Back to Journals » OncoTargets and Therapy » Volume 11
Midazolam activates caspase, MAPKs and endoplasmic reticulum stress pathways, and inhibits cell cycle and Akt pathway, to induce apoptosis in TM3 mouse Leydig progenitor cells
Authors Kang FC ![]() , Wang SC, Chang MM
, Wang SC, Chang MM ![]() , Pan BS, Wong KL, Cheng KS
, Pan BS, Wong KL, Cheng KS ![]() , So EC
, So EC ![]() , Huang BM
, Huang BM ![]()
Received 18 October 2017
Accepted for publication 15 January 2018
Published 15 March 2018 Volume 2018:11 Pages 1475—1490
DOI https://doi.org/10.2147/OTT.S154442
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Fu-Chi Kang,1,* Shu-Chun Wang,2,* Ming-Min Chang,2 Bo-Syong Pan,3 Kar-Lok Wong,4 Ka-Shun Cheng,4,5 Edmund Cheung So,4,6 Bu-Miin Huang2,7
1Department of Anesthesia, Chi Mei Medical Center, Chiali, Tainan, Taiwan, Republic of China; 2Department of Cell Biology and Anatomy, College of Medicine, National Cheng Kung University, Tainan, Taiwan, Republic of China; 3Department of Cancer Biology, Wake Forest University School of Medicine, Winston Salem, NC, USA; 4Department of Anesthesia, China Medical University, Taichung, Taiwan, Republic of China; 5Department of Anesthesiology, The Qingdao University Yuhuangding Hospital, Yantai, Shandong, China; 6Department of Anesthesia, An Nan Hospital, China Medical University, Tainan, Taiwan, Republic of China; 7Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, Taiwan, Republic of China
*These authors contributed equally to this work
Background: Midazolam (MDZ) has powerful hypnosis, amnesia, anti-anxiety and anticonvulsant effects. Studies have shown that prenatally developmental toxicity of diazepam can be observed in many organs/tissues. However, it remains elusive in male reproductive system.
Materials and methods: TM3 mouse Leydig progenitor cell line was used to determine whether MDZ has any unfavorable effects.
Results: Midazolam significantly decreased cell viability in dose- and time-dependent manners in TM3 cells. In flow cytometry analysis, midazolam significantly increased subG1 phase cell numbers, and annexin V/PI double staining assay further confirmed that MDZ induced apoptosis in TM3 cells. Moreover, MDZ significantly induced the expression of caspase-8 and -3 proteins and the phosphorylation of JNK, ERK1/2 and p38. Besides, MDZ didn’t activate Akt pathway in TM3 cells. Furthermore, the expressions of p-EIF2α, ATF4, ATF3 and CHOP were induced by midazolam, suggesting that midazolam could induce apoptosis through endoplasmic reticulum (ER) stress in TM3 cells. Additionally, the expressions of cyclin A, cyclin B and CDK1 were inhibited by midazolam through the regulation of p53 in TM3 cells, indicating that midazolam could regulate cell cycle to induce apoptosis.
Conclusion: Midazolam could activate caspase, MAPKs and ER stress pathways and impede Akt pathway and cell cycle to induce apoptosis in TM3 mouse Leydig progenitor cells.
Keywords: midazolam, TM3, Leydig progenitor cells, apoptosis, caspase, MAPKs, ER stress, cell cycle
Introduction
Midazolam (MDZ), a general benzodiazepine sedative and anesthetic agent, has powerful hypnosis, amnesia, anti-anxiety, anticonvulsant and skeletal muscle relaxant effects by improving Cl− channel gated through the γ-aminobutyric acid type A receptor in the central nervous system.1 However, potential prenatal developmental toxicity of diazepam related to neurogenesis has been assessed in several studies.2–5 A previous study has also illustrated that MDZ could suppress cell viability and osteogenic differentiation in human mesenchymal stem cells.6 It is highly possible that reproductive system will also be affected by anesthetic agent during fetal period, which should be profoundly investigated. In males, Leydig stem and progenitor cells are very important in testis development during the fetal period for normal reproductive functions.7 Studies have demonstrated that ontogenesis of Leydig cell function involves at least two generations of cells. The first generation develops during fetal life, and these fetal Leydig cells are responsible for the masculinization of the male urogenital system.7–9 These cells regress thereafter, although fetal Leydig cells persevere in adult life and their number remains constant at least until 90 days post-partum in the rat.8,9 The second Leydig cell population appears during puberty and produces the hormone testosterone, which is required for the onset of spermatogenesis and maintenance of male reproductive functions.8,9 It is well known that various factors could affect Leydig stem/progenitor cell development during embryo and fetal periods, which would result in male infertility.9 In fact, studies have shown that MDZ could regulate Leydig cell steroidogenesis and apoptosis.10–14
Apoptosis is characterized by some features, such as nuclear chromatin condensation, cytoplasmic shrinkage, dilated endoplasmic reticulum and membrane blebbing.15 The biochemical symbol of apoptosis is the activation of caspases (cysteine aspartate-specific proteases), which are the members of the family of cysteine proteases related to ced-3, the “death gene” of the nematode Caenorhabditis elegans.16,17 There are at least two regulatory pathways for the caspase activation: extra-mitochondrial pathway (extrinsic pathway), which is mediated by death receptors,18 and intra-mitochondrial pathway (intrinsic pathway), which is mediated by cytochrome c.19 Studies have shown that receptor-associated adaptor proteins, such as FADD (Fas-associated death domain), facilitate close association of certain caspases to promote caspase auto-processing.20–22 Similar adaptor molecules, such as CED-4 homologue and Apaf-1 (Apoptotic protease-activating factor), may play key roles in promoting apoptosis by clustering caspases at intracellular sites.23 On the other hand, mitochondria can sense apoptotic signals and convey them to activate adaptor Apaf-1 via the release of cytochrome c. Cytochrome c, Apaf-1 and procaspase-9 form a complex called apoptosome. The apoptosome formation can cause the autolytical cleavage of procaspase-9 to form active caspase-9, which can further cleave caspase-3, -6 and -7.24
A defect in cell division can lead to developmental abnormalities as well as cancerous growth, and the eukaryotic cell cycle is controlled by the activities of cyclin-dependent kinases (CDKs) and cyclins.25 A previous study has demonstrated that a correlation between the G2/M arrest and the induction of mitogen-activated protein kinases (MAPKs) is related to apoptosis.26 MAPK pathway plays a key role in the progression of cancer, and can regulate cell growth, proliferation, differentiation, migration, apoptosis and so on.27,28 MAPKs are protein-serine/threonine kinases, including ERK1/2, JNK and p38. Each subgroup of MAPKs is activated through a cascade of phosphorylation events, beginning with the activation of MAPK kinase kinases (MAP3Ks) to regulate cell fate.29 Study has demonstrated that MDZ triggers apoptosis by activating JNK and p38 in MA-10 mouse Leydig tumor cells.12–14
Another cell-fate-regulating pathway, the Akt signaling pathway, is a pro-survival pathway and acts by inhibiting apoptotic signal cascades and activating pro-survival signal.30 The activation of Akt inhibits a number of pro-apoptotic Bcl-2 family members, including Bad, Bax and Bim.31 Akt also positively regulates anti-apoptotic pathways, inducing the activation of NF-κB transcription factor, which promotes the transcription of a wide range of anti-apoptotic genes, in particular Bcl-2 and Bcl-x-L.31 Scientific data have indicated that the attenuation of Akt has induced apoptosis in the human malignant testicular germ cells.32
In protein homeostasis, unfolded protein response (UPR) will be stimulated in response to ER stress in cells.33 Three signaling pathways, protein kinase RNA (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring protein-1α (IRE1α), will then be activated by UPR.34 Study has shown that PERK activation could phosphorylate eukaryotic translation initiation factor 2α (eIF2α) to selectively induce the transcription factor ATF4, which could further increase the expression of pro-apoptotic CCAAT/enhancer-binding protein-homologous protein (CHOP) to induce cell apoptosis.35 In addition, ATF6 transited to the Golgi will be cleaved by site-1 protease (S1P) and site-2 protease (S2P) to become an activated transcription factor ATF6.35 Moreover, activated-IRE1 could remove a small intron from the mRNA encoding X-box binding protein 1 (XBP1), which will become an active transcription factor. Combination of ATF6 and IRE1 pathways can then induce CHOP expression to trigger apoptosis.36 In addition, caspase-12 pathway could be stimulated by ER stress through an interaction with IRE1 to stimulate apoptosis.37
The p53 protein is activated in response to numerous factors, including DNA damage, oxidative stress and osmotic shock, and deregulated expression of oncogenes.38 p53 induces expression of genes involved in both death receptor-dependent (CD95 and DR5) and mitochondrion-dependent (PUMA, Bax, Bid) apoptosis pathways. The p53 protein can also inhibit the Bcl-2 family of proteins, which are involved in promoting cell survival.39 Cyclin-dependent kinase inhibitor 1A (CDKN1A, also known as p21) is an important part of the p53 response. In the nucleus, p21 inhibits CDK1 and 2 and thereby blocks mitosis in response to DNA damage. In the cytoplasm, p21 blocks apoptosis by inhibiting CASP3.40 It has been shown that TGFβ signals are transmitted through type I and II transmembrane serine/threonine receptors and inhibit cell proliferation by causing a G1 cell-cycle arrest through the activities of SMAD family member 2, 3, and 4 (SMAD2-4). This signaling pathway also regulates the expression of c-Myc, p15 and p21.41
TM3 cells are developed from neonatal BALB/c mouse testicular interstitial cells with LH receptor and can respond to trophic hormones to produce testosterone.42–44 Also, TM3 cells have similar progenitor cell characteristics.9,44 Thus, TM3 cell line is suitable to study the apoptotic effect of MDZ and investigate the associated mechanisms. In the present study, we have observed that MDZ induced cell rounding up and detachment phenomena; decreased cell number; stimulated cell death; regulated cell cycle; and activated caspase MAPK, Akt, ER stress and p53 pathways in TM3 mouse Leydig progenitor cells. This study revealed how MDZ could regulate mouse Leydig progenitor cell development, thus enriching the knowledge on how exposure to anesthetic agents during the fetal period could possibly affect testicular normal development resulting in male abnormal reproductive functions.
