Back to Journals » OncoTargets and Therapy » Volume 9
Midazolam regulated caspase pathway, endoplasmic reticulum stress, autophagy, and cell cycle to induce apoptosis in MA-10 mouse Leydig tumor cells
Authors So EC ![]() , Chen Y
, Chen Y ![]() , Wang S, Wu C, Huang M, Lai M, Pan B, Kang F
, Wang S, Wu C, Huang M, Lai M, Pan B, Kang F ![]() , Huang B
, Huang B ![]()
Received 1 December 2015
Accepted for publication 16 February 2016
Published 27 April 2016 Volume 2016:9 Pages 2519—2533
DOI https://doi.org/10.2147/OTT.S101671
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Faris Farassati
Edmund Cheung So,1,2 Yung-Chia Chen,3 Shu-Chun Wang,4 Chia-Ching Wu,4 Man-Chi Huang,4 Meng-Shao Lai,4 Bo-Syong Pan,4,5 Fu-Chi Kang,6 Bu-Miin Huang4
1Department of Anesthesia, An Nan Hospital, China Medical University, Tainan, Taiwan, Republic of China; 2Department of Anesthesia, School of Medicine, China Medical University, Taichung, Taiwan; Republic of China; 3Department of Anatomy, School of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan, Republic of China; 4Department of Cell Biology and Anatomy, College of Medicine, National Cheng Kung University, Tainan, Taiwan, Republic of China; 5Department of Cancer Biology, Wake Forest University School of Medicine, Winston Salem, NC, USA; 6Department of Anesthesia, Chi Mei Medical Center, Chiali, Tainan, Taiwan, Republic of China
Purpose: Midazolam is widely used as a sedative and anesthetic induction agent by modulating the different GABA receptors in the central nervous system. Studies have also shown that midazolam has an anticancer effect on various tumors. In a previous study, we found that midazolam could induce MA-10 mouse Leydig tumor cell apoptosis by activating caspase cascade. However, the detailed mechanism related to the upstream and downstream pathways of the caspase cascade, such as endoplasmic reticulum (ER) stress, autophagy, and p53 pathways plus cell cycle regulation in MA-10 mouse Leydig tumor cells, remains elusive.
Methods: Flow cytometry assay and Western blot analyses were exploited.
Results: Midazolam significantly decreased cell viability but increased sub-G1 phase cell numbers in MA-10 cells (P<0.05). Annexin V/propidium iodide double staining further confirmed that midazolam induced apoptosis. In addition, expressions of Fas and Fas ligand could be detected in MA-10 cells with midazolam treatments, and Bax translocation and cytochrome c release were also involved in midazolam-induced MA-10 cell apoptosis. Moreover, the staining and expression of LC3-II proteins could be observed with midazolam treatment, implying midazolam could induce autophagy to control MA-10 cell apoptosis. Furthermore, the expressions of p-EIF2α, ATF4, ATF3, and CHOP could be induced by midazolam, indicating that midazolam could stimulate apoptosis through ER stress in MA-10 cells. Additionally, the expressions of cyclin A, cyclin B, and CDK1 could be inhibited by midazolam, and the phosphorylation of p53, P27, and P21 could be adjusted by midazolam, suggesting that midazolam could manage cell cycle through the regulation of p53 pathway to induce apoptosis in MA-10 cells.
Conclusion: Midazolam could induce cell apoptosis through the activation of ER stress and the regulation of cell cycle through p53 pathway with the involvement of autophagy in MA-10 mouse Leydig tumor cells.
Keywords: midazolam, apoptosis, ER stress, autophagy, cell cycle, MA-10 cells, caspase, tumor
Introduction
Midazolam is widely used as a sedative and anesthetic induction agent, and it is a derivative of benzodiazepine. Midazolam impairs motor performance in experimental animals and probably exhibits a muscle relaxant effect similar to the other benzodiazepines,1 which has powerful anxiolytic, hypnotic, and sedative properties by modulating the GABA receptors in the central nervous system.2,3 In a previous study, we illustrated that midazolam could stimulate steroidogenesis in MA-10 mouse Leydig tumor cells via PKA and PKC pathways along with the expression of peripheral benzodiazepine receptor and StAR proteins,4 and could induce MA-10 cell apoptosis through caspase and MAPK signaling pathways.5 In the present study, we further investigated the detailed mechanism of midazolam-induced apoptosis on MA-10 cells.
Apoptosis plays a pivotal role in the elimination of unwanted, damaged, or infected cells in multicellular organisms and also in diverse biological processes, including development, cell differentiation, and proliferation.6 There are two general types of signaling pathways leading from a triggering event to the activation of an initiator caspase and from there to apoptotic death. The first of these depends upon the participation of mitochondria, and the second involves death receptors.7 The mitochondrial pathway of apoptosis is the major mechanism of physiological cell death in vertebrates. In this pathway, pro-apoptotic members of the Bcl-2 family cause mitochondrial outer membrane permeabilization, allowing the release of cytochrome c, which interacts with Apaf-1 to trigger caspase activation and apoptosis.8 The death receptor pathway would be initiated through the activation of death receptors by pro-apoptotic ligands, including FasL, TNF-α, and TRAIL. After binding, the death domains of these receptors bind to FADD, resulting in the recruitment of DISC, and hence lead to the activation of CASP8 which triggers downstream effector caspase, CASP3 or 7.9
Autophagy is the process whereby organelles and other cell components are degraded by lysosomes. There are various types of autophagy, including macro-autophagy, micro-autophagy, and chaperone-mediated autophagy.10 Autophagy can be divided into three distinct stages: vesicle nucleation (formation of the so-called phagophore), vesicle elongation (growth and closure of the autophagosome), and fusion of the autophagosome with a lysosome to form an autolysosome. This process is controlled by the Atg. Autophagy can act independently of the apoptotic signaling pathways.
The endoplasmic reticulum (ER) is a dynamic organelle of fundamental importance in all eukaryotic cells, and it is central for translocation, folding, and posttranslational modification of newly synthesized proteins.11 Physiological and pathological conditions, such as hypoxia, nutrient deprivation, and pro-inflammatory cytokines, can disturb ER homeostasis and thus negatively impact upon protein folding, resulting in an accumulation of unfolded proteins, and causing ER stress.12 In response to ER stress, cells stimulate the unfolded protein response (UPR) to restore protein homeostasis. If ER stress is chronically prolonged, cell death often occurs through apoptosis.12 The UPR is distinguished by three signaling proteins named PERK, ATF6, and IRE1α.13 In response to ER stress, activation of PERK leads to phosphorylation of eIF2α, and then selectively induces a transcription factor ATF4, causing the up-regulated expression of pro-apoptotic CCAAT/CHOP to induce apoptosis.14 On the other hand, ATF6 transits to the Golgi where it is cleaved by S1P and S2P, yielding an activated transcription factor, ATF6.14 In addition, activated IRE1 catalyzes removal of a small intron from the messenger RNA of the gene encoding XBP1 to produce an active transcription factor. The ATF6 and the IRE1 pathways may also induce the expression of CHOP to trigger apoptosis.15 ER stress can also activate CASP12 localized at the ER membrane through an interaction with IRE1, leading cells to undergo apoptosis.16
Cell cycle is an important player regulating the cell growth progression, and the complex CDKs and cyclins can drive cells from one stage to another stage during cell proliferation.17,18 Positive stimulation may promote cell progression and help cell proliferation. However, the abnormal regulation of cell cycle could transform normal cells into cancer cells.18 The p53 gene is a suppressor gene which can regulate cell proliferation, cell growth, DNA repair, and apoptosis. Studies have demonstrated that p53 could be regulated by cell cycle progression and apoptosis.19,20 However, another study showed that p53 was present in malignancy and associated with cell proliferation as expressed by PCNA and c-myc.21
In fact, we have found that midazolam could stimulate caspase and MAPK, but inhibit Akt pathways to induce apoptosis in MA-10 cells.5 In the present study, we would like to further investigate the detailed mechanism activated by midazolam in MA-10 cells, and will focus on the apoptotic pathway, autophagy, ER stress, and cell cycle-related pathway.
