Back to Journals » OncoTargets and Therapy » Volume 12
MicroRNA-765 sensitizes osteosarcoma cells to cisplatin via downregulating APE1 expression
Authors Liang W, Li C, Li M, Wang D, Zhong Z ![]()
Received 15 November 2018
Accepted for publication 18 May 2019
Published 3 September 2019 Volume 2019:12 Pages 7203—7214
DOI https://doi.org/10.2147/OTT.S194800
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Wei Liang,1,2,* Chongyi Li,1,* Mengxia Li,1 Dong Wang,1 Zhaoyang Zhong1
1Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing 400042, People’s Republic of China; 2Department of Oncology, The Third Affiliated Hospital of Chongqing Medical University (Gener Hospital), Chongqing 401120, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhaoyang Zhong
Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, No. 10 Changjiang Zhi Rd, Yuzhong District, Chongqing 400042, People’s Republic of China
Tel +860 236 875 7183
Fax +860 236 889 4062
Email [email protected]
Objectives: Osteosarcoma (OS) is the most common bone cancer diagnosed in children and adolescents. Expression of APE1 is commonly increased in OS, and this is negatively correlated with a sensitivity to platinum and a favorable prognosis. However, the mechanism underlying high APE1 expression in OS is not fully understood.
Methods: A bioinformatics analysis of the APE1 3’-UTR combined with previous microarray data was used to identify miRNAs that regulate APE1 expression. The effects of miR-765 on cisplatin (cDDP) sensitivity were estimated in OS cell lines (9901 and HOS) and BALB/c mice (n=4 per group). The relative expression and association between miR-765 and APE1 were assessed in a cohort of OS patients (n=43 in total) with Kaplan-Meier and Cox proportional hazards regression. All statistical tests were two-sided and p<0.05 was considered significant.
Results: Bioinformatics analysis implied that miR-765 may target APE1. Luciferase assay and WB showed that miR-765 bound directly to the 3’-UTR of APE1 and downregulated APE1 expression in OS cells. Further experiments revealed that miR-765 sensitized OS cells to cisplatin and was associated with decreased DNA repair activity. In vivo analyses suggested the sensitivity of cisplatin in xenograft OS tissues was increased after injection with miR-765 agomir. The clinical data showed a negative correlation between miR-765 and APE1 expression (r=0.307, p=0.045). Log-rank test revealed that OS patients with positive expression of miR-765 obtained a significantly longer survival than those with negative expression (22.0 vs. 9.0 months, p=0.001), which is just the opposite with respect to APE1 expression (12.00 vs. 22.00 months, p=0.039). The Cox regression analysis found miR-765 may be an independent prognostic factor for OS survival (p=0.007, HR=0.389, 95% CI: 0.196-0.772).
Conclusion: miR-765 sensitizes OS cells to cisplatin and impedes DNA damage repair through the downregulation of APE1. High expression of miR-765 may benefit OS patient survival, making it a viable target for reversing cisplatin-induced resistance in OS patients.
Keywords: miR-765, APE1, osteosarcoma, chemosensitivity, DNA damage repair
Introduction
Osteosarcoma (OS) is the most common type of bone cancer diagnosed in children and adolescents. The incidence of OS increases starting at the age of 5 years and remains elevated during childhood and adolescence, and then the incidence decreases in adulthood.1 Since the introduction of standard systemic therapy consisting of neoadjuvant chemotherapy followed by surgery and adjuvant therapy, the overall survival of OS patients has improved greatly.2 However, chemoresistance was still a limitation for extending the overall survival of OS patients.3 Thus, identifying potential factors involved in OS chemoresistance and elucidating their underlying mechanisms was pivotal for the development of new therapeutic strategies.4
MicroRNAs (miRNAs), which were short noncoding RNAs with a length of 21–23 nucleotides, participate in regulating multiple cell functions. These small molecules inhibit translation and induce degradation of target messenger RNAs (mRNAs) by binding to their 3ʹ untranslated regions (3ʹ UTRs), thus resulting in posttranscriptional downregulation of gene expression.5 Recently, miRNAs have displayed great potential in regulating tumor biology, including tumor proliferation, invasion, angiogenesis, and chemosensitivity by regulating downstream oncogenes or tumor suppressors. APE1, a key protein of the base excision repair (BER) pathway, plays a pivotal role in regulating chemosensitivity of OS.4 A previous study demonstrated that downregulation of APE1 contributed to enhanced chemosensitivity of OS cells to chemotherapeutic agents and enabled a favorable prognosis for OS patients. Likewise, the overexpression of APE1 in tumor tissues and cancer cells lead to chemoresistance and poor prognoses for OS patients.6 Thus, exploring factors that downregulate APE1 expression might provide promising strategies for overcoming the chemoresistance of OS patients.
Given the powerful role of APE1 in triggering chemoresistance in OS patients, we rationalized that miRNA-suppressed APE1 expression might improve the chemosensitivity of OS. In this study, we aim to explore the mechanism by which miR-765 sensitizes osteosarcoma cells to cisplatin.
Materials and methods
Cell lines and cell culture
Saos-2, U2OS, MG63, HOS and hFOB 1.19 cell lines were purchased from the American Tissue Culture Collection (Manassas, VA, USA). 9901 cells were donated by Fourth Military Medical University, Xian, China. All of the osteosarcoma cell lines were cultured as recommended by the suppliers and was approved by ethics committee of Daping Hospital.
Reagents
miRNA mimics and their matched miRNA NC, agomiR-765 and matched scramble NC were purchased from RiboBio Co. Ltd (Guangzhou, China). Cisplatin was purchased from Sigma-Aldrich (Wisconsin, USA)
miRNA/vector transfections
Cells were plated without antibiotics approximately 24 h before transfection. Transient transfection of miRNA mimics/inhibitor (Ribobio, GuangZhou, China) or siRNA (Sigma, USA) were carried out by using LipofectamineTM 2000 (Invitrogen, CA, USA) according to the manufacturer’s protocol. All miRNA/siRNA were transfected for 48 hrs.
The custom APE1-siRNA (5ʹ-GUCUGGUACGACUGGAGUACC-3ʹ,5ʹ-UACUCCAGUCGUACCAGACCU-3ʹ) and the negative control (5ʹ-CCAUGAGGUCAGCAUGGUCUG-3ʹ, 5ʹ-GACCAUGCUGACCUCAUGGAA-3ʹ) were devised according to Wang et al6,21 studies.