Materials and methods
Chemicals
MDZ was purchased from Roche Products Ltd (Welwyn Garden City, United Kingdom). Waymouth MB 752/1 medium, propidium iodide (PI), penicillin-streptomycin, RNase A, Folin and Ciocalteu’s phenol regent, ethylene diamine tetraacetic acid (EDTA), 30% acrylamide/bis-acrylamide solution, MTT and monoclonal antibody against β-actin were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Fetal bovine serum (FBS), Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 and trypsin-EDTA were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Gentamycin sulfate was purchased from A.G. Scientific (San Diego, CA, USA). Sodium chloride, potassium chloride, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid and Tris base were purchased from JT Bacer (Phillipsburg, NJ, USA). Disodium hydrogen phosphate, potassium dihydrogen phosphate, and tissue culture grade sodium bicarbonate were purchased from Riedel-deHaen (Seelze, Germany). Sucrose was purchased from Panreac (Barcelona, Spain). Hydrochloric acid, sodium dodecyl sulfate (SDS), Tween 20 and dimethyl sulfoxide (DMSO) were purchased from Merck (Darmstadt, Germany). Isoton II was purchased from Beckman Coulter (Brea, CA, USA). Micro BCA protein assay kit was purchased from Thermo Fisher Scientific. Donkey anti-rabbit IgG conjugated with horseradish peroxidase and donkey anti-mouse IgG conjugated with horseradish peroxidase were purchased from PerkinElmer Inc. (Waltham, MA, USA). Annexin V-FITC apoptosis detection kit was purchased from Strong Biotech (Taipei, Taiwan). Polyclonal antibodies against cleaved caspase-8, cleaved caspase-9, phosphor-ERK1/2, ERK1/2, phosphor-JNK, JNK, phosphor-p38 MAPK, p38 MAPK, phosphor-Akt, Akt, cleaved caspase-12, Bax, cytochrome C and COX IV were purchased from Cell Signaling (Beverly, MA, USA). Monoclonal antibodies against cleaved caspase-3, p27, p53, phosphor-p53 (Ser 15), IRE1α, phosphor-EIF2α, PERK and ATF4 were purchased from Cell Signaling. Polyclonal antibodies against ATF3, CHOP, CDK1, cyclin A and phosphor-p21 (Thr 145) were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Polyclonal antibody against phosphor-IRE1α was purchased from Abcam (Cambridge, UK). Polyclonal antibodies against ATF6β, EIF2α and XBP1 were purchased from Abgent (San Diego, CA, USA). Polyclonal antibodies against phosphor-p27 (Ser 10), cyclin B and p21 were purchased from Gene Tex (Irvine, CA, USA). Enhanced chemiluminescence (ECL) detection kit was purchased from EMD Millipore (Billerica, MA, USA).
Cell culture
TM3 mouse Leydig progenitor cells were purchased from ATCC (Manassas, VA, USA) and maintained in DMEM/F12 medium supplemented with 10% FBS at 37°C in a humidified environment containing 95% air and 5% CO2 for all the following experiments.
Morphology observation
TM3 cells were seeded at a concentration of 6×105/mL in 6 cm Petri dishes with 2 mL culture medium and treated without or with different concentrations of MDZ (150 and 300 μM) for 24 hours. Cell morphology was observed under Olympus CK40 light microscopy and images were recorded by Olympus DP20 digital camera (Olympus, Tokyo, Japan).
MTT viability test
MTT assay is a colorimetric assay for assessing cell viability.45 TM3 cells were seeded at a concentration of 8×103 cell/well. After 70%–80% confluence was obtained, cells were treated without or with different concentrations of MDZ (6, 30, 150 or 300 μM) for 1, 3, 6, 12 and 24 hours. The selected concentrations of MDZ were in accordance with those used regularly in clinical applications. At the end of experiments, each well had MTT (0.5 mg/mL) added and plates were incubated at 37°C for 4 hours. Then, the medium was discarded and 50 μL of DMSO was added into each well to dissolve the crystals by keeping the plates on a shaker at 37°C for 20 minutes in the dark. The cell viability was detected at a wavelength of 570 nm by VersaMax ELISA reader (Molecular Devices, Sunnyvale, CA, USA).45
Cell cycle analysis
To investigate whether MDZ could induce death in TM3 mouse Leydig progenitor cells through apoptosis, their DNA contents were examined by PI staining through flow cytometric analysis. TM3 cells were seeded at a concentration of 6×105 in 6 cm Petri dishes with 2 mL culture medium and treated without or with different concentrations of MDZ (150 and 300 μM) for 12 and 24 hours. Cells were harvested by trypsin digestion, centrifuged and then washed with Isoton II and fixed with 70% ethanol for at least 2 hours at −20°C. After fixation, cells were washed with cold Isoton II and then collected after centrifugation. Cell suspensions were mixed with 100 μg/mL RNase and stained with 40 μg/mL PI solution for 30 minutes. The stained cells were analyzed at a wavelength of 488 nm for PI detection by FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Cells in subG1 phase have less DNA contents on cell cycle distribution, which is considered to be a result of DNA fragmentation and an outcome of cell apoptosis.
Annexin V/PI double staining assay
After harvesting the cells with trypsin and washing with 2 mL of culture medium, cell suspensions were centrifuged at 300× g for 10 minutes at 4°C. The pellets were resuspended with cold Isoton II and centrifuged again. The pellets were mixed with 100 μL staining solution for 15 minutes according to the user’s manual of Annexin V-FITC apoptosis detection kit from Strong Biotech. The stained cells were analyzed at 488 nm excitation, using 515 nm band pass filter for FITC detection and >600 nm band pass filter for PI detection, by FACScan flow cytometer (Becton Dickinson). The double-negative cells (viable), annexin V single-positive cells (early apoptotic), PI single-positive cells (necrotic), and double-positive cells (late apoptotic) could be illustrated in four quadrants.46
Protein extraction and Western blot
Cells were seeded in 6 cm Petri dishes. After treatments, medium was transferred to 15 mL tubes and cells were washed with cold PBS, and then suspensions were centrifuged at 600× g for 10 minutes at 4°C. Attached cells were lysed by using 20 μL of lysis buffer with proteinase inhibitor. The pellets were resuspended with 10 μL of lysis buffer and mixed with cell lysates, and then the suspension was centrifuged at 12,000× g for 12 minutes at 4°C. The supernatants were collected and stored at −80°C. Protein concentrations of cell lysates were determined by the Lowry assay.47
For Western blot, cell lysates were dissolved in 12% SDS-PAGE gel with standard running buffer at room temperature and electrophoretically transferred to polyvinyldifluoride membrane at 4°C. After blocking the membrane and incubating it with primary antibodies overnight at 4°C, the membrane was washed and incubated with HRP-conjugated secondary antibodies, and then detected by ECL kit through UVP EC3 BioImaging Systems (UVP, Upland, CA, USA).
Statistical analysis
The data are expressed as mean ± standard error of the mean (SEM) of three separate experiments. Statistical significance of differences between control and treatment groups was determined by one-way analysis of variance (ANOVA) and then LSD comparison. Statistical significance was considered as p<0.05 in all experiments.
Results
MDZ induced cell death through apoptosis in TM3 cells
TM3 cells were treated without or with different concentrations of MDZ (30 and 150 μM) for 24 hours, and results showed that cell shrinkage with membrane blebbing could be observed by 150 μM MDZ treatment (Figure 1A), indicating that MDZ could induce TM3 cell death possibly through apoptosis. To confirm the cell death effect of MDZ on TM3 cells, MTT viability test was performed. TM3 cells were treated with 6, 30, 150 and 300 μM concentrations for 1, 3, 6, 12 and 24 hours, and results demonstrated that MDZ from 150 to 300 μM for 3 to 24 hours significantly decreased cell viability (Figure 1B) (p<0.05). After treatment with 150 μM MDZ for 24 hours, cell viability of TM3 cells decreased to 74%±5.6% (Figure 1B).
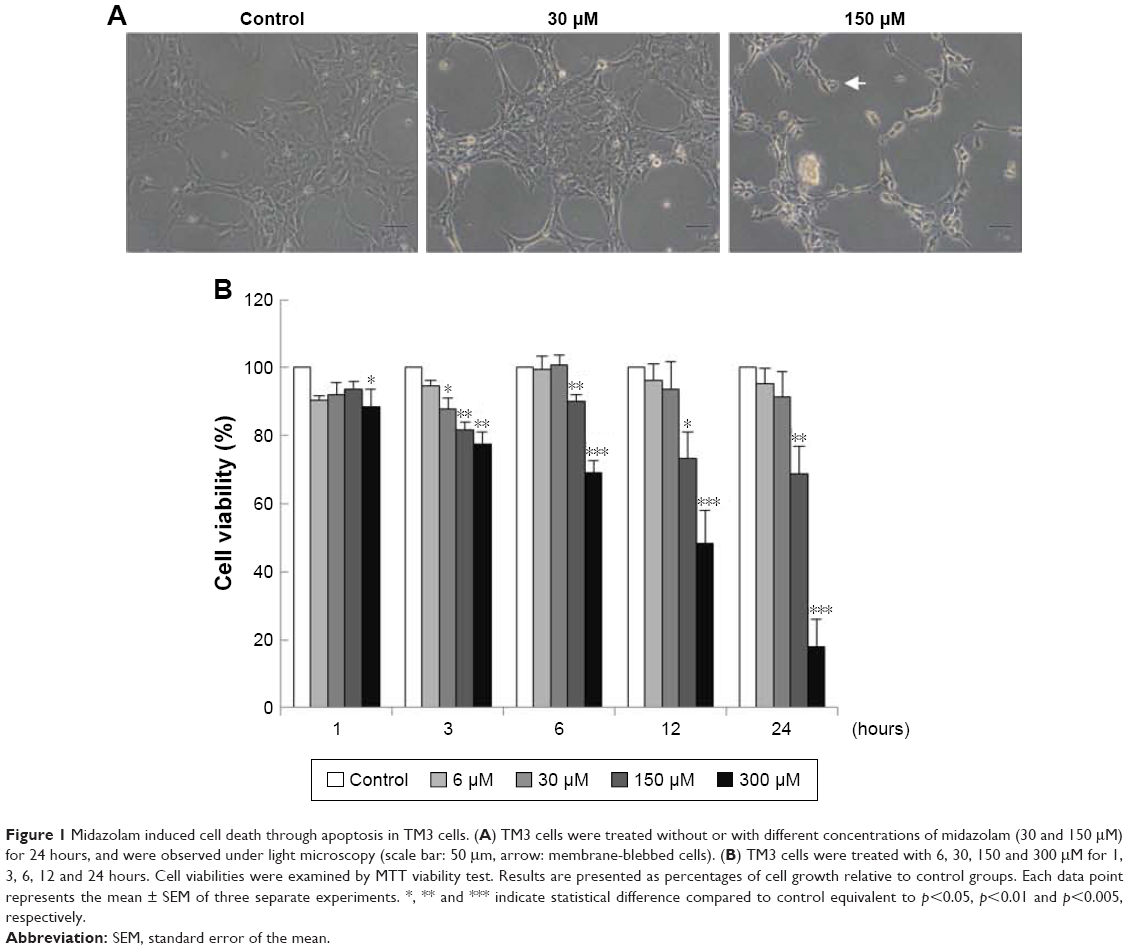 | Figure 1 Midazolam induced cell death through apoptosis in TM3 cells. (A) TM3 cells were treated without or with different concentrations of midazolam (30 and 150 μM) for 24 hours, and were observed under light microscopy (scale bar: 50 μm, arrow: membrane-blebbed cells). (B) TM3 cells were treated with 6, 30, 150 and 300 μM for 1, 3, 6, 12 and 24 hours. Cell viabilities were examined by MTT viability test. Results are presented as percentages of cell growth relative to control groups. Each data point represents the mean ± SEM of three separate experiments. *, ** and *** indicate statistical difference compared to control equivalent to p<0.05, p<0.01 and p<0.005, respectively. |
MDZ regulated cell cycle to induce apoptosis in TM3 cells
To investigate whether MDZ could affect cell cycle to cause apoptosis, TM3 cells were treated with MDZ and the DNA contents were examined by flow cytometry. Results showed that treatment with 300 μM MDZ for 24 hours significantly increased cell percentage of subG1 phase, a sign of DNA fragmentation related to apoptosis, in TM3 cells (Figure 2A) (p<0.05). In addition, treatments with 150 μM MDZ for 12 hours and 300 μM MDZ for 24 hours significantly increased the cell percentage of G2/M phase in TM3 cells (Figure 2B) (p<0.05), implying a G2/M phase arrest. These data demonstrated that MDZ could regulate the distribution of cell cycle by increasing subG1 phase with G2/M phase arrest to induce apoptosis in TM3 cells.
