Back to Journals » OncoTargets and Therapy » Volume 12
MicroRNA-652 suppresses malignant phenotypes in glioblastoma multiforme via FOXK1-mediated AKT/mTOR signaling pathway
Authors Yang H, Song Z, Wu X, Wu Y, Liu C
Received 9 February 2019
Accepted for publication 8 May 2019
Published 10 July 2019 Volume 2019:12 Pages 5563—5575
DOI https://doi.org/10.2147/OTT.S204715
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
This paper has been retracted.
Huimei Yang, 1 Zhenzhen Song, 1 Xia Wu, 2 Yilei Wu, 2 Chengxia Liu 3
1Department of Laboratory, The Third People’s Hospital of Linyi, Linyi, Shandong 276023, People’s Republic of China; 2Department of Oncology, The Third People’s Hospital of Linyi, Linyi, Shandong 276023, People’s Republic of China; 3Department of Pathology, Linyi Cancer Hospital, Linyi, Shandong 276023, People’s Republic of China
Purpose: An increasing number of studies have documented that dysregulation of microRNAs (miRNAs) is common in glioblastoma multiforme (GBM). miR-652 is aberrantly expressed in various human cancers and plays important roles in numerous cancer-related processes. However, the expression profiles and potential roles of miR-652 in GBM remain largely unknown.
Patients and methods: Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed to determine miR-652 expression in GBM tissues and cell lines. The effects of miR-652 upregulation on GBM cell proliferation, clone formation, apoptosis, migration and invasion were measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, clone formation, flow cytometry and Transwell® migration and invasion assays, respectively. In vivo xenotransplantation was utilized to determine the effect of miR-652 on GBM tumor growth in vivo. Of note, the molecular mechanisms underlying the tumor-suppressing activity of miR-652 upregulation in GBM cells were also investigated using a series of experiments, including bioinformatics analysis, luciferase reporter assay, RT-qPCR and Western blot analysis.
Results: miR-652 expression was considerably downregulated in GBM tissues and cell lines. Low miR-652 expression was strongly correlated with Karnofsky performance score and tumor size. Overall survival duration was shorter in GBM patients with low miR-652 expression than in those with high miR-652 expression. miR-652 resumption considerably suppressed the proliferation, clone formation, migration, and invasion and promoted the apoptosis of GBM cells in vitro. In addition, forkhead-box k1 (FOXK1) was demonstrated as the direct target gene of miR-652 in GBM cells. FOXK1 downregulation led to a tumor-suppressing activity similar to that of miR-652 upregulation. Restoration of FOXK1 expression partially neutralized the influence of miR-652 overexpression on GBM cells. Furthermore, ectopic miR-652 expression deactivated the AKT/mTOR pathway in GBM cells via FOXK1 regulation. Moreover, miR-652 impaired GBM tumor growth in vivo, probably caused by miR-652-mediated suppression of FOXK1/AKT/mTOR signaling.
Conclusion: miR-652 inhibits FOXK1 and deactivates the AKT/mTOR pathway, thereby resulting in the suppression of malignant phenotypes of GBM cells in vitro and in vivo.
Keywords: glioblastoma multiforme, microRNA, Forkhead-box K1, malignant development
Introduction
Glioma originates from the neural ectoderm and is the most frequent subtype of primary human brain malignant tumors.1 Based on the degree of malignancy, gliomas can be subdivided into four histopathological grades, ie WHO grades I–IV.2 Glioblastoma multiforme (GBM), a WHO grade IV glioma, accounts for approximately 70% of the total glioma incidence.3 Rapid growth and high invasiveness are the favorable characteristics that enable GBM cells to surround normal brain cells, making it difficult to completely resect tumors via surgery.4 Despite substantial advances in the treatment modalities, including surgical resection, radiotherapy, and chemotherapy, the therapeutic outcomes of patients with GBM remain poor, with a 5-year survival rate of <3%.5 The median survival period of patients with GBM is only 12–14 months after diagnosis.6 Therefore, investigating the mechanisms responsible for GBM pathogenesis is important for identifying novel and effective targets for early diagnosis and treatment.
microRNAs (miRNAs) are highly conserved, single-stranded, and short RNA molecules that are 18–24 nucleotides long7 and negatively modulate gene expression by inducing mRNA cleavage or translational inhibition of mRNAs via complete or incomplete complementary binding to the 3′-untranslated region (UTR) of their target genes in a base-pairing manner.8,9 By regulating the protein expression of their target genes, an increasing number of miRNAs have been shown to be differently expressed in human malignancies and to be closely related with carcinogenesis and cancer progression.10–12 Numerous miRNAs are significantly upregulated or downregulated in GBM and contribute to the regulation of various cellular biological processes, such as cell proliferation, apoptosis, cycle, differentiation, metastasis, and angiogenesis.13–15 Weakly expressed miRNAs exhibit tumor-suppressing roles,16 whereas overexpressed miRNAs exhibit oncogenic roles with respect to GBM occurrence and development.17 Therefore, miRNAs can serve as therapeutic targets and diagnostic tools for patients with GBM.
To date, approximately 1572 miRNAs have been identified in the human genome; however, only a small number of these miRNAs have been well studied.18 miR-652 is aberrantly expressed in various human cancers and plays important roles in numerous cancer-related processes;19–21 however, its expression pattern and potential roles in GBM remain largely unknown. Thus, the present study aimed to detect miR-652 expression in GBM and to determine its specific roles in the malignant phenotypes of GBM both in vitro and in vivo. In addition, the molecular mechanisms underlying miR-652 activity in GBM cells were investigated. Our results not only provide novel insights into the mechanisms underlying GBM progression but also highlight novel therapeutic targets for treating patients with this malignancy.
Materials and methods
Collection of tissue specimens
In total, 47 pairs of GBM tissues and adjacent normal brain tissues were collected from patients with GBM who underwent surgical resection at The Third People’s Hospital of Linyi. None of these patients had been treated with preoperative radiotherapy or chemotherapy. The collected tissues were immediately frozen in liquid nitrogen and stored at −80 °C. The use of clinical tissue specimens was approved by the Ethics Committee of The Third People’s Hospital of Linyi, and written informed consent was obtained from all patients prior to surgery.
Cell culture, oligonucleotides, plasmids, and transfection
Normal human astrocytes (NHAs) were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) and cultured in astrocyte medium (ScienCell Research Laboratories) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). In total, four human GBM cell lines (U251, U138, T98, and LN229), were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All GBM cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). All cells were maintained at 37 °C in a humidified incubator supplied with 5% CO2.
AgomiR-652 and agomiR-NC were purchased from GenePharma (Shanghai, China). Specific small interfering (si)RNAs targeting FOXK1 expression (siFOXK1) and the scrambled negative control (siNC) were synthesized by Wuhan Genesil Biotechnology Co., Ltd. (Wuhan, China). The FOXK1-overexpression plasmid pcDNA3.1-FOXK1 (pc-FOXK1) and empty plasmid pcDNA3.1 were generated by the Chinese Academy of Sciences (Changchun, China). Cells were seeded into 6-well plates the night before transfection. All oligonucleotides and plasmids were introduced into cells using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), following the manufacturer’s instructions.
