Back to Journals » OncoTargets and Therapy » Volume 10
Microtubule-targeting agents can sensitize cancer cells to ionizing radiation by an interphase-based mechanism
Authors Markowitz D, Ha G, Ruggieri R, Symons M
Received 2 June 2017
Accepted for publication 29 September 2017
Published 24 November 2017 Volume 2017:10 Pages 5633—5642
DOI https://doi.org/10.2147/OTT.S143096
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Daniel Markowitz,1,2 Grace Ha,2 Rosamaria Ruggieri,1,2 Marc Symons1,2
1Hofstra Northwell School of Medicine, Hempstead, 2Karches Center for Oncology Research, Feinstein Institute for Medical Research, Manhasset, NY, USA
Background: The cytotoxic effects of microtubule-targeting agents (MTAs) are often attributed to targeted effects on mitotic cells. In clinical practice, MTAs are combined with DNA-damaging agents such as ionizing radiation (IR) with the rationale that mitotic cells are highly sensitive to DNA damage. In contrast, recent studies suggest that MTAs synergize with IR by interfering with the trafficking of DNA damage response (DDR) proteins during interphase. These studies, however, have yet to demonstrate the functional consequences of interfering with interphase microtubules in the presence of IR. To address this, we combined IR with an established MTA, mebendazole (MBZ), to treat glioma cells exclusively during interphase.
Materials and methods: To test whether MTAs can sensitize interphase cells to IR, we treated GL261 and GBM14 glioma cells with MBZ during 3–9 hours post IR (when the mitotic index was 0%). Cell viability was measured using a WST-1 assay, and radiosensitization was quantified using the dose enhancement factor (DEF). The effect of MBZ on the DDR was studied via Western blot analysis of H2AX phosphorylation. To examine the effects of MTAs on intracellular transport of DDR proteins, Nbs1 and Chk2, cytoplasmic and nuclear fractionation studies were conducted following treatment of glioma cells with MBZ.
Results: Treatment with MBZ sensitized interphase cells to the effects of IR, with a maximal DEF of 1.34 in GL261 cells and 1.69 in GBM14 cells. Treatment of interphase cells with MBZ led to more sustained γH2AX levels post IR, indicating a delay in the DDR. Exposure of glioma cells to MBZ resulted in a dose-dependent sequestration of Chk2 and Nbs1 in the cytoplasm.
Conclusion: This study demonstrates that MBZ can sensitize cancer cells to IR independently of the induction of mitotic arrest. In addition, evidence is provided supporting the hypothesis that MTA-induced radiosensitization is mediated by inhibiting DDR protein accumulation into the nucleus.
Keywords: microtubules, mebendazole, ionizing radiation, radiosensitization, interphase, DNA damage response
Introduction
Microtubule-targeting agents (MTAs) have been used for a long time against a wide range of malignancies. It is generally believed that MTAs kill cancer cells by causing cell cycle arrest in M-phase, followed by activation of apoptotic pathways and cell death.1–4 Many studies have used this rationale to explain the potent radiosensitization effects exerted by MTAs,5,6 ie, by inducing mitotic arrest, MTAs increase the proportion of tumor cells in a phase of the cell cycle that is very susceptible to DNA damage.7,8 Currently, chemoradiotherapy regimens including MTAs have been proven effective for the treatment of breast cancer, esophageal cancer and a variety of other neoplasms.9,10
Support for a mitosis-based mechanism for the therapeutic effects of MTAs has been derived from the characteristic side effects of these drugs, which include hair loss, neutropenia and gastrointestinal upset. These deleterious effects demonstrate the profound sensitivity of rapidly dividing tissues to MTAs. However, the action of MTAs on mitotic cells fails to explain their clinical efficacy against many slow-growing solid tumors with exceptionally low mitotic indices.11 Most human tumors have a doubling time of 30–60 days or longer, making it unlikely that mitotic arrest serves as a critical mechanism of MTA-induced therapeutic benefit.12 A prime example of this “proliferation rate paradox” is the significant activity of MTAs against adenocarcinoma of the prostate, a highly indolent cancer.13,14 Thus, a number of interphase-based mechanisms for the efficacy of MTAs in cancer therapy have been proposed, although not without controversy.15,16
A recent study has shown that the MTAs, vincristine (VCR) and paclitaxel, can delay DNA damage repair.17 These MTAs were also shown to interfere with the trafficking of DNA damage response (DDR) proteins, including ATM, ATR and p53, from the cytosol to the nucleus, strongly suggesting that MTAs can sensitize cells to radiation by blocking microtubule-based transport of DDR proteins into the nucleus during interphase.
It is challenging to physically separate mitotic from interphase cells in the presence of an MTA, as this results in a steady accumulation of new mitotic cells as long as the MTA is present. Thus, the question remains as to what extent the role of MTAs in radiosensitization is caused by interference with microtubule-facilitated nuclear import. To address this question, we took advantage of the fact that ionizing radiation (IR) treatment induces G2–M cell cycle arrest, thereby transiently eliminating the mitotic cell population and strongly enriching for interphase cells. Using glioblastoma cells and the MTA, mebendazole (MBZ), as a model system, we show that the effect of MBZ in interphase is responsible for the majority of the radiosensitization effect of this MTA.
