Back to Journals » ImmunoTargets and Therapy » Volume 13
Microbiome Differences in Colorectal Cancer Patients and Healthy Individuals: Implications for Vaccine Antigen Discovery
Authors Ibeanu GC, Rowaiye AB, Okoli JC, Eze DU
Received 26 July 2024
Accepted for publication 2 December 2024
Published 13 December 2024 Volume 2024:13 Pages 749—774
DOI https://doi.org/10.2147/ITT.S486731
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Flavio Salazar-Onfray
Gordon C Ibeanu,1 Adekunle B Rowaiye,1,2 Joy C Okoli,1 Daniel U Eze1
1Department of Pharmaceutical Sciences, North Carolina Central University, Durham, NC, 27707, USA; 2Department of Agricultural Biotechnology, National Biotechnology Development Agency, Abuja, Nigeria
Correspondence: Adekunle B Rowaiye, Department of Pharmaceutical Sciences, North Carolina Central University, Durham, NC, 27707, USA, Tel +1-9193812004, Email [email protected]
Background: Colorectal cancer (CRC) is the third most prevalent cancer worldwide, with numerous risk factors contributing to its development. Recent research has illuminated the significant role of the gut microbiota in CRC pathogenesis, identifying various microbial antigens as potential targets for vaccine development.
Aim: This review aimed at exploring the potential sources of microbial antigens that could be harnessed to create effective CRC vaccines and understand the role of microbiome-CRC interactions in carcinogenesis.
Methods: A comprehensive search of original research and review articles on the pathological links between key microbial candidates, particularly those more prevalent in CRC tissues, was conducted. This involved extensive use of the PubMed and Medline databases, as well as the Google Scholar search engine, utilizing pertinent keywords. A total of one hundred and forty-three relevant articles in English, mostly published between 2018 and 2024, were selected.
Results: Numerous microbes, particularly bacteria and viruses, are significantly overrepresented in CRC tissues and have been shown to promote tumorigenesis by inducing inflammation and modulating the immune system. This makes them promising candidates for antigens in the development of CRC vaccines.
Conclusion: The selection of microbial antigens focuses on their capacity to trigger a strong immune response and their link to tumor presence and progression. Identifying and validating these antigens through preclinical testing is essential in developing a CRC vaccine.
Plain Language Summary: Colorectal cancer (CRC) is a common and serious disease that affects many people worldwide. Research has shown that the bacteria and other microorganisms in our gut play a significant role in the development of CRC. This review aimed to identify specific microbial targets that could be used to develop effective vaccines against CRC.
The researchers searched through numerous scientific articles and found that certain bacteria and viruses are more commonly found in CRC tissues and promote tumor growth by causing inflammation and affecting the immune system. These microbes have the potential to be used as antigens in CRC vaccines.
The identification of these antigens is a crucial step towards developing a vaccine that can prevent or treat CRC. Further research is needed to validate these findings and move closer to creating an effective vaccine against this disease.
In simpler terms, this research is exploring how the gut microbiome contributes to colorectal cancer and searching for specific targets to develop a vaccine that can help prevent or treat the disease.
Keywords: colorectal cancer, microbe, antigen, vaccine, tumor
Introduction
Colorectal cancer (CRC) ranks as the third most prevalent cancer and the second-leading cause of cancer-related morbidity and mortality globally.1
The World Cancer Fund International report that in 2022, there were 1,926,425 newly diagnosed cases of colorectal cancer.2 Despite progress in screening and treatment, CRC continues to present significant public health challenges due to its high incidence and mortality rates.3
Emerging research has increasingly highlighted the critical role of the gut microbiome in CRC pathogenesis. Empirical evidence from preclinical and clinical studies suggests that gut microbiota plays a vital role in physiological development and maintaining immunological balance. Through extensive research, disruptions in this microbial community, known as dysbiosis, have been linked to various cancers, including CRC.4
The connection between gut microbiota and CRC has become a focal area for potential interventions, including vaccine development targeting specific pathogenic bacterial species. Research underscores that certain microbes play critical roles in CRC by promoting chronic inflammation, DNA damage, and immune evasion, providing a basis for targeting these organisms in potential therapeutic strategies. Key areas of research support this rationale include microbiome and immune modulation in CRC;5 carcinogenic effects of pathogenic bacteria;6 restoration of microbial balance;7 and microbial metabolic pathways and CRC8 (Figure 1).