Plasmid construction and luciferase assay
The full-length 3ʹ-UTR of human APE1 was amplified by PCR from genomic DNA of 9901 cells, and the potential miR-765 binding site in the 3ʹ-UTR of APE1 was mutated by the overlap extension PCR method. Both wild-type and mutant 3ʹ-UTRs were subcloned into the pMIR-Report plasmid (Promega, Wisconsin, USA) directly downstream of the renilla luciferase coding sequence. The authenticity and orientation of the inserts were confirmed by sequencing. Luciferase assays were performed as previously described riefly, 5×103 cells per well were seeded in 96-well plates 24 h before transfection. Cells were co-transfected with miRNA mimics or NC plus wild-type or mutant APE1 3ʹ-UTR plasmids using LipofectamineTM 2000 (Invitrogen, CA, USA). 36 h post-transfection, cells were assayed for both firefly and renilla luciferase using Dual-GloTM Luciferase Assay System (Promega, Wisconsin, USA) according to the manufacturer’s instruction.
Quantitative real time qRT-PCR analysis
Total RNAs, including miRNAs, were extracted by using GeneJET RNA Purification Kit (Thermo, CA, USA) from cultured cells following the manufacturer’s instructions. Expression of mature miR-765 was determined by Bulge-Loop™ miRNA qRT-PCR Primer Set (Ribobio, Guangzhou, China) with SYBR Green quantitive real time-PCR (qRT-PCR), and U6 snRNA was used as an internal control. Gene mRNA expression analyses were performed by qRT-PCR assays using the PrimeScript RT reagent Kit (TaKaRa, Dalian, China) and SYBR Premix Ex Taq (TaKaRa, Dalian, China) according to the manufacturer’s instructions. The comparative Ct method was used to calculate the relative changes in gene expression.
Western blot
Total protein was collected by using RIPA buffer containing protease inhibitors (Sigma, Wisconsin, USA). The supernatant protein concentration was measured using a BCA kit (Beijing Dingguo, Beijing, China). Western blot was done as described previously.22 Primary antibodies included mouse monoclonal antibody anti-APE1 (1:8000) and mouse monoclonal antibody anti-β-Actin (Proteintech, Wuhan, China), The HRP-conjugated Goat anti mouse secondary antibody was used, and the bands were visualized by chemiluminescence. Protein expression was quantified using Image-Lad 5.0 software (Bio-Rad, USA) normalized against internal control (β-actin).
Assessing chemosensitivity to cisplatin
To assess the sensitivity to cisplatin in vitro, cell viability assays were performed by CCK-8 (Beyotime, Beijing, China). Logarithmically growing cells which were transfected with miR-765 mimics or miR NC were seeded at 5×103 cells/well in a 96-well plate. Cells were then treated for the indicated time with cisplatin within a suitable concentration range, and then 10 μl of CCK-8 solution was added to each well, and the plate was incubated for 2 h in a humidified incubator. The absorbance of each well was measured at 450 nm using a Model 550 series microplate reader. The assay was performed using three replicates. Cell viability was expressed as the ratio of treated cells to that of untreated controls at each dose or concentration. The IC50 value for each cell line was determined by nonlinear regression analysis using GraphPad Prism (GraphPad Software Inc., San Diego, CA). Statistical differences between IC50 values for the various groups were determined by one-way ANOVA analysis.
γ-H2AX immunofluorescence microscopy
The 9901 cells were plated on 4-well chamber slides, allowed to attach overnight, and exposed to 4 µg/mL CISPLATIN for different time points. After treatment, cells were washed with ice-cold phosphate-buffered saline (PBS), blocked in 5% bovine serum albumin for 1 hr at room temperature, incubated with the antibody (fluorescein isothiocyanate [FITC]-conjugated anti-phospho-histone γ-H2AX [Millipore, Billerica, MA, USA]) for 16 hrs at 4 °C in the dark, washed with PBS, and mounted in Vectashield mounting medium containing diamidino-2-phenylindole (Vector Laboratories, Burlingame, USA). γ-H2AX foci were examined using a Zeiss Axio Imager A1 fluorescence microscope.
Tumor xenograft studies
This research was approved by the Research Ethics Board of Daping Hospital. We subcutaneously injected 5×106 9901 cells resuspended with phosphate-buffered saline into the right flanks of 5-week-old BALB/C athymic mice (HFK Bioscience, Beijing). Mice were housed and maintained under specific pathogen-free conditions. All animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals of Daping Hospital. When mice had palpable tumors, the mice were randomly assigned, in order to avoid treatment bias, to treatment groups (n=4 mice per group). Cisplatin (5 mg/kg) was administered intraperitoneally every 4 days for 28 days. For agomiR treatment, agomiR-765 or agomiR-NC (RiboBio, Guangdong, China) was directly injected intratumorally at the dose of 1 nmol (diluted in 20 μL phosphate buffered saline) per mouse every 4 days for 7 total injections. Tumor volumes were calculated as length × (square of width)/2. After the initial treatment, the tumor size was measured every day. Mice were sacrificed by cervical dislocation under anesthesia. Investigators were blinded to the treatment groups.
Tissue samples
Forty-three patient samples were collected between September 2010 and June 2013. All the patients never received preoperative adjuvant chemotherapy. The tumor content of the specimens was assessed by hematoxylin and eosin staining in the pathology units. Only specimens containing more than 60% of tumor tissue were used. All samples were routinely fixed immediately after surgery in 10% formalin. After fixation, samples were dehydrated, incubated in xylene, infiltrated with paraffin, and finally embedded in paraffin. Samples were the diagnoses performed by a sarcoma pathologist. This research was approved by the Research Ethics Board of Daping Hospital. All patients who provided tissues were provided with written informed consent, and all of them agreed to the use of their samples in scientific research.
Tissue microarray construction
Tissue Microarray Construction has been described previously .23 Briefly, areas containing viable tumor were marked on the paraffin wax tissue blocks. Duplicate 2.0 mm tissue cores taken from different areas of the same tissue block for each case (three cores per case) were used to construct the tissue microarrays using an arraying machine from Beecher Instruments (Sun Prairie, WI). Array blocks were sectioned to produce 4 μm sections.
In situ hybridization (ISH) and analysis
The cellular expression of miR-765 was determined by an in-situ hybridization technique. Briefly, tissue microarrays were deparaffinized and rehydrated. After Proteinase-K incubation for 15 min at 37 °C, tissue sections were hybridized with 3ʹ,5ʹ-DIG-labeled LNA miR-765 antisense probeor LNA scrambled control probe (Ruibo, Guangzhou, China) at 55 °C for 1 hr. Slides were then washed and blocked with DIG blocking buffer (Roche, Switzerland, Basel), incubated with anti-DIG antibody(Roche, Switzerland, Basel)diluted 1:1000 in blocking solution containing 2% sheep serum and incubated for 60 min at RT. Color development was achieved by applying freshly prepared BCIP (Amrescon) to the sections and incubating slides for 2 hrs at 30 °C in the humidifying chamber. Nuclear Fast Red (Amrescon) was applied for nuclear counterstaining.