Materials and methods
Chemicals
Midazolam was purchased from Hoffman-La Roche Ltd. (Basel, Switzerland). Waymouth MB 752/1 medium, propidium iodide (PI), penicillin–streptomycin, RNase A, Folin and Ciocalteu’s phenol regent, ethylenediaminetetraacetic acid (EDTA), 30% acrylamide/bis-acrylamide solution, MTT, and monoclonal antibody against β-actin were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Fetal bovine serum, Dulbecco’s Modified Eagle’s Medium/F12, and trypsin-EDTA were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Gentamycin sulfate was purchased from A.G. Scientific Inc. (San Diego, CA, USA). NaCl, KCl, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and Tris base were purchased from JT Baker (Phillipsburg, NJ, USA). Na2HPO4, KH2PO4, and tissue culture grade NaHCO3 were purchased from Riedel-de Haen (Seelze, Germany). Sucrose was purchased from Panreac (Barcelona, Spain). HCl, sodium dodecyl sulfate (SDS), Tween 20, and DMSO were purchased from EMD Millipore (Billerica, MA, USA). Isoton II was purchased from Beckman Coulter (Brea, CA, USA). Micro BCA Protein Assay Kit was purchased from Thermo Fisher Scientific. Donkey anti-rabbit IgG conjugated with horseradish peroxidase (HRP) and donkey anti-mouse IgG conjugated with HRP were purchased from PerkinElmer Inc. (Waltham, MA, USA). Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit was purchased from Strong Biotech (Taipei, Taiwan). Polyclonal antibodies against cleaved CASP12, Bax, cytochrome c, and COX IV were purchased from Cell Signaling Technology (Beverly, MA, USA). Monoclonal antibody against cleaved p27, p53, phosphor-p53 (Ser 15), IRE1α, phosphor-EIF2α, PERK, and ATF4 were purchased from Cell Signaling Technology. Polyclonal antibody against Fas, FasL, ATF3, CHOP, CDK1, cyclin A, and phosphor-p21 (Thr 145) were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Polyclonal antibody against phosphor-IRE1α and truncated Bid were purchased from Abcam (Cambridge, UK). Polyclonal antibodies against ATF6β, EIF2α, and XBP1 were purchased from Abgent (San Diego, CA, USA). Polyclonal antibodies against phosphor-p27 (Ser 10), cyclin B, and p21 were purchased from GeneTex (Irvine, CA, USA). Enhanced chemiluminescence detection kit was purchased from Merck Millipore (Billerica, MA, USA).
Cell culture
The MA-10 cell line, a gift from Dr Mario Ascoli (University of Iowa, Iowa City, IA, USA), is a mouse Leydig tumor cell, which can be cultured in the Waymouth medium containing 10% FBS. Cells were incubated in a humidified atmosphere containing 95% air and 5% CO2 at 37°C.
Morphological observation
MA-10 cells were seeded at a concentration of 6×105/mL in a 6 cm Petri dish with 2 mL culture medium, and treated without or with different concentrations of midazolam (30 and 150 μM) for 24 hours, respectively. Cell morphology was then observed with Olympus CK40 light microscopy and images recorded with an Olympus DP20 digital camera (Olympus Corporation, Tokyo, Japan).
MTT viability test
The MTT assay is a colorimetric assay for assessing cell viability.22 MA-10 cells were seeded in 96-well plates containing 1.2×104 cells per well. After 70%–80% confluence, cells were treated without or with different concentrations of midazolam (6, 30, and 150 μM) for 1, 3, 6, 12, and 24 hours, respectively. After treatment, MTT (0.5 mg/mL) was added to each well at different time points and incubated at 37°C for 4 hours. The medium was discarded, and 50 μL DMSO was added into each well to dissolve the crystals by shaking the plate at 37°C for 20 minutes in the dark. The cell viability was detected at λ=570 nm by VersaMax ELISA reader (Molecular Devices LLC, Sunnyvale, CA, USA).22
Cell cycle analysis
To investigate whether midazolam could induce MA-10 cell death through apoptosis, their DNA contents were then examined by PI staining through flow cytometric analysis. MA-10 cells were seeded at a concentration of 6×105 in a 6 cm Petri dish with 2 mL culture medium, and treated without or with different concentrations of midazolam (30 and 150 μM) for 12 and 24 hours, respectively. Cells were harvested by trypsin digestion and centrifugation, and then washed with isoton II and fixed with 70% ethanol for at least 2 hours at −20°C. After fixation, cells were washed with cold isoton II and then collected by centrifugation. Cell suspensions were mixed with 100 μg/mL RNase, and stained with 40 μg/mL PI solution for 30 minutes. The stained cells were analyzed at λ=488 nm for PI detection with a FACScan flow cytometer (BD, Franklin Lakes, NJ, USA). Cells in sub-G1 phase have less DNA contents on cell cycle distribution, which is considered to be DNA fragmentation, and as a result of cell apoptosis.4,5
Annexin V/PI double staining assay
After harvesting cells with trypsin and washing it with 2 mL culture medium, cell suspensions were centrifuged at 1,000 rpm for 10 minutes at 4°C. The pellets were resuspended with cold isoton II and centrifuged again. The pellets were mixed with 100 μL staining solution for 15 minutes according to the user’s manual of Annexin V-FITC apoptosis detection kit from Srong Biotech (Taipei, Taiwan). The stained cells were analyzed at λ=488 nm excitation using a 515 nm band pass filter for FITC detection and >600 nm band pass filter for PI detection by FACScan flow cytometer. The double-negative cells (viable), annexin V single-positive cells (early apoptotic), PI single positive cells (necrotic), and double positive cells (late apoptotic) could be illustrated in four quadrants.4,5
Mitochondrial protein isolation
MA-10 cells (1.7×107 cells/dish) were seeded in a 6 cm Petri dish. After treatments, cells were then collected from the dishes by scraping in 4 mL cold phosphate-buffered saline. The cell suspensions were centrifuged at 600× g for 10 minutes at 4°C. The pellets were resuspended with mitochondrial isolation buffer consisting of 10 mM Tris, 0.25 M sucrose, 0.1 mM EDTA, with pH 7.4. The cells were homogenized at 1,000 rpm for 22 strokes using a motorized glass homogenizer fitted with a serrated Teflon pestle. The homogenates were centrifuged at 600× g for 30 minutes, and the resultant supernatants were centrifuged at 12,000× g for another 30 minutes. The pellets were resuspended in 50 μL lysis buffer with proteinase inhibitor, which were considered as mitochondrial fractions; and the supernatants were collected as cytosolic fractions. Both fractions were analyzed by Western blotting as previously mentioned.5
Immunofluorescent staining
MA-10 cells were seeded in 12-well plates containing 6×104 cells with 2 mL culture medium per well. After 70%–80% confluence, cells were treated without or with midazolam (150 μM) for 24 hours. For double-immunolabeling studies, the MA-10 cells were stained with primary mouse antibody against LC3-I/II (1:250; Abgent, St Louis, MO, USA) with Alexa-543-conjugated goat anti-mouse secondary antibody (Thermo Fisher Scientific). The confocal images were obtained using an excitation wavelength of 488 nm (for Enhanced Green Fluorescent Protein) and 543 nm (for Alexa-543), respectively (model SP2 TCS; Leica Microsystems, Wetzlar, Germany).
Protein extraction and Western blot
MA-10 cells were seeded in a 6 cm Petri dish. After treatments, medium was transferred to a 15 mL tube and cells were washed in cold phosphate-buffered saline, then, suspensions were centrifuged at 3,200 rpm for 10 minutes at 4°C. Attached cells were lysed with 20 μL lysis buffer with proteinase inhibitor. The pellets were resuspended in 10 μL lysis buffer and mixed with cell lysates, and then centrifuged at 12,000× g for 12 minutes at 4°C. The supernatants were collected and stored at −80°C. Protein concentrations of cell lysates were determined by Lowry assay through VersaMax ELISA reader.23
For Western blot, cell lysates were resolved by 12% SDS-polyacrylamide gel electrophoresis with standard running buffer at room temperature, and electrophoretically transferred to a polyvinyl difluoride membrane at 4°C. After blocking membranes and incubating it with primary antibodies overnight at 4°C, the membrane was washed and incubated with HRP-conjugated secondary antibodies, and then detected with enhanced chemiluminescence kit (UVP EC3 BioImaging Systems, Upland, CA, USA).4,5
Statistics
The data are expressed as mean ± standard error of the mean of three separate experiments. Statistical significance of differences between control and treatment groups were determined by one-way analysis of variance and then least significant difference comparison. Statistical significance was considered as P<0.05 in all experiments.
Results
Midazolam induced cell death through apoptosis in MA-10 cells
We have previously demonstrated that midazolam could induce cell death through apoptosis.5 In the present experiments, we double checked these phenomena. MA-10 cells were treated without or with different concentrations of midazolam (30 and 150 μM) for 24 hours, and the morphological changes were examined under light microscopy. MA-10 cells without midazolam treatment firmly attached with polygonal shapes, and became rounded-up with obvious blebbing in the plasma membrane after 150 μM midazolam treatment for 24 hours (Figure 1A). MTT assay was further used to confirm the effect of midazolam on cell viability. MA-10 cell viability was significantly decreased by treatment with 30 and 150 μM midazolam for 12 and 24 hours, respectively (P<0.05) (Figure 1B). These results validate that midazolam did induce cell death in MA-10 cells.