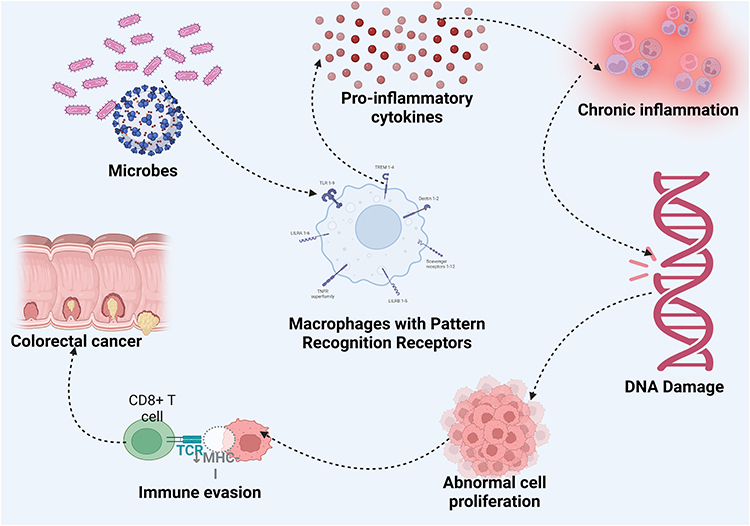 |
Figure 1 Microbes and Colorectal carcinogenesis. |
Recently, the microbiota’s influence on the effectiveness of anti-tumor treatments has also been observed in digestive cancers. For example, several bacterial species such as F. nucleatum, B. fragilis, and colibactin-producing E. coli (CoPEC), are involved in these processes. CoPEC bacteria are commonly found in the colonic mucosa of CRC patients and have been shown to promote colorectal carcinogenesis in susceptible mouse models of CRC.9 Created in BioRender. Rowaiye, A. (2024) https://BioRender.com/i16a085.
Various other microbial species, especially bacteria and viruses, have been found to be overrepresented in CRC tissues and have been linked to CRC pathogenesis through sequencing studies in CRC patients as well as functional studies in cell cultures and animal models.10 These microbes release antigens in the gut, which are recognized by antigen-presenting cells (APCs) through pattern recognition receptors, such as toll-like receptors. This triggers the release of inflammatory cytokines, such as IL-6, and TNF-α, initiating an immune response.11
Chronic inflammation driven by these cytokines can result in DNA damage and mutations in both epithelial and immune cells, creating conditions conducive to tumorigenesis. Host DNA is damaged through genotoxic compounds, and this may indirectly contribute to CRC by influencing signaling pathways such as E-cadherin/β-catenin, TLR4/MYD88/NF-κB, and SMO/RAS/p38 MAPK. Furthermore, these microbes support CRC progression by helping tumor cells evade immune detection—achieved by suppressing immune cell activity, maintaining a pro-inflammatory environment, or interfering with autophagy.12
Given this intricate relationship between microbes and CRC, there is a growing interest in exploring microbial antigens as potential targets for CRC vaccine development. Vaccines designed to elicit immune responses against specific microbial antigens could offer a novel approach to preventing or treating CRC. This article delves into the potential sources of microbial antigens that could be leveraged for CRC vaccine development, examining the existing evidence on microbial involvement in CRC and evaluating the feasibility of these antigens as vaccine candidates. By identifying and characterizing these microbial antigens, we can pave the way for innovative strategies that harness the immune system to combat CRC effectively.