Reverse transcription-quantitative polymerase chain reaction
Total RNA was extracted from cells or tissue specimens using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and then placed in an ND-2000 spectrophotometer (NanoDrop Technologies, Houston, TX, USA) for the quantification of total RNA. Reverse transcription was performed using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The synthesized complementary DNA (cDNA) was then used for the quantification of miR-652 expression using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). To analyze FOXK1 mRNA expression level, cDNA was prepared from total RNA using the Prime-Script RT reagent Kit (TaKaRa, Dalian, China). Subsequently, quantitative PCR was performed using the SYBR Premix Ex Taq™ II kit (TaKaRa, Dalian, China). U6 small nuclear RNA (snRNA) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used to normalize miR-652 and FOXK1 mRNA expressions, respectively. All data were analyzed using the 2−ΔΔCt method.22 The primers were designed as follows: miR-652, 5′-ACACTCCAGCTGGGCAACCCTAGGAGAGGGTGC-3′ (forward) and 5′-GTGTCGTGGAGTCGGCAATTC-3′ (reverse); U6, 5′-GCTTCGGCAGCACATATACTAAAAT-3′ (forward) and 5′-CGCTTCACGAATTTGCGTGTCAT-3′ (reverse); FOXK1, 5′-GCCTCCTTGACAATACCGCT-3′ (forward) and 5′-TTCCAAACCCTCCCTCTGGT-3′ (reverse); and GAPDH, 5′-AATGGGCAGCCGTTAGGAAA-3′ (forward) and 5′- GCGCCCAATACGACCAAATC −3′ (reverse).
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay
Twenty-four hours after culture, the transfected cells were harvested and prepared as a cell suspension. Next, 100 µl of the cell suspension containing 3×103 cells was inoculated into each well of a 96-well plate. The cells were then incubated at 37 °C with 5% CO2 for 0, 24, 48, and 72h. MTT assay was performed at the indicated time points to determine cell proliferation. Briefly, 20 µl of MTT reagent (5 mg/ml; Sigma-Aldrich, St. Louis, MO, USA) was added to each well; after 4 h of incubation, the culture medium in each well was replaced with 100 µl of dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO, USA), which helped dissolve the formazan precipitate. Absorbance at 490 nm was recorded using a microplate reader (SpectraMax M5; Molecular Devices, CA, USA).
Clone formation assay
The transfected cells were trypsinized after 24 h of incubation and inoculated into 6-well plates at a density of 1000 cells/well. The cells were then incubated at 37°C in a humidified incubator supplied with 5% CO2 for 2 weeks. At the end of the experiment, the cells were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde, and stained with methyl violet. Finally, the number of colonies containing ≥50 cells was counted under an inverted microscope (CKX41; Olympus Corporation, Tokyo, Japan).
Flow cytometry assay
After 48 h of the transfection, the apoptosis rate was examined using the Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (BioLegend, Inc., San Diego, CA, USA). The cells were harvested and then washed with ice-cold PBS. After suspension in 100 µl of binding buffer, the cells were double-stained with 5 µl annexin V-FITC and 5 µl propidium iodide at 37 °C for 30 min under darkness. Finally, the cells were analyzed using flow cytometry (FACScan™; BD Biosciences) for the measurement of the apoptosis rate. Data were analyzed using CellQuest™ software version 5.1 (BD Biosciences, San Jose, CA, USA).
Transwell® migration and invasion assays
A total of 48 h after the transfection, the cells were collected and used for Transwell® migration and invasion assays. Briefly, the upper compartment of a 24-well Transwell® chamber (BD Biosciences, Bedford, MA) was loaded with 200 µl of cell suspension containing 5×104 cells/well. The lower compartments were filled with 600 µl of DMEM containing 10% FBS. After 24 h of incubation, non-migrated cells were gently wiped off using a cotton swab. Cells that migrated through the membranes were fixed with 4% paraformaldehyde, stained with 0.1% crystal violet, and washed with PBS. Cell migratory ability was evaluated by counting the average number of migrated cells in five randomly selected fields per chamber under an inverted CKX41 microscope. Except that the Transwell chambers were precoated with Matrigel (BD Biosciences, Bedford, MA), the experimental procedure of the Transwell invasion assay was the same as that of the Transwell migration assay.
In vivo xenotransplantation
All protocols involving animals were approved by the Ethics Committee of The Third People’s Hospital of Linyi and were performed in accordance with the Animal Protection Law of the People’s Republic of China-2009. BALB/c male nude mice obtained from Shanghai Biomodel Organism Science & Technology Development Co., Ltd. (Shanghai, China) were subcutaneously inoculated with cells transfected with agomiR-652 or agomiR-NC. Tumor length and width were recorded every week. The volume of each tumor xenograft was analyzed using the following formula: Volume = (length × width2)/2. All nude mice were euthanized 4 weeks after the injection after which the tumor xenografts were excised, weighed, and stored for further use.
Bioinformatics analysis
The putative target genes of miR-652 were predicted using TargetScan (http://www.targetscan.org/vert_71/) and miRanda (http://www.microrna.org/microrna/home.do).
Luciferase reporter assay
Fragments of FOXK1 3′-UTR containing wild-type (Wt) miR-652 binding sequences and mutant (Mut) 3′-UTR containing mutations of the miR-652 binding sequences were chemically synthesized by GenePharma and inserted into the pMIR-REPORT vector (Promega, Madison, WI, USA) to generate the pMIR-FOXK1-3′-UTR-Wt and pMIR-FOXK1-3′-UTR-Mut plasmids, respectively. For reporter assays, the pMIR-FOXK1-3′-UTR-Wt or pMIR-FOXK1-3′-UTR-Mut along with agomiR-652 or agomiR-NC, respectively, was transfected into the cells using Lipofectamine 2,000, following the manufacturer’s protocol. The transfected cells were harvested at 48 h post-transfection and assayed using the Dual-Luciferase Reporter Assay system (Promega Corporation, Madison, WI, USA), following the manufacturer’s protocol. Renilla luciferase activity was maintained as a normalized control.
Western blot analysis
Cells or homogenized tissues were lysed with ice-cold radioimmunoprecipitation assay lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China). Total protein concentration was detected using the Enhanced BCA Protein Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China). Equal amounts of protein were separated using10% sodium dodecyl sulfate-polyacrylamide electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were then blocked at room temperature for 2 h with 5% fat-free milk prepared in Tris-buffered saline containing 0.1% Tween-20 (TBST). The membrane was incubated overnight at 4 °C with primary antibodies against FOXK1 (cat. no. sc-373810; 1:1000; Santa Cruz Biotechnology, CA, USA), p-AKT (cat. no. sc-81433; 1:1000 dilution; Santa Cruz Biotechnology), AKT (cat. no. sc-56878; 1:1000 dilution; Santa Cruz Biotechnology), p-mTOR (cat. no. ab137133; 1:1,000; Abcam, Cambridge, UK), mTOR (cat. no. ab134903; 1:1,000; Abcam), or GAPDH (ab128915; 1:1000; Abcam). After washing thrice with TBST, the membranes were further incubated with a goat anti-mouse (cat. no.ab6789) or goat anti-rabbit (cat. no.ab6721) horseradish peroxidase-conjugated secondary antibody (1:5000; Abcam, Cambridge, UK) at room temperature for 2 h. The protein signals were visualized using an enhanced chemiluminescence detection reagent (Pierce Biotechnology, Inc., Rockford, IL, USA). GAPDH was used as the loading control.
