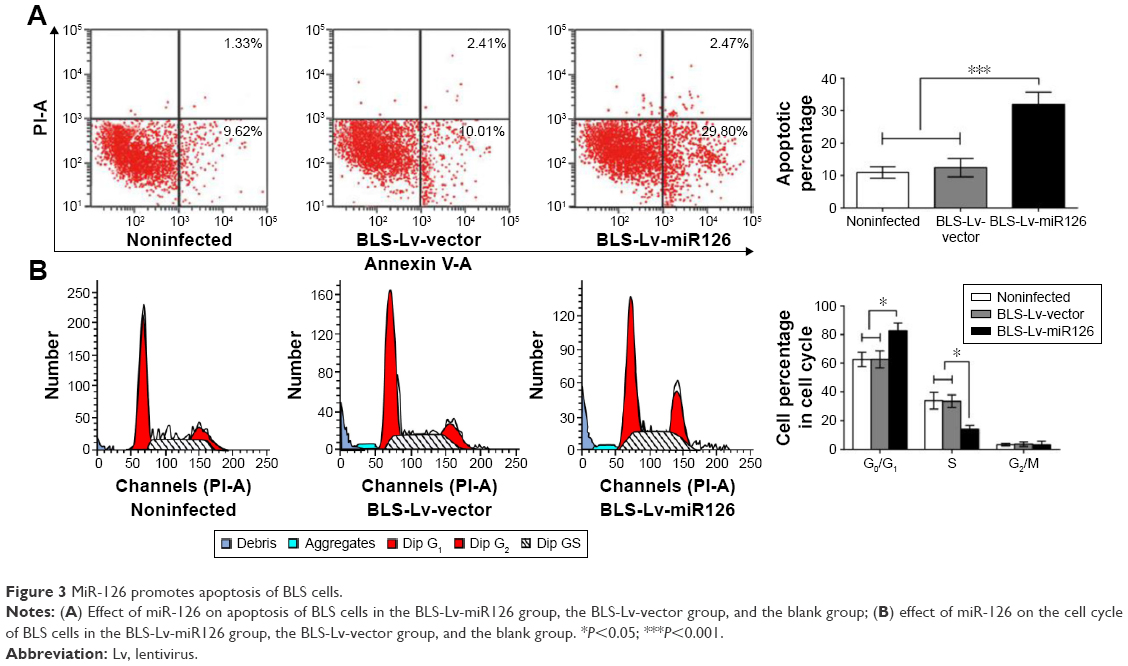 | Figure 3 MiR-126 promotes apoptosis of BLS cells. |
MiR-126 inhibits migration and invasion of BLS cells
Transwell migration and invasion assays were performed, and the numbers of cells migrating through the membranes in the BLS-Lv-miR126 group, the BLS-Lv-vector group, and the noninfected group were 34.35±6.21, 65.15±8.82, and 68.33±9.74, respectively (Figure 4A); the numbers of cells invading through the membranes were 31.28±4.67, 59.38±8.65, and 60.45±7.28, respectively (Figure 4B). The numbers of cells migrating/invading through the membranes in the BLS-Lv-miR126 group were significantly lower than those in the BLS-Lv-vector group and the noninfected group (all P<0.05). The results showed that miR-126 could inhibit migration and invasion of the BLS cells.