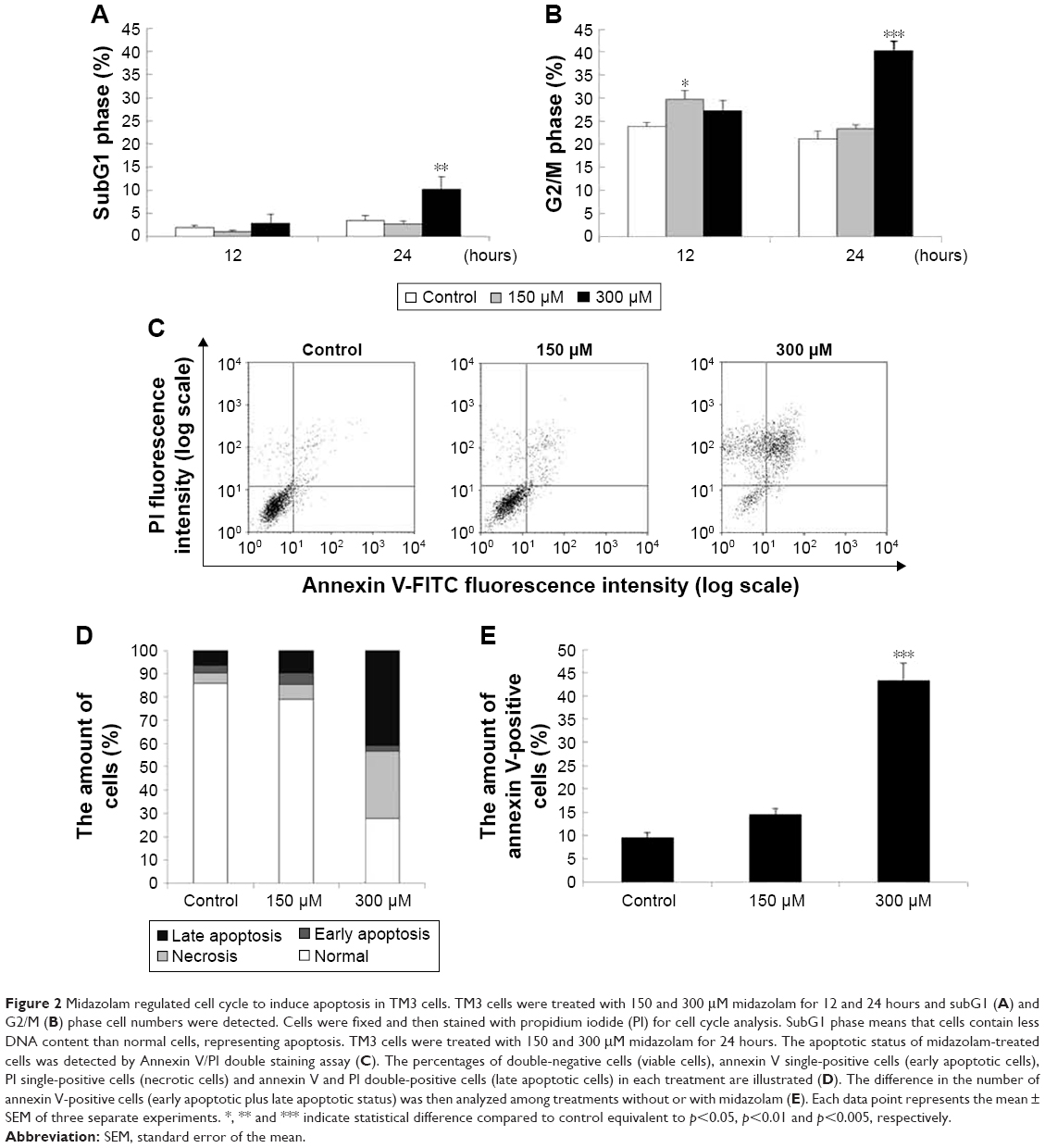 | Figure 2 Midazolam regulated cell cycle to induce apoptosis in TM3 cells. TM3 cells were treated with 150 and 300 μM midazolam for 12 and 24 hours and subG1 (A) and G2/M (B) phase cell numbers were detected. Cells were fixed and then stained with propidium iodide (PI) for cell cycle analysis. SubG1 phase means that cells contain less DNA content than normal cells, representing apoptosis. TM3 cells were treated with 150 and 300 μM midazolam for 24 hours. The apoptotic status of midazolam-treated cells was detected by Annexin V/PI double staining assay (C). The percentages of double-negative cells (viable cells), annexin V single-positive cells (early apoptotic cells), PI single-positive cells (necrotic cells) and annexin V and PI double-positive cells (late apoptotic cells) in each treatment are illustrated (D). The difference in the number of annexin V-positive cells (early apoptotic plus late apoptotic status) was then analyzed among treatments without or with midazolam (E). Each data point represents the mean ± SEM of three separate experiments. *, ** and *** indicate statistical difference compared to control equivalent to p<0.05, p<0.01 and p<0.005, respectively. |
To further confirm that MDZ induced TM3 cell apoptosis, annexin V/PI double staining assay followed by flow cytometry were performed. Data showed that the number of annexin V-positive (early and late apoptosis) cells significantly increased with 300 μM MDZ treatment for 24 hours in TM3 cells (Figure 2C–E) (p<0.05). These results confirm that MDZ induced TM3 cell death through apoptosis.
MDZ activated caspase cascade to induce apoptosis in TM3 cells
Caspase cascade, including extrinsic and intrinsic pathways, is an important inducer of the apoptotic pathway.48 Our data have shown that MDZ could induce apoptosis in TM3 cells. Thus, Western blotting analysis was used to determine whether MDZ could induce apoptosis through caspase extrinsic and intrinsic pathways. Results showed that treatment with 150 μM MDZ for 24 hours significantly increased the expression of cleaved caspase-8 and -3 (Figure 3A, B and D) (p<0.05), while the expressions of cleaved caspase-9 and PARP were not affected in TM3 cells (Figure 3A, C and E) (p>0.05). These results showed that MDZ could activate caspase-8 and -3, but not caspase-9 and PARP, to induce apoptosis in TM3 cells.