Statistical analysis
Data were expressed as mean ± standard deviation and analyzed using the Statistical Package for Social Sciences version 19.0 (IBM SPSS, Inc., Armonk, NY, USA). Spearman’s correlation analysis was used to assess the associations between miR-652 and FOXK1 mRNA levels in GBM tissues. Differences between two groups were analyzed using the two-tailed Student’s t-test, whereas those between multiple groups were analyzed using one-way ANOVA followed by the Bonferroni post-hoc test. The associations between miR-652 expression and clinicopathological parameters in patients with GBM were explored using the Chi-squared test. The Kaplan–Meier method was used to evaluate survival. Differences in survival were evaluated using the log-rank test. P<0.05 was considered statistically significant.
Results
miR-652 was downregulated in GBM tissues and cell lines
To illustrate the potential relevance of miR-652 in GBM, we first detected miR-652 expression in 47 pairs of GBM tissues and adjacent normal brain tissues. RT-qPCR results revealed that miR-652 expression was significantly lower in GBM tissues than in adjacent normal brain tissues (Figure 1A; P<0.05). miR-652 expression in various GBM cell lines was also determined using RT-qPCR. NHAs were used as the control. miR-652 expression was significantly lower in all four tested GBM cell lines (U251, U138, T98, and LN229) than in NHAs (Figure 1B; P<0.05).
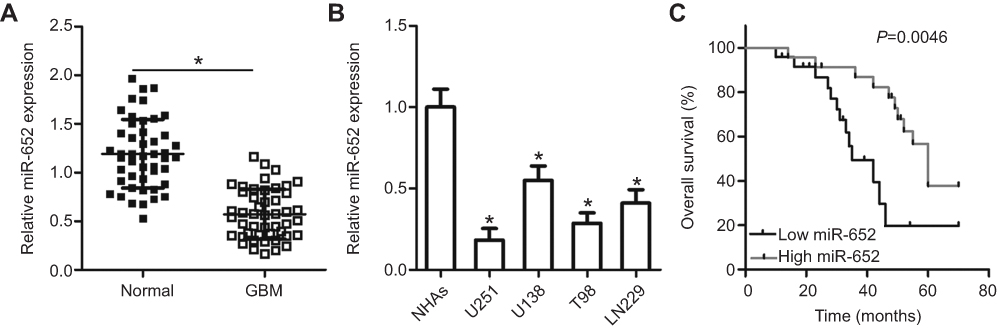 |
Figure 1 Expression of miR-652 is decreased in glioblastoma multiforme (GBM) tissues and cell lines. (A) RT-qPCR analysis was performed to analyze miR-652 expression in 47 pairs of GBM tissues and adjacent normal brain tissues. *P<0.05 vs normal brain tissues. (B) Expression level of miR-652 was also detected in four human GBM cell lines (U251, U138, T98, and LN229) and in normal human astrocytes (NHAs). *P<0.05 vs NHAs. (C) Patients with GBM harboring low miR-652 expression had shorter overall survival rate than those patients with high miR-652 level. *P<0.05 vs high miR-652 level group. |
The clinical significance of miR-652 was investigated in patients with GBM. Based on the median miR-652 expression in GBM tissues as a cutoff point, all 47 patients with GBM were divided into the miR-652 low- (n=24) and high-expression group (n=23). Low miR-652 expression was strongly associated with Karnofsky performance score (KPS; P = 0.041) and tumor size (P = 0.017) (Table 1). Furthermore, the overall survival duration was shorter in patients with GBM with a low miR-652 expression than in those with a high miR-652 expression (Figure 1C; P = 0.0046). These results suggest that miR-652 is associated with the development and progression of GBM.
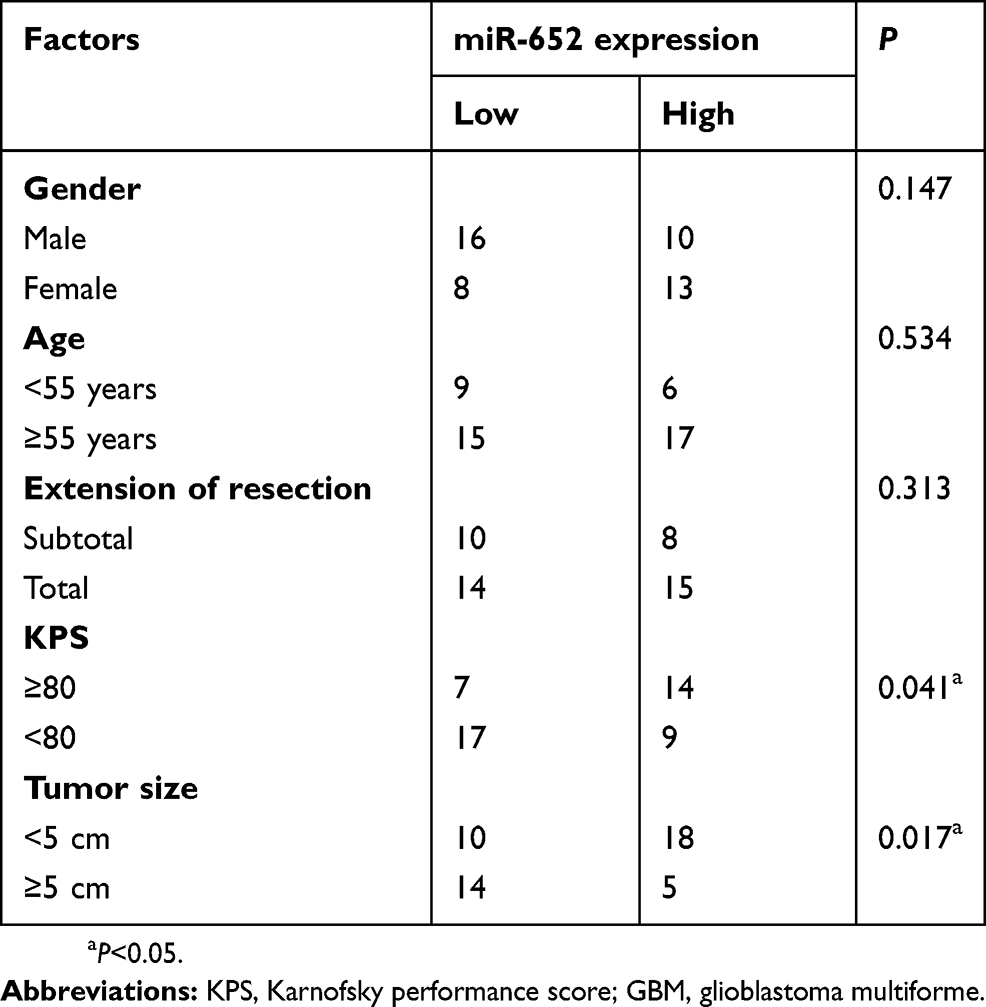 |
Table 1 Correlation of miR-652 relative expression level with clinicopathological factors of patients with GBM |
miR-652 overexpression inhibited cell proliferation, clone formation, and metastasis as well as promoted cell apoptosis in GBM
Among the four GBM cell lines, U251 and T98 exhibited the lowest levels of miR-652. Therefore, these two cell lines were chosen for subsequent functional assays. To explore the biological roles of miR-652 in GBM, we transfected U251 and T98 cells with agomiR-652 or agomiR-NC. Figure 2A indicates that agomiR-652 increased miR-652 expression in U251 and T98 cells (P<0.05). MTT and clone formation assays were performed to determine the influence of miR-652 upregulation on the proliferative and clone formative capacities of GBM cells. The results indicated that cell proliferation (Figure 2B; P<0.05) and clone formation (Figure 2C; P<0.05) was significantly inhibited in U251 and T98 cells transfected with agomiR-652 than in those transfected with agomiR-NC. Next, we performed flow cytometry analysis to examine the effects of miR-652 upregulation on GBM cell apoptosis. The results revealed that exogenous miR-652 expression promoted the apoptosis of U251 and T98 cells (Figure 2D; P<0.05). Furthermore, Transwell migration and invasion assays were performed to measure the migration and invasion of U251 and T98 cells transfected with agomiR-652 or agomiR-NC. The results indicated that ectopic miR-652 expression significantly suppressed the migration (Figure 2E; P<0.05) and invasion (Figure 2F; P<0.05) of U251 and T98 cells relative to that in the agomiR-NC group. These results suggest that miR-652 functions as a tumor-suppressing miRNA in GBM, apparently inhibiting the growth and metastasis of GBM cells in vitro.