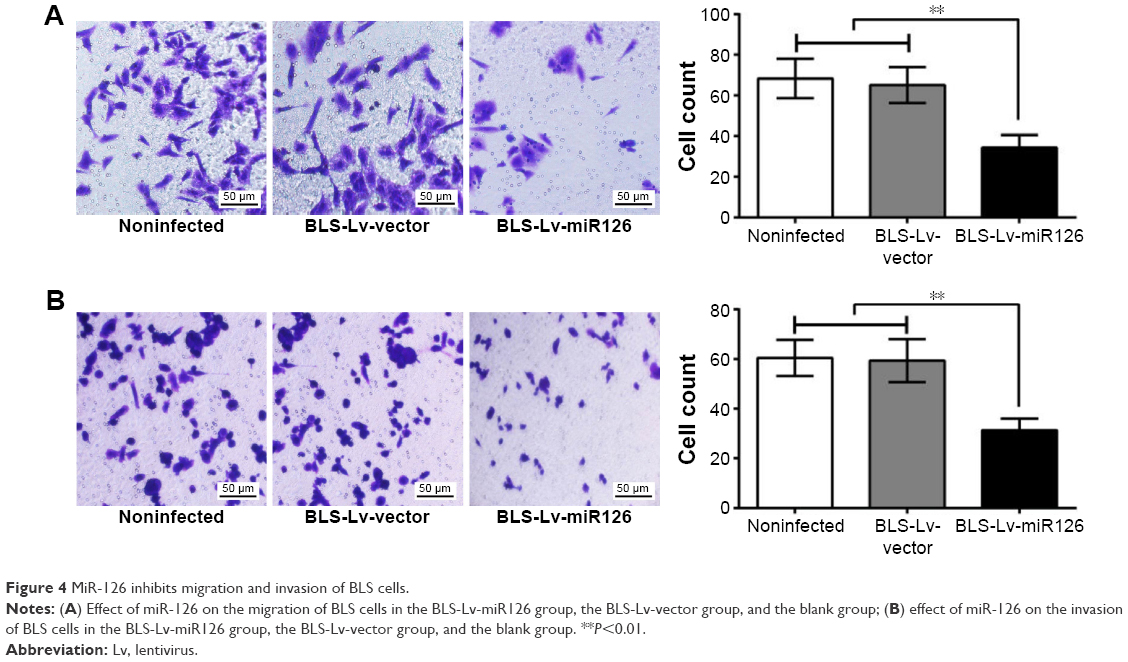 | Figure 4 MiR-126 inhibits migration and invasion of BLS cells. |
PIK3R2 is a target gene of miR-126
PIK3R2 and corresponding miR-126-binding target sites were identified using online prediction software (TargetScan [http://www.targetscan.org/vert_61/]), and the putative binding sequence for miR-126 in 3′UTR of the PIK3R2 mRNA is shown in Figure 5A. To verify that miR-126 affects the predicted binding sites and luciferase activity, mutated sequences were designed. Wild-type sequences and mutant sequences derived from the PIK3R2 3′UTR with a deletion in the predicted miR-126-binding sites were inserted into a luciferase reporter plasmid. BLS cells were cotransfected with miR-126 mimics and wild-type (Wt-miR-126 PIK3R2) or mutant (Mut-miR-126/PIK3R2) recombinant plasmids, and the luciferase activity assays showed that the luciferase activity of the Wt-miR-126 PIK3R2 plasmid significantly decreased by 60% (P<0.05), while no significant effect of the miR-126 mimics on the luciferase activity of the Mut-miR-126/PIK3R2 plasmid was found (P>0.05) (Figure 5B).
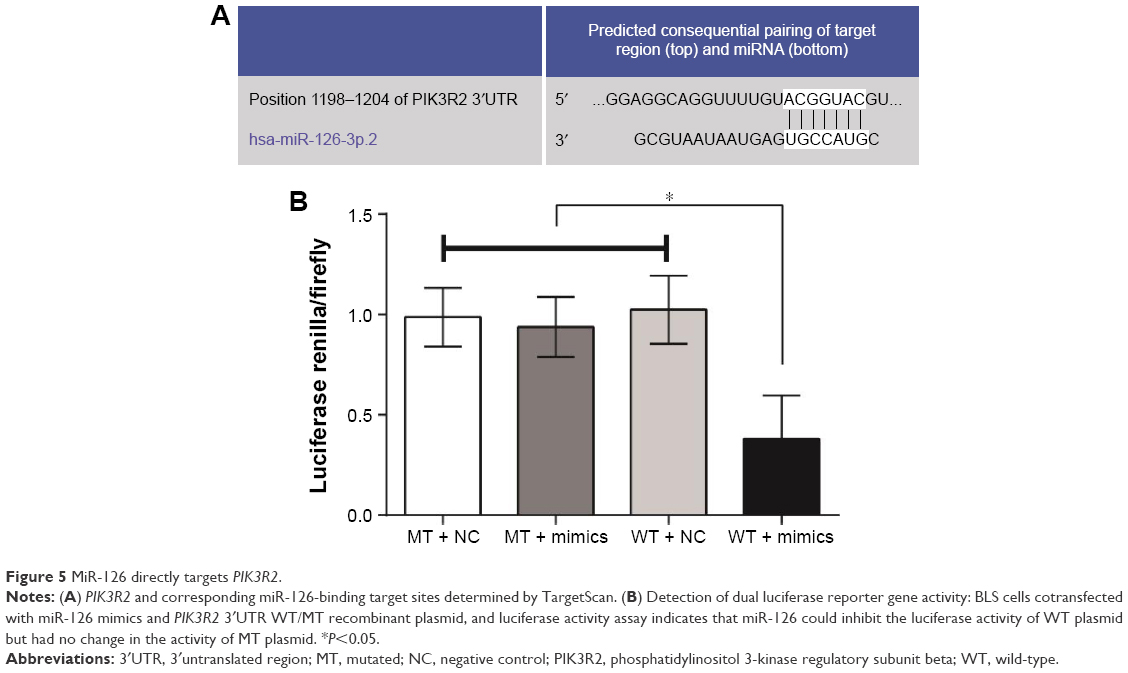 | Figure 5 MiR-126 directly targets PIK3R2. |
MiR-126 inhibits the expression of PIK3R2 and the PI3K/Akt signaling pathway
Compared to the BLS-Lv-vector group and the noninfected group, PIK3R2 mRNA (Figure 6A) and protein levels (Figure 6B) in the BLS-Lv-miR126 group were significantly decreased (both P<0.001), indicating targeted inhibition of PIK3R2 by miR-126 in the BLS cells. As a key protein of the PI3K/Akt signaling pathway, PIK3R2 