Methods
A thorough search of original research and review articles, primarily published between 2018 and 2024, was conducted to investigate the pathological links between key microbial candidates, particularly those more prevalent in CRC tissues. This search utilized the PubMed and Medline databases, along with the Google Scholar search engine, employing keyword combinations such as “Colorectal cancer AND Fusobacterium nucleatum”, “Colorectal cancer AND Streptococcus gallolyticus”, “Colorectal cancer AND Escherichia coli”, “Colorectal cancer AND Bacteroides fragilis”, “Colorectal cancer AND Human Papilloma Virus”, “Colorectal cancer AND Epstein-Barr Virus”, and more. All relevant articles were selected and reviewed, resulting in a total of one hundred and forty-three articles published in English being included in the analysis.
The inclusion criteria for articles in this review encompassed original research and review papers mostly published between 2018 and 2024, written in English, and focusing specifically on CRC and its association with certain microbial candidates, including Fusobacterium nucleatum, Streptococcus gallolyticus, Escherichia coli, Bacteroides fragilis, Human Papilloma Virus (HPV), and Epstein-Barr Virus (EBV). Articles were selected based on their relevance, specifically if they discussed the pathological role of these microbes in the development, progression, or other mechanisms related to CRC.
The exclusion criteria included articles that did not focus on the association between CRC and the specified microbial candidates, non-English language publications, duplicates from different databases, and or non-peer-reviewed articles.
In this review, several confounding factors were considered to contextualize findings accurately, including prior treatments and vaccination status. Many CRC patients undergo treatments such as chemotherapy and antibiotics, which can influence microbial colonization and impact the role of microbes like Fusobacterium nucleatum or Escherichia coli in tumor progression. Studies that did not account for these treatments were evaluated cautiously (Figure 2).
 |
Figure 2 Flow diagram for the article selection process. |
Sources of Microbial Antigen
Microbial antigens for CRC vaccine development may be obtained from bacterial, viral, fungal, or protozoan proteins (Figure 3).
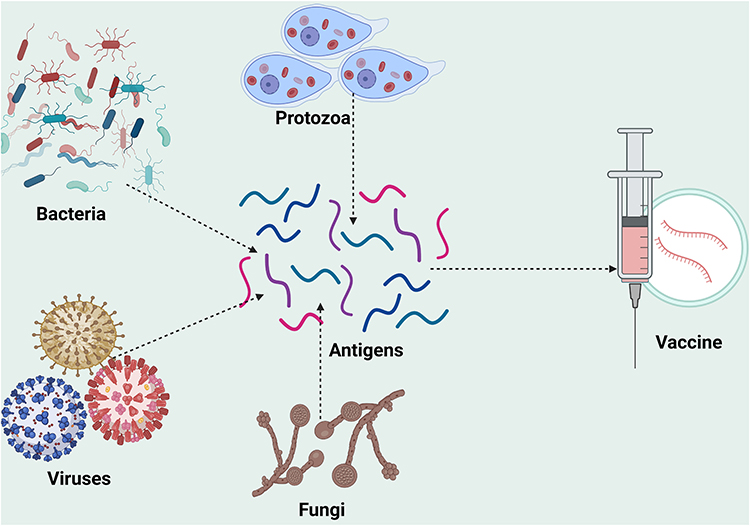 |
Figure 3 Sources of microbial antigen for CRC vaccine development. Created in BioRender. Rowaiye, A. (2024) https://BioRender.com/e00z426. |
Bacterial
Bacteria associated with CRC often share several common properties that contribute to tumorigenesis (Table 1). These properties include the induction of chronic inflammation, immune system modulation, genotoxic effects, alteration of tumor microenvironment, disruption of host signaling pathways, biofilm formation, metabolic interference, and synergistic interaction with other microbes.10,13,14
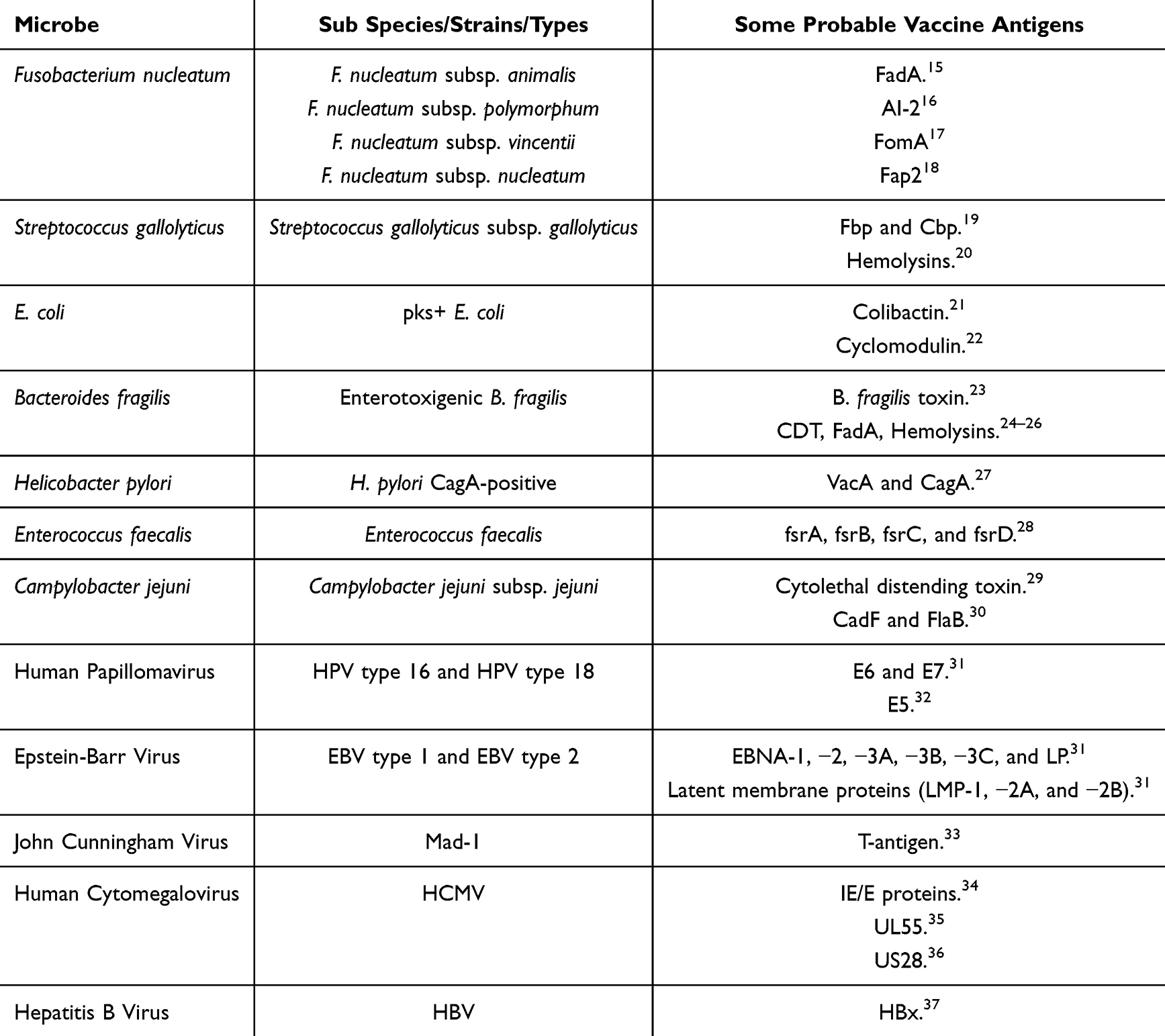 |
Table 1 Microbes and Their Probable Vaccine Antigens |
Fusobacterium Nucleatum
Fusobacterium nucleatum (F. nucleatum) is a Gram-negative obligate anaerobic bacterium commonly found in the oral cavity and is implicated in various oral diseases, including periodontitis and gingivitis.38 Capable of colonizing the colon tissues through blood stream infection, F. nucleatum is enriched in both stool and tumor tissues of CRC patients.12,39 This bacterium is known to adhere to and invade colorectal epithelial cells promoting inflammation, the activation of oncogenic pathways, and contributing to CRC tumorigenesis and chemoresistance.40