Immunohistochemical staining and analysis
Immunohistochemical staining was performed according to standard procedures. Sections were stained with mouse monoclonal anti-APE1 antibody (1:8000) and mouse monoclonal anti-Ki67 antibody (1:100,zhongsanjinqiao,Beijing,China) overnight at 4 °C. Negative controls were treated identically but without the primary antibody. The results for APE1(the same as Western Blot)staining were scored on the basis of the percentage of positively stained cells (score 0: no positive cells; score 1: ≤10% positive cells; score 2: 11%-25% positive cells; score 3: 26–50% positive cells; and score 4: ≥51% positive cells). For further statistical analysis, scores 0 and 1 were categorized as negative expression, and scores 2 and 3 were categorized as positive expression as described previously.24 The staining was simultaneously determined separately by two independent experts under the same conditions. In rare cases, discordant scores were reevaluated and scored on the basis of consensual opinion.
Statistical analysis
All p-values less than 0.05 were considered statistically significant. All assays were independently performed at least three times. All values are presented as mean ± standard deviation (SD). One-way ANOVA was used for multiple group comparisons.
Spearman rank correlation was used to determine the correlation between APE1 and miR-765 expression. The overall survival rate of patients with Osteosarcoma (OS) was calculated using a Kaplan- Meier survival analysis. The significance of multiple predictors of survival was assessed using Cox regression analysis.
Results
miR-765 targeting of APE1 in osteosarcoma cells
The qRT-PCR results showed that intrinsic APE1 levels were negatively associated with miR-765 in a panel of osteosarcoma cells (Saos2, 9901, MG63, U2OS, hFOB1.19, and HOS) (Figure 1A). Moreover, using several different databases and software programs, including DIANA-microT, TargetScan, and RNA Hybrid, our data showed that the APE1 gene is a potential miR-765 target in silico. The mature sequence of miR-765 and its seed region are shown in Figure 1B.
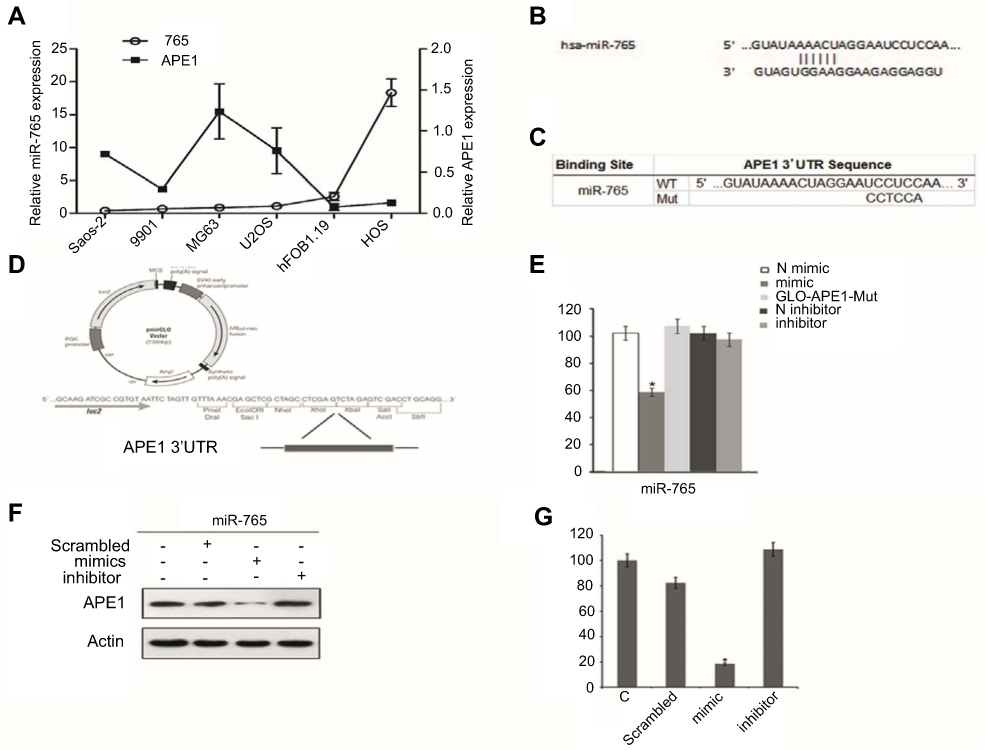 |
Figure 1 MiR-765 targets APE1 in OS cells. (A) Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) analysis in five osteosarcoma cancer cell lines showed an inverse expression trend between miR-765 and APE1 expression. The expression levels of miR-765 and APE1 were normalized to hFOB1.19. (B) The predicted duplex formation between human WT APE1 3ʹ-UTR and miR-765 is shown. (C) The WT andMut APE1 3ʹ-UTR sequences are shown. (D) Schematic diagram showing luciferase reporter constructs containing the APE1 3ʹ-UTR sequences. (E) Effects of miR-765 mimics (inhibitors) on APE1 3ʹ-UTR luciferase reporters in HOS cells. Empty vector pmirGLO-Report and negative control mimics (N) were included as controls. Luciferase activities were calculated as the ratio of firefly/renilla activities and normalized to the negative control mimics group. The results were obtained from three independent experiments, with each experiment conducted in triplicate. Data are shown as the mean ± SD. (F and G) Western blot analysis was performed to quantify the intracellular APE1 levels. Actin was used as an internal control. |
To determine whether miR-765 is capable of regulating APE1 expression via the binding sites in its 3ʹ-UTR, we cloned the APE1 3ʹ-UTR containing the predicted miR-765 binding site and placed it downstream of the firefly luciferase coding region in the pMIR-REPORT luciferase vector, resulting in wild type luciferase reporter pMIR-APE1-wt-3ʹ-UTR (Figure 1C and D). The putative miR-765 binding sites were mutated, which generated mutant luciferase reporter pMIR-APE1-mut-3ʹ-UTR (Figure 1C and D).