was directly regulated by miR-126. Therefore, whether the PI3K/Akt signal pathway was regulated by miR-126 was verified using Western blot analysis. Compared with the BLS-Lv-vector group and the noninfected group, the phosphorylation of Akt and PI3K in BLS cells in the BLS-Lv-miR126 group was decreased (all P<0.05), which is consistent with the changes in the PIK3R2 protein (Figure 6C). Total Akt and PI3K protein levels were not significantly altered.
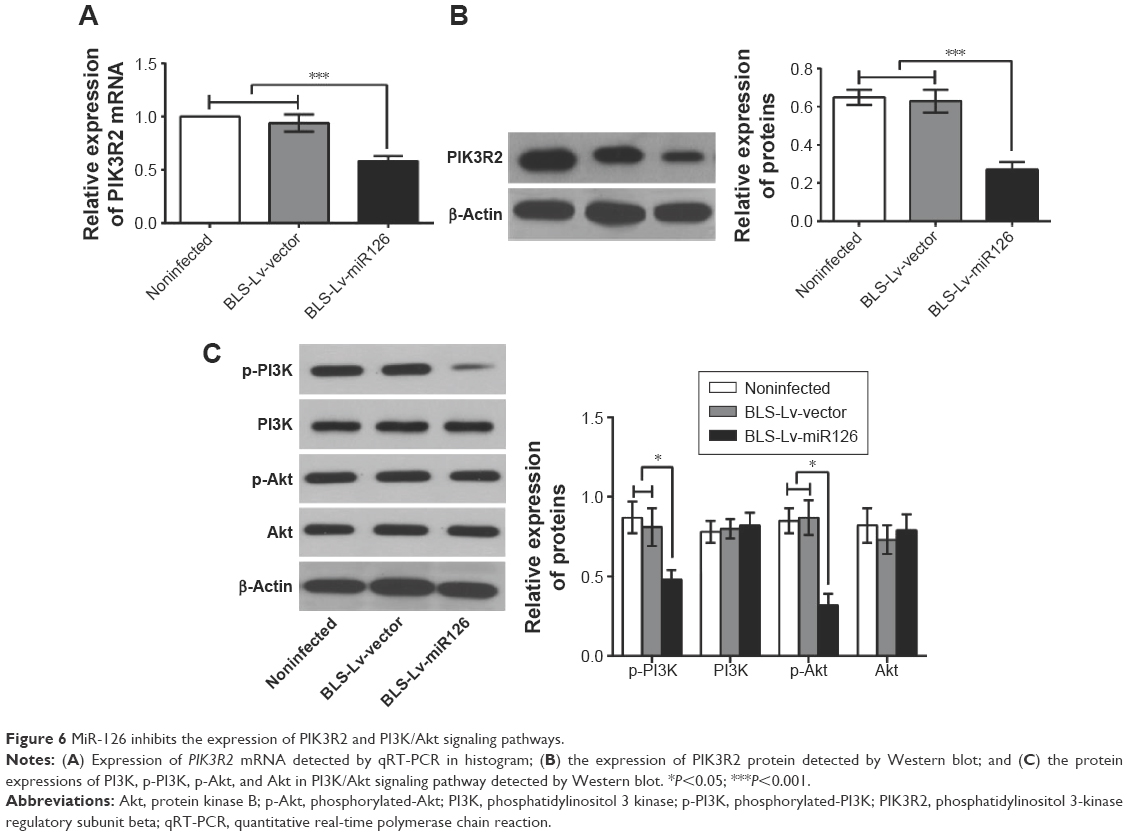 | Figure 6 MiR-126 inhibits the expression of PIK3R2 and PI3K/Akt signaling pathways. |
PIK3R2 is involved in miR-126-induced BLS cell proliferation and apoptosis
To determine whether PIK3R2 was involved in the miR-126-induced changes in BLS cells, anti-miR-126, PIK3R2 siRNA, or anti-miR-126 + PIK3R2 siRNA were transfected into BLS cells, and changes in the protein expressions of PI3K, phosphorylated PI3K (p-PI3K), phosphorylated Akt (p-Akt), and Akt in the PIK3R2/PI3K/Akt signaling pathway were determined. Western blot analysis revealed that cells in the anti-miR-126 group had significantly increased PIK3R2 protein expression compared with that of the NC group (P<0.01). PIK3R2 protein expression and p-PI3K and p-Akt levels decreased significantly in the PIK3R2 siRNA group and the anti-miR-126 + PIK3R2 siRNA group compared with the anti-miR-126 group (all P<0.001), while no significant difference was observed between the NC group and the anti-miR-126 + PIK3R2 siRNA group (P>0.05). The expressions of PIK3R2, p-Akt, and p-PI3K in the PIK3R2 siRNA group were significantly lower than those in the NC group and the anti-miR-126 + PIK3R2 siRNA group (all P<0.01) (Figure 7A).