Accumulating evidence from several metagenomic studies and other empirical data establishes a strong association between an enrichment of F. nucleatum in the colorectal carcinoma tissues, tumor microenvironment and fecal samples of CRC patients, suggesting it as a potential risk factor in CRC initiation and progression1,41 These studies indicate that tumor formation is associated with the colonization by F. nucleatum which subsequently aids cancer cells in developing a supportive tumor microenvironment and confers chemoresistance.39
Yamaoka et al (2018) conducted a study to compare the abundance of F. nucleatum in colorectal tissues with clinicopathologic and molecular features of CRC. Droplet digital polymerase-chain reaction (PCR) was used to measure the absolute copy numbers of F. nucleatum in 100 CRC tissues and 72 matched normal-appearing mucosal tissues. In normal-appearing mucosal tissues and CRC tissue samples, respectively, the detection rates of F. nucleatum were 63.9 and 75.0%, and the median copy number of F. nucleatum was 0.4 and 1.9/ng DNA, respectively. The levels of F. nucleatum are significantly elevated in human colorectal adenomas and carcinomas compared to adjacent normal tissue.42
Using arbitrarily primed PCR (AP-PCR) as the strain typing method, Komiya et al (2019), identified four subspecies of Fusobacterium nucleatum—F. nucleatum subsp. animalis, F. nucleatum subsp. nucleatum, F. nucleatum subsp. polymorphum, and F. nucleatum subsp. Vincentii from both the CRC tissue and saliva samples. Identical F. nucleatum strains were found in both CRC tissue and saliva samples from 42.9% of the patients. Notably, 75% of patients who were F. nucleatum-positive in both CRC and saliva specimens had identical strains. These results suggest that F. nucleatum in CRC originates in the oral cavity.43
F. nucleatum infection results in microsatellite instability (MSI) triggering a microenvironment that contributes to the chemoresistance of tumor cells.39 Lee et al (2018) identified unique mutational signatures in CRC tissue based on F. nucleatum levels. Using quantitative PCR (qPCR), the study measured the amount of F. nucleatum in tumor tissue and adjacent normal tissue in adjuvant (N = 128) and metastatic (N = 118) cohorts and categorized patients into F. nucleatum-high and F. nucleatum-low groups based on the amount of F. nucleatum. Furthermore, the next-generation sequencing of 40 genes within the 5 critical pathways (P53, PI3K, RTK-RAS, WNT, and TGF-β) performed in the adjuvant cohort revealed that patients with MSI-H and CIMP-H exhibited higher levels of F. nucleatum in tumor tissue. F. nucleatum-high patients showed higher rates of transition mutations and C to T (G to A) nucleotide changes, regardless of MSI status.
That patients with microsatellite instability-high (MSI-H) and CpG island methylator phenotype-high (CIMP-H) CRC exhibited higher levels of F. nucleatum in their tumor tissues. Additionally, these F. nucleatum-high patients demonstrated increased rates of transition mutations, specifically cytosine (C) to thymine (T) and G to guanine (G) to adenine (A) mutations. Additionally, in F. nucleatum-high patients, there are increased mutation rates in the AMER1 and ATM genes, as well as in the TGF-β signaling pathway.40