Each reporter was cotransfected with miR-765 mimics (inhibitors) into HOS cells, and luciferase assays were completed to determine the effects of miR-765 on the expression of APE1. Real-time PCR analysis verified the efficacy of transfection by revealing markedly increased expression of mature miR-765 in HOS after transfection. For the wild type APE1 reporter, overexpression of miR-765 mimics significantly reduced luciferase activities (p<0.05) compared to nontarget negative control miRNA mimics, whereas this effect was abolished in the case of the mutant reporter in which the miR-765 binding site was mutated (Figure 1E). These results strongly suggest that miR-765 downregulates the expression of APE1 by directly targeting its 3ʹ-UTR.
To further validate the finding that miR-765 regulates expression of APE1, we assessed its protein expression level using Western blot analysis in HOS cells transfected with miR-765 mimics (inhibitors) or nontarget negative controls. Compared to negative controls, miR-765 mimics significantly reduced the protein level of APE1 (p<0.05) (Figure 1F and G).
miR-765 sensitized osteosarcoma cells to cisplatin through APE1
APE1 is an integral component of the cellular DNA damage response. Thus, miR-765–mediated APE1 downregulation is hypothesized to increase the sensitivity of cells to cisplatin. The CCK-8 results showed that osteosarcoma cells overexpressing miR-765 were more sensitive to cisplatin compared to control cells (Figure 2A and B). Furthermore, osteosarcoma cells cotransfected with miR-765 mimics and APE1 siRNA showed an increased sensitivity to cisplatin in comparison with cells co-transfected with miR-765 mimics and control siRNA (Figure 2C and D). Taken together, these results suggest that miR-765 could sensitize osteosarcoma cells to cisplatin through APE1.
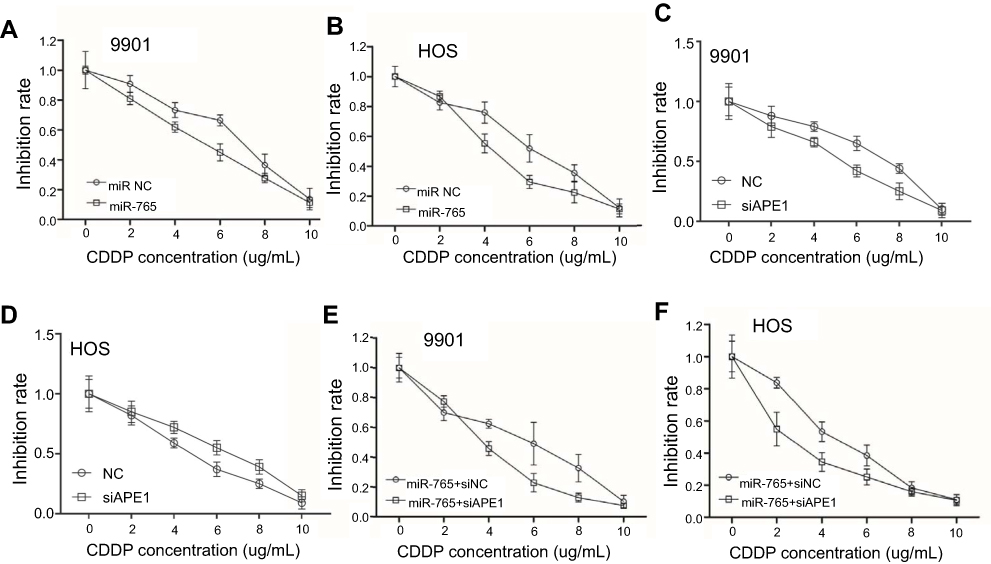 |
Figure 2 Effect of miR-765 on cisplatin sensitivity through APE1 inhibition. (A and B) 9901/HOS cells were transfected with miR-765 mimics or the miR mimic negative control (miR NC) for 48 hrs. Cell viability was assayed by CCK-8 after treatment with increasing concentrations of cisplatin for 48 hrs. Data are shown as the mean ±95% confidence interval from three independent experiments. (C and D) 9901/HOS cells were transfected with APE1 siRNA or the siRNA negative control (miR NC) for 48 hrs. Cell viability was assayed by CCK-8 after treatment with increasing concentrations of cisplatin for 48 hrs. Data are shown as the mean ±95% confidence interval from three independent experiments. (E and F) 9901/HOS cells were cotransfected with miR-765 mimics and siAPE1 (siNC) for 48 hrs. Cell viability was assayed by CCK-8 after treatment with increasing concentrations of cisplatin for 48 hrs. Data are shown as the mean ±95% confidence interval from three independent experiments. |
miR-765 increased the DNA damage of osteosarcoma cells treated with cisplatin
We subsequently investigated whether miR-765 downregulation of APE1 modulates unrepaired DNA damage: this was accomplished via the assessment of phosphorylated histone H2AX (γ-H2AX), which forms foci at double-stranded DNA breaks and recruits DSB repair proteins.7–9 Correlating with the amount of unrepaired damage, the number of γ-H2AX foci was quantified at different time points after cisplatin treatment. Compared with cells treated only with cisplatin, the combination of miR-765 mimics and cisplatin induced a significant increase of γ-H2AX foci (Figure 3A and B). Moreover, the addition of APE1 siRNA to OS cells treated with miR-765 mimics and cisplatin led to retention of these foci. Taken together, these results indicate that miR-765 increases the DNA damage of osteosarcoma cells partially through downregulating APE1.