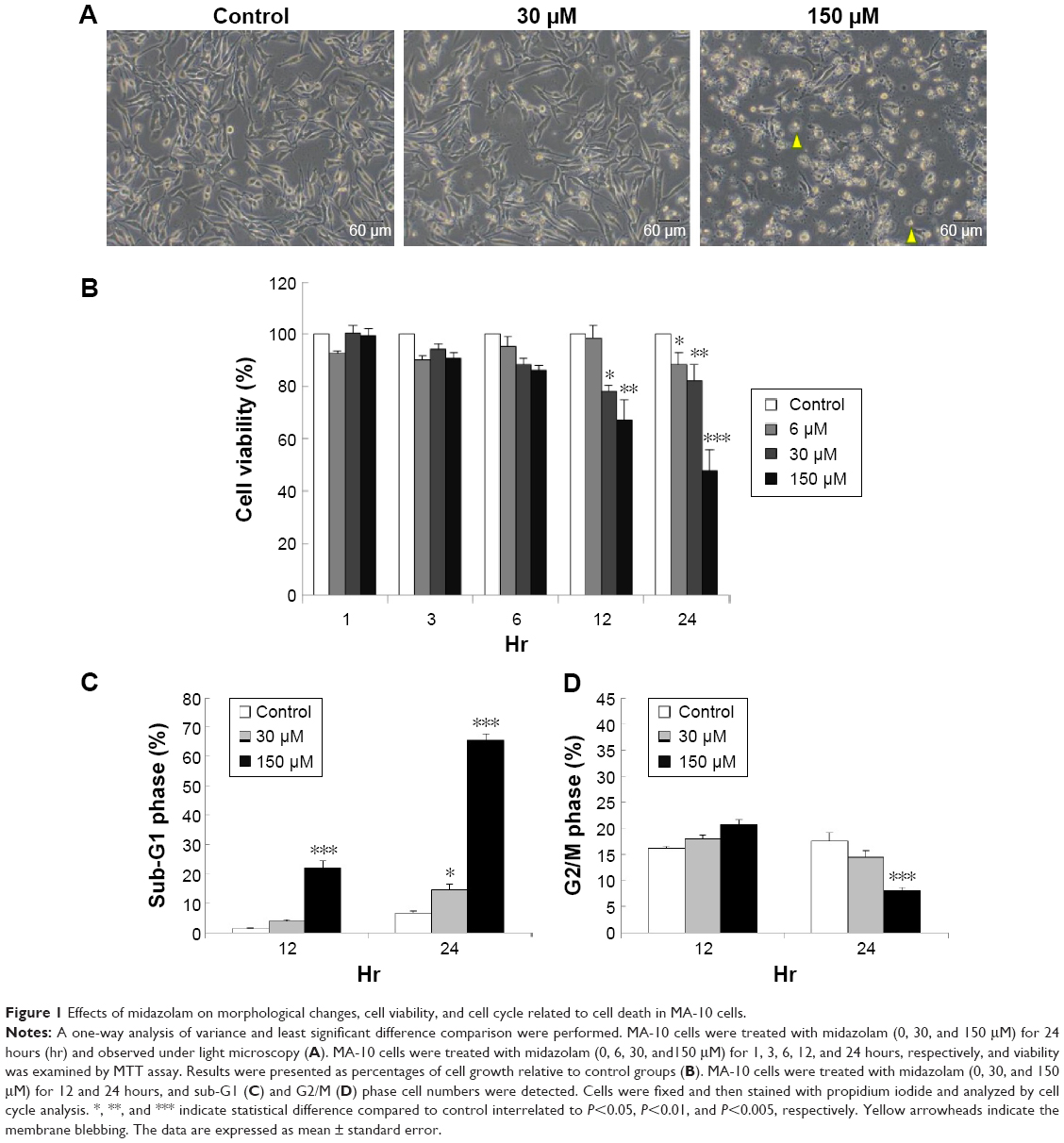 | Figure 1 Effects of midazolam on morphological changes, cell viability, and cell cycle related to cell death in MA-10 cells. |
Studies have shown that the increase of sub-G1 phase and G2/M phase arrest could cause cell death through apoptosis.24,25 To investigate whether midazolam could affect MA-10 cell cycle and result in apoptosis, cells were treated with midazolam (30 and 150 μM) for 12 and 24 hours, respectively, and their DNA contents were examined by flow cytometry. The increase of sub-G1 phase cell number was observed in MA-10 cells treated with 150 μM midazolam for 12 and 24 hours, respectively (Figure 1C) (P<0.05). However, cell number in G2/M phase significantly decreased in MA-10 cells treated with 150 μM midazolam for 24 hours (Figure 1D) (P<0.05). These data demonstrated that midazolam could regulate cell cycle by increasing sub-G1 phase with the decrease of G2/M phase to induce apoptosis in MA-10 cells.
To further verify that midazolam could induce MA-10 cell apoptosis, annexin V/PI double staining assay followed by flow cytometry analysis was used (Figure 2A). Results showed that the number of annexin V-positive (early and late apoptosis) MA-10 cells significantly increased after 24 hours of 150 μM midazolam treatment (Figure 2B and C) (P<0.05). These results again demonstrated that midazolam did induce apoptosis in MA-10 cells.
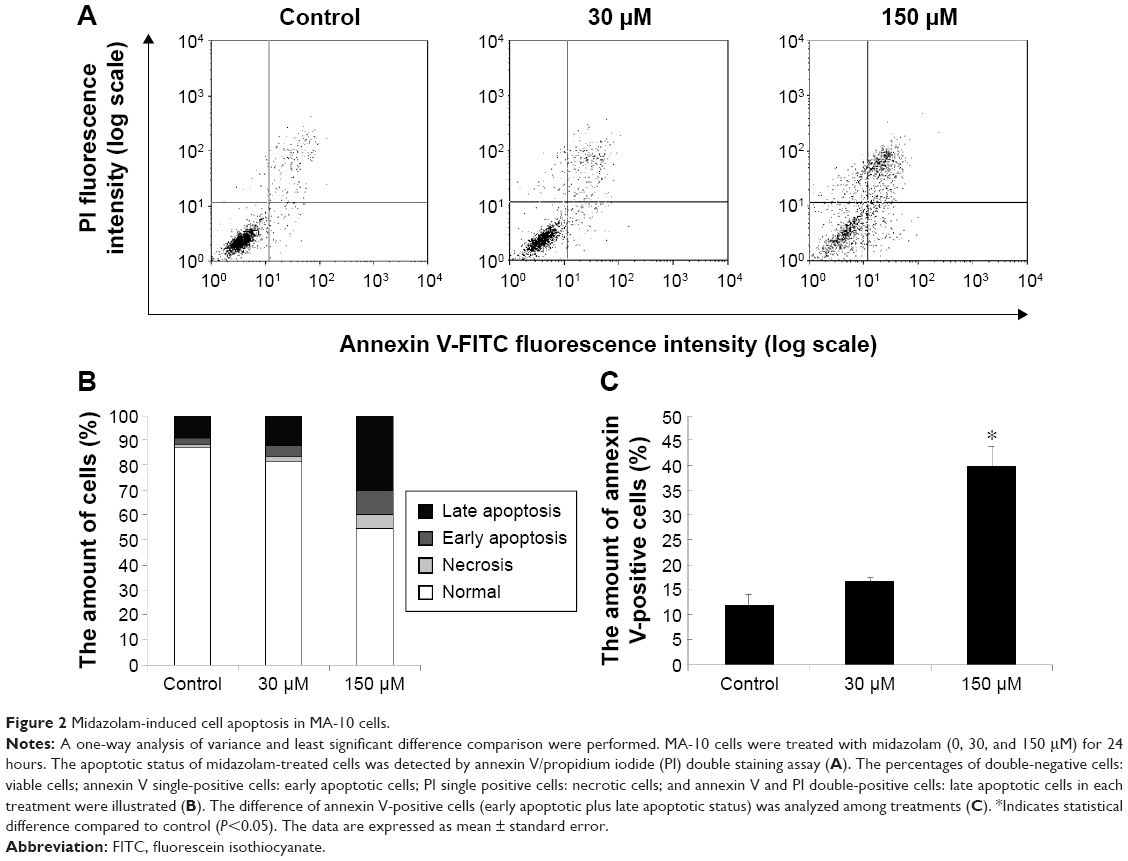 | Figure 2 Midazolam-induced cell apoptosis in MA-10 cells. |
Midazolam induced extrinsic and intrinsic caspase pathways related to apoptosis in MA-10 cells
We have previously demonstrated that midazolam can induce apoptosis by activating CASP8 and 9 pathways.5 However, whether midazolam could regulate the upstream and/or downstream pathways of CASP8 and 9 remains indefinable. Studies have shown that the association of FasL and Fas could activate death receptor pathway, which in turn could stimulate the activation of CASP8 to induce cell apoptosis.9 Thus, the expressions of Fas ligand and Fas were examined. Results showed that 12 and 24 hours 150 μM midazolam treatments significantly decreased the expressions of FasL (Figure 3A and B), and 12 hours 150 μM midazolam treatment decreased the expression of Fas (Figure 3A and C), respectively (P<0.05). These data suggested that midazolam could regulate the expressions of Fas and FasL to activate death receptor pathway and to further induce MA-10 cell apoptosis.