Beyond being a significant risk factor, F. nucleatum is a potential biomarker for CRC progression.12 F. nucleatum has been detected from stages 0 to IV, indicating that F. nucleatum can adhere to CRC tissue from an early stage of tumorigenesis.43 Yamaoka et al (2018) discovered that F. nucleatum copy numbers in stage IV CRC tissues were significantly higher than those in the normal-appearing mucosa of the same patients. The abundance of F. nucleatum in CRC tissues correlated with tumor size and KRAS mutation and was significantly associated with shorter overall survival times, particularly in patients with stage IV CRC. Additionally, in the normal-appearing mucosa of patients with CRC, the F. nucleatum copy number was significantly higher in those with stage IV disease compared to those with stages I–III. These results suggest that determining F. nucleatum levels may help predict clinical outcomes in CRC patients.42
Using 16S rRNA sequencing, De Carvalho et al (2019) investigated the abundance of the microbial community in a series of colorectal tumor samples treated at Barretos Cancer Hospital in Brazil. The presence and impact of F. nucleatum DNA (Fn DNA) on CRC prognosis was further evaluated by qPCR in 152 CRC cases. The study observed an enrichment of Fn DNA in tumor tissue that was significantly associated with proximal tumors, greater depth of invasion, higher clinical stages, poor differentiation, MSI-positive status, BRAF-mutated tumors, and the loss of expression of mismatch-repair proteins MLH1, MSH2, and PMS2. Furthermore, the presence of Fn DNA in CRC tissue was associated with worse cancer-specific survival (69.9% vs 82.2% at 5 years) and overall survival (63.5% vs 76.5%).44
F. nucleatum has been implicated in both the development and metastasis of CRC through several mechanisms, which include virulence factors, intestinal metabolites, DNA mutation, and immunomodulation.1,12 F. nucleatum also recruits tumor-infiltrating immune cells, creating a pro-inflammatory microenvironment that promotes colorectal neoplasia progression. Furthermore, F. nucleatum potentiates CRC development through toll-like receptor 2 (TLR2)/toll-like receptor 4 (TLR4) signaling and microRNA (miRNA)-21 expression. It also increases the CRC recurrence and chemoresistance by mediating a molecular network involving miRNA-18a*miRNA-4802, and autophagy components.41
F. nucleatum contains virulence factors such as FadA, Fap2, FomA, and AI-2, which influences its adhesion, invasion, and mutagenicity.15,17,18,45 FadA enhances the harmful effects of F. nucleatum on CRC by activating the chk2 protein, resulting in DNA damage and tumor progression.15 Facilitated by the FadA protein, F. nucleatum encourages the growth and development of CRC by altering the signaling pathway of E-Cadherin and beta-Catenin.46 The Fap2 protein on the surface of F. nucleatum helps the bacterium to preferentially colonize and enrich CRC tissues by binding to specific sugar molecules (Gal-GalNAc) present on the surface of cancer cells.18 Additionally, Fap2 also binds immune cells, causing immunosuppression.16 The AI-2 protein triggers macrophages to switch to the M1 type, which leads to a strong pro-inflammatory response, releasing cytokines like IL-1β, IL-8, and TNF-α, by activating the TNFSF9/IL-1β pathway. Therefore, AI-2 could be a potential new target for treating diseases linked to an imbalance of gut microbiota through immunotherapy.16
Streptococcus Gallolyticus