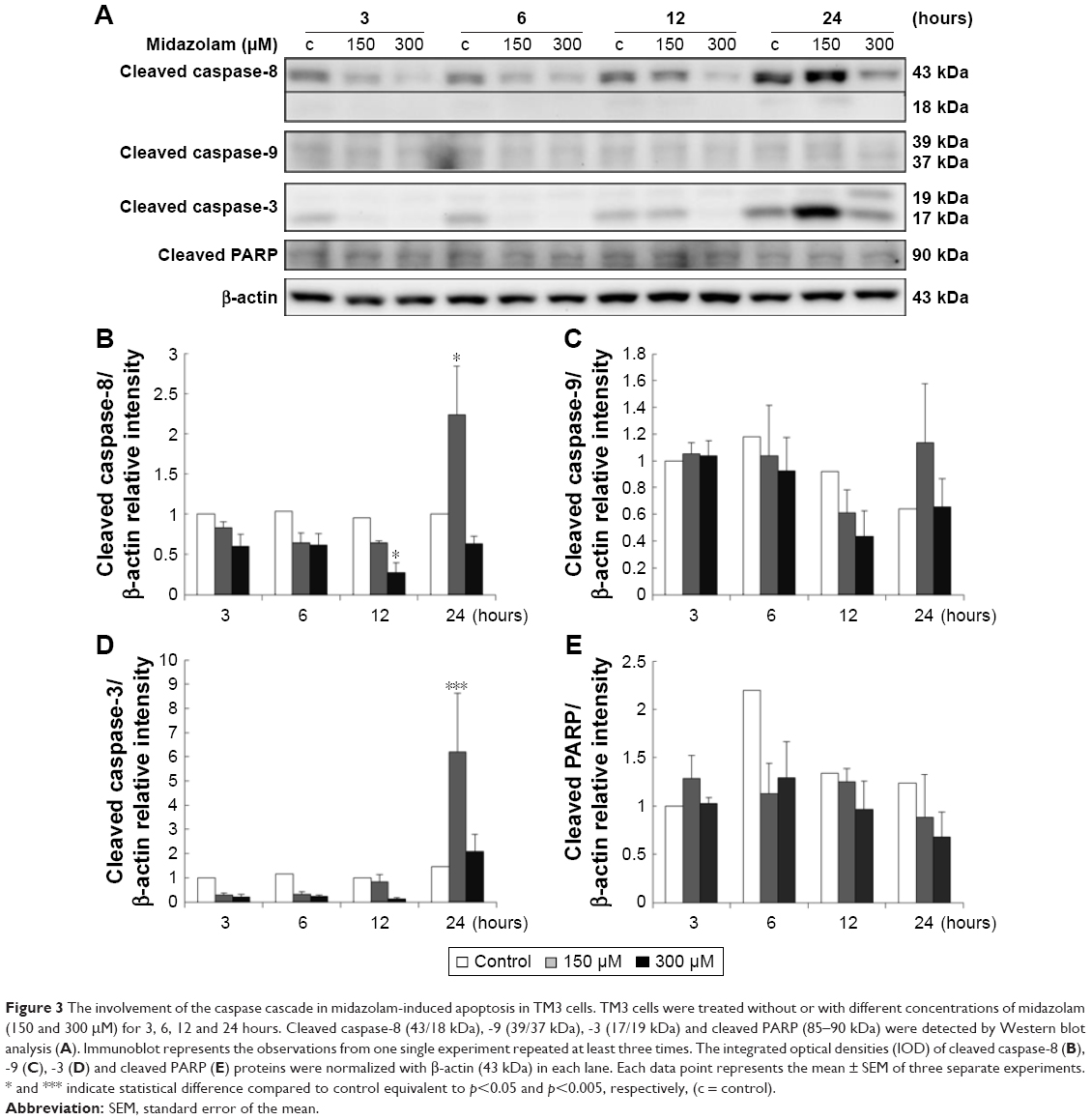 | Figure 3 The involvement of the caspase cascade in midazolam-induced apoptosis in TM3 cells. TM3 cells were treated without or with different concentrations of midazolam (150 and 300 μM) for 3, 6, 12 and 24 hours. Cleaved caspase-8 (43/18 kDa), -9 (39/37 kDa), -3 (17/19 kDa) and cleaved PARP (85–90 kDa) were detected by Western blot analysis (A). Immunoblot represents the observations from one single experiment repeated at least three times. The integrated optical densities (IOD) of cleaved caspase-8 (B), -9 (C), -3 (D) and cleaved PARP (E) proteins were normalized with β-actin (43 kDa) in each lane. Each data point represents the mean ± SEM of three separate experiments. * and *** indicate statistical difference compared to control equivalent to p<0.05 and p<0.005, respectively, (c = control). |
MDZ stimulated MAPK pathways to induce apoptosis in TM3 cells
Studies have shown that the MAPK pathways could regulate cell growth, proliferation, apoptosis, and other biological processes.27–29 To determine whether MDZ-induced TM3 cell apoptosis would be regulated by MAPK pathways, the related proteins were analyzed by Western blot. Results showed that treatment with 300 μM MDZ for 24 hours significantly stimulated the expressions of phosphor-JNK (Figure 4A and B), phosphor-ERK (Figure 4A and C) and phosphor-p38 (Figure 4A and D) in TM3 cells (p<0.05). Moreover, treatment with 150 μM MDZ for 6 and 24 hours significantly induced the expression of phosphor-p38 is expressed at 6 hours and phosphor-JNK is expressed at 24 hours, respectively (Figure 4A, B and D) (p<0.05). These results showed that MDZ could stimulate MAPK pathways to induce apoptosis in TM3 cells.
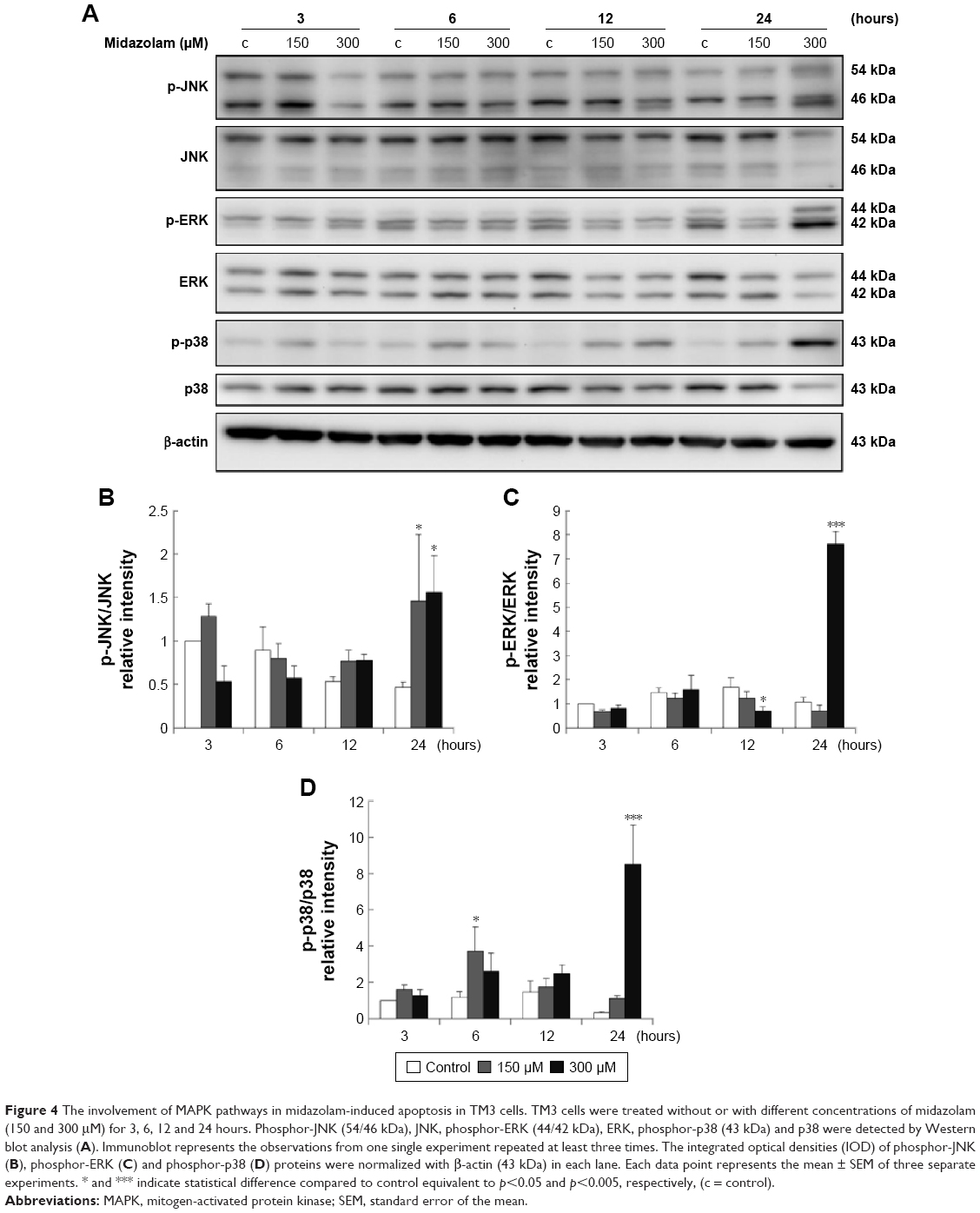 | Figure 4 The involvement of MAPK pathways in midazolam-induced apoptosis in TM3 cells. TM3 cells were treated without or with different concentrations of midazolam (150 and 300 μM) for 3, 6, 12 and 24 hours. Phosphor-JNK (54/46 kDa), JNK, phosphor-ERK (44/42 kDa), ERK, phosphor-p38 (43 kDa) and p38 were detected by Western blot analysis (A). Immunoblot represents the observations from one single experiment repeated at least three times. The integrated optical densities (IOD) of phosphor-JNK (B), phosphor-ERK (C) and phosphor-p38 (D) proteins were normalized with β-actin (43 kDa) in each lane. Each data point represents the mean ± SEM of three separate experiments. * and *** indicate statistical difference compared to control equivalent to p<0.05 and p<0.005, respectively, (c = control). |
Involvement of Akt pathways in MDZ-induced apoptosis in TM3 cells
Previous studies have indicated that Akt pathway is a pro-survival pathway and acts by inhibiting apoptotic signal cascades and activating pro-survival signal cascades.30–32 To investigate whether MDZ could induce apoptosis in TM3 cells by inhibiting Akt pathway, the expressions of Akt, phosphor-Akt, mTOR and phosphor-mTOR (downstream target protein of Akt) were analyzed by Western blot. Results showed that 300 μM MDZ treatment for 24 hours significantly induced the expression of phosphor-Akt (Figure 5A and B) (p<0.05). However, MDZ could not induce the expression of phosphor-mTOR (Figure 5A and C) (p>0.05). These results illustrated the stimulation of Akt, but not mTOR, by MDZ, suggesting that the activation of Akt pathway could not be turned on to protect TM3 cells.