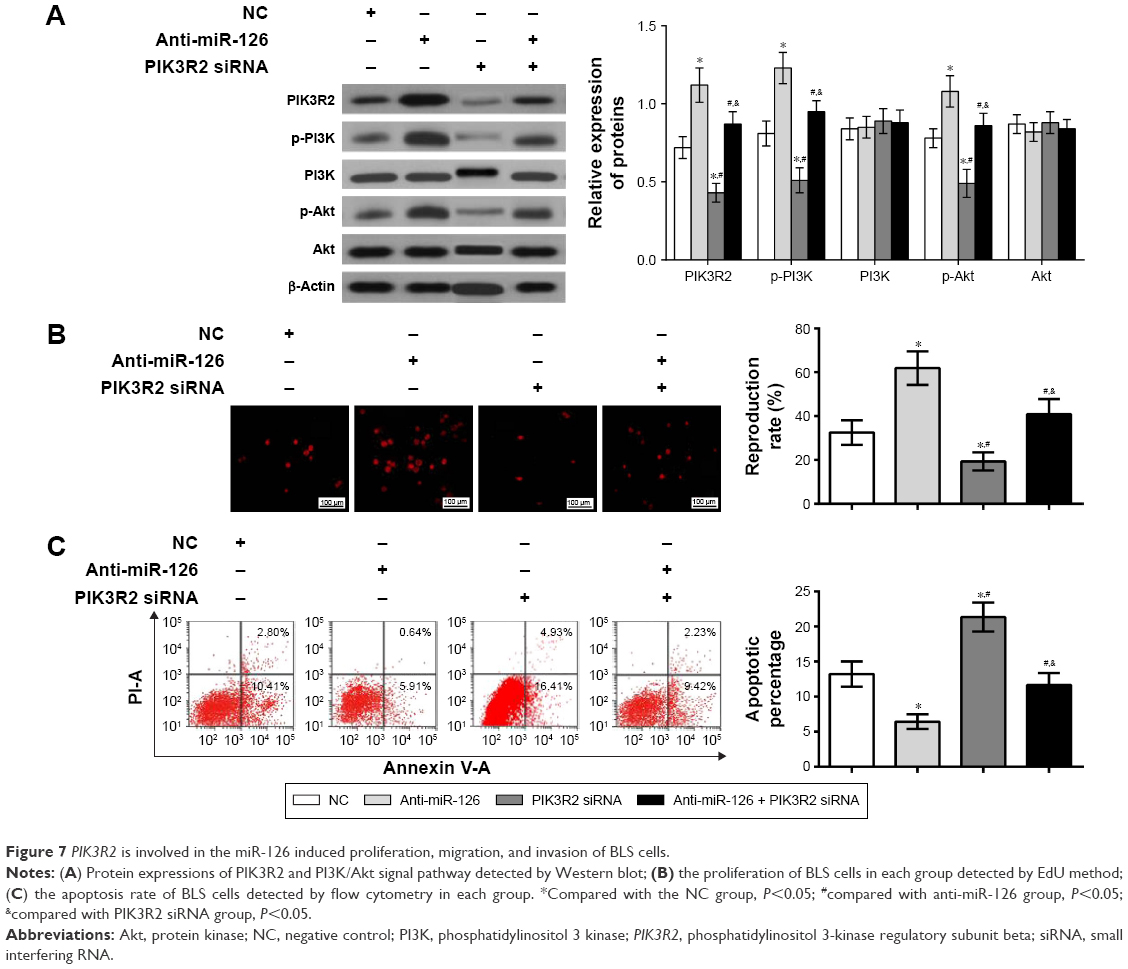 | Figure 7 PIK3R2 is involved in the miR-126 induced proliferation, migration, and invasion of BLS cells. |
To determine whether PIK3R2 was directly involved in miR-126-induced proliferation and apoptosis of BLS cells, EdU assays were performed and showed increased proliferation in the anti-miR-126 group relative to the NC group (P<0.01) and significantly decreased proliferation in the PIK3R2 siRNA group and the anti-miR-126 + PIK3R2 siRNA group compared with the anti-miR-126 group (all P<0.05). No difference in proliferation was found between the NC group and the anti-miR-126 + PIK3R2 siRNA group (P>0.05). The proliferation of the PIK3R2 siRNA group was significantly lower than that of the NC group and the anti-miR-126 + PIK3R2 siRNA group (all P<0.05; Figure 7B). Flow cytometry analysis showed a lower apoptosis rate in BLS cells from the anti-miR-126 group than the NC group (6.43%±1.05% vs 13.21%±1.81%, respectively, P<0.01), and the PIK3R2 siRNA group and the anti-miR-126 + PIK3R2 siRNA group had significantly increased apoptosis of BLS cells compared with the anti-miR-126 group (6.43%±1.05% vs 21.34%±2.06%; 6.43%±1.05% vs 11.65%±1.72%, all P<0.05). No significant difference was found between the NC group and the anti-miR-126 + PIK3R2 siRNA group (P>0.05). The apoptosis rate in the PIK3R2 siRNA group was significantly higher than that of the NC group and the anti-miR-126 + PIK3R2 siRNA group (all P<0.05; Figure 7C). Therefore, PIK3R2 was directly involved in the miR-126-induced BLS proliferation and apoptosis.