Materials and methods
Cell lines and reagents
GL261 (glioma) cells were obtained from the National Cancer Institute (Frederick, MD, USA). Cells were cultured in macrophage serum-free medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum, 1% penicillin/streptomycin and 1 mM L-glutamine. GBM14 cells have been described previously,18 and they were obtained by Dr J Sarkaria at Mayo Clinic (Rochester, MN, USA) and cultured in StemPro media (Thermo Fisher Scientific), as directed by the manufacturer.
Drug treatment
For each experiment, GL261 and GBM14 cells were cultured without drug or treated with MBZ (Sigma-Aldrich Co., St Louis, MO, USA). MBZ stocks were prepared by dissolving the drug in dimethyl sulfoxide (DMSO; Sigma-Aldrich Co.). About 24 hours after plating, glioma cells were treated with MBZ, while control cells were treated with 0.01% DMSO.
Irradiation procedure
For all experiments requiring radiation, cells were irradiated using a biological irradiator (RS2000; Rad Source Technologies, Buford, GA, USA).
Antibodies
Phospho-MPM2 was obtained from EMD Millipore (Billerica, MA, USA), and γH2AX, H2AX, Chk2, Nbs1 and GAPDH were purchased from Cell Signaling (Danvers, MA, USA).
Immunofluorescence
GL261 cells were seeded at a density of 30,000 cells/well in 24-well plates containing cover slips coated with laminin (Sigma-Aldrich Co.). The following day, cells were exposed to 6 Gy of IR. At designated time points post IR, cells were washed in PBS and fixed in 4% formaldehyde in PBS at room temperature. Once the cells were fixed, immunofluorescence was performed using an MPM2 antibody (EMD Millipore) and counterstained with 4,6-Diamidino-2-phenylindole, dihydrochloride (DAPI). For each condition, a total of 10 fluorescence micrograph images were taken with a Zeiss Axiovert 200M inverted microscope (Thornwood, NY, USA), running on Axiovision software. For each image field, the total number of cells and the number of MPM2-positive cells were quantified. The mitotic index was calculated at each time point using the number of MPM2-positive cells as a percentage of the total number of cells counted in all 10 fields.
Cell viability assays
GL261 cells were seeded in 96-well plates at a density of 1,000 cells/well. GBM14 cells were seeded at a density of 10,000 cells/well. About 24 hours after seeding, cells were treated with 25–150 nM MBZ and irradiated with 3, 6 or 9 Gy. Drug treatment was applied for a 6-hour time window beginning either 6 hours pre IR or 3 hours post IR. Following the 6-hour window, the drug was washed out and replaced with fresh medium. Control cells were either left nonirradiated or irradiated with 3, 6 or 9 Gy of IR. Cell viability was examined 72 hours post IR using a WST-1 cell viability assay (Abcam, Cambridge, MA, USA) at an absorbance of 450 nm. For any given radiation dose–response, data were normalized by the fraction of viable cells treated with a given dose of drug in the absence of radiation. The radiosensitizing effect of MBZ and VCR was quantified using the dose enhancement factor (DEF) at the point of 50% (DEF50) cell viability. The DEF was calculated for each MBZ concentration using the following formula: (surviving fraction with radiation alone)/(surviving fraction with radiation + MBZ).
Assessment of the DDR
GL261 cells were seeded at a density of 800,000 cells/well in 6-cm dishes. About 24 hours after seeding, cells were irradiated with 6 Gy of IR, followed by treatment with 150 nM MBZ during 3–9 hours post IR. Control cells were either left nonirradiated or irradiated with 6 Gy. All cells were harvested in lysis buffer at 0.5, 3, 6 and 9 hours post IR. The lysis buffer was composed of 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 10% Nonidet P-40, 1× protease inhibitor (Thermo Fisher Scientific), phosphatase inhibitor (Hoffman-La Roche Ltd., Basel, Switzerland) and 0.4 U/mL Benzonase (EMD Millipore). For each condition, 60 μg of cell lysate was diluted in 1× NuPAGE Sample Reducing Agent (Thermo Fisher Scientific) and 1× NuPAGE LDS Sample Buffer (Thermo Fisher Scientific). All samples were loaded onto a NuPAGE, 4%–12% Bis-Tris Protein Gel (Thermo Fisher Scientific) for Western blot analysis. Histone H2AX and γH2AX were detected by Western blot using anti-histone H2AX or anti-γ-H2AX (Ser139) monoclonal antibodies (Cell Signaling).
Nuclear and cytoplasmic fractionation
GL261 cells were seeded at a density of 500,000 cells in 6-cm dishes. The following day, cells were treated with 3 Gy. MBZ treatment (25–250 nM) was performed for a 6-hour time window beginning 3 hours post IR. Irradiated control cells were treated with 3 Gy followed by 0.01% DMSO in medium during the same time period. Nonirradiated control cells were treated with 0.01% DMSO in medium for a total of 6 hours. After the 6-hour period of MBZ treatment, cells were harvested for protein analysis in cytoplasmic (C) and nuclear (N) fractions. The C and N fractions were collected according to the protocol of the NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Fisher Scientific). For each sample, 20% of the final volume of the C or N fractions was diluted in 1× NuPAGE Sample Reducing Agent and 1× NuPAGE LDS Sample Buffer. All samples were loaded onto a NuPAGE, 4%–12% Bis-Tris Protein Gel for Western blot analysis. Blots were incubated in antibodies: Nbs1, Chk2, GAPDH and H2AX monoclonal antibodies (all from Cell signaling). Cytoplasmic and nuclear fractions were normalized by GAPDH and H2AX levels, respectively. The percentage of cytoplasmic retention of DDR proteins was calculated by the following formula: [C/(C + N)] × 100%.