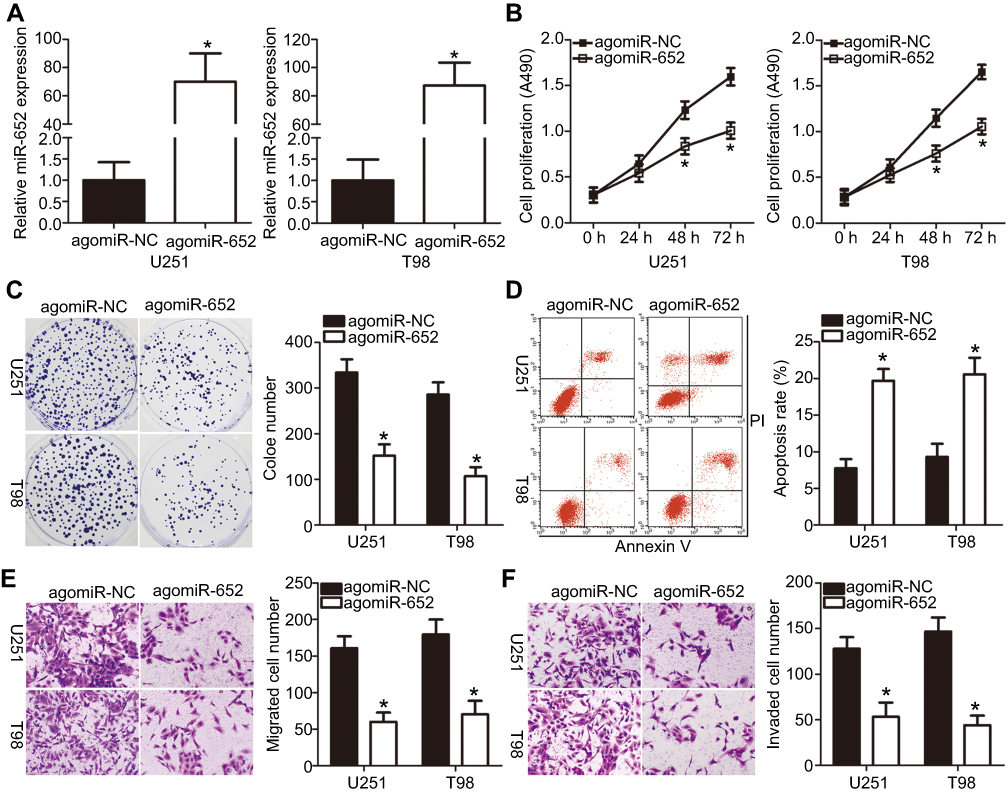 |
Figure 2 Upregulation of miR-652 inhibits proliferation, clone formation, migration, and invasion but promotes apoptosis of U251 and T98 cells. (A) U251 and T98 cells were transfected with agomiR-652 or agomiR-NC; the latter functions as the control for agomiR-652. After transfection for 48 h, miR-652 expression was determined using RT-qPCR analysis. *P<0.05 vs agomiR-NC. (B and C) 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide and clone formation assays were employed to investigate the influence of miR-652 overexpression on U251 and T98 cell proliferative and clone formative capacities. *P<0.05 vs agomiR-NC. (D) The proportions of apoptotic U251 and T98 cells after agomiR-652 or agomiR-NC transfection were examined using flow cytometry assay. *P<0.05 vs agomiR-NC. (E and F) Transwell migration and invasion assays were performed to evaluate the migration and invasion of U251 and T98 cells transfected with agomiR-652 or agomiR-NC. *P<0.05 vs agomiR-NC. |
FOXK1 was a direct target gene of miR-652 in GBM cells
To elucidate the mechanisms underlying the functional effects of miR-652 on GBM cells, bioinformatics analysis was employed to search for the putative targets of miR-652. As indicated in Figure 3A, miR-652 is partially complementary to the 3′-UTR of FOXK1. FOXK1 was selected for further identification considering its crucial roles in GBM development and progression.23 Luciferase reporter assay was performed to determine whether miR-652 could directly bind to the 3′-UTR of FOXK1. The results revealed that the luciferase activity of the plasmid carrying the wild-type FOXK1 3′-UTR was significantly reduced by miR-652 upregulation (P<0.05); however, the inhibitory effects were abolished when the miR-652 binding site in the 3′-UTR of FOXK1 was mutated (Figure 3B).
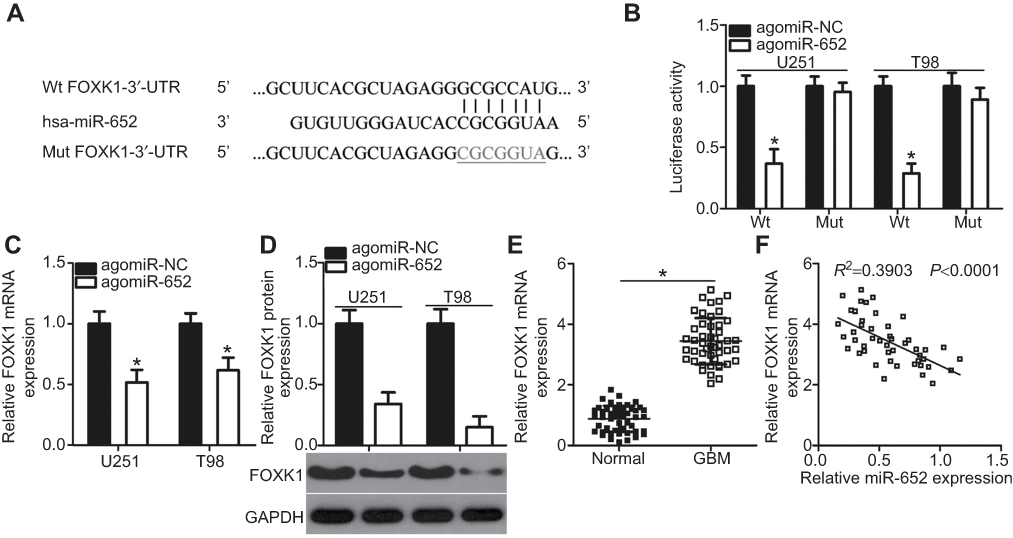 |
Figure 3 FOXK1 is a direct target gene of miR-652 in glioblastoma multiforme (GBM) cells. (A) Predicted wild-type (Wt) miR-652 binding sequences in the 3′-UTR of FOXK1 and the mutant containing altered nucleotides in the 3′-UTR of FOXK1. (B) U251 and T98 cells were cotransfected with pMIR-FOXK1-3′-UTR-Wt or pMIR-FOXK1-3′-UTR-Mut and agomiR-652 or agomiR-NC. Luciferase activities were detected at 48 h post-transfection. *P<0.05 vs agomiR-NC. (C and D) Expression levels of FOXK1 mRNA and protein in U251 and T98 cells transfected with agomiR-652 or agomiR-NC were measured by RT-qPCR and Western blot analysis, respectively. *P<0.05 vs agomiR-NC. (E) The expression level of FOXK1 mRNA in 47 pairs of GBM tissues and adjacent normal brain tissues was determined by RT-qPCR. *P<0.05 vs normal brain tissues. (F) The expression relationship between miR-652 and FOXK1 mRNA levels in GBM tissues was analyzed by Spearman’s correlation analysis. R2=0.3903, P<0.0001. |
We measured FOXK1 expression in miR-652–overexpressing U251 and T98 cells to further demonstrate that endogenous FOXK1 expression could be negatively regulated by miR-652 in GBM cells. The expression levels of FOXK1 mRNA (Figure 3C; P<0.05) and protein (Figure 3D; P<0.05) were both decreased by agomiR-652 transfection in U251 and T98 cells. Furthermore, FOXK1 was upregulated in GBM tissues compared with that in normal brain tissues (Figure 3E; P<0.05) and the upregulation of FOXK1 exhibited an inverse correlation with miR-652 in GBM tissues (Figure 3F; R2=0.3903, P<0.0001). Taken together, these results demonstrate that FOXK1 is a direct target gene of miR-652 in GBM.