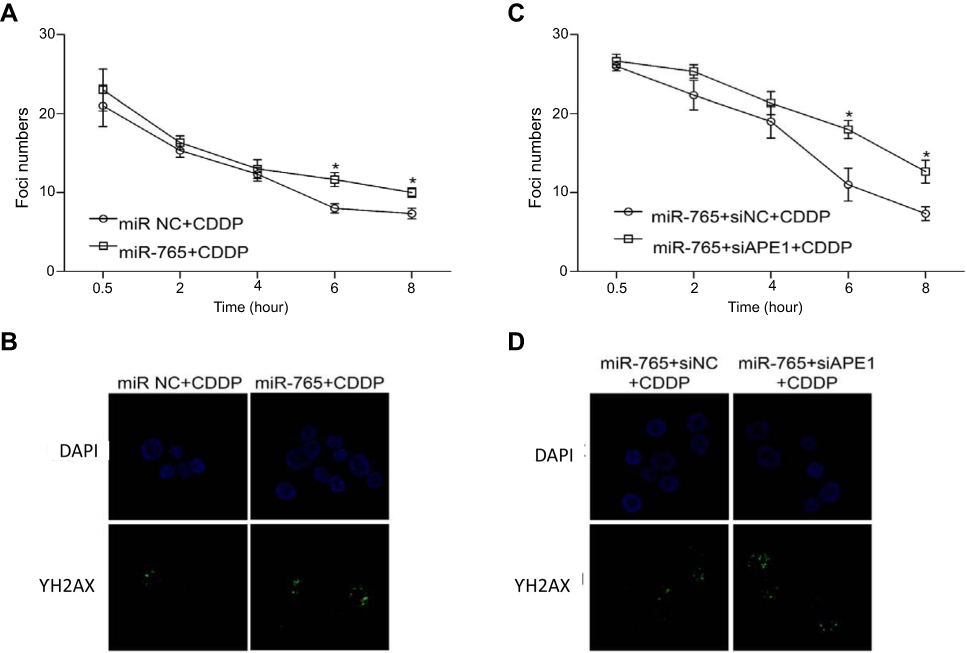 |
Figure 3 Effect of miR-765 on DNA repair through APE1 inhibition. The number of γ-H2AX foci/cell was counted in a minimum of 200 cells per treatment group. The average number of γ-H2AX foci/cell is shown. Error bars indicate S.D. * p<0.05. (A and B) The representative images of 9901 cells transfected with miR-765 mimics or the miR NC and treated with cisplatin (5 ug/ml) from the 6 hr groups are shown. (C and D) The representative images of 9901 cells cotransfected with miR-765 mimics and siAPE1 (siNC) and treated with cisplatin (5 µg/ml) from the 6 hr groups are shown. |
miR-765 combined with cisplatin enhanced the tumor growth inhibition in xenograft models
To further demonstrate the role of miR-765 in chemosensitization, we used miR-765 agomiR, a cholesterol-conjugated 2ʹ-O-methyl-modified miR-765 exhibiting suitable pharmacokinetic properties for in vivo studies.10 Mice were treated with vehicle alone, cisplatin plus miR-765 agomiR, or cisplatin plus scrambled agomiR. Cisplatin plus scrambled agomiR had no statistically significant effects on tumor growth. However, in mice treated with cisplatin plus miR-765 agomiR, tumor growth was statistically significantly delayed, with 59.8% (p=0.0041) and 47.1% (p=0.0044) reductions in tumor volume at day 35 compared with vehicle or cisplatin plus scramble agomiR, respectively (cisplatin plus miR-765 agomiR: mean tumor volume =259.2 mm3, 95% CI =169.0–349.4 mm3; vehicle alone: mean tumor volume =644.0 mm3, 95% CI =386.8–901.3 mm3; cisplatin plus scrambled agomiR: mean tumor volume =489.8 mm3, 95% CI =350.8–628.8 mm3) (Figure 4A and B). Moreover, tumor weights on day 35 after tumor cell injection with cisplatin plus miR-765 agomiR were statistically significantly lower than vehicle or cisplatin plus scramble agomiR, with 62.6% (p=0.0034) and 51.7% (p=0.0027) reductions, respectively (cisplatin plus miR-765 agomiR: mean tumor weight =0.2190 g, 95% CI =0.1364–0.3016 g; vehicle alone: mean tumor weight =0.5850 g, 95% CI =0.3495–0.8205 g; cisplatin plus scrambled agomiR: mean tumor weight =0.4530 g, 95% CI =0.3258–0.5802 g) (Figure 4C). These data support the notion that miR-765 is a potent cisplatin sensitizer in vivo.
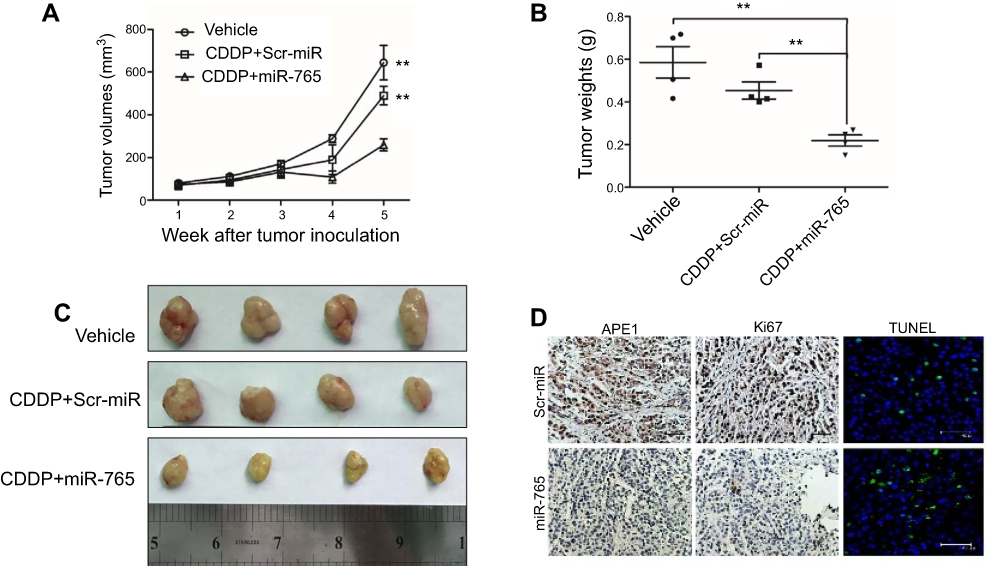 |
Figure 4 Effect of miR-765 on tumor growth combined with cisplatin in the xenograft model. (A) Growth curves of 9901 subcutaneous xenograft tumors treated with vehicle, cisplatin (5 mg/kg, intraperitoneally every 4 days) plus agomiR-765 (1 nmol, intratumoral injection, every 4 days), or cisplatin (5 mg/kg, intraperitoneally every 4 days) plus agomiR-scramble-NC (1 nmol, intratumoral injection, every 4 days) are shown. Tumor volumes were calculated as length × (square of width)/2. Upwards and downwards arrows indicate the start and end of treatment, respectively. n=4 per group (**p<0.005, two-sided Student t-test). (B) The gross morphology of tumors measured on day 35 after tumor cell injection. (C) The tumors were weighed immediately after isolation from mice (**p<0.005, two-sided Student t-test). (D) The immunohistochemistry analyses for APE1 and Ki67, along with TUNEL staining, were carried out on 9901 xenograft tumor sections collected from mice treated with the indicated treatments. Representative staining samples are shown. |
Tumor sections were analyzed using immunohistochemistry with anti-APE1 and anti-Ki67 antibodies to measure the expression of APE1 and the cellular proliferation levels. Tumors overexpressing miR-765 had a lower level of APE1 expression and proliferation potential than control tumors (Figure 4D). Furthermore, miR-765-overexpressing tumors had a higher apoptosis index according to TUNEL staining. Therefore, these data suggest that miR-765-dependent downregulation of APE1 plays a potential role in the treatment of osteosarcoma.