Statistical analysis
All analyses were performed using Graphpad Prism 7 software. Radiosensitization experiments were analyzed using the two-way ANOVA method to compare the mean cell viability between treatment groups. The interaction between the MBZ treatment group and radiation dose was also examined. One-way ANOVA was used to compare the concentration of a drug that gives half-maximal response (EC50) values for each effect as outlined in Table 1. If a significant difference between means was found by ANOVA, then multiple comparisons between treatment groups were conducted. The Tukey–Kramer method was used to adjust for multiple comparisons. Studies examining the effect of MBZ on intracellular transport of DDR proteins were analyzed with unpaired two-tailed Student’s t-test for direct comparisons of means. The same method was used to compare means in the analysis of H2AX phosphorylation following exposure to IR. For all studies, results were considered statistically significant for values of P<0.05.
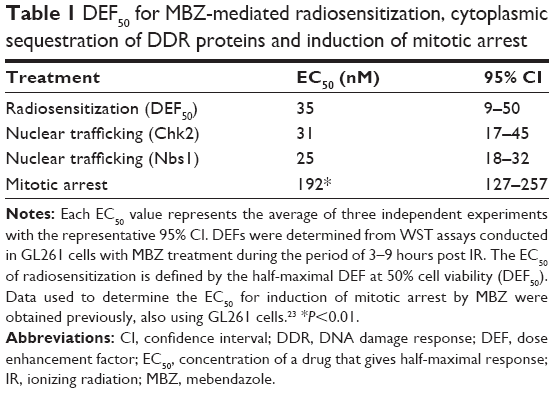 | Table 1 DEF50 for MBZ-mediated radiosensitization, cytoplasmic sequestration of DDR proteins and induction of mitotic arrest |
Results
IR transiently eliminates the mitotic cell population
To select for a population composed entirely of interphase cells, we took advantage of the fact that IR treatment induces G2–M cell cycle arrest and transiently eliminates the mitotic cell population. Glioma cells were treated with 6 Gy of IR. To monitor the mitotic index, we quantified the proportion of MPM2-positive cells by immunofluorescence at several time points post IR. MPM2 is an antibody that recognizes a phosphorylated serine/threonine epitope found in proteins that are phosphorylated at the onset of mitosis.19 MPM2 is recognized as a reliable mitotic marker in the literature and has been used to assess the mitotic index in cells exposed to radiation and a number of chemotherapeutic agents.20–22 The baseline mitotic index of the glioma cell population was 3.5% and was strongly inhibited during a period of 3–10 hours after exposure to 6 Gy of IR. Recovery of the mitotic cell population begins after 10 hours post IR and completes by 24 hours post IR. To confirm that the mitotic index remained strongly inhibited even in the presence of MBZ, we applied MBZ during 3–9 hours post IR. In the presence of MBZ, the mitotic index remained suppressed at 4 hours (mitotic index =0.58%), 6 hours (mitotic index =0.12%) and 9 hours (mitotic index =0.28%) post IR. Thus, the treatment of GL261 glioma cells with 6 Gy of IR eliminated the mitotic cell population for a time period of ~6 hours (Figure 1).
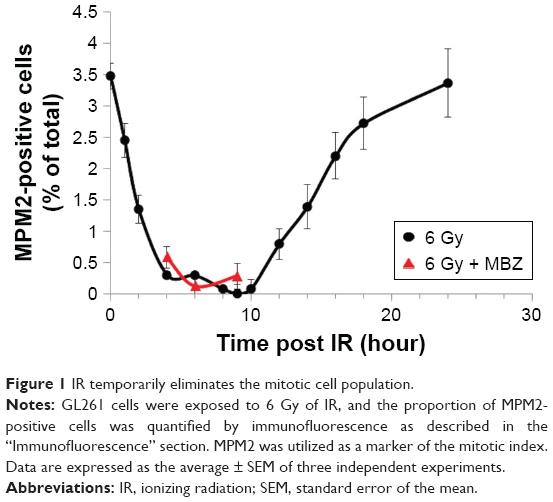 | Figure 1 IR temporarily eliminates the mitotic cell population. |
MTAs sensitize interphase cells to IR
In the following experiments, we compared the radiosensitizing effect of MTAs when applied during different time periods with respect to IR, using both murine GL261 cells and primary patient-derived GBM14 cells. A simplified schematic of all three treatment conditions is shown in Figure 2. To examine whether MTAs can sensitize glioma cells to IR, we treated GL261 cells with MBZ (Figures 3 and 4) or VCR (Figure 5). In order to test, whether MTAs can sensitize interphase cells to IR, we treated glioma cells with 25–150 nM MBZ during 3–9 hours post IR (Figures 3A and 4A). The use of MBZ after exposure to IR made it possible to study the impact of MBZ independent of its effect on mitotic cells. Cells were exposed to MBZ after different doses of IR. After this 6-hour time frame, the drug was washed out and replaced with a drug-free medium. The radiosensitizing effect of MBZ on interphase cells was compared with the effect of MBZ on a cell population composed of both interphase and mitotic cells. Thus, cells were treated with MBZ for a 6-hour time window immediately prior to irradiation, and subsequently the drug was either washed out and replaced with drug-free medium (Figures 3B and 4B) or left in the original medium until the end of the assay, 72 hours post IR (Figures 3C and 4C). Treatment with MBZ post IR sensitized GL261 cells to IR with a maximal DEF at 50% viability (DEF50) of 1.34 (Figure 3D). Treatment for a 6-hour time window pre IR, which increased the mitotic index to 9.2% from a baseline value of 2.8%,23 sensitized glioma cells to IR with a maximal DEF50 of 1.2 in GL261 cells (Figure 3E) and 1.33 in GBM14 cells (Figure 4E). In the GL261 cell line, maximal radiosensitization was observed when the drug was applied pre IR and left in the medium for 72 hours, DEF50 =1.41 (Figure 3F). In the GBM14 cell line, the application of the drug pre IR followed by 72-hour incubation sensitized cells to IR with a DEF50 =1.60 (Figure 4F). Maximal radiosensitization in the GBM14 cell line was achieved when MBZ was applied post IR, DEF50 =1.69 (Figure 4D). These observations show a maximal or near-maximal radiosensitization effect of MBZ when applied to cells during interphase.