Inhibition of FOXK1 has roles similar to those of miR-652 upregulation in GBM cells
To explore the role of FOXK1 in GBM development, loss of function assays were performed in U251 and T98 cells by transfecting the cells with siFOXK1. FOXK1 expression was significantly knocked down in U251 and T98 cells after transfection with siFOXK1 (Figure 4A; P<0.05). MTT and clone formation assays revealed that silencing FOXK1 expression restricted the proliferation (Figure 4B; P<0.05) and clone formation (Figure 4C; P<0.05) of U251 and T98 cells. In addition, the flow cytometry assay demonstrated that the levels of apoptotic U251 and T98 cells were significantly higher in the siFOXK1 group than in the siNC group (Figure 4D; P<0.05). Furthermore, Transwell migration and invasion assays were conducted to determine whether FOXK1 is implicated in the regulation of GBM metastasis, The results showed that FOXK1 knockdown suppressed the migratory (Figure 4E; P<0.05) and invasive (Figure 4F; P<0.05) capacities of U251 and T98 cells. Hence, FOXK1 inhibition exhibited roles similar to those exhibited by miR-652 overexpression in GBM cells, thereby suggesting that FOXK1 is a downstream target of miR-652 in GBM cells.
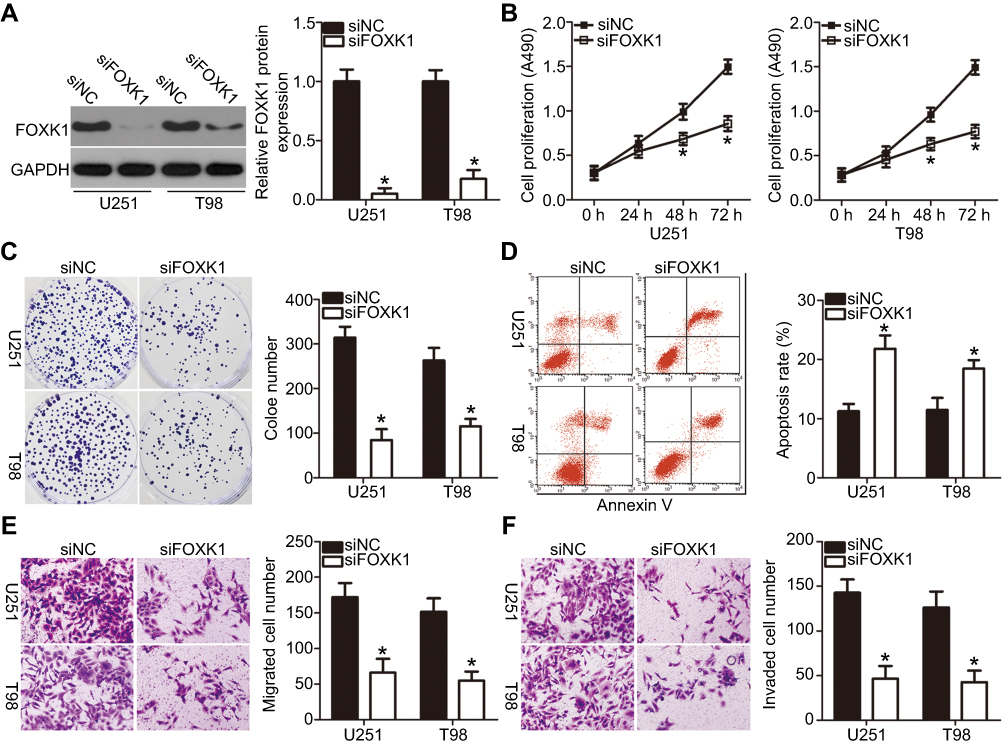 |
Figure 4 Decreasing FOXK1 expression suppresses glioblastoma multiforme (GBM) cell proliferation, clone formation, and metastasis and induces cell apoptosis in vitro. siFOXK1 or siNC was introduced into U251 and T98 cells. Transfected cells were collected and used for following assays. (A) Western blot analysis was used to detect FOXK1 protein expression in the indicated cells. *P<0.05 vs siNC. (B–D) 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, clone formation, and flow cytometry assays were performed to assess the effect of FOXK1 knockdown on the proliferation, clone formation, and apoptosis of the U251 and T98 cells. *P<0.05 vs siNC. (E and F) The migratory and invasive abilities of U251 and T98 cells treated as described above were explored by Transwell migration and invasion assays. *P<0.05 vs siNC. |
FOXK1 inhibited the miR-652-induced tumor-suppressing roles of GBM cells
Having proven that FOXK1 is a direct target of miR-652, we determined if the tumor-suppressing roles of miR-652 in GBM progression could be achieved by inhibiting reductions in FOXK1. AgomiR-652 combined with either pcDNA3.1 or pc-FOXK1 was transfected into U251 and T98 cells. After 72 h of transfection, the total protein was extracted and the FOXK1 protein level was detected using Western blot analysis. The results indicated that co-transfection with pc-FOXK1 inhibited the decreases in FOXK1 protein levels in U251 and T98 cells induced by miR-652 overexpression (Figure 5A; P<0.05). Of note, the restoration of FOXK1 expression abolished the influence of miR-652 upregulation on U251 and T98 cell proliferation (Figure 5B; P<0.05), clone formation (Figure 5C; P<0.05), apoptosis (Figure 5D; P<0.05), migration (Figure 6A; P<0.05), and invasion (Figure 6B; P<0.05). These results support the idea that the tumor-suppressive roles of miR-652 in GBM progression mainly depend on its regulation of FOXK1 expression.