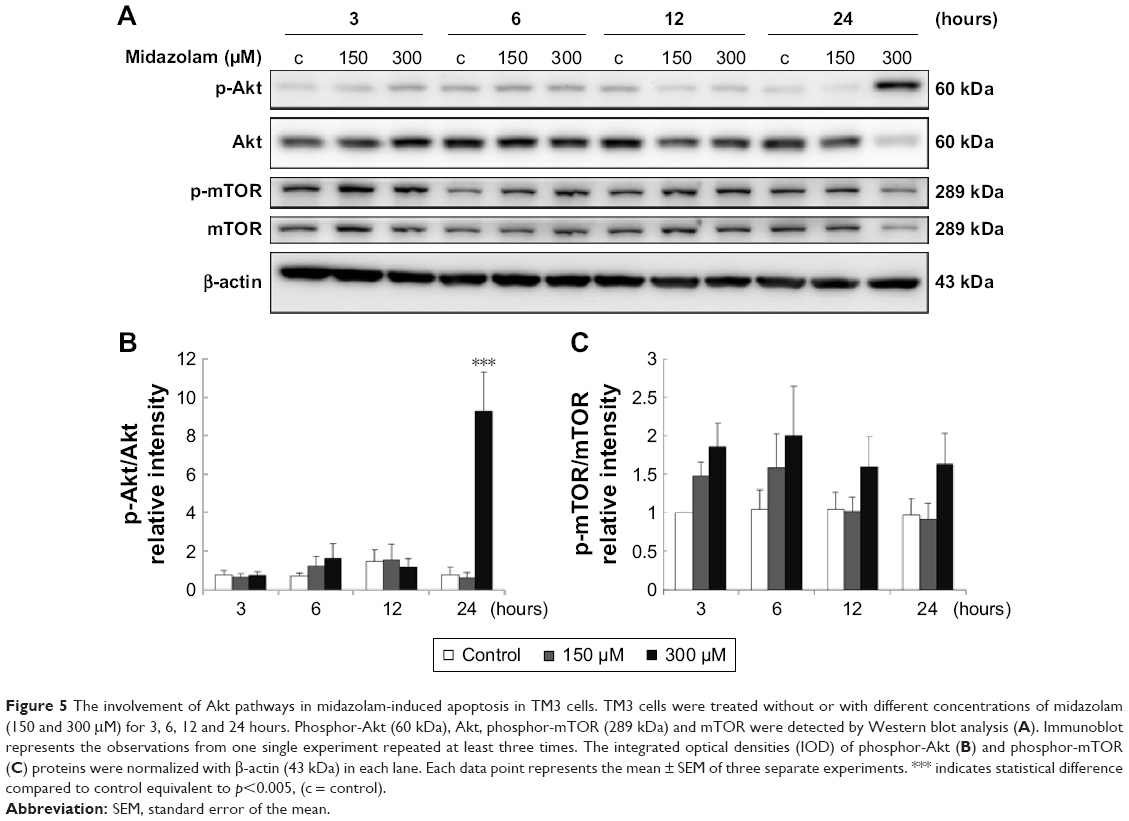 | Figure 5 The involvement of Akt pathways in midazolam-induced apoptosis in TM3 cells. TM3 cells were treated without or with different concentrations of midazolam (150 and 300 μM) for 3, 6, 12 and 24 hours. Phosphor-Akt (60 kDa), Akt, phosphor-mTOR (289 kDa) and mTOR were detected by Western blot analysis (A). Immunoblot represents the observations from one single experiment repeated at least three times. The integrated optical densities (IOD) of phosphor-Akt (B) and phosphor-mTOR (C) proteins were normalized with β-actin (43 kDa) in each lane. Each data point represents the mean ± SEM of three separate experiments. *** indicates statistical difference compared to control equivalent to p<0.005, (c = control). |
Involvement of ER stress pathways in MDZ-induced apoptosis in TM3 cells
Studies have shown that the misfolded proteins could induce ER stress to restore protein homeostasis, and if the stress is prolonged, apoptotic cell death ensues.33–37 To examine whether MDZ could also regulate ER stress pathways to induce apoptosis in TM3 cells, the ER stress-related proteins, including ATF6, IRE1α, cleaved caspase-12, PERK and other downstream proteins, were analyzed by Western blot analysis.
The expressions of ATF6, p-IRE1α and XBP1 were not affected by MDZ (Figure 6A–D) (p>0.05), but the expression of cleaved caspase-12 was reduced by treatment with 300 μM MDZ for 3, 12 and 24 hours in TM3 cells (Figure 6A and E) (p<0.05). We further observed the involvement of PERK-related pathway in TM3 cells. The expression of PERK was decreased by MDZ at 300 μM concentration for 12 and 24 hours, and 150 μM for 24 hours (Figure 7A and B) (p<0.05). However, the expression of phosphor-EIF2α was significantly increased after treatment with 300 μM MDZ for 3, 6, 12 and 24 hours (Figure 7A and C) (p<0.05). MDZ at 150 μM concentration significantly elevated the expression of ATF4 at 3, 6 and 12 hours (Figure 7A and D) (p<0.05) and ATF3 at 6 and 12 hours (Figure 7A and E) (p<0.05). MDZ at 150 μM from 3 to 24 hours significantly elevated the expression of CHOP (Figure 7A and F) (p<0.05), and at 300 μM increased the expression of CHOP in 6 hours treatment (Figure 7A and F) (p<0.05).
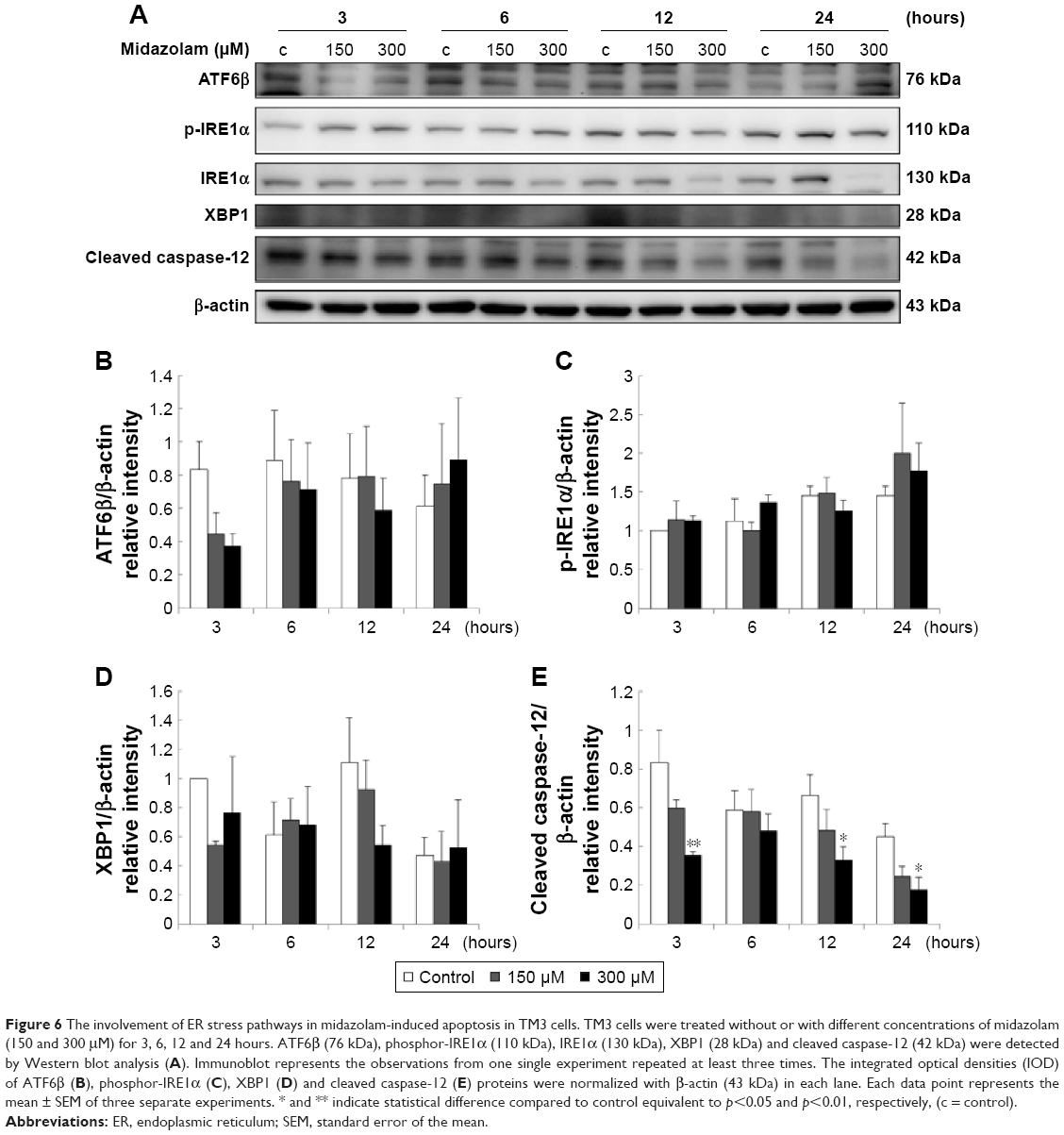 | Figure 6 The involvement of ER stress pathways in midazolam-induced apoptosis in TM3 cells. TM3 cells were treated without or with different concentrations of midazolam (150 and 300 μM) for 3, 6, 12 and 24 hours. ATF6β (76 kDa), phosphor-IRE1α (110 kDa), IRE1α (130 kDa), XBP1 (28 kDa) and cleaved caspase-12 (42 kDa) were detected by Western blot analysis (A). Immunoblot represents the observations from one single experiment repeated at least three times. The integrated optical densities (IOD) of ATF6β (B), phosphor-IRE1α (C), XBP1 (D) and cleaved caspase-12 (E) proteins were normalized with β-actin (43 kDa) in each lane. Each data point represents the mean ± SEM of three separate experiments. * and ** indicate statistical difference compared to control equivalent to p<0.05 and p<0.01, respectively, (c = control). |
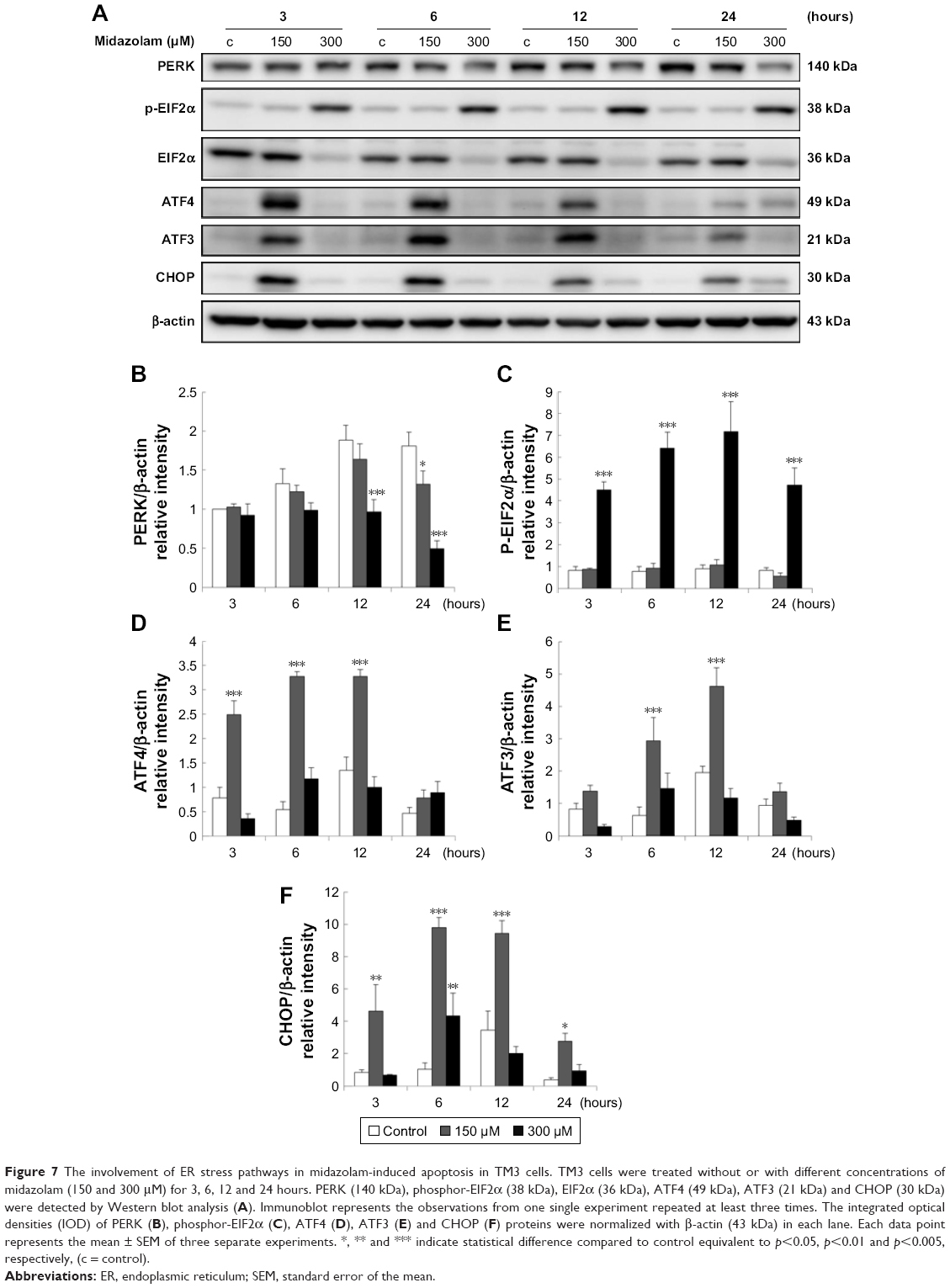 | Figure 7 The involvement of ER stress pathways in midazolam-induced apoptosis in TM3 cells. TM3 cells were treated without or with different concentrations of midazolam (150 and 300 μM) for 3, 6, 12 and 24 hours. PERK (140 kDa), phosphor-EIF2α (38 kDa), EIF2α (36 kDa), ATF4 (49 kDa), ATF3 (21 kDa) and CHOP (30 kDa) were detected by Western blot analysis (A). Immunoblot represents the observations from one single experiment repeated at least three times. The integrated optical densities (IOD) of PERK (B), phosphor-EIF2α (C), ATF4 (D), ATF3 (E) and CHOP (F) proteins were normalized with β-actin (43 kDa) in each lane. Each data point represents the mean ± SEM of three separate experiments. *, ** and *** indicate statistical difference compared to control equivalent to p<0.05, p<0.01 and p<0.005, respectively, (c = control). |