Prognostic role of miR-765 in osteosarcoma patients
The physiological relevance of the APE1/miR-765 interaction was further established by evaluating the endogenous expression patterns of miR-765 and APE1 in osteosarcoma patients. The demographic and clinical characteristics of patients are shown in Table 1. APE1 level was quantified using immunohistochemistry (IHC), while miR-765 level was assessed using fluorescent in situ hybridization (FISH). Among 43 samples, the average APE1 level was higher (approximately twofold; p=0.0034) in the miR-765 negative group than that in the miR-765 positive group (Figure 5A). The representative images are shown in Figure 5B. Using Spearman correlation analysis, an inverse correlation, with R2=0.307, was observed between miR-765 and APE1 (p<0.001); this suggests miR-765-dependent regulation of APE1 (Table 2).
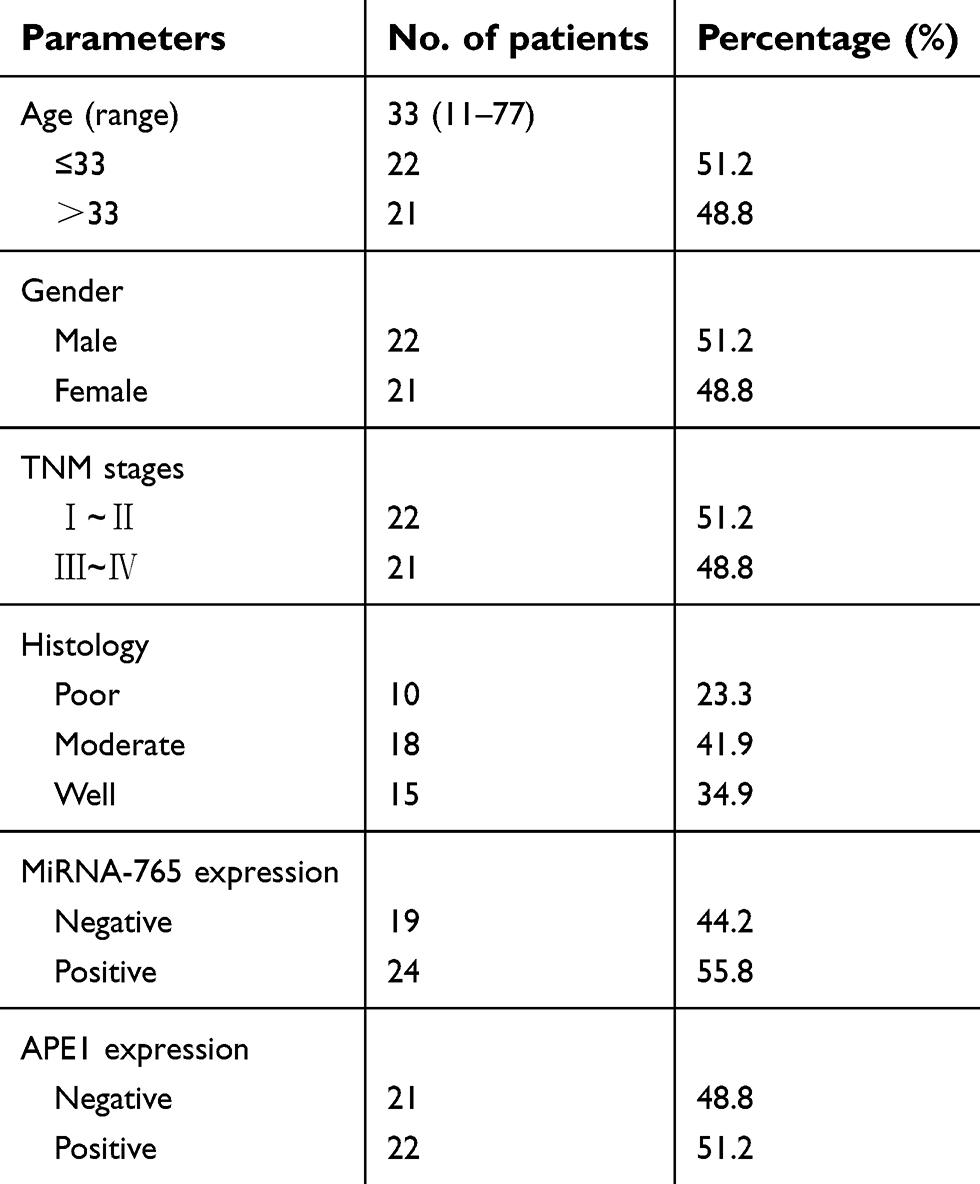 |
Table 1 The demographic and clinical characteristics for the research group |
 |
Table 2 The correlation between miR-765 and APE1 expression in OS patients |
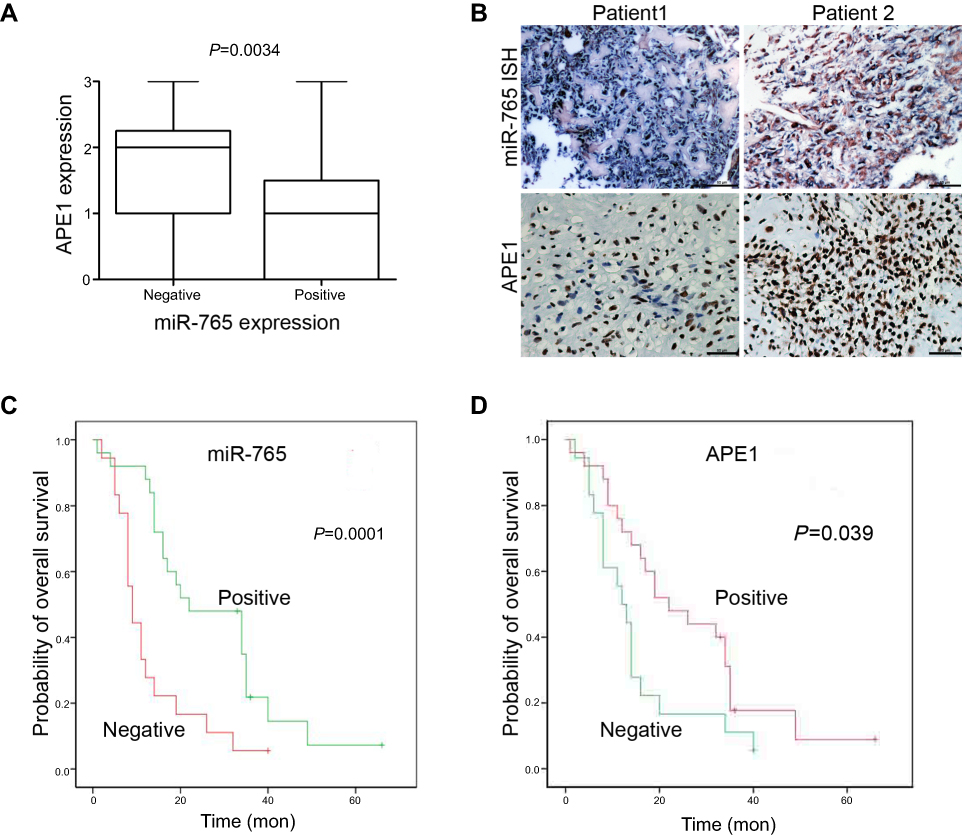 |
Figure 5 Clinical validation of miR-765 and APE1. (A) APE1 and miR-765 expression are inversely correlated in osteosarcoma samples. Patients were divided equally into two groups according to miR-765 expression level, and APE1 expression showed considerable differences between the miR-765 low and miR-765 high groups (p=0.0034, Mann–Whitney U test). (B) Paraffin-embedded, formalin-fixed osteosarcoma cancer tissues were incubated with a locked nucleic acid anti-miR-765 probe for in situ hybridization (ISH) and anti-APE1 antibody for immunohistochemical (IHC) analysis with scrambled probe and phosphate-buffered saline as negative controls, respectively. Representative photographs are shown. Scale bars =50 μm. (C and D) A Kaplan–Meier analysis of overall survival for osteosarcoma patients with the corresponding expression profiles of APE1 (D) and miR-765 (C) is shown (p=0.039 and p=0.001, respectively, log-rank test). |
Kaplan-Meier survival analysis was used to assess the relationship between miR-765 expression and patient survival. Similarly, to negative APE1 expression (negative vs positive APE1 expression: median overall survival =22.00 months vs median overall survival=12.00 months, p=0.039), positive expression of miR-765 is associated with longer OS (positive vs negative miR-765 expression, median overall survival =22.00 months vs median overall survival =9.00 months, p=0.001) (Figure 4C and D). The COX hazard probability regression models revealed that only miR-765 was an independent prognostic factor for osteosarcoma patients (HR =0. 389, 95% CI =0.196 to 0. 772, p=0. 007) (Table 3).
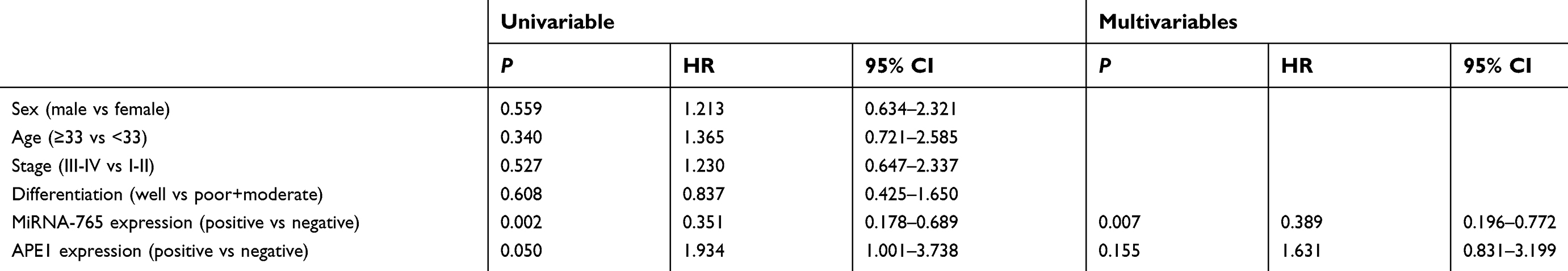 |
Table 3 The univariate and multivariate analyses of prognostic factors in 43 patients with osteosarcoma for overall survival |
Discussion