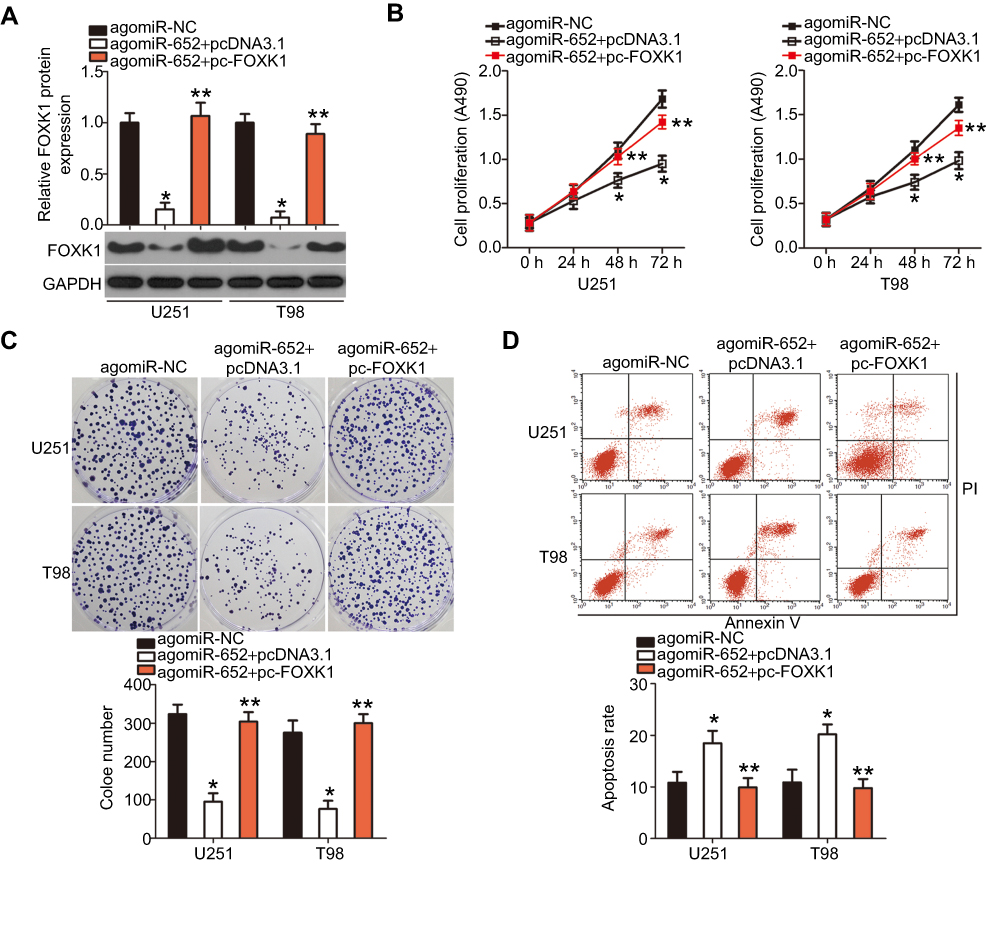 |
Figure 5 FOXK1 is involved in miR-652 regulation of glioblastoma multiforme (GBM) cell proliferation, clone formation, and apoptosis. FOXK1 protein expression was restored in agomiR-652-transfected U251 and T98 cells through cotransfection with pc-FOXK1. (A) The transfected cells were harvested after 72 h of incubation and then subjected to Western blot analysis to quantify FOXK1 protein expression. *P<0.05 vs agomiR-NC. **P<0.05 vs agomiR-652+ pcDNA3.1. (B–D) Determination of the proliferation, clone formation, and apoptosis of the aforementioned cells was performed with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, clone formation, and flow cytometry assays, respectively. *P<0.05 vs agomiR-NC. **P<0.05 vs agomiR-652+ pcDNA3.1. |
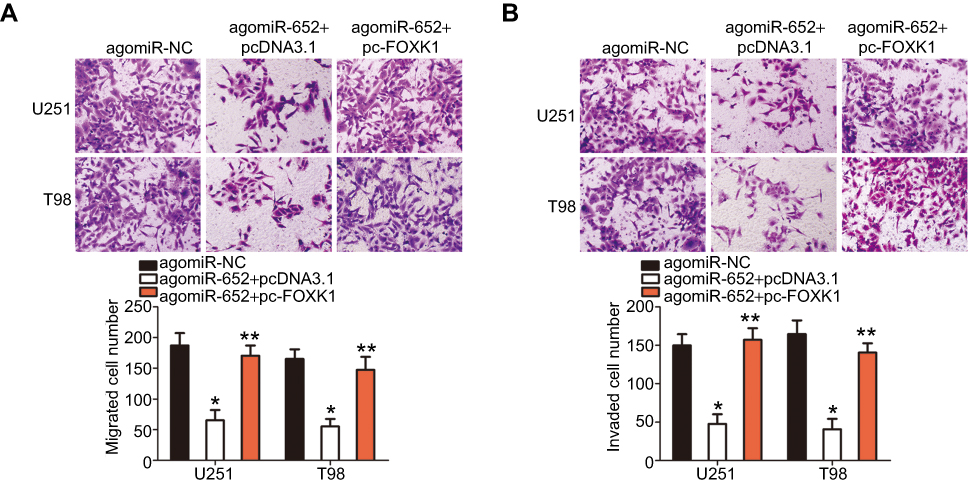 |
Figure 6 Reintroduction of FOXK1 abolishes the inhibitory effects of miR-652 on the migration and invasion of glioblastoma multiforme (GBM) cells. (A and B) AgomiR-652 was cotransfected with pcDNA3.1 or pc-FOXK1 into U251 and T98 cells. After 48 h culture, Transwell migration and invasion assay was used for the determination of cell migratory and invasive abilities. *P<0.05 vs agomiR-NC. **P<0.05 vs agomiR-652+ pcDNA3.1. |
miR-652 inhibited the AKT/mTOR signaling pathway in GBM cells by regulating FOXK1
Previous studies have shown that the AKT/mTOR pathway could be regulated by FOXK1.24,25 Therefore, we attempted to determine whether miR-652 is involved in regulating the AKT/mTOR pathway in GBM cells. The expression of ectopic miR-652 significantly decreased the expressions of p-AKT and p-mTOR in U251 and T98 cells, thereby indicating that miR-652 deactivates the AKT/mTOR pathway in GBM. In addition, the suppressive effects of miR-652 overexpression on the AKT/mTOR pathway in U251 and T98 cells were reversed via cotransfection with pc-FOXK1 (Figure 7). These results suggest that miR-652 inhibits the AKT/mTOR pathway activation in GBM cells by downregulating FOXK1 expression.