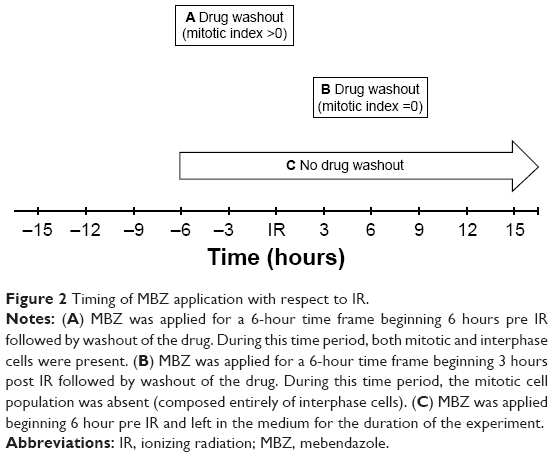 | Figure 2 Timing of MBZ application with respect to IR. |
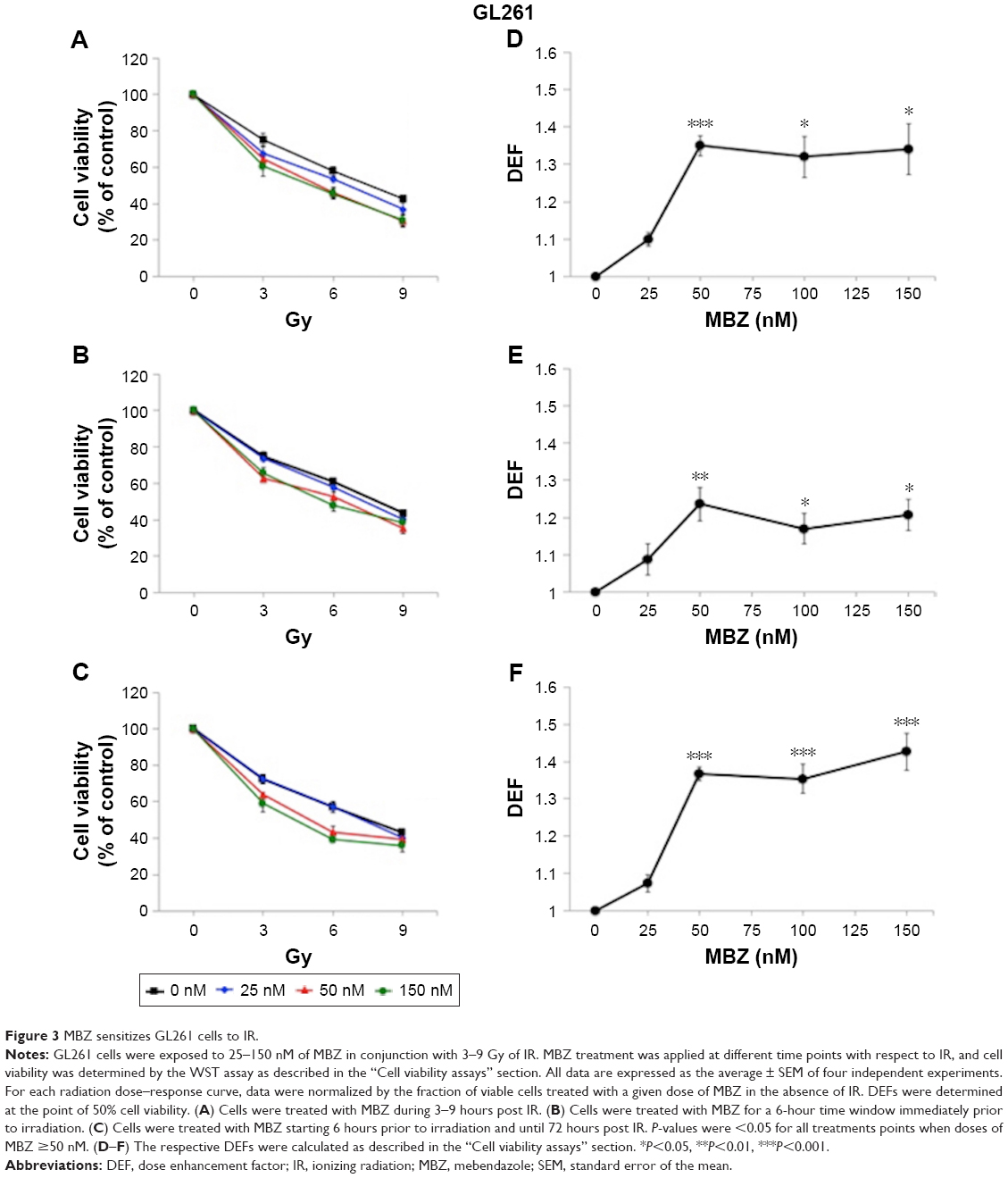 | Figure 3 MBZ sensitizes GL261 cells to IR. |
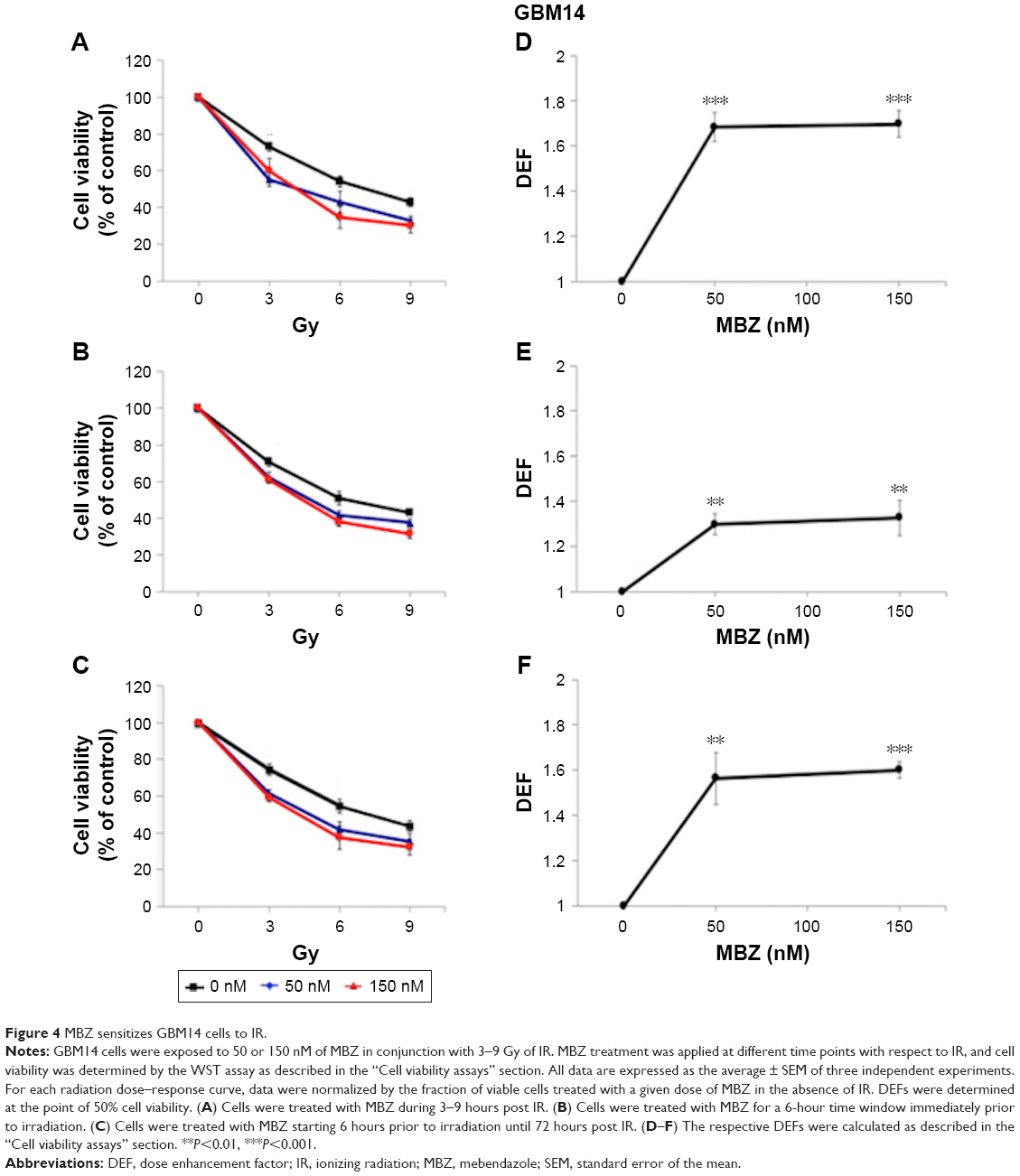 | Figure 4 MBZ sensitizes GBM14 cells to IR. |
 | Figure 5 VCR sensitizes GL261 cells to IR. |
To investigate whether these findings can be generalized to other MTAs, we performed an identical set of experiments in GL261 cells using VCR, an established MTA that is frequently used in clinical practice. The radiosensitizing effect of VCR was greatest when applied post IR, DEF50 =1.53 (Figure 5A and D). The application of VCR for a 6-hour period pre IR radiosensitized GL261 cells with a DEF50 of 1.34 (Figure 5B and E). When VCR was applied pre IR and left in the medium for the remaining 72 hours, the magnitude of the radiosensitizing effect was quite similar, DEF50 =1.30 (Figure 5C and F). Thus, it appears that the radiosensitizing effect of both MBZ and VCR is largely determined by the impact of these agents on interphase cells.