PIK3R2 is involved in the miR-126-induced migration and invasion of BLS cells
Whether PIK3R2 directly participated in the migration and invasion of BLS cells induced by miR-126 was further explored. Transwell migration assay indicated that the migration of BLS cells in the anti-miR-126 group was significantly higher than that in the NC group (P<0.01). The migratory ability of BLS cells in the PIK3R2 siRNA group and the anti-miR-126 + PIK3R2 siRNA group decreased significantly compared with the anti-miR-126 group (all P<0.05). No difference in migration was found between the NC group and the anti-miR-126 + PIK3R2 siRNA group (P>0.05). The migration of the PIK3R2 siRNA group was significantly lower than that of the NC group and the anti-miR-126 + PIK3R2 siRNA group (all P<0.05) (Figure 8A). Transwell invasion assays showed that invasion of BLS cells in the anti-miR-126 increased significantly compared with that in the NC group (P<0.05). The invasive ability of BLS cells in the PIK3R2 siRNA group and the anti-miR-126 + PIK3R2 siRNA group decreased significantly compared with the anti-miR-126 group (all P<0.05). No significant difference was found between the NC group and the anti-miR-126 + PIK3R2 siRNA group (P>0.05). The invasion in the PIK3R2 siRNA group was significantly lower than the NC group and the anti-miR-126 + PIK3R2 siRNA group (all P<0.05) (Figure 8B). In conclusion, PIK3R2 was directly involved in the miR-126-induced invasion and migration of BLS cells.