Streptococcus gallolyticus is a Gram-positive bacterium mostly found in the rumen of herbivores and the digestive tract of birds, and less commonly in the human intestinal tract, Streptococcus gallolyticus subsp. Gallolyticus (SGG), formerly known as S. bovis type I, is a leading cause of septicemia and infective endocarditis in elderly and immunocompromised individuals.47 A meta-analysis study spanning a 40-year period reveals that 65% of patients with invasive SGG infections also had concomitant colorectal neoplasia. Also, molecular analyses suggest potential zoonotic transmission of SGG.47
Using both cell culture and molecular methods (PCR with specific primers for the sodA gene), Sheikh et al, (2020), detected SGG in fecal samples of CRC patients. In Ahvaz, southwest Iran, a total of 106 fecal samples were collected from 22 CRC patients and 40 healthy individuals. The stool culture results showed SGG in 9% of these samples from CRC patients and no SGG was isolated from the healthy individuals. Additionally, in CRC patients, the sodA gene was detected in 40.9%, while 5% of the specimens in the control group had the sodA gene.48
Guven et al (2019) examined the prevalence and quantity of Streptococcus gallolyticus (Sg) in the saliva of CRC patients compared to controls. PCR analyses were conducted on saliva samples from 71 CRC patients and 77 controls. The results showed that CRC patients had significantly higher amounts of Sg compared to controls. Additionally, the levels of Sg were lower in the MSI+ group. Receiver operating characteristic (ROC) analysis revealed that the salivary amount of Sg had diagnostic value for CRC, with an area under the curve (AUC) of 0.84. This study revealed that higher amounts of Sg was found in the saliva of CRC patients, and salivary Sg may be useful in distinguishing CRC.49
Using genetically susceptible CRC (Apc+/− Notch-inducible) mice, Aymeric et al (2018) found SGG colonization to be significantly higher in tumor-bearing mice, displacing native resident enterococci. This ecological shift is due to the activation of a specific SGG bacteriocin, termed “gallocin”, capable of eliminating enterococci. Notably, the presence of bile acids significantly enhanced this bacteriocin activity in vivo, facilitating greater SGG colonization. The continuous activation of the Wnt pathway, an early CRC signaling alteration, and reduced expression of the bile acid transporter gene Slc10A2 due to the Apc mutation contribute to sustained SGG colonization.50
Zhang et al (2018) explored the carcinogenic effects of S. gallolyticus and its potential mechanisms in azoxymethane and dextran sodium sulfate-induced colitis-associated CRC in mice. Oral pretreatment with S. gallolyticus was used to evaluate its detrimental effects. The results revealed that S. gallolyticus is enriched in colonic carcinoma compared to adenoma and healthy mice. Pretreatment with S. gallolyticus aggravated tumor formation, resulting in more colonic obstruction, a larger number of tumor nodes, and increased tumor node diameter. Furthermore, S. gallolyticus selectively recruited tumor-infiltrating myeloid cells (not mast cells) and induced the production of myeloid cell-derived proinflammatory cytokines, generating an immune-suppressive microenvironment and promoting neoplasia progression.51