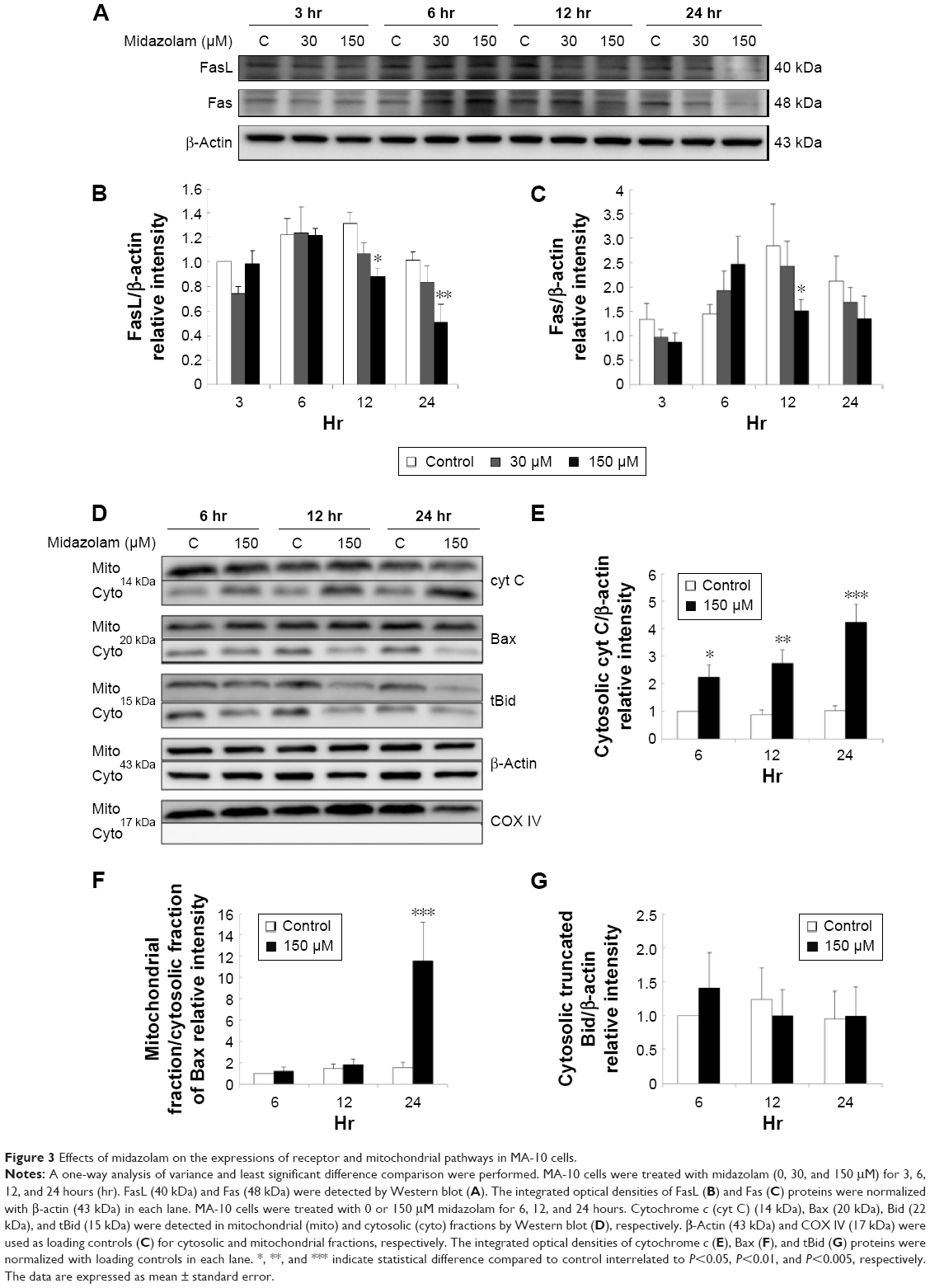 | Figure 3 Effects of midazolam on the expressions of receptor and mitochondrial pathways in MA-10 cells. |
A study has shown that mitochondrial pathway is controlled by a number of pro- and anti-apoptotic members of Bcl-2 family molecules, which in turn could modulate the activation of CASP9 to regulate cell apoptosis.26 We have observed the activation of CASP9 in midazolam-induced apoptosis in MA-10 cells.5 To further investigate the role between intrinsic pathway and Bcl-2 family proteins in midazolam-induced MA-10 cell apoptosis, the expressions of cytochrome c, Bax, and truncated Bid were examined by Western blot. Results showed that 150 μM midazolam treatment of 6, 12, and 24 hours significantly induced the release of cytochrome c in a time-dependent manner in MA-10 cells (Figure 3D and E) (P<0.05). In addition, 150 μM midazolam treatment for 24 hours significantly increased the ratio of mitochondrial and cytosolic Bax (Figure 3D and F) (P<0.05), indicating a translocation of Bax from cytosol to mitochondria. These results suggested that midazolam could significantly activate CASP9 through the release of cytochrome c and the induction of Bax translocation to induce MA-10 cell apoptosis. Moreover, the expression of truncated Bid in cytosolic fraction was not significantly affected by 150 μM midazolam treatments for 6, 12, and 24 hours (Figure 3D and G) (P>0.05), showing that midazolam might not activate the cross talk between CASP8 and 9 pathways.
Midazolam-induced autophagy related to apoptosis in MA-10 cells
Autophagy can act independently upon the apoptotic signaling pathways, and LC3-II is the first mammalian protein identified that is specifically associated with autophagosome membranes correlating with autophagosome formation.27 To determine whether midazolam could induce autophagy, MA-10 cells were stably transfected with GFP-LC3 and then treated without or with 150 μM midazolam for 24 hours. GFP-LC3 puncta formations were observed in midazolam-treated MA-10 cells (Figure 4A), suggesting that autophagosome formation was induced by midazolam. To confirm the immunofluorescence results obtained earlier, the expression of LC3-II in MA-10 cells was further assessed by Western blot. Data showed that LC3-II was significantly expressed after treatment with 150 μM midazolam for 6 and 12 hours, respectively (P<0.05) (Figure 4B).