These results suggest that MDZ could induce apoptosis by activating ER stress-related PERK pathway in TM3 cells. Interestingly, these data showed that the activation of caspase cascade and MAPK pathways was observed at 24 hours treatment of MDZ, and the activation of ER stress pathways occurred at 3 hours treatment, which was earlier compared to caspase cascade and MAPK pathways, implying that MDZ could activate ER stress pathways prior to the caspase cascade and MAPK pathways to induce apoptosis in TM3 cells.
Involvement of cell cycle in MDZ-induced apoptosis in TM3 cells
Previous study has demonstrated a correlation between the G2/M arrest and the induction of apoptosis.26 According to previous experiments, MDZ at 300 μM for 24 hours could regulate G2/M arrest in TM3 cells (Figure 2B). Thus, whether MDZ could induce apoptosis through cell cycle regulation or the expression of cyclin A, cyclin B and CDK1 proteins was examined by Western blot analysis.
Results showed that MDZ at 150 and 300 μM concentrations for 6 hours decreased the expression of cyclin A (Figure 8A and B) (p<0.05), but 150 μM for 12 hours increased the expression of cyclin A (Figure 8A and B) (p<0.05). Interestingly, MDZ at 150 μM for 12 hours increased the expression of cyclin B (Figure 8A and C) (p<0.05), but 300 μM for 12 hours decreased the expression of cyclin B (Figure 8A and C) (p<0.05). Moreover, MDZ at 300 μM for 12 and 24 hours significantly decreased the expression of CDK1 (Figure 8A and D) (p<0.05). These data showed that MDZ could regulate the expressions of cyclin A and cyclin B to reduce the expression of CDK1, suggesting that MDZ could actually induce G2/M arrest to induce apoptosis in TM3 cells.
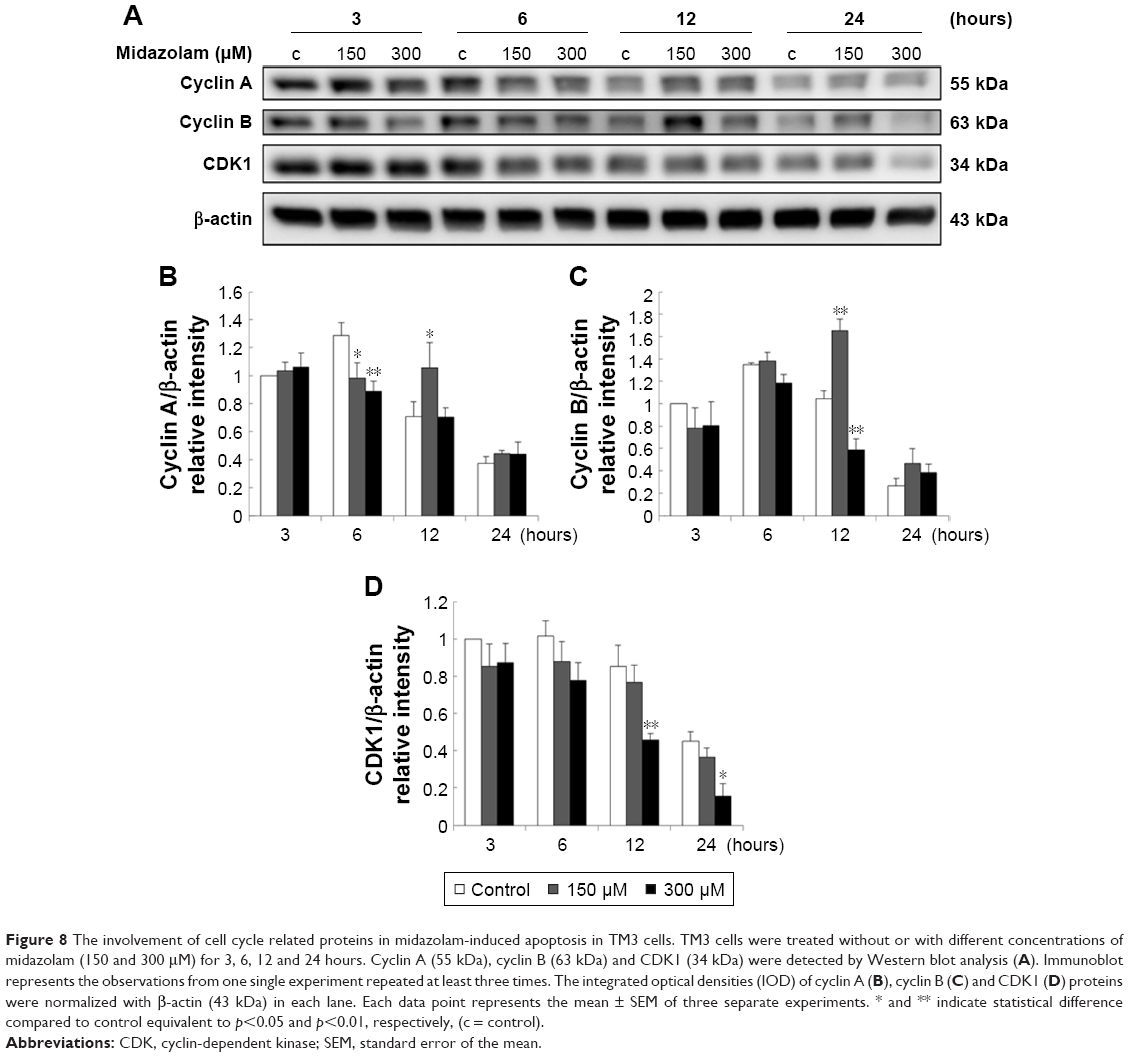 | Figure 8 The involvement of cell cycle related proteins in midazolam-induced apoptosis in TM3 cells. TM3 cells were treated without or with different concentrations of midazolam (150 and 300 μM) for 3, 6, 12 and 24 hours. Cyclin A (55 kDa), cyclin B (63 kDa) and CDK1 (34 kDa) were detected by Western blot analysis (A). Immunoblot represents the observations from one single experiment repeated at least three times. The integrated optical densities (IOD) of cyclin A (B), cyclin B (C) and CDK1 (D) proteins were normalized with β-actin (43 kDa) in each lane. Each data point represents the mean ± SEM of three separate experiments. * and ** indicate statistical difference compared to control equivalent to p<0.05 and p<0.01, respectively, (c = control). |
We further explored the possible role of p53 pathway upstream to the cell cycle regulation in MDZ-treated TM3 cells. MDZ at 300 μM for 6 hours significantly induced the expression of phosphor-p53 (Figure 9A and B) (p<0.05), but 300 μM MDZ for 12 hours decreased its expression (Figure 9A and B) (p<0.05). The expression of phosphor-p27 was reduced by 300 μM MDZ at 6, 12 and 24 hour treatments (Figure 9A and C) (p<0.05). However, the expression of phosphor-p21 was not affected by MDZ (Figure 9A and D) (p>0.05). These data suggested that MDZ could possibly regulate p53 pathway to cause G2/M arrest, and then induce apoptosis in TM3 cells.