Through decades of progress, the survival rate of OS patients with localized disease has improved greatly. However, for patients with metastatic or recurrent disease, the long-term survival situation remains unsatisfactory, largely due to chemoresistance. In this study, we demonstrated that miR-765-mediated APE1 suppression sensitized OS cells to cisplatin chemotherapy. Thus, our findings provided a potential strategy for the management of chemoresistant OS patients.
We focused on the regulation of APE1 expression by miRNAs, since APE1 was a well-documented initiator of chemoresistance in OS patients by mediating base excision repair. Utilizing bioinformatic analysis, we successfully selected one miRNA (miR-765) as a direct inhibitor of APE1. Additional miRNAs could be involved in the regulation of APE1, and there could also be additional chemoresistance-related targets besides APE1 regulated by miR-765. Therefore, high throughput methods are necessary in future studies to identify more OS chemoresistance-related miRNAs and genes.
MiRNAs are a class of small noncoding regulatory RNA molecules involved in a wide array of biological processes. The abnormal expression of miRNA is associated with various cancers.4,11–14 Our data demonstrated that miR-765 was highly expressed in OS cells, and an increasing sensitivity to cisplatin was observed both in cell lines and xenograft tumors. The cytotoxicity of cisplatin primarily results from adduct formation ability with DNA. These DNA distortions could effectively block the progression of DNA replication and activate the cell cycle checkpoint.16–19 Multiple pathways are involved in the repair of DNA damage induced by cisplatin. The nucleotide excision repair pathway participates in the removal of DNA intrastrand crosslinks,15,16 while the HR pathway is involved in removing interstrand crosslinks.17,18 During DNA damage, single and double strand breaks are generated, and other repair systems are involved, including the base excision repair (BER) pathway. We found that DNA repair capability was decreased in OS cells with elevated expression of miR-765 when treated with cisplatin, which indicated that miR-765 may sensitize OS cells to cisplatin through decreasing DNA repair capabilities.
APE1 is a multifunctional protein with both DNA repair and redox activity. APE1 was found to be rate limiting for the repair of DNA damage induced by hydrogen peroxide (presumably 3ʹ-phosphate) and bleomycin (3ʹ-phosphoglycolates)19 and was a predominant enzyme for the excision of 3ʹ-phosphoglycolate residues from a single nucleotide gap.20 These findings suggested the essential role for APE1 in the BER pathway. A previous study demonstrated that RNA-mediated APE1 suppresses sensitivity of OS to DNA damaging agents.6 In accord with our current study, the suppression of APE1 by miR-765 resulted in identical effects in OS chemotherapy. These results suggested that APE1 was a powerful and promising target in OS chemotherapy.