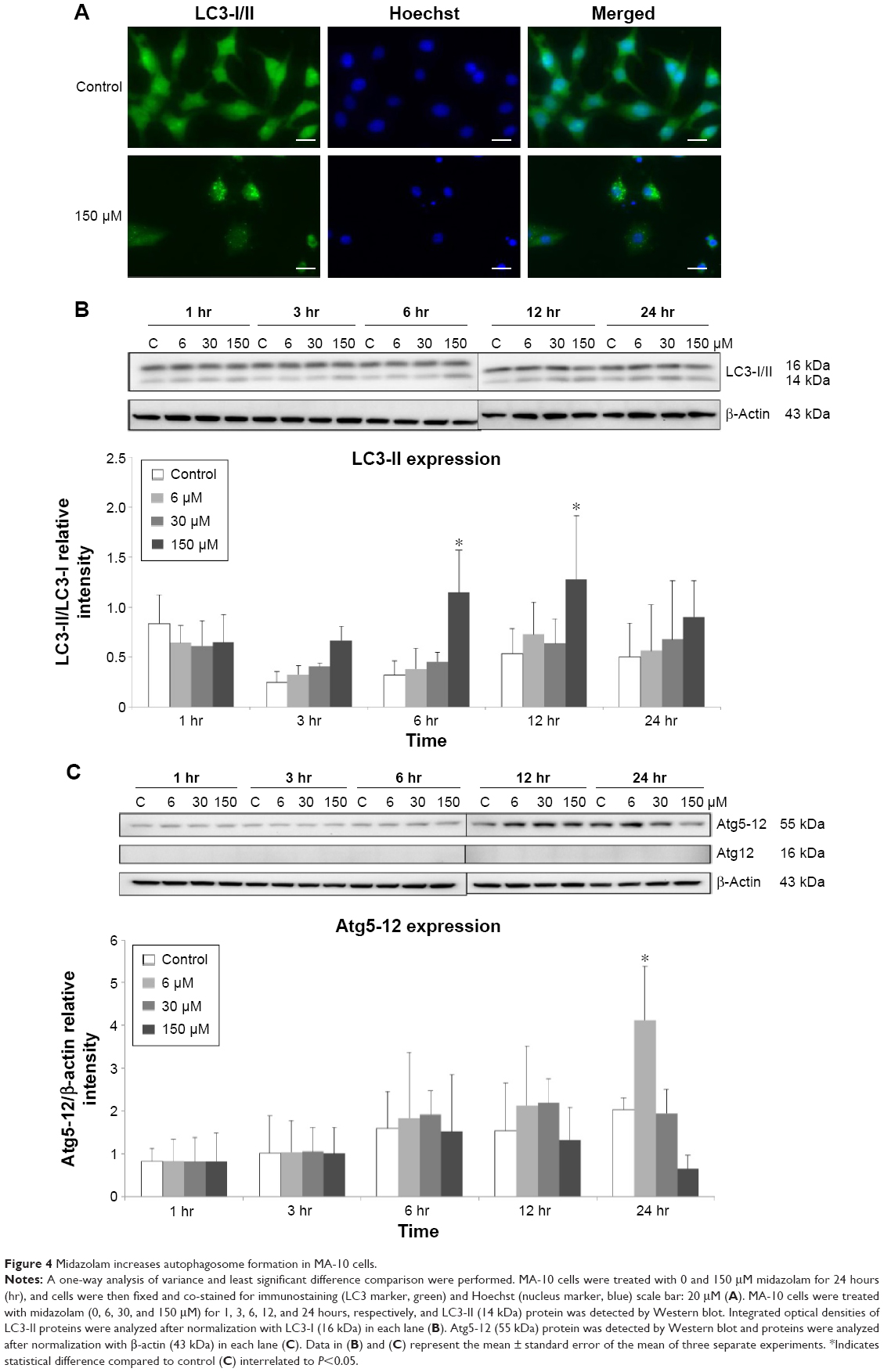 | Figure 4 Midazolam increases autophagosome formation in MA-10 cells. |
During autophagy, LC3-I is converted to LC3-II through lipidation by an Atg ubiquitin-like system that allows LC3 to associate with autophagic vesicles. Thus, the expressions of Atg5-12 were detected by Western blot in MA-10 cells treated without or with 6, 30, and 150 μM midazolam for 1, 3, 6, 12, and 24 hours, respectively. Results showed that 24-hour treatment with 6 μM midazolam significantly increased the ratio of Atg5-12 expression (P<0.05) (Figure 4C). These results strongly suggested that midazolam could activate autophagy to induce apoptosis in MA-10 cells.
Midazolam-induced ER stress pathways related to apoptosis in MA-10 cells
Previous studies have shown that the misfolded proteins could induce ER stress to restore protein homeostasis, and if the stress is prolonged, apoptotic cell death ensues.12 To examine whether midazolam could regulate ER stress pathways to induce apoptosis in MA-10 cells, the ER stress related proteins, including ATF6, IRE1α, CASP12, PERK and downstream proteins (XBP1, EIF2α, ATF4, ATF3, and CHOP), were analyzed by Western blot.
The expression of ATF6 was significantly inhibited by 24 hours 150 μM midazolam treatment (Figure 5A and B) (P<0.05), while the expressions of p-IRE1α, XBP1, and cleaved CASP12 were not affected by midazolam (Figure 5A, C–E) (P>0.05).
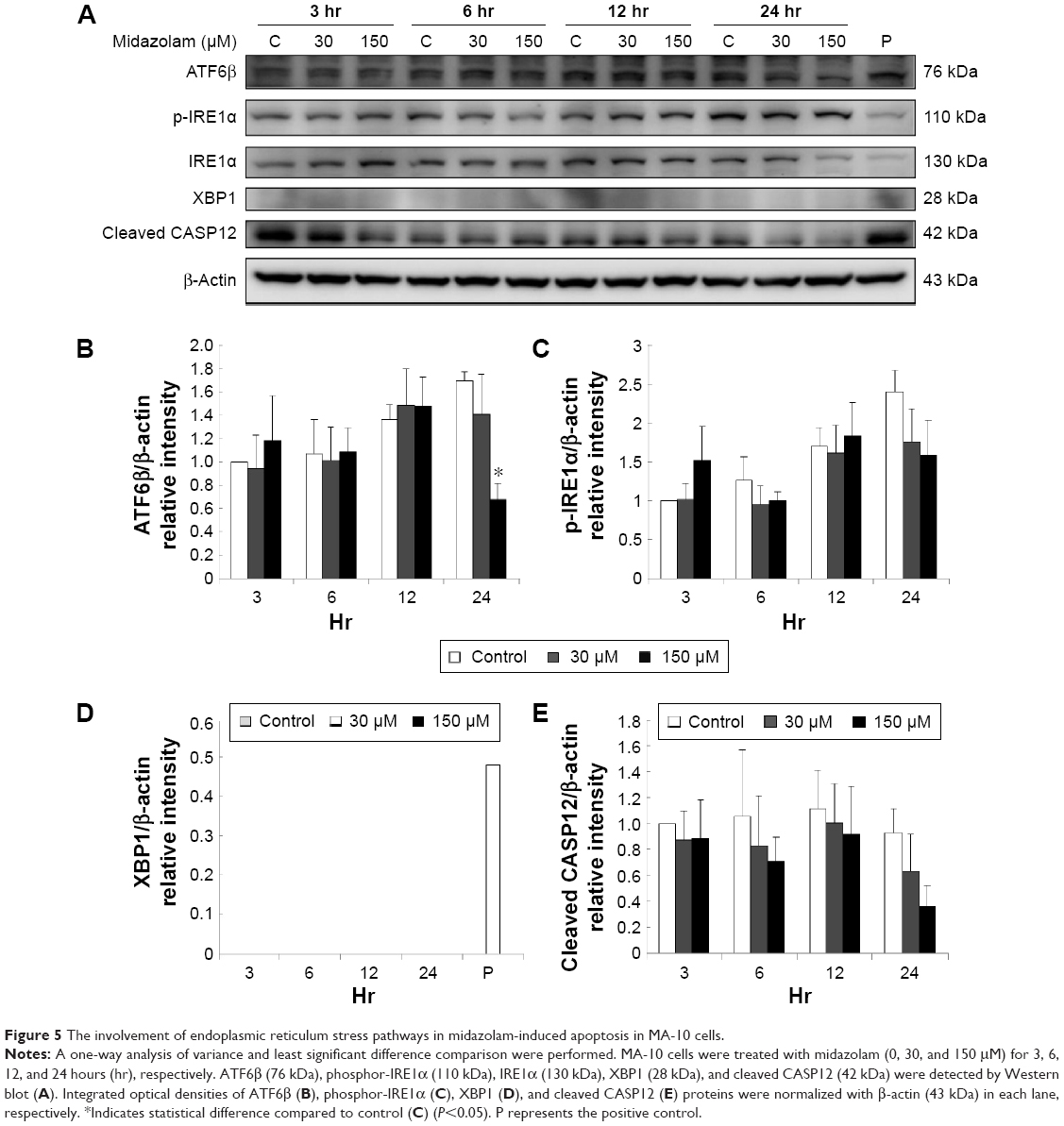 | Figure 5 The involvement of endoplasmic reticulum stress pathways in midazolam-induced apoptosis in MA-10 cells. |
We further observed the involvement of PERK-related pathway in MA-10 cells. The expression of PERK was significantly decreased by 12 and 24 hours 150 μM midazolam treatments (Figure 6A and B) (P<0.05). In addition, the expression of phosphor-EIF2α was significantly elevated by 3 hours 150 μM midazolam treatment (Figure 6A and C) (P<0.05), while the expression of ATF4 was significantly elevated by 150 μM midazolam treatment for 3 and 6 hours, respectively (Figure 6A and D) (P<0.05). Moreover, the downstream molecules, ATF3 and CHOP, were activated by 150 μM midazolam treatment for 6 and 24 hours, respectively (Figure 6A, E, and F) (P<0.05).