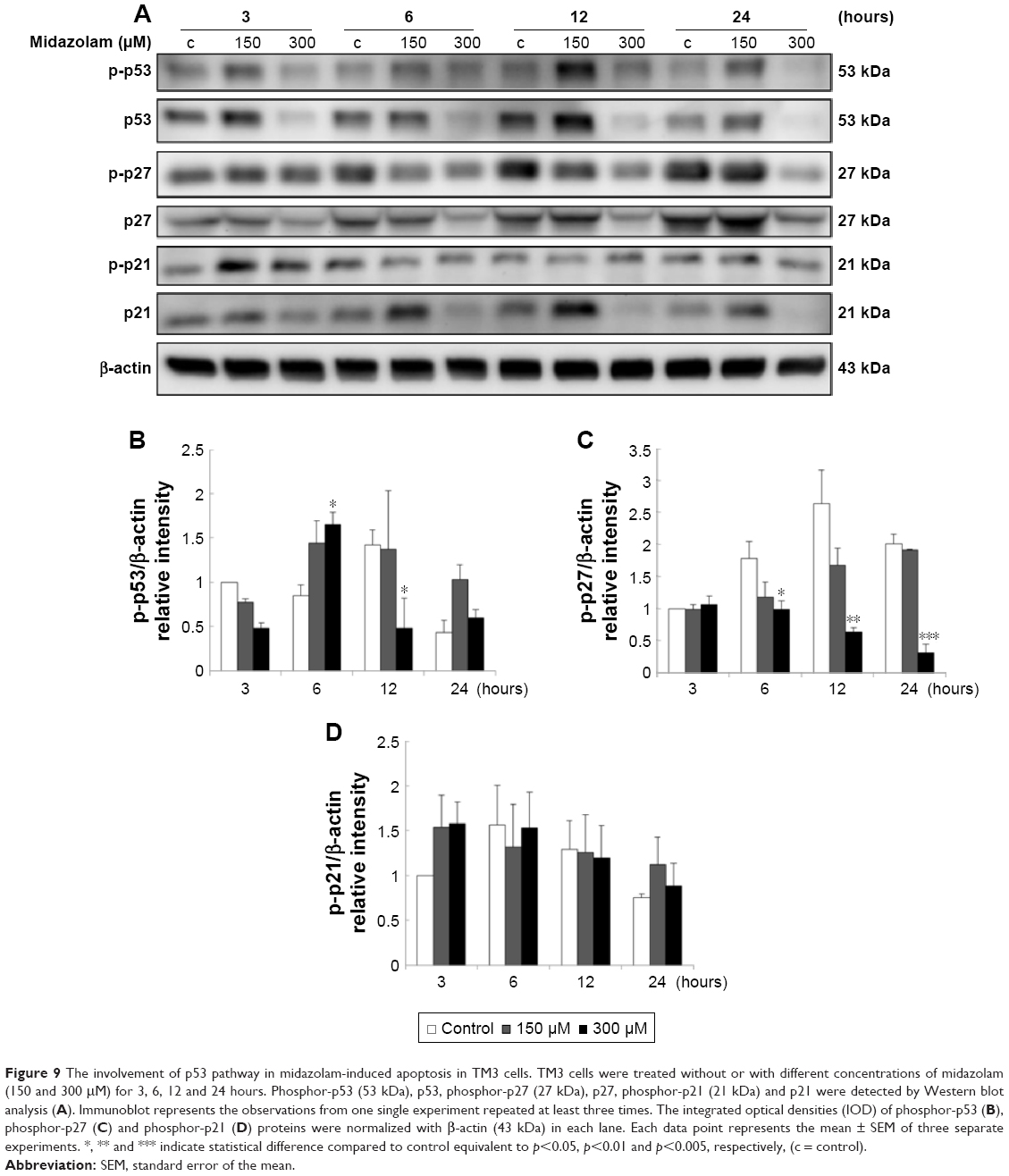 | Figure 9 The involvement of p53 pathway in midazolam-induced apoptosis in TM3 cells. TM3 cells were treated without or with different concentrations of midazolam (150 and 300 μM) for 3, 6, 12 and 24 hours. Phosphor-p53 (53 kDa), p53, phosphor-p27 (27 kDa), p27, phosphor-p21 (21 kDa) and p21 were detected by Western blot analysis (A). Immunoblot represents the observations from one single experiment repeated at least three times. The integrated optical densities (IOD) of phosphor-p53 (B), phosphor-p27 (C) and phosphor-p21 (D) proteins were normalized with β-actin (43 kDa) in each lane. Each data point represents the mean ± SEM of three separate experiments. *, ** and *** indicate statistical difference compared to control equivalent to p<0.05, p<0.01 and p<0.005, respectively, (c = control). |
Discussion
Apoptosis is characterized by a series of dramatic transformations of cellular architecture, and the weakening of cell cytoskeleton is regulated by the activation of caspases, causing morphology changes such as cell shrinkage and membrane blebbing.49 Our morphological results showed that MDZ could induce cell shrinkage and membrane blebbing, indicating MDZ could influence cytoskeleton and morphology changes to induce cell death related to apoptosis in TM3 Leydig progenitor cells.
The significant increase of subG1 phase after treatment with MDZ indicated that MDZ could cause DNA fragmentation to induce apoptosis in TM3 cells. Interestingly, MDZ could also induce G2/M phase arrest in TM3 cells. Studies have shown that the increase of subG1 phase and G2/M phase arrest could cause cell death through apoptosis.6,50 Hence, our data suggested that MDZ-induced apoptosis was associated with the regulation of cell cycle. The annexin V/PI double staining assay also indicated that the induction of apoptosis by MDZ occurs in a dose-dependent manner in TM3 cells, suggesting that MDZ could truly induce cell apoptosis. According to these observations, it is highly possible that MDZ would induce TM3 cell apoptosis through cell cycle arrest.
It is well known that apoptosis is mainly initiated by extrinsic and intrinsic signals, and then by the activation of the caspase cascade.51 We have illustrated that MDZ could activate caspase pathways to induce MA-10 cell apoptosis.13,14 In the present study, we found that MDZ could also induce apoptosis in TM3 cells with the activation of caspase-8 and -3, but not the cleavages of caspase-9 and PARP. Interestingly, the higher dosage of MDZ (300 μM) did not affect the expressions of caspases and PARP. It has been shown that PARP cleavage can be used as a phenotype for cell apoptosis and an indirect evidence of the activation of caspase-3 to induce apoptosis through DNA fragmentation.52 Therefore, these data suggested that TM3 cells might need a longer time with a higher dosage of MDZ to activate the caspase cascade, which should be investigated in detail to reveal an accurate mechanism.
Apoptotic pathways are regulated by many pathways, and one among them is MAPK pathway including JNK, ERK1/2 and p38 MAPK. MAPK pathways may respond to cellular stress to regulate cell survival or apoptosis.27 Regulation of MAPK pathways by MDZ remains unclear in previous studies, although one of the studies has shown that MDZ diminishes phosphorylation of p38 MAPK in neutrophils.53 We have illustrated that MDZ could activate JNK and p38 to induce MA-10 cell apoptosis.13,14 In the present study, activities of JNK, ERK and p38 were elevated by MDZ in TM3 cells. Our data suggested that MDZ could activate MAPK pathways to induce apoptosis in TM3 cells. Interestingly, MDZ could reduce the expression of total MAPKs, suggesting that MDZ could quickly induce the phosphorylation of MAPKs, lead to instability of MAPKs, and then result in degradation of MAPKs, or MDZ could affect the expression of MAPKs at translational level.
Akt signaling pathway is a pro-survival pathway and acts by inhibiting apoptotic signal cascades and activating pro-survival signal.30 It has been reported that the activation of PI3K/Akt pathway could be observed in normal cells and the formation of some cancers.54 In addition, activation of Akt not only inhibits pro-apoptotic ability of Bad, Bax and Bim, but also enhances anti-apoptotic ability of NF-κB transcription factor promoting the transcription of anti-apoptotic genes to inhibit apoptosis.31 Our previous study has shown that MDZ could induce MA-10 cell apoptosis by inhibiting Akt pathway.13,14 However, present results showed that MDZ could elevate the phosphorylation of Akt in TM3 cells, and the G2/M arrest of cells was observed at the same time. Study has illustrated that the activation of Akt pathway occurs in response to DNA damage and aids in DNA repair and cell survival.31 According to these observations, it is highly possible that MDZ would induce TM3 cell apoptosis through cell cycle arrest, and a portion of cells might survive by activating Akt pathway. It is reported that MDZ protects neuroblastoma cells through Akt-phosphorylation in reactive oxygen species derived cellular injury.55 However, our data showed that the expression of phosphor-mTOR, the downstream target protein of Akt, was not changed by MDZ treatment, implying that the protective effect of Akt pathway on TM3 cells could not be activated and, hereafter, cell apoptosis occurred.
Different physiological and pathological perturbations lead to accumulation of unfolded or misfolded proteins in the ER lumen, which is termed ER stress.33,56 ER stress triggers UPR to increase protein folding capacity and decrease unfolded protein load.33,56 If ER stress is chronically prolonged, cell death often occurs through apoptosis which is induced by PERK, ATF6 and IRE1α.33 In the present study, MDZ did induce the activation of PERK downstream proteins, including EIF2α, ATF4, ATF3 and CHOP, in TM3 cells. Thus, MDZ could stimulate MAPK pathways and ER stress pathway, but not Akt pathway, to induce TM3 cell apoptosis.
Eukaryotic cell cycle is tightly controlled by cyclins and CDKs, and imbalance in the regulation of cell cycle would cause developmental abnormalities and cancer growth.25 It was reported that chemo-drugs caused DNA damage in cancer cells, leading to cell cycle arrest, such as G2/M arrest, and then inducing cell apoptosis.57,58 In our study, MDZ could induce G2/M phase arrest in TM3 cells. In addition, MDZ modulated cyclin A and cyclin B, and then caused the inactivation of CDK1, suggesting that MDZ could regulate cell cycle-related proteins to induce G2/M arrest in TM3 cells. Our data further illustrated that MDZ elevated the phosphorylation of p53 in TM3 cells. These data suggested that p53 activation could facilitate MDZ-induced TM3 cell apoptosis through G2/M arrest.