The interactions between several clinical isolates of Streptococcus gallolyticus (Sg) and human colon cancer cells, as well as their effects in mouse models has been investigated. It was observed that certain Sg strains could stimulate host cell proliferation (proliferation-promoting Sg, PP-Sg), while others could not (non-proliferation-promoting Sg, NP-Sg). Compared to NP-Sg strains, the PP-Sg strains adhered significantly better to colon cancer cells, better colonization of colon tissues in A/J mice, promoted tumor development in an azoxymethane-induced mouse model of CRC. These findings provide insights into the mechanisms underlying the Sg-CRC association and have important implications for clinical studies aiming to correlate Sg with clinical and pathological features of CRC.52 Furthermore, evidence suggests that these bacteria may contribute to CRC through a genomic passenger mechanism.53
A prospective nested case–control study demonstrated a positive association between antibody responses to SGG and the development of CRC in serum samples taken before the onset of evident disease. Pre-diagnostic serum samples from 485 first incident CRC cases (with a mean time between blood draw and diagnosis of 3.4 years) and 485 matched controls from the European Prospective Investigation into Nutrition and Cancer (EPIC) study for antibody responses to 11 SGG proteins using multiplex serology. These 11 SSG proteins (structural and non-structural) are Gallo0112A, Gallo0112B, Gallo0272* Gallo0577, Gallo0748* Gallo0933, Gallo1570, Gallo1675* Gallo2018* Gallo2178* and Gallo2179 (*antigens included in 6-marker panel). In this study, antibody positivity for any of the 11 SGG proteins evaluated was significantly associated with CRC risk, with 56% of controls and 63% of cases testing positive. Additionally, positivity for two or more proteins from a previously identified SGG 6-marker panel, which has greater CRC specificity, was observed in 9% of controls compared to 17% of CRC cases, indicating a significantly increased CRC risk.54
Antibody responses to Streptococcus gallolyticus subspecies gallolyticus (SGG) proteins, particularly the pilus protein Gallo2178, have been consistently associated with an increased risk of CR.54 Using multiplex serology, Butt et al (2018) measured antibody responses to nine SGG proteins in participants from 10 prospective US cohorts, including 4,063 incident CRC cases and 4,063 matched controls. The results established the association between antibody responses to SGG Gallo2178 and CRC risk for individuals diagnosed within 10 years after the blood draw.55 In a similar study, Genua et al, (2023), assessed the association of immune responses to bacterial exposure with advancing stages of colorectal neoplasia. It was observed that IgA sero-positivity to any SGG protein, specifically Gallo0272 and Gallo1675, was associated with an increased occurrence of advanced adenoma. This finding indicates that antibody responses to SGG are associated with the occurrence of colorectal adenomas.56
Several antigenic proteins from Streptococcus gallolyticus contribute to CRC development by promoting chronic inflammation, modulating the gut microbiota, enhancing tumor cell proliferation and migration, and suppressing host immune responses. Some of these proteins include - Fibronectin-binding protein (Fbp) and Collagen-binding protein (Cbp) which facilitate bacterial adhesion to host cells, and hemolysins, which damages host cells and tissues.19,20
Escherichia Coli
Escherichia coli (E. coli) is a Gram-negative bacterium that is commonly found in the intestines of humans and other warm-blooded animals. While many strains of E. coli are harmless and part of the normal gut flora, certain strains can be pathogenic.57