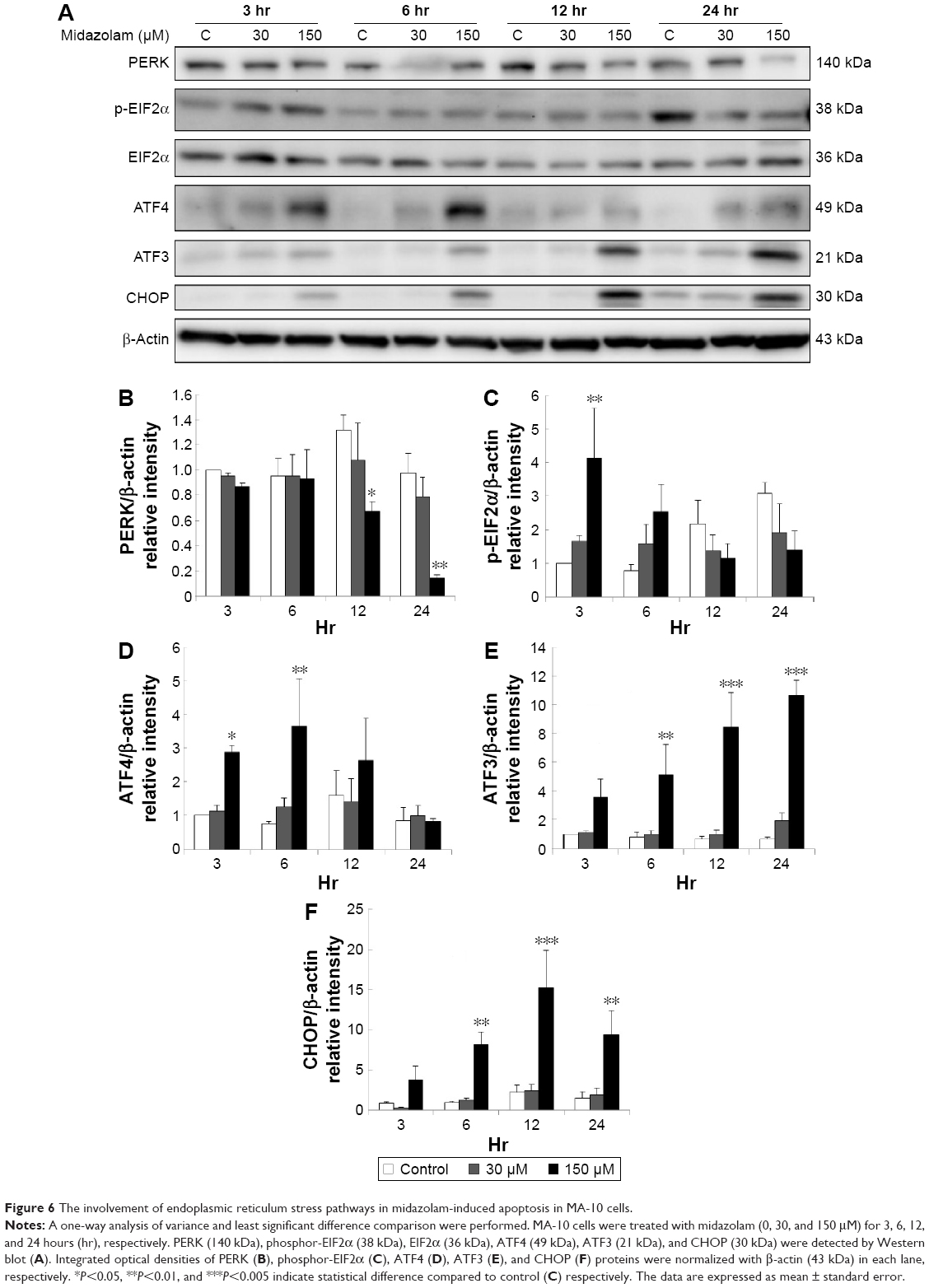 | Figure 6 The involvement of endoplasmic reticulum stress pathways in midazolam-induced apoptosis in MA-10 cells. |
These results suggest that midazolam could induce apoptosis by activating ER stress related-PERK pathway in MA-10 cells.
Midazolam-induced cell cycle and p53 pathway related to apoptosis in MA-10 cells
A previous study has demonstrated a correlation between the G2/M arrest and the induction of apoptosis.28 According to the previous experiments, 300 μM midazolam treatment for 24 hours could increase sub-G1 phase cell number with the decrease of G2/M arrest in MA-10 cells (Figure 1C and D). It is highly possible that midazolam could regulate cell cycle to induce apoptosis in MA-10 cells. Thus, the expressions of cyclin A, cyclin B, CDK1, p-53, phosphor-p53, p27, phosphor-p27, p21 and phosphor-p21 proteins, after midazolam treatment, were examined by Western blot.
Midazolam treatment of 30 μM for 24 hours and 150 μM for 6, 12, and 24 hours, respectively, could significantly reduce the expressions of cyclin A and cyclin B (Figure 7A to C) (P<0.05). Midazolam treatment of 30 μM for 3 hours and 150 μM for 3, 6, 12, and 24 hours, correspondingly, could also reduce the expression of CDK1 in MA-10 cells (Figure 7A and D) (P<0.05).
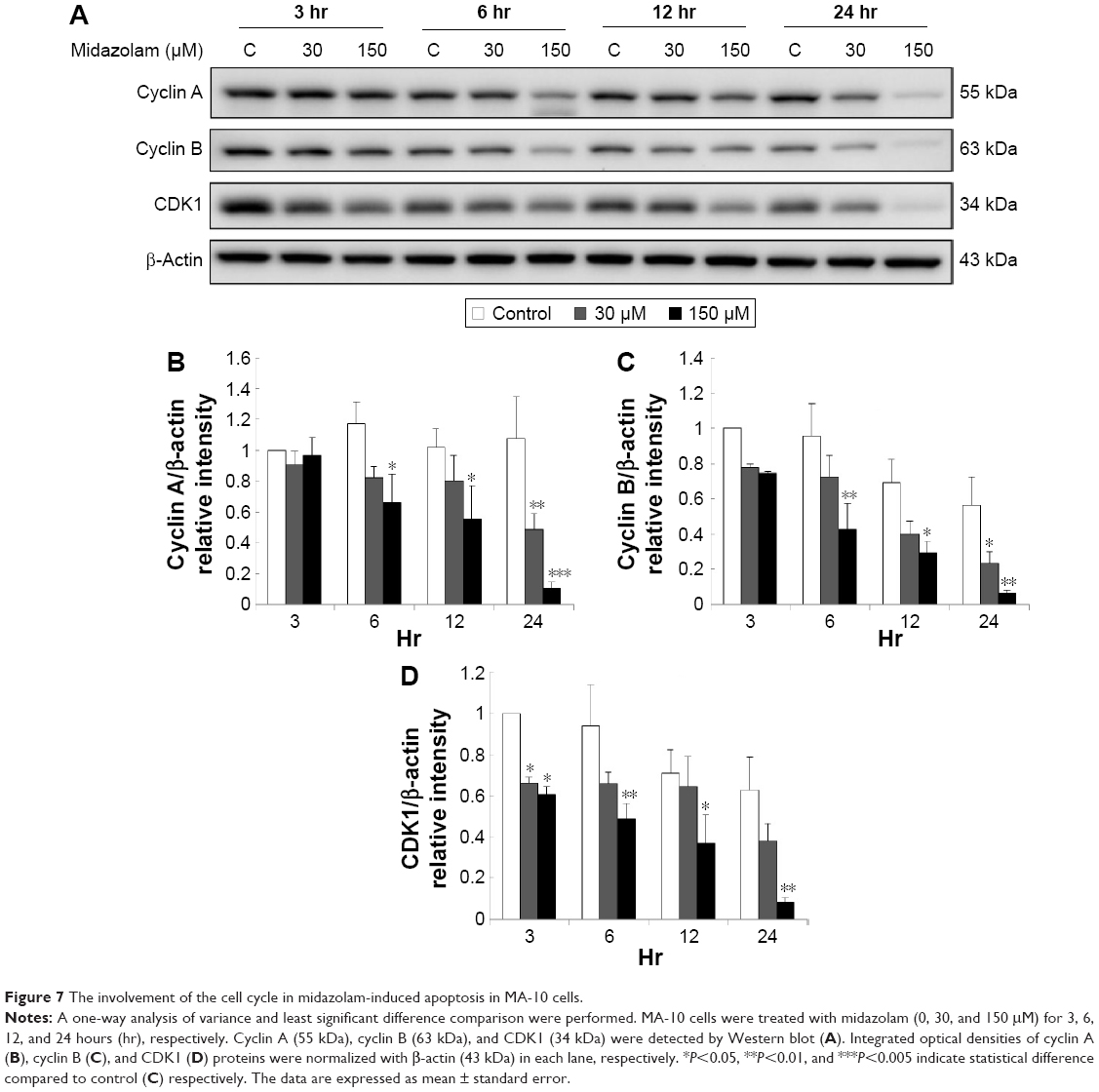 | Figure 7 The involvement of the cell cycle in midazolam-induced apoptosis in MA-10 cells. |
We further explored the possible role of p53 pathway, which is upstream to the cell cycle regulation, in midazolam-treated MA-10 cells. Midazolam treatment of 30 and 150 μM for 6, 12, and 24 hours significantly decreased the expression of phosphor-p53, respectively (Figure 8A and B) (P<0.05). However, midazolam treatment of 30 μM for 24 hours induced the expression of phosphor-p27 (Figure 8A and C) (P<0.05). Moreover, midazolam treatment of 150 μM for 6 hours significantly decreased the expression of phosphor-p21 (Figure 8A and D) (P<0.05).