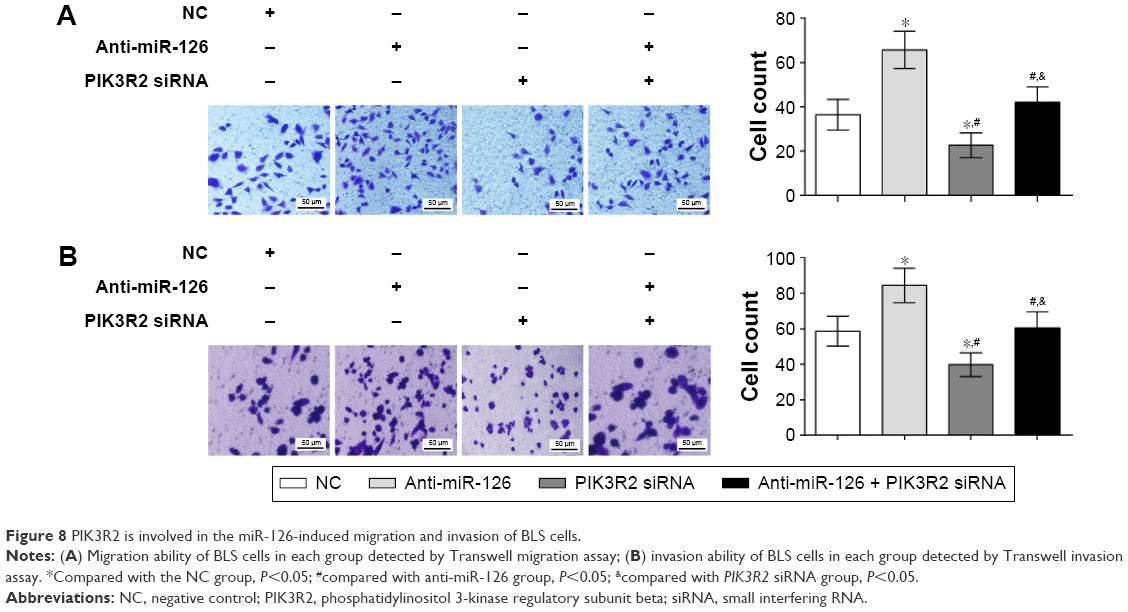 | Figure 8 PIK3R2 is involved in the miR-126-induced migration and invasion of BLS cells. |
Discussion
The role of miR-126 in BLS cells was investigated using transfection of recombinant Lv-miR126, and we found that miR-126 inhibited the proliferation, migration, and invasion and promoted apoptosis of the BLS cells. Previous evidence showed that the tumor suppressor miR-126 was down-regulated in cancer cell lines and in primary bladder and prostate tumors.16 Additionally, miR-126 is related to urinary bladder cancer and may serve as a novel RNA-based tumor marker.23 The potential role of miR-126 in targeting the PIK3R2-mediated PI3K/Akt signaling pathway was further evaluated. The luciferase reporter assay showed that PIK3R2 was a direct target of miR-126 in bladder cancer, which is consistent with previous evidence suggesting that PI3KR2 is a target of miR-126.19,24
In addition, it was found that miR-126 inhibited the expression of PIK3R2 and the PI3K/Akt signaling pathway, indicating that miR-126 could negatively regulate the target gene and inhibit the PIK3R2/PI3K/Akt signaling pathway. PIK3R2 is a key protein in the PI3K/Akt signaling pathway, and miR-126 plays various roles through regulation of the PIK3R2/PI3K/Akt pathway.25,26 As previously described, PI3K/Akt pathway activation occurs in bladder urothelial carcinomas, and the role of the PI3K/Akt/mTOR pathway in bladder cancer oncogenesis has been elucidated.21,27 Furthermore, the involvements of miR-126 in the PIK3R2/PI3K/Akt signaling pathway has been evaluated, and potential therapeutic roles have been suggested through targeting miR-126-regulated pathways.28,29
Another important finding was the involvement of PIK3R2 in the miR-126-induced proliferation, apoptosis, migration, and invasion of BLS cells, suggesting that the miR-126/PIK3R2/PI3K/Akt signaling pathway may be a potential therapeutic target for bladder cancer treatment. MiR-126 regulates target gene-mediated signaling pathways involved in adhesion, cell growth, proliferation, migration and invasion, as well as tumorigenicity and metastasis in human cancers.30,31 Interestingly, attenuated invasive potential of bladder cancer cells can result from the restoration of miR-126 levels, indicating that miR-126 inhibits invasion in bladder cancer.18 It has been reported that PIK3R2 was a direct target of miR-126, and miR-126 can inhibit the proliferation and migration of cancer cells via the PIK3R2/PI3K/Akt signaling pathway.32,33 Furthermore, altered protein glycosylation and the activation of the PI3K/Akt/mTOR pathway may be potential therapeutic targets.7,34 Our results indicated that overexpression of miR-126 negatively regulated the target gene PIK3R2 and further inhibited the PI3K/Akt signaling pathway. However, inconsistent results have been described for miR-126 concerning apoptosis in different cell types. miR-126 overexpression was found to inhibit vascular endothelial cell apoptosis by targeting the anti-apoptotic PI3K/Akt pathway via PIK3R2.35 In addition, miR-126 can activate the PI3K/Akt pathway, and miR-126 may play a role in tumorigenesis and growth by regulating the VEGF/PI3K/Akt signaling pathway.26,36 The complex regulation of the PI3K/Akt pathway, along with the multiple mechanisms to activate or inhibit this pathway, makes it a highly challenging pathway to target.7 Therefore, the therapeutic value of miR-126 in bladder cancer needs to be further confirmed due to the diverse effects of miR-126 on the PIK3R2/PI3K/Akt pathway.