Among the numerous factors that may eventually lead to CRC, certain strains of Escherichia coli (E. coli) from the B2 phylogenetic group have been implicated, particularly those that produce colibactin from their polyketide synthesis (pks) locus. This genomic island codes for the synthesis of colibactin, a genotoxin which alkylates DNA at adenine residues, and induces double-strand breaks, cell cycle arrest, mutations, and chromosomal instability in eukaryotic cells.21,58,59
In a study by Pleguezuelos-Manzano et al (2020), human intestinal organoids were repeatedly exposed to genotoxic pks+ E. coli via luminal injection over a period of 5 months. Whole-genome sequencing of clonal organoids before and after this exposure revealed a unique mutational signature that was not present in organoids injected with isogenic pks-mutant bacteria. This specific mutational signature was also found in a subset of human cancer genomes from two independent cohorts, predominantly in CRC. The results reveal a distinct mutational signature in CRC, suggesting that it directly results from previous exposure to bacteria carrying the colibactin-producing pks pathogenicity island.59
Another study that was conducted by Iyadoraiet al (2020) to detect and compare the prevalence of pks+ E. coli in CRC patients and healthy controls was followed by an investigation of the virulence triggered by pks+ E. coli using an in-vitro model. Results showed that 16.7% of the CRC patients and 4.3% of the healthy controls harbored pks+ E. coli. Furthermore, primary colon epithelial cells displayed syncytia and swelling, while colorectal carcinoma HCT116 cells exhibited megalocytosis in response to treatment with the isolated pks+ E. coli strains. These findings suggest that pks+ E. coli was more frequently isolated from the tissues of CRC patients compared to healthy individuals, and that the in-vitro assays showed that the isolated strains may be involved in the initiation and development of CRC.21
Given the growing evidence of the role of the immune environment, particularly antitumor T-cells, in CRC development, a study investigated the impact of colibactin-producing E. coli (CoPEC) on these cells in human CRC and APCMin/+ mice. The study found that CRC patients colonized by CoPEC had reduced levels of tumor-infiltrating T lymphocytes (CD3+ T-cells). This decrease was also observed in mice with chronic CoPEC infection, where CD3+ and CD8+ T-cells decreased while colonic inflammation increased. The mesenteric lymph nodes (MLNs) of CoPEC-infected mice showed a significant reduction in antitumor T-cells compared to controls, and anti-mouse PD-1 immunotherapy was less effective in CoPEC-infected mice with the MC38 tumor model. These findings suggest that CoPEC may create a procarcinogenic immune environment by suppressing the antitumor T-cell response, potentially leading to resistance to immunotherapy.60
E. coli produces a bacterial toxin known as cyclomodulin, which inhibits the host cell cycle by inducing G2 phase arrest without triggering the DNA-damage checkpoint-signaling pathway. This toxin causes cytopathic effects, such as the recruitment of focal adhesion plates and the formation of stress fibers. These actions lead to an irreversible halt in the cell cycle at the G2/M transition and sustained inhibitory phosphorylation of CDK1, a key regulator of mitosis.22 Other virulence factors include adhesins and invasins that facilitate