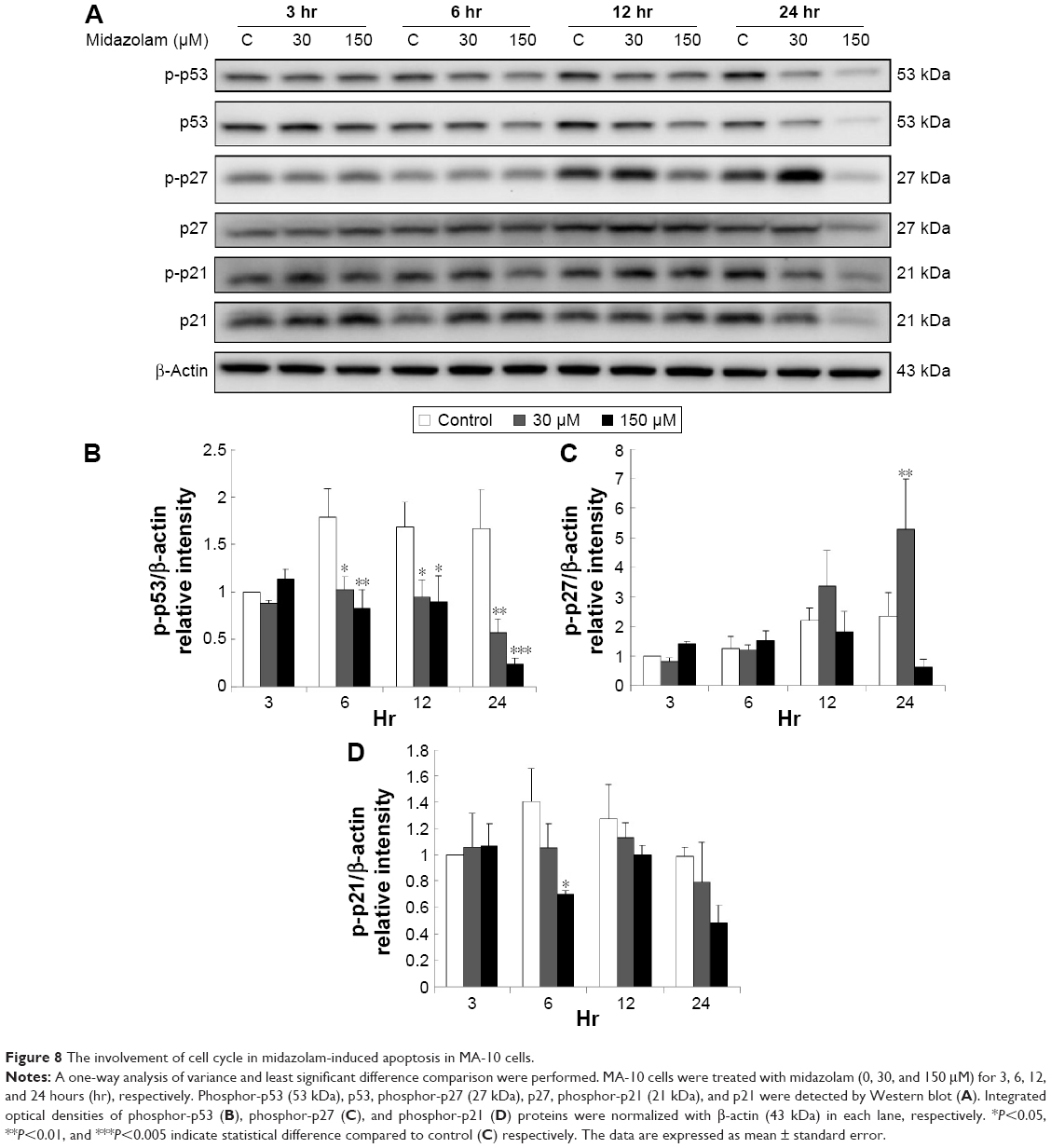 | Figure 8 The involvement of cell cycle in midazolam-induced apoptosis in MA-10 cells. |
These data strongly suggest that midazolam could possibly regulate p53 pathway to downregulate cell cycle progress and then to induce apoptosis in MA-10 cells.
Discussion
Previous studies on sedative drugs’ effects have mainly focused on its clinical dosages and side effects,29,30 and some investigations have indicated that midazolam could induce neurotoxicity.31 A recent study showed that midazolam can induce cancer cell apoptosis.32 In fact, our earlier studies have shown that midazolam could briefly induce MA-10 mouse Leydig tumor cell apoptosis by activating caspase and MAPK pathways.4,5 However, the detailed mechanism related to the upstream and downstream pathways of the caspase cascade and MAPK pathways in MA-10 cells remains elusive. Therefore, we explored whether midazolam could control ER stress, autophagy, and p53 pathways plus cell cycle regulation to induce apoptosis in MA-10 cells. As predicted, midazolam significantly reduced cell viability in a dose- and time-dependent manner, and increased sub-G1 phase in MA-10 cells in the present experiments, which is consistent with our published data.4,5 Indeed, these data also suggested that midazolam-induced apoptosis was associated with the regulation of cell cycle. Moreover, the annexin V/PI double staining assay also showed the induction of apoptosis by midazolam in a dose-dependent manner, suggesting that midazolam did truly induce cell apoptosis in MA-10 cells.
Cancer cells have different properties from normal cells to promote tumor progression, including evasion of apoptosis causing the reduction of cell death, and there are numerous anticancer drugs with the ability to induce apoptosis for therapies.33 It is well-known that apoptosis is mainly initiated by extrinsic and intrinsic signaling, and then activation of caspase cascade.34 We have illustrated that midazolam could activate CASP8 and 9 pathways to induce MA-10 cell apoptosis.5 We further determined the role of extrinsic pathway, and data showed that the expressions of Fas and FasL significantly decreased with 12-hour midazolam treatment in MA-10 cells. It is possible that midazolam could be associated with Fas and/or FasL in the early stage to stimulate CASP8 pathway inducing MA-10 apoptosis while the expression of Fas and FasL started to decrease. Thus, the decreased expressions of Fas and FasL with 12-hour midazolam treatment in MA-10 cells were observed. This postulation will need experiments to be confirmed. In fact, some studies have found that Fas ligand-independent pathways could activate the Fas death pathway by recruiting the adaptor protein FADD or depletion of membrane cholesterol.35,36 Therefore, it is also possible that midazolam might be able to activate Fas extrinsic pathway in a ligand-independent manner to induce CASP8 for MA-10 cell apoptosis. Consequently, midazolam might activate other death receptors or pro-apoptotic ligands to induce cell apoptosis. This assumption also needs further investigations of other possible extrinsic pathways.
It is well-known that mitochondrial pathway is controlled by a number of pro- and anti-apoptotic members of Bcl-2 family molecules.26 Pro-apoptotic proteins include Bid, Bim, Bax, and Bad, which are associated with insertion and oligomerization on outer mitochondrial membranes to trigger apoptosis through the induction of cytochrome c release.37 Besides, studies have shown that midazolam could initiate the mitochondrial pathway by inducing the release of cytochrome c, and then activating CASP9 in neuron cells.38 Our data showed that midazolam could induce Bax translocation and cytochrome c release in MA-10 cells, which are parallel to those studies. In addition, studies have demonstrated that CASP8 could induce the cleavage of Bid (tBid formation), and lead to cytochrome c release with the activation of CASP9 pathway in cell apoptosis.39,40 In the present study, we observed that midazolam activated CASP8, but the formation of tBid was not affected, suggesting that midazolam could induce cytochrome c release through mitochondrial pathway without involvement of CASP8 to induce MA-10 cell apoptosis.