Conclusion
In conclusion, overexpression of miR-126 negatively regulated the target gene PIK3R2 and further inhibited the PI3K/Akt signaling pathway, thereby reducing proliferation, migration, and invasion of BLS cells and promoting cell apoptosis. These results suggest that miR-based targeted therapy for bladder cancer may be a promising strategy. Because PIK3R2/PI3K/Akt is a key component of multiple pathways, miR-126 may also be involved in the regulation of proliferation and invasion-related pathways of bladder cancer in BLS cells by regulating the PIK3R2/PI3K/Akt pathway. Further studies are needed to elucidate the relationship between miR-126 and the proliferation and invasion of bladder cancer cells to improve our preliminary work.
Acknowledgments
This research was funded by the Anhui Natural Science Foundation (1608085 MH166). We would like to acknowledge the helpful comments on this paper received from our reviewers.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, et al. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Ploeg M, Aben KK, Kiemeney LA. The present and future burden of urinary bladder cancer in the world. World J Urol. 2009;27(3):289–293. | ||
Chen WQ, Zeng HM, Zheng RS, et al. Cancer incidence and mortality in China, 2007. Chin J Cancer Res. 2012;24(1):1–8. | ||
The American Cancer Society. Cancer Facts and Figures. 2012. | ||
Cheng L, Zhang S, MacLennan GT, et al. Bladder cancer: translating molecular genetic insights into clinical practice. Hum Pathol. 2011;42(4):455–481. | ||
McConkey DJ, Lee S, Choi W, et al. Molecular genetics of bladder cancer: emerging mechanisms of tumor initiation and progression. Urol Oncol. 2010;28(4):429–440. | ||
Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28(3–4):305–316. | ||
Thomas S, Overdevest JB, Nitz MD, et al. Src and caveolin-1 reciprocally regulate metastasis via a common downstream signaling pathway in bladder cancer. Cancer Res. 2011;71(3):832–841. | ||
Seront E, Pinto A, Bouzin C, et al. PTEN deficiency is associated with reduced sensitivity to mTOR inhibitor in human bladder cancer through the unhampered feedback loop driving PI3K/Akt activation. Br J Cancer. 2013;109(6):1586–1592. | ||
Catto JW, Alcaraz A, Bjartell AS, et al. MicroRNA in prostate, bladder, and kidney cancer: a systematic review. Eur Urol. 2011;59(5):671–681. | ||
Noguchi S, Yasui Y, Iwasaki J, et al. Replacement treatment with microRNA-143 and -145 induces synergistic inhibition of the growth of human bladder cancer cells by regulating PI3K/Akt and MAPK signaling pathways. Cancer Lett. 2013;328:353–361. | ||
Han Y, Chen J, Zhao X, et al. MicroRNA expression signatures of bladder cancer revealed by deep sequencing. PLoS One. 2011;6(3):e18286. | ||
Mlcochova H, Hezova R, Stanik M, et al. Urine microRNAs as potential noninvasive biomarkers in urologic cancers. Urol Oncol. 2014;32(1):e41–e49. | ||
Huang L, Luo J, Cai Q, et al. MicroRNA-125b suppresses the development of bladder cancer by targeting E2F3. Int J Cancer. 2011;128(8):1758–1769. | ||
Zaravinos A, Radojicic J, Lambrou GI, et al. Expression of miRNAs involved in angiogenesis, tumor cell proliferation, tumor suppressor inhibition, epithelial-mesenchymal transition and activation of metastasis in bladder cancer. J Urol. 2012;188(2):615–623. | ||
Saito Y, Friedman JM, Chihara Y, et al. Epigenetic therapy upregulates the tumor suppressor microRNA-126 and its host gene EGFL7 in human cancer cells. Biochem Biophys Res Commun. 2009;379(3):726–731. | ||
Snowdon J, Boag S, Feilotter H, et al. A pilot study of urinary microRNA as a biomarker for urothelial cancer. Can Urol Assoc J. 2013;7(1–2):28–32. | ||