It has been shown that MDZ in different forms or in different culture media among various cell types would have different cytotoxicity effects.59 Interestingly, we have illustrated that MDZ at 150 μM concentration could significantly reduce MA-10 cell viability to induce apoptosis,13,14 while higher dosage of MDZ (300 μM) in the present study was needed to induce the same effect in TM3 cells. Besides, cell viability of MA-10 cells decreased to 47% with 150 μM MDZ for 24-hour treatment, but cell viability of TM3 cells only decreased to 26%.13,14 Therefore, MA-10 mouse Leydig tumor cells were more sensitive to MDZ than TM3 mouse Leydig progenitor cells. The different phenomena as to how and why MDZ has different effects on MA-10 mouse Leydig tumor cells and TM3 mouse Leydig progenitor cells need further investigations.
Conclusion
MDZ could induce cell apoptosis by activating ER stress (at 3 hours), caspases (at 24 hours) and MAPK pathways (at 24 hours), and by impeding cell cycle (at 6 hours) and Akt pathway (at 24 hours) in TM3 mouse Leydig progenitor cells. These results imply the misuse of MDZ could cause Leydig progenitor cell death affecting male reproductive functions.
Acknowledgments
This work was supported by Chi Mei-NCKU Hospital grant (CMNCKU10621) (FCK and BMH) and Ministry of Science and Technology (MOST 105-2320-B-006-028) (BMH), Taiwan, Republic of China.
Disclosure
The authors report no conflicts of interest in this work.
References
Reves JG, Fragen RJ, Vinik HR, Greenblatt DJ. Midazolam: pharmacology and uses. Anesthesiology. 1985;62(3):310–324. | ||
Laegreid L. Clinical observations in children after prenatal benzodiazepine exposure. Dev Pharmacol Ther. 1990;15(3–4):186–188. | ||
Laegreid L, Olegård R, Conradi N, Hagberg G, Wahlström J, Abrahamsson L. Congenital malformations and maternal consumption of benzodiazepines: a case-control study. Dev Med Child Neurol. 1990;32(5):432–441. | ||
Dolovich LR, Addis A, Vaillancourt JM, Power JD, Koren G, Einarson TR. Benzodiazepine use in pregnancy and major malformations or oral cleft: meta-analysis of cohort and case-control studies. BMJ. 1998;317(7162):839–843. | ||
Suehara T, Morishita J, Ueki M, Ueno M, Maekawa N, Mizobuchi S. Effects of sevoflurane exposure during late pregnancy on brain development of offspring mice. Paediatr Anaesth. 2016;26(1):52–59. | ||
Zhang T, Shao H, Xu KQ, Kuang LT, Chen RF, Xiu HH. Midazolam suppresses osteogenic differentiation of human bone marrow-derived mesenchymal stem cells. Eur Rev Med Pharmacol Sci. 2014;18(9):1411–1418. | ||
Chen YC, Chen YH, Pan BS, Chang MM, Huang BM. 2017. Functional study of Cordyceps sinensis and cordycepin in male reproduction – a review. J Food Drug Anal. 2017;25(1):197–205. | ||
Habert R, Lejeune H, Saez JM. Origin, differentiation and regulation of fetal and adult Leydig cells. Mol Cell Endocrinol. 2001;179(1–2):47–74. | ||
Chen H, Ge RS, Zirkin BR. Leydig cells: From stem cells to aging. Mol Cell Endocrinol. 2009;306(1–2):9–16. | ||
Braestrup C, Squires R. Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H) diazepam binding. Proc Natl Acad Sci U S A. 1977;74(9):3805–3809. | ||
Papadopoulos V, Brown AS. Role of the peripheral-type benzodiazepine receptor and the polypeptide diazepam binding inhibitor in steroidogenesis. J Steroid Biochem Mol Biol. 1995;53(1–6):103–110. | ||
So EC, Chang YT, Hsing CH, Poon PW, Leu SF, Huang BM. The effect of midazolam on mouse Leydig cell steroidogenesis and apoptosis. Toxicol Lett. 2010;192(2):169–178. | ||
So EC, Lin YX, Tseng CH, et al. Midazolam induces apoptosis in MA-10 mouse Leydig tumor cells through caspase activation and the involvement of MAPK signaling pathway. OncoTargets Ther. 2014;7:211–221. | ||
So EC, Chen YC, Wang SC, et al. Midazolam regulated caspase pathway, endoplasmic reticulum stress, autophagy, and cell cycle to induce apoptosis in MA-10 mouse Leydig tumor cells. OncoTargets Ther. 2016;9:2519–2533. | ||
Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284(5756):555–556. | ||
Cerretti DP, Kozlosky CJ, Mosley B, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science. 1992;256(5053):97–100. | ||
Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75(4):641–652. | ||
Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. | ||
Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–489. | ||
Ahmad M, Srinivasula SM, Wang L, et al. CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res. 1997;57(4):615–619. | ||
Yang X, Chang HY, Baltimore D. Autoproteolytic activation of pro-caspases by oligomerization. Mol Cell. 1998;1(2):319–325. | ||
Stennicke HR, Ryan CA, Salvesen GS. Reprieval from execution: the molecular basis of caspase inhibition. Trends Biochem Sci. 2002;27(2):94–101. | ||
Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1(7):949–957. | ||
Gupta S. Molecular steps of tumor necrosis factor receptor-mediated apoptosis. Curr Mol Med. 2001;1(3):317–324. | ||
Boxem M. Cyclin-dependent kinases in C. elegans. Cell Div. 2006;1:6. | ||
Wang Y, Ji P, Liu J, Broaddus RR, Xue F, Zhang W. Centrosome-associated regulators of the G(2)/M checkpoint as targets for cancer therapy. Mol Cancer. 2009;8:8. | ||
Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298(5600):1911–1912. | ||
Lee SH, Jaganath IB, Atiya N, Manikam R, Sekaran SD. Suppression of ERK1/2 and hypoxia pathways by four Phyllanthus species inhibits metastasis of human breast cancer cells. J Food Drug Anal. 2016;24(4):855–865. | ||
Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J Signal Transduct. 2011;2011:792639. | ||
Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway – beyond rapalogs. Oncotarget. 2010;1(7):530–543. | ||
Hein AL, Ouellette MM, Yan Y. Radiation-induced signaling pathways that promote cancer cell survival (review). Int J Oncol. 2014;45(5):1813–1819. | ||
Zhou C, Zhao XM, Li XF, et al. Curcumin inhibits AP-2gamma-induced apoptosis in the human malignant testicular germ cells in vitro. Acta Pharmacol Sin. 2013;34(9):1192–1200. | ||
Luo K, Cao SS. Endoplasmic reticulum stress in intestinal epithelial cell function and inflammatory bowel disease. Gastroenterol Res Pract. 2015;2015:328791. | ||
Sanderson TH, Gallaway M, Kumar R. Unfolding the unfolded protein response: unique insights into brain ischemia. Int J Mol Sci. 2015;16(4):7133–7142. | ||
Logue SE, Cleary P, Saveljeva S, Samali A. New directions in ER stress-induced cell death. Apoptosis. 2013;18(5):537–546. | ||
Kania E, Pająk B, Orzechowski A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. Biomed Res Int. 2015;2015:352794. | ||
Kitamura M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am J Physiol Renal Physiol. 2008;295(2):F323–F334. | ||
Han ES, Muller FL, Pérez VI, et al. The in vivo gene expression signature of oxidative stress. Physiol Genomics. 2008;34(1):112–126. | ||
Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28(3):128–136. | ||
Cmielová J, Rezáčová M. p21Cip1/Waf1 protein and its function based on a subcellular localization [corrected]. J Cell Biochem. 2011;112(12):3502–3506. | ||
Kamato D, Burch ML, Piva TJ, et al. Transforming growth factor-β signalling: role and consequences of Smad linker region phosphorylation. Cell Signal. 2013;25(10):2017–2024. | ||
Mather JP. Establishment and characterization of two distinct mouse testicular epithelial cell lines. Biol Reprod. 1980;23(1):243–252. | ||
Goldenberg RC, Fortes FS, Cristancho JM, et al. Modulation of gap junction mediated intercellular communication in TM3 Leydig cells. J Endocrinol. 2003;177(2):327–335. | ||
Geigerseder C, Doepner RF, Thalhammer A, Krieger A, Mayerhofer A. Stimulation of TM3 Leydig cell proliferation via GABA(A) receptors: a new role for testicular GABA. Reprod Biol Endocrinol. 2004;2:13. | ||
Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. | ||
van Engeland M, Ramaekers FC, Schutte B, Reutelingsperger CP. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry. 1996;24(2):131–139. | ||
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. | ||
Creagh EM, Martin SJ. Caspases: cellular demolition experts. Biochem Soc Trans. 2001;29(Pt 6):696–702. | ||
Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. | ||
Paul-Samojedny M, Suchanek R, Borkowska P, et al. Knockdown of AKT3 (PKBgamma) and PI3KCA suppresses cell viability and proliferation and induces the apoptosis of glioblastoma multiforme T98G cells. Biomed Res Int. 2014;2014:768181. | ||
Oliveira JB, Gupta S. Disorders of apoptosis: mechanisms for autoimmunity in primary immunodeficiency diseases. J Clin Immunol. 2008;28:S20–S28. | ||
Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371(6495):346–347. | ||
Ghori K, O’Driscoll J, Shorten G. The effect of midazolam on neutrophil mitogen-activated protein kinase. Eur J Anaesthesiol. 2010;27(6):562–565. | ||
Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. | ||
Chong WS, Hyun CL, Park MK, et al. Midazolam protects B35 neuroblastoma cells through Akt-phosphorylation in reactive oxygen species derived cellular injury. Korean J Anesthesiol. 2012;62(2):166–171. | ||
Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev. 2011;91(4):1219–1243. | ||
Koromilas AE. Roles of the translation initiation factor eIF2alpha serine 51 phosphorylation in cancer formation and treatment. Biochim Biophys Acta. 2015;1849(7):871–880. | ||
Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–320. | ||
Shityakov S, Sohajda T, Puskas I, Roewer N, Forster C, Broscheit JA. Ionization states, cellular toxicity and molecular modeling studies of midazolam complexed with trimethyl-beta-cyclodextrin. Molecules. 2014;19(10):16861–16876. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.