MBZ treatment prolongs DDR after irradiation
To confirm that MTAs sensitize interphase cells to IR by interfering with the DDR, we examined the level of IR-induced phosphorylated H2AX (γH2AX), which is a highly sensitive marker of DNA damage.24,25 We exposed glioma cells to 6 Gy of IR, followed by treatment with 150 nM MBZ during 3–9 hours post IR. Cells were harvested at different time points post IR, and γH2AX was quantified by Western blot analysis. As shown in Figure 6, treatment with MBZ led to more sustained γH2AX levels in response to IR, indicating a delay in the DDR. This finding demonstrated that MTAs sensitize interphase cells to IR by interfering with the DDR.
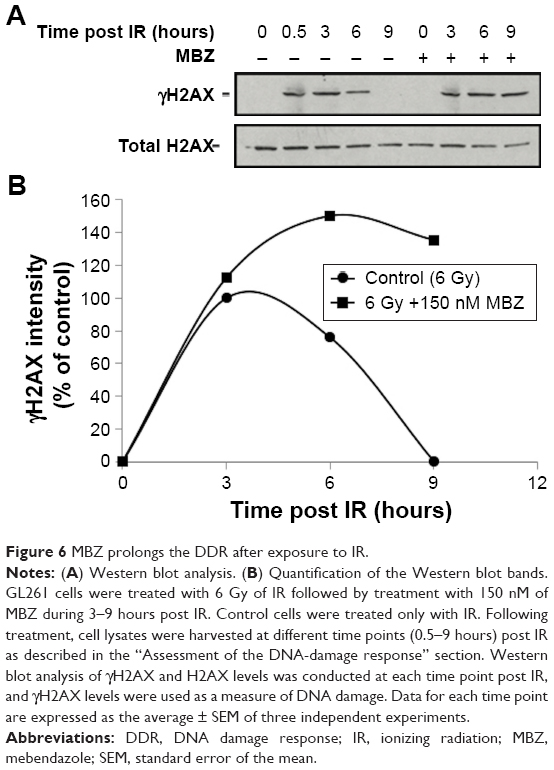 | Figure 6 MBZ prolongs the DDR after exposure to IR. |
MBZ interferes with the trafficking of DDR proteins
To investigate whether MTAs synergize with IR by disrupting intracellular transport of DDR proteins, we performed cytoplasmic and nuclear (C/N) fractionation studies with glioma cells that had been exposed to IR followed by treatment with MBZ. Similar to the cell viability studies, MBZ was applied during 3–9 hours post IR (only when the mitotic cell population was absent). Glioma cells were treated with different concentrations of MBZ followed by C/N fractionation. Each fraction was analyzed by Western blot. We selected two DDR proteins, Chk2 and Nbs1, to serve as potential targets of MTA-mediated toxicity. Chk2 is a key protein kinase involved in the DDR that is responsible for cell cycle checkpoint activation and DNA repair following DNA damage induced by IR.26 Nbs1 is a crucial component of an enzymatic complex that repairs double strand breaks (DSBs) following irradiation or heat shock.27
In control cells, which had not been treated with MBZ, Chk2 and Nbs1 were localized entirely in the nucleus (Figure 7A). Radiation treatment did not significantly alter the intracellular distribution of DDR proteins (Figure 7A). Treatment of glioma cells with MBZ sequestered Chk2 and Nbs1 in the cytoplasm in a dose-dependent manner (Figure 7B and C). These results demonstrate that MBZ, like other MTAs,17 inhibits the trafficking of DDR proteins from the cytoplasm to the nucleus.