Jia AY, Castillo-Martin M, Bonal DM, et al. MicroRNA-126 inhibits invasion in bladder cancer via regulation of ADAM9. Br J Cancer. 2014;110(12):2945–2954. | ||
Liu LY, Wang W, Zhao LY, et al. Mir-126 inhibits growth of SGC-7901 cells by synergistically targeting the oncogenes PI3KR2 and Crk, and the tumor suppressor PLK2. Int J Oncol. 2014;45(3):1257–1265. | ||
Qin A, Wen Z, Zhou Y, et al. MicroRNA-126 regulates the induction and function of CD4(+) Foxp3(+) regulatory T cells through PI3K/AKT pathway. J Cell Mol Med. 2013;17(2):252–264. | ||
Calderaro J, Rebouissou S, de Koning L, et al. PI3K/AKT pathway activation in bladder carcinogenesis. Int J Cancer. 2014;134(8):1776–1784. | ||
Ribeiro J, Marinho-Dias J, Monteiro P, et al. MiR-34a and miR-125b expression in HPV infection and cervical cancer development. Biomed Res Int. 2015;2015:304584. | ||
Hanke M, Hoefig K, Merz H, et al. A robust methodology to study urine microRNA as tumor marker: microRNA-126 and microRNA-182 are related to urinary bladder cancer. Urol Oncol. 2010;28(6):655–661. | ||
DA Silva ND Jr, Fernandes T, Soci UP, et al. Swimming training in rats increases cardiac MicroRNA-126 expression and angiogenesis. Med Sci Sports Exerc. 2012;44(8):1453–1462. | ||
Gao J, Zhou XL, Kong RN, et al. MicroRNA-126 targeting PIK3R2 promotes rheumatoid arthritis synovial fibro-blasts proliferation and resistance to apoptosis by regulating PI3K/AKT pathway. Exp Mol Pathol. 2015;100(1):192–198. | ||
Zhu N, Zhang D, Xie H, et al. Endothelial-specific intron-derived miR-126 is down-regulated in human breast cancer and targets both VEGFA and PIK3R2. Mol Cell Biochem. 2011;351(1–2):157–164. | ||
Ching CB, Hansel DE. Expanding therapeutic targets in bladder cancer: the PI3K/Akt/mTOR pathway. Lab Invest. 2010;90(10):1406–1414. | ||
Zhang J, Zhang Z, Zhang DY, et al. MicroRNA 126 inhibits the transition of endothelial progenitor cells to mesenchymal cells via the PIK3R2-PI3K/Akt signalling pathway. PLoS One. 2013;8(12):e83294. | ||
Banerjee N, Kim H, Talcott S, et al. Pomegranate polyphenolics suppressed azoxymethane-induced colorectal aberrant crypt foci and inflammation: possible role of miR-126/VCAM-1 and miR-126/PI3K/AKT/mTOR. Carcinogenesis. 2013;34(12):2814–2822. | ||
Liu B, Peng XC, Zheng XL, et al. MiR-126 restoration down-regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer. 2009;66(2):169–175. | ||
Feng R, Chen X, Yu Y, et al. MiR-126 functions as a tumour suppressor in human gastric cancer. Cancer Lett. 2010;298(1):50–63. | ||
Crawford M, Brawner E, Batte K, et al. MicroRNA-126 inhibits invasion in non-small cell lung carcinoma cell lines. Biochem Biophys Res Commun. 2008;373(4):607–612. | ||
Nie ZC, Weng WH, Shang YS, et al. MicroRNA-126 is down-regulated in human esophageal squamous cell carcinoma and inhibits the proliferation and migration in EC109 cell via PI3K/AKT signaling pathway. Int J Clin Exp Pathol. 2015;8(5):4745–4754. | ||
Costa C, Pereira S, Lima L, et al. Abnormal protein glycosylation and activated PI3K/Akt/mTOR pathway: role in bladder cancer prognosis and targeted therapeutics. PLoS One. 2015;10(11):e0141253. | ||
Chen L, Wang J, Wang B, et al. MiR-126 inhibits vascular endothelial cell apoptosis through targeting PI3K/Akt signaling. Ann Hematol. 2016;95(3):365–374. | ||
Sui XQ, Xu ZM, Xie MB, et al. Resveratrol inhibits hydrogen peroxide-induced apoptosis in endothelial cells via the activation of PI3K/Akt by miR-126. J Atheroscler Thromb. 2014;21(2):108–118. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.