We designed the in vivo and clinical research to confirm the roles of miR-765 and APE1 in OS chemotherapy. The in vivo experiments revealed that miR-765 could enhance the therapeutic effect of cisplatin by increasing apoptosis in the OS xenograft tumor. The clinical investigation also confirmed the negative correlation between miR-765 and APE1. Further studies were performed to determine the significance of miR-765 with respect to prognosis. The results showed that patients with high expression of miR-765 or low expression of APE1 exhibited a relatively longer survival and a better prognosis than those with the opposite expression. The multivariate analysis confirmed that miR-765 was independent prognostic factors for osteosarcoma. These findings were consistent with our in vitro results. However, due to the retrospective design and sample size limitation, our findings require further confirmation by large scale prospective trials.
We concluded that the expression of miR-765 was positively related to both cisplatin sensitivity and favorable prognosis in osteosarcoma, and we partially confirmed that the mechanism was via interference with the DNA repair capability, which occurs because of decreased APE1 expression. Chemotherapy with an auxiliary miRNA might represent a promising intervention for improving OS patient prognosis (Figure 6).
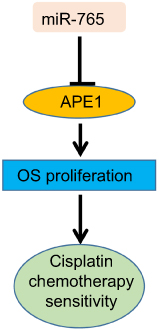 |
Figure 6 Graphic summary of this study. |
Acknowledgment
This work was supported by grant (No. 81172117) from National Natural Science Foundation of China (NSFC).
Disclosure
Authors have no potential conflicts of interest to disclose in this work.
References
1. Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):83–103. doi:10.3322/caac.21219
2. Chou AJ, Gorlick R. Chemotherapy resistance in osteosarcoma: current challenges and future directions. Expert Rev Anticancer Ther. 2006;6(7):1075–1085. doi:10.1586/14737140.6.7.1075
3. Wagner ER, Luther G, Zhu G, et al. Defective osteogenic differentiation in the development of osteosarcoma. Sarcoma. 2011;2011:325238. doi:10.1155/2011/325238
4. Zhou Y, Huang Z, Wu S, Zang X, Liu M, Shi J. miR-33a is up-regulated in chemoresistant osteosarcoma and promotes osteosarcoma cell resistance to cisplatin by down-regulating TWIST. J Exp Clin Cancer Res. 2014;33:12. doi:10.1186/1756-9966-33-12
5. Kong YW, Ferland-McCollough D, Jackson TJ, Bushell M. microRNAs in cancer management. Lancet Oncol. 2012;13(6):e249–e258. doi:10.1016/S1470-2045(12)70073-6
6. Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3(6):679–686.
7. Bassing CH, Alt FW. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle. 2004;3(2):149–153. doi:10.4161/cc.3.2.689
8. Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst). 2004;3(8–9):959–967. doi:10.1016/j.dnarep.2004.03.024
9. Liu WL, Gao M, Tzen KY, et al. Targeting phosphatidylinositide3-kinase/Akt pathway by BKM120 for radiosensitization in hepatocellular carcinoma. Oncotarget. 2014;5(11):3662–3672. doi:10.18632/oncotarget.1978
10. Sun C, Li N, Yang Z, et al. miR-9 regulation of BRCA1 and ovarian cancer sensitivity to cisplatin and PARP inhibition. Journal of the National Cancer Institute. 2013;105(22):1750–1758. doi:10.1093/jnci/djt302
11. He H, Ni J, Huang J. Molecular mechanisms of chemoresistance in osteosarcoma (Review). Oncol Lett. 2014;7(5):1352–1362. doi:10.3892/ol.2014.1935
12. Mezzanzanica D, Bagnoli M, De Cecco L, Valeri B, Canevari S. Role of microRNAs in ovarian cancer pathogenesis and potential clinical implications. Int J Biochem Cell Biol. 2010;42(8):1262–1272. doi:10.1016/j.biocel.2009.12.017
13. Xu K, Liang X, Shen K, et al. miR-297 modulates multidrug resistance in human colorectal carcinoma by down-regulating MRP-2. Biochem J. 2012;446(2):291–300. doi:10.1042/BJ20120386
14. Zhao X, Yang Z, Li G, et al. The role and clinical implications of microRNAs in hepatocellular carcinoma. Sci China Life Sci. 2012;55(10):906–919. doi:10.1007/s11427-012-4384-x
15. Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478(1–2):23–43.
16. Moggs JG, Szymkowski DE, Yamada M, Karran P, Wood RD. Differential human nucleotide excision repair of paired and mispaired cisplatin-DNA adducts. Nucleic Acids Res. 1997;25(3):480–491. doi:10.1093/nar/25.3.480
17. Tompkins JD, Wu X, Her C. MutS homologue hMSH5: role in cisplatin-induced DNA damage response. Mol Cancer. 2012;11:10. doi:10.1186/1476-4598-11-10
18. McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2(8):483–490. doi:10.1016/S1470-2045(01)00454-5
19. Izumi T, Hazra TK, Boldogh I, et al. Requirement for human AP endonuclease 1 for repair of 3ʹ-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis. 2000;21(7):1329–1334.
20. Parsons JL, Dianova II, Dianov GL. APE1 is the major 3ʹ-phosphoglycolate activity in human cell extracts. Nucleic Acids Res. 2004;32(12):3531–3536. doi:10.1093/nar/gkh676
21. Ren T, Qing Y, Dai N, et al. Apurinic/apyrimidinic endonuclease 1 induced upregulation of fibroblast growth factor 2 and its receptor 3 induces angiogenesis in human osteosarcoma cells. Cancer Sci. 2014;105(2):186–194. doi:10.1111/cas.12334
22. Gu X, Cun Y, Li M, et al. Human apurinic/apyrimidinic endonuclease siRNA inhibits the angiogenesis induced by X-ray irradiation in lung cancer cells. Int J Med Sci. 2013;10(7):870–882. doi:10.7150/ijms.5727
23. Cai Z, Zeng Y, Xu B, et al. Galectin-4 serves as a prognostic biomarker for the early recurrence/metastasis of hepatocellular carcinoma. Cancer Sci. 2014;105(11):1510–1517. doi:10.1111/cas.12536
24. Li Z, Qing Y, Guan W, et al. Predictive value of APE1, BRCA1, ERCC1 and TUBB3 expression in patients with advanced non-small cell lung cancer (NSCLC) receiving first-line platinum-paclitaxel chemotherapy. Cancer Chemother Pharmacol. 2014;74(4):777–786. doi:10.1007/s00280-014-2562-1
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.