Different physiological and pathological perturbations could lead to accumulation of unfolded or misfolded proteins in the ER lumen, which is termed ER stress. ER stress triggers the UPR to increase protein folding capacity and decrease unfolded protein load.41 If ER stress is chronically prolonged, cell death often occurs through apoptosis, which is induced by PERK, ATF6, and IRE1α.12 In the present study, we found that midazolam could induce the activations of PERK downstream proteins, including EIF2α, ATF4, ATF3, and CHOP in MA-10 cells. The activation of PERK-related pathways occurred at 3 hours, and was prolonged for 24 hours in MA-10 cells, indicating that midazolam might induce ER stress for a long period of time to induce apoptosis by activating PERK-related pathway. Interestingly, these data showed that the activations of caspase cascade and MAPK pathways were observed at 24-hour midazolam treatment,5 and the activation of ER stress pathways occurred at 3-hour treatment, which was earlier compared to caspase cascade and MAPK pathways, indicating that midazolam probably activated ER stress pathways prior to the caspase cascade and MAPK pathways to induce apoptosis in MA-10 cells.
The defect of cell division can lead to developmental abnormalities as well as cancerous growth, and the eukaryotic cell cycle is controlled by the activities of CDKs and cyclins. The blocking of CDKs and/or cyclins would obstruct cell cycle to provoke cell death.17,42 In the present study, midazolam was able to reduce activities of cyclin A, cyclin B, and CDK1 in MA-10 cells, illustrating our observations are not unprecedented compared to other studies. However, midazolam decreased the phosphorylation of p53 and p21 but increased the phosphorylation of p27, implying that midazolam could regulate p53 pathway to interfere with cell cycle inducing MA-10 apoptosis. In general, p53 pathway, including p21 and p27, would impede the activities of CDKs and/or cyclins to block cell cycle for meiosis inducing apoptotic cell death.43–45 In spite of these paradoxical observations, midazolam reduced phosphorylation of p53 and p21, we will need to under go further investigation to clarify the authentic mechanisms.
Conclusion
Midazolam could induce cell apoptosis through the activation of ER stress and regulation of cell cycle through p53 pathway with the involvement of autophagy in MA-10 mouse Leydig tumor cells. These results suggested that midazolam could be a good anticancer therapeutic drug in Leydig/testicular cancers.
Acknowledgments
This work was supported by grants from Chi Mei-NCKU hospital (CMNCKU10425) (to FCK and BMH) and the Ministry of Science and Technology (MOST 104-2320-B-006-012) (to BMH) and (MOST 104-2320-B-006-041) (to BMH), Taiwan, Republic of China.
Disclosure
The authors report no conflicts of interest in this work.
References
Reves JG, Fragen RJ, Vinik HR, Greenblatt DJ. Midazolam: pharmacology and uses. Anesthesiology. 1985;62(3):310–324. | ||
García-Pedrajas F, Arroyo J. Midazolam in anesthesiology. Rev Med Univ Navarra.1989;33(4):211–221. | ||
Olkkola KT, Ahonen J. Midazolam and other benzodiazepines. Handb Exp Pharmacol. 2008;(182):335–360. | ||
So EC, Chang YT, Hsing CH, et al. The effect of midazolam on mouse Leydig cell steroidogenesis and apoptosis. Toxicol Lett. 2010;192(2):169–178. | ||
So EC, Lin YX, Tseng CH, et al. Midazolam induces apoptosis in MA-10 mouse Leydig tumor cells through caspase activation and the involvement of MAPK signaling pathway. Onco Targets Ther. 2014;7:211–221. | ||
Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem. 2011;351(1–2):41–58. | ||
Zimmermann KC, Bonzon C, Green DR. The machinery of programmed cell death. Pharmacol Ther. 2001;92(1):57–70. | ||
Bender CE, Fitzgerald P, Tait SW, et al. Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc Natl Acad Sci U S A. 2012;109(13):4904–4909. | ||
Lewis-Wambi JS, Jordan VC. Estrogen regulation of apoptosis: how can one hormone stimulate and inhibit? Breast Cancer Res. 2009;11(3):206. | ||
Esteve JM, Knecht E. Mechanisms of autophagy and apoptosis: Recent developments in breast cancer cells. World J Biol Chem. 2011;2(10):232–238. | ||
Kim SR, Lee YC. Endoplasmic reticulum stress and the related signaling networks in severe asthma. Allergy Asthma Immunol Res. 2015;7(2):106–117. | ||
Luo K, Cao SS. Endoplasmic reticulum stress in intestinal epithelial cell function and inflammatory bowel disease. Gastroenterol Res Pract. 2015;2015:328791. | ||
Sanderson TH, Gallaway M, Kumar R. Unfolding the unfolded protein response: unique insights into brain ischemia. Int J Mol Sci. 2015;16(4):7133–7142. | ||
Logue SE, Cleary P, Saveljeva S, Samali A. New directions in ER stress-induced cell death. Apoptosis. 2013;18(5):537–546. | ||
Kania E, Pajak B, Orzechowski A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. Biomed Res Int. 2015;2015:352794. | ||
Kitamura M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am J Physiol RenaL Physiol. 2008;295(2):F323–F334. | ||
Boxem M. Cyclin-dependent kinases in C. elegans. Cell Div. 2006;1:6. | ||
Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266(5192):1821–1828. | ||
Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4(1):1–7. | ||
Bates S, Vousden KH. p53 in signaling checkpoint arrest or apoptosis. Curr Opin Genet Dev. 1996;6(1):12–18. | ||
Korkolopoulou P, Oates J, Kittas C, Crocker J. p53, c-myc p62 and proliferating cell nuclear antigen (PCNA) expression in non-Hodgkin’s lymphomas. J Clin Pathol. 1994;47(1):9–14. | ||
Green LM, Reade JL, Ware CF. Rapid colorimetric assay for cell viability: application to the quantitation of cytotoxic and growth inhibitory lymphokines. J Immunol Methods. 1984;70(2):257–268. | ||
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. | ||
Paul-Samojedny M, Suchanek R, Borkowska P, et al. Knockdown of AKT3 (PKBgamma) and PI3KCA suppresses cell viability and proliferation and induces the apoptosis of glioblastoma multiforme T98G cells. Biomed Res Int. 2014;2014:768181. | ||
Zhang C, Chen Z, Zhou X, et al. Cantharidin induces G/M phase arrest and apoptosis in human gastric cancer SGC-7901 and BGC-823 cells. Oncol Lett. 2014;8(6):2721–2726. | ||
Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. | ||
Hung SY, Huang WP, Liou HC, et al. Autophagy protects neuron from Aβ-induced cytotoxicity. Autophagy. 2009;5(4):502–510. | ||
Wang Y, Ji P, Liu J, et al. Centrosome-associated regulators of the G(2)/M checkpoint as targets for cancer therapy. Mol Cancer. 2009;8:8. | ||
Harris CE, Grounds RM, Murray AM, et al. Propofol for long-term sedation in the intensive care unit. A comparison with papaveretum and midazolam. Anaesthesia. 1990;45(5):366–372. | ||
Radke J. Analgesia and sedation in intensive care patients. Anaesthesist 1992;41(12):793–808. | ||
Yilmaz E, Hough KA, Gebhart GF, Williams BA, Gold MS. Mechanisms underlying midazolam-induced peripheral nerve block and neurotoxicity. Reg Anesth Pain Med. 2014;39(6):525–533. | ||
Mishra SK, Kang JH, Lee CW, et al. Midazolam induces cellular apoptosis in human cancer cells and inhibits tumor growth in xenograft mice. Mol Cells. 2013;36(3):219–226. | ||
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. | ||
Oliveira JB, Gupta S. Disorders of apoptosis: mechanisms for autoimmunity in primary immunodeficiency diseases. J Clin Immunol. 2008;28 Suppl 1:S20–S28. | ||
Micheau O, Solary E, Hammann A, Dimanche-Boitrel MT. Fas ligand-independent, FADD-mediated activation of the Fas death pathway by anticancer drugs. J Biol Chem. 1999;274(12):7987–7992. | ||
Gniadecki R. Depletion of membrane cholesterol causes ligand-independent activation of Fas and apoptosis. Biochem Biophys Res Commun. 2004;320(1):165–169. | ||
Sarosiek KA, Chi X, Bachman JA, et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell. 2013;51(6):751–765. | ||
Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135(3):815–827. | ||
Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. | ||
Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta. 2011;1813(4):558–563. | ||
Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev. 2011;91(4):1219–1243. | ||
Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28(33):2925–2939. | ||
Somasundaram K. Tumor suppressor p53: regulation and function. Front Biosci. 2000;5:D424–D437. | ||
Fabris L, Berton S, Pellizzari I, et al. p27kip1 controls H-Ras/MAPK activation and cell cycle entry via modulation of MT stability. Proc Natl Acad Sci U S A. 2015;112(45):13916–13921. | ||
Pan BS, Wang YK, Lai MS, Mu YF, Huang BM. Cordycepin induced MA-10 mouse Leydig tumor cell apoptosis by regulating p38 MAPKs and PI3K/AKT signaling pathways. Sci Rep. 2015;5:13372. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.