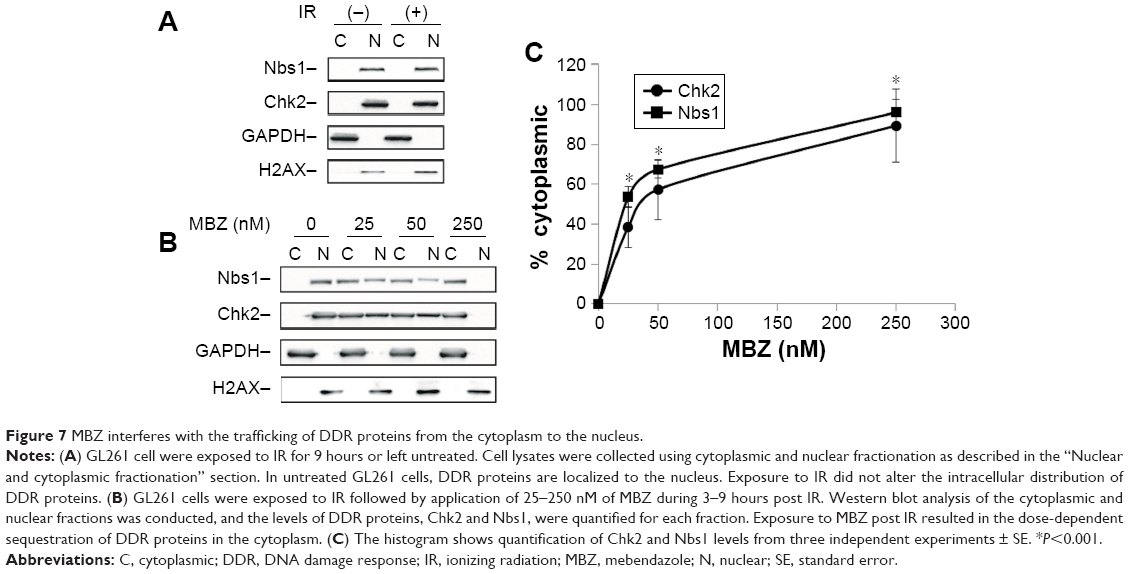 | Figure 7 MBZ interferes with the trafficking of DDR proteins from the cytoplasm to the nucleus. |
MBZ sensitizes glioma cells to IR by interfering with the trafficking of DDR proteins
To more closely examine the relationship between the effects of MBZ on intracellular trafficking of DDR proteins and IR sensitization, we compared the EC50 values for radiosensitization (DEF), cytoplasmic sequestration of DDR proteins and induction of mitotic arrest by MBZ (Table 1) in GL261 cells. The EC50 was defined as the concentration of MBZ required to achieve a half-maximal effect. The EC50 of cytoplasmic sequestration for Chk2 (31 nM) and Nbs1 (25 nM) was significantly lower than the EC50 of mitotic arrest (192 nM). Most notably, the EC50 of radiosensitization (35 nM) was very similar to the EC50 for cytoplasmic sequestration of DDR proteins, but significantly lower than the EC50 of mitotic arrest (P<0.01). Similarly, the EC50 for radiosensitization by VCR (<0.5 nM) is significantly lower than the EC50 for mitotic arrest in GL261 cells (2.5 nM).23 These findings support the hypothesis that MBZ indeed radiosensitizes GL261 cells in large part by interfering with intracellular trafficking of DDR proteins during interphase.
Discussion
Studying the mechanisms of action of MTAs in cell toxicity, and in particular whether MTAs target cells in mitotic phase or interphase, has been hampered by the difficulty in separating these two cell populations. In this study, we transiently eliminated the mitotic cell population by irradiating glioblastoma cells, which allowed us to examine the extent to which the MTA, MBZ, can radiosensitize these cells in a cell population that is essentially made up of interphase cells only. Our results show that in this system, the effect of MBZ in interphase is responsible for the majority of the radiosensitization effect caused by this MTA.
For a long time, MTAs have been believed to inhibit tumor growth primarily by targeting mitotic cells, but this hypothesis has come under considerable scrutiny.11,12,15,16 We examined the role of interphase microtubules by determining the effect of MBZ as a radiosensitizer, when present during a time window in which mitosis is prevented by G2–M cell cycle arrest. We showed that this regimen is better at radiosensitization of tumor cells than when MBZ is present before IR administration, leading to an increase in the number of mitotic cells, and is very similar to that caused by chronic treatment with MBZ, strongly indicating that interphase microtubules are indeed the targets of MBZ. In addition, the EC50 of MBZ for radiosensitization is much lower than that for inducing mitotic arrest, further supporting the notion that the radiosensitizing effect of MBZ is independent of its effect on mitosis. We found essentially the same results for VCR. Thus, most likely this conclusion holds for a wide range of MTAs.
We also observed that MBZ, even when applied for only 6 hours, leads to a strong delay in the DDR. Thus, our results strongly support the hypothesis that MTAs prolong DNA damage repair by interfering with the trafficking of DDR proteins from the cytosol to the nucleus.17
Interestingly, the EC50 for radiosensitization by MBZ (35 nM) is very similar to that of cytoplasmic sequestration of DDR proteins by MBZ (25 nM), which is much lower than the EC50 of MBZ for the induction of mitotic arrest (184 nM), further supporting the notion that MTAs radiosensitize by blocking trafficking of DDR proteins to the nucleus. The low EC50 of MBZ for the inhibition of DDR protein trafficking is surprising, because it is also much lower than the EC50 for microtubule depolymerization (132 nM) that we determined recently.23 This EC50 largely reflects that of the depolymerization of interphase microtubules, as most of the cells are in interphase.
Although the mechanistic basis for the relatively low EC50 of MBZ for the inhibition of DDR protein trafficking remains to be determined, it will be of great interest to examine whether our findings with MBZ extend to other MTAs. Our observation that radiosensitization can be accomplished at a concentration of MBZ that is significantly lower than the concentration needed for cell killing on its own also has important clinical implications, as it suggests the possibility to utilize a dose of MBZ that minimizes toxicity. Importantly, our results also have implications for the optimal timing of administration of MTAs when used as radiosensitizers. Indeed, we show that optimal radiosensitization is obtained when inhibition of microtubule formation is achieved during the post-IR DNA repair period.
This study also underlines the critical role of microtubule-based transport in the response to DNA damage. Thus, further elucidation of these mechanisms may lead to the identification of novel therapeutic targets for radiosensitization.