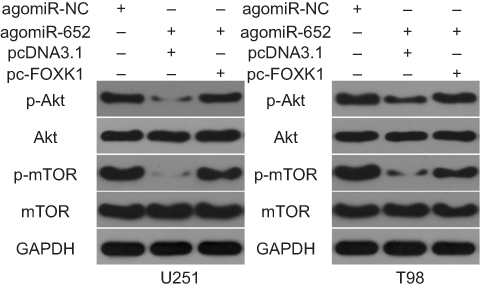 |
Figure 7 Activation of the AKT/mTOR signaling pathway is suppressed by miR-652 in glioblastoma multiforme (GBM) cells. Western blot analysis was used to measure the protein expression levels of important molecules associated with the AKT/mTOR pathway in U251 and T98 cells post agomiR-652 and pc-FOXK1 or pcDNA3.1 cotransfection. |
miR-652 overexpression decreased GBM cell growth in vivo
In vivo xenotransplantation was performed to determine whether miR-652 affected GBM cell growth in vivo. U251 cells transfected with agomiR-652 or agomiR-NC were subcutaneously implanted into nude mice. The tumor volume (Figure 8A and B; P<0.05) and weight (Figure 8C; P<0.05) were significantly lower in the agomiR-652 group than in the agomiR-NC group. These results were confirmed via detection of miR-652 expression in the xenografts. miR-652 expression was significantly upregulated in tumor xenografts via infection with agomiR-652 (Figure 8D; P<0.05). Western blot analysis revealed that FOXK1, p-AKT, and p-mTOR were expressed at lower levels in tumor xenografts from mice in the agomiR-652 group than in those from the mice in the agomiR-NC group (Figure 8E; P<0.05). Taken together, these results indicate that miR-652 has an inhibitory role on GBM cell growth in vivo and that this role may be related to inhibition of the FOXK1/AKT/mTOR pathway.
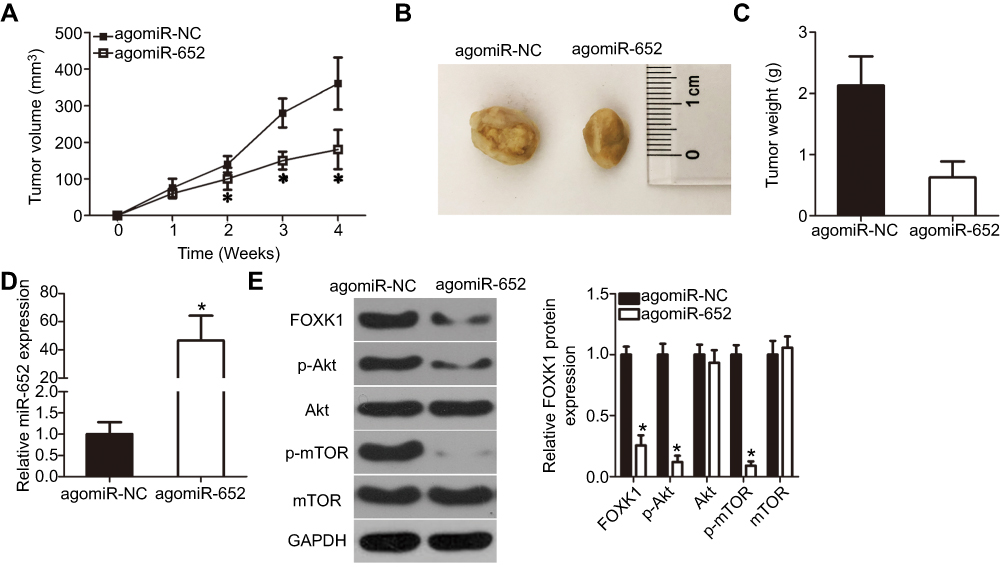 |
Figure 8 miR-652 inhibits tumor growth of glioblastoma multiforme (GBM) in vivo. (A) Growth curve for tumor volumes in xenografts derived from agomiR-652 or agomiR-NC-transfected U251 cells were determined on the basis of the tumor volume detected every week for 4 weeks. *P<0.05 vs agomiR-NC. (B) Photographs of tumor xenografts from the agomiR-652 and agomiR-NC groups. (C) The tumor xenografts were resected 4 weeks after inoculation. The weights of tumor xenografts were measured. *P<0.05 vs agomiR-NC. (D) RT-qPCR analysis was performed to quantify miR-652 expression in the tumor xenografts. *P<0.05 vs agomiR-NC. (E) The expression levels of FOXK1, p-AKT, AKT, p-mTOR, and mTOR in the tumor xenografts were measured by Western blot analysis. *P<0.05 vs agomiR-NC. |
Discussion
An increasing number of studies have documented that miRNAs dysregulation is a common event in GBM.26,27 Dysregulated miRNAs play pivotal roles in GBM development and progression.28 In particular, miRNAs have significant potential as therapeutic targets for patients with GBM.29 Thus, dysregulated miRNAs in GBM should be investigated to identify the valuable therapeutic methods for patients with this aggressive cancer. To the best of our knowledge, our study is the first to report on the detection of miR-652 expression in GBM tissues and in various GBM cell lines. In addition, the present study examined the clinical importance of aberrant miR-652 expression in patients with GBM. Of note, the molecular mechanisms by which miR-652 affected GBM development and progression in vitro and in vivo were particularly investigated. Our study unveiled the tumor-suppressive roles of miR-652 in GBM progression by directly targeting FOXK1 and deactivating the AKT/mTOR pathway.
miR-652 is evaluated in endometrial cancer, and increased miR-652 expression is reportedly closely associated with shorter overall survival durations and earlier recurrence in patients with endometrial cancer.19 In addition, miR-652 is upregulated in non-small–cell lung cancer tissues and cell lines. Reportedly, a high miR-652 expression is significantly correlated with lymph node metastasis, TNM stage, and prognosis in patients with non-small–cell lung cancer.20 In contrast, miR-652 expression has been shown to be decreased in pancreatic cancer, and this decrease is reportedly associated with unfavorable clinicopathological characteristics of patients with pancreatic cancer.21 However, the expression profile of miR-652 in GBM has been rarely reported to date. In the present study, we showed that miR-652 was clearly downregulated in GBM tissues and cell lines. miR-652 downregulation was significantly correlated with KPS and tumor size in patients with GBM. Patients with GBM harboring a low miR-652 expression had shorter overall survival duration than that of patients with a high miR-652 expression. These findings suggest that miR-652 is a promising indicator for predicting the prognosis of GBM patients.
miR-652 has tumor-promoting or -suppressing roles in carcinogenesis and cancer progression. For instance, resumption of miR-652 expression facilitates cell proliferation and metastasis in endometrial cancer in vitro and in vivo.19 Upregulation of miR-652 promotes cell proliferation and migration invasion as well as inhibits cell apoptosis in non-small–cell lung cancer.20 In contrast, miR-652 plays a tumor-suppressive role in pancreatic cancer21 and pediatric acute lymphoblastic leukemia.30 However, the specific roles of miR-652 in GBM progression are still unknown. Hence, the effects of miR-652 on the malignant phenotypes of GBM were analyzed in this study. In vitro and in vivo functional assays revealed that ectopic miR-652 expression attenuated GBM cell proliferation, clone formation, migration, and invasion in vitro as well as promoted cell apoptosis and decreased tumor growth in vivo. These findings suggest that miR-652 is a potential therapeutic marker for patients with GBM.
Multiple genes, including retinoic acid–related orphan receptor-A,19 lethal(2) giant larvae 1,20 and zinc finger E-box binding homeobox 1,21 have been recognized as the direct downstream targets of miR-652. FOXK1, a forkhead family transcription factor31 shown to be a novel direct target of miR-652 in GBM, has been implicated in development and metabolism and is overexpressed in colorectal,32 liver,24 gastric,33 and esophageal cancers.34 FOXK1 is also expressed at high levels in GBM tissues and cell lines.23 FOXK1 behaves as an aggressive oncogene by regulating cell proliferation, cycle, and apoptosis.23 We successfully demonstrated that FOXK1 expression was negatively modulated by miR-652 and that miR-652 overexpression significantly restricted the progression and development of GBM in vitro and in vivo. Thus, decreasing FOXK1 expression or restoring miR-652 expression might be effective therapeutic approaches for treating patients with GBM.