Conclusion
We have shown that MBZ sensitizes cancer cells to IR in a manner that is largely independent of the induction of mitotic arrest by this microtubule inhibitor. We also provide evidence that MBZ-induced radiosensitization is mediated by inhibiting DDR protein accumulation into the nucleus. Thus, this study strongly supports a critical role for DDR protein trafficking in the response to radiation, suggesting that elements of the protein trafficking machinery can be mined for additional radiosensitization targets.
Acknowledgments
GBM14 cells were kindly provided by Dr J Sarkaria, Mayo Clinic, Rochester, MN, USA. All data generated and analyzed during this study are included in this publication. This study was supported by grants to MS from the Swim Across America Foundation and the Project To Cure Foundation and to RR from the Zankel Foundation.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Wang T, Wang H, Soong Y. Paclitaxel-induced cell death. Cancer. 2000;88(11):2619–2628. | ||
Bhalla K. Microtubule-targeted anticancer agents and apoptosis. Oncogene. 2003;22:9075–9086. | ||
Tu Y, Cheng S, Zhang S, Sun H, Xu Z. Vincristine induces cell cycle arrest and apoptosis in SH-SY5Y human neuroblastoma cells. Int J Mol Med. 2013;31(1):113–119. | ||
Jordan MA, Thrower D, Wilson L. Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J Cell Sci. 1992;102:401–406. | ||
Gabikian P, Tyler B, Zhang I, Li KW, Brem H, Walter K. Radiosensitization of malignant gliomas following intracranial delivery of paclitaxel biodegradable polymer microspheres. J Neurosurg. 2014;120(5):1078–1085. | ||
Tishler R, Geard C, Hall E, Schiff P. Taxol sensitizes human astrocytoma cells to radiation. Cancer Res. 1992;52:3495–3497. | ||
Sinclair WK. Cyclic X-ray responses in mammalian cells in vitro. 1968. Radiat Res. 2012;178(2):AV112–AV124. | ||
Elkind M, Sutton H. Radiation response of mammalian cells grown in culture: I. Repair of X-Ray damage in surviving Chinese hamster cells. Radiat Res. 1960;13(4):556–593. | ||
Neuner G, Patel A, Suntharalingam M. Chemoradiotherapy for esophageal cancer. Gastrointest Cancer Res. 2009;3:57–65. | ||
Mandilaras V, Bouganim N, Spayne J, et al. Concurrent chemoradiotherapy for locally advanced breast cancer-time for a new paradigm? Curr Oncol. 2015;22:25–32. | ||
Mitchison T. Proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23:1–6. | ||
Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol. 2011;8:244–250. | ||
Mancuso A, Oudard S, Sternberg C. Effective chemotherapy for hormone-refractory prostate cancer (HRPC): present status and perspectives with taxane-based treatments. Crit Rev Oncol Hematol. 2007;61:176–185. | ||
Tannock I, de Wit R, Berry W, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. | ||
Ogden A, Padmashree R, Reid M, Aneja R. Interphase microtubules: chief casualties in the war on cancer. Drug Discov Today. 2014;19(7):824–829. | ||
Field J, Kanakkanthara A, Miller J. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg Med Chem. 2014;22(18):5050–5059. | ||
Poruchynsky M, Komlodi-Pasztor E, Trostel S, et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc Natl Acad Sci U S A. 2015;112(5):1571–1576. | ||
Carlson B, Grogan P, Mladek A, et al. Radiosensitizing effects of temozolomide observed in vivo only in a subset of O6-methylguanine-DNA methyltransferase methylated glioblastoma multiforme xenografts. Int J Radiat Oncol Biol Phys. 2009;75(1):212–219. | ||
Tapia C, Kutzner H, Mentzel T, Savic S, Baumhoer D, Glatz K. Two mitosis-specific antibodies, MPM-2 and Phospho-Histone H3 (Ser28), allow rapid and precise determination of mitotic activity. Am J Surg Pathol. 2006;30(1):83–89. | ||
Tao Y, Leteur C, Calderaro J, et al. The Aurora B kinase inhibitor AZ1152 sensitizes cancer cells to fractionated irradiation and induces mitotic catastrophe. Cell Cycle. 2009;8(19):3172–3181. | ||
Akihito A, Chano T, Futami K, et al. RECQL1 and WRN proteins are potential therapeutic targets in head and neck squamous cell carcinoma. Cancer Res. 2011;71(13):4598–4607. | ||
Lindgren T, Stigbrand T, Johansson L, Riklund K, Eriksson D. Alterations in gene expression during radiation-induced mitotic catastrophe in HeLa Hep2 cells. Anticancer Res. 2014;34(8):3875–3880. | ||
De Witt M, Gamble A, Hanson D, et al. Repurposing mebendazole as a replacement for vincristine for the treatment of brain tumors. Mol Med. Epub 2017 Apr 5. doi:10.2119/molmed.2017.00011. | ||
Mah L, El-Osta A, Karagiannis T. γH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–686. | ||
Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol. 2012;920:613–626. | ||
Zannini L, Delia D, Buscemi G. Chk2 kinase in the DNA damage response and beyond. J Mol Cell Biol. 2014;6(6):442–457. | ||
Lamarche B, Orazio N, Weitzman M. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010;584(17):2682–2695. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.