Conclusion
In conclusion, miR-652 directly decreased FOXK1 expression and deactivated the AKT/mTOR pathway in GBM, thereby stunting GBM progression and development. These results provide novel insights into the malignant development of GBM, thereby providing new options for GBM therapy under miR-652/FOXK1/AKT/mTOR regulation. However, due to the limitation of obtained information, we could not analyze the correlation between miR-652 expression and molecular subtypes, recurrence tumors, IDH1 mutated tumors. Therefore, future studies should be performed to determine this.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hirst TC, Vesterinen HM, Conlin S, et al. A systematic review and meta-analysis of gene therapy in animal models of cerebral glioma: why did promise not translate to human therapy? Evidence Based Preclin Med. 2014;1:e00006. doi:10.1002/ebm2.6
2. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–820. doi:10.1007/s00401-016-1545-1
3. Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi:10.2353/ajpath.2007.070011
4. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi:10.1056/NEJMra0708126
5. Komotar RJ, Otten ML, Moise G, Connolly ES
6. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi:10.1056/NEJMoa043330
7. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297.
8. German MA, Pillay M, Jeong DH, et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008;26:941–946. doi:10.1038/nbt1417
9. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi:10.1038/nrg1379
10. Zhou L, Liu F, Wang X, Ouyang G. The roles of microRNAs in the regulation of tumor metastasis. Cell Biosci. 2015;5:32. doi:10.1186/s13578-015-0028-8
11. Aakula A, Kohonen P, Leivonen SK, et al. Systematic identification of microRNAs that impact on proliferation of prostate cancer cells and display changed expression in tumor tissue. Eur Urol. 2016;69:1120–1128. doi:10.1016/j.eururo.2015.09.019
12. Zhang J, Lv J, Zhang F, et al. MicroRNA-211 expression is down-regulated and associated with poor prognosis in human glioma. J Neurooncol. 2017;133:553–559. doi:10.1007/s11060-017-2464-2
13. Huang SW, Ali ND, Zhong L, Shi J. MicroRNAs as biomarkers for human glioblastoma: progress and potential. Acta Pharmacol Sin. 2018. doi:10.1038/aps.2017.173
14. Gomez Zubieta DM, Hamood MA, Beydoun R, Pall AE, Kondapalli KC. MicroRNA-135a regulates NHE9 to inhibit proliferation and migration of glioblastoma cells. CCS. 2017;15:55. doi:10.1186/s12964-017-0209-7
15. Zhang Z, Lei B, Wu H, Zhang X, Zheng N. Tumor suppressive role of miR-194-5p in glioblastoma multiforme. Mol Med Rep. 2017;16:9317–9322. doi:10.3892/mmr.2017.7826
16. Wang N, Zhang Y, Liang. H. microRNA-598 inhibits cell proliferation and invasion of glioblastoma by directly targeting metastasis associated in colon cancer-1. Oncol Res. 2018;26(8):1275–1283.
17. Gu JJ, Fan KC, Zhang JH, Chen HJ, Wang SS. Suppression of microRNA-130b inhibits glioma cell proliferation and invasion, and induces apoptosis by PTEN/AKT signaling. Int J Mol Med. 2018;41:284–292. doi:10.3892/ijmm.2017.3233
18. Chen L, Wang X, Wang H, et al. miR-137 is frequently down-regulated in glioblastoma and is a negative regulator of Cox-2. Eur J Cancer. 2012;48:3104–3111. doi:10.1016/j.ejca.2012.02.007
19. Sun X, Dongol S, Qiu C, et al. miR-652 promotes tumor proliferation and metastasis by targeting RORA in endometrial cancer. MCR. 2018;16:1927–1939. doi:10.1158/1541-7786.MCR-18-0267
20. Yang W, Zhou C, Luo M, et al. MiR-652-3p is upregulated in non-small cell lung cancer and promotes proliferation and metastasis by directly targeting Lgl1. Oncotarget. 2016;7:16703–16715. doi:10.18632/oncotarget.7697
21. Deng S, Li X, Niu Y, et al. MiR-652 inhibits acidic microenvironment-induced epithelial-mesenchymal transition of pancreatic cancer cells by targeting ZEB1. Oncotarget. 2015;6:39661–39675. doi:10.18632/oncotarget.v6i37
22. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi:10.1006/meth.2001.1262
23. Ji ZG, Jiang HT, Zhang PS. FOXK1 promotes cell growth through activating wnt/beta-catenin pathway and emerges as a novel target of miR-137 in glioma.American. J Trans Res. 2018;10:1784–1792.
24. Cui H, Gao Q, Zhang L, Han F, Wang L. Knockdown of FOXK1 suppresses liver cancer cell viability by inhibiting glycolysis. Life Sci. 2018;213:66–73. doi:10.1016/j.lfs.2018.10.018
25. Zhang P, Tang WM, Zhang H, et al. MiR-646 inhibited cell proliferation and EMT-induced metastasis by targeting FOXK1 in gastric cancer. Br J Cancer. 2017;117:525–534. doi:10.1038/bjc.2017.181
26. Quan J, Qu J, Zhou L. MicroRNA-539 inhibits glioma cell proliferation and invasion by targeting DIXDC1. Biomed Pharmacother. 2017;93:746–753. doi:10.1016/j.biopha.2017.06.097
27. Nie E, Jin X, Wu W, et al. MiR-198 enhances temozolomide sensitivity in glioblastoma by targeting MGMT. J Neurooncol. 2017;133:59–68. doi:10.1007/s11060-017-2425-9
28. Luo JW, Wang X, Yang Y, Mao Q. Role of micro-RNA (miRNA) in pathogenesis of glioblastoma. Eur Rev Med Pharmacol Sci. 2015;19:1630–1639.
29. Babashah S, Soleimani M. The oncogenic and tumour suppressive roles of microRNAs in cancer and apoptosis. Eur J Cancer. 2011;47:1127–1137. doi:10.1016/j.ejca.2011.02.008
30. Jiang Q, Lu X, Huang P, et al. Expression of miR-652-3p and effect on apoptosis and drug sensitivity in pediatric acute lymphoblastic leukemia. Biomed Res Int. 2018;2018:5724686. doi:10.1155/2018/5724686
31. Garry DJ, Meeson A, Elterman J, et al. Myogenic stem cell function is impaired in mice lacking the forkhead/winged helix protein MNF. Proc Natl Acad Sci U S A. 2000;97:5416–5421. doi:10.1073/pnas.100501197
32. Lu SR, Li Q, Lu JL, Liu C, Xu X, Li JZ. Long non-coding RNA LINC01503 promotes colorectal cancer cell proliferation and invasion by regulating miR-4492/FOXK1 signaling. Exp Ther Med. 2018;16:4879–4885. doi:10.3892/etm.2018.6775
33. Dong L, Hong H, Chen X, Huang Z, Wu W, Wu F. LINC02163 regulates growth and epithelial-to-mesenchymal transition phenotype via miR-593-3p/FOXK1 axis in gastric cancer cells. Artif Cells Nanomed Biotechnol. 2018;46(sup2):607–615..
34. Chen D, Wang K, Li X, et al. FOXK1 plays an oncogenic role in the development of esophageal cancer. Biochem Biophys Res Commun. 2017;494:88–94. doi:10.1016/j.bbrc.2017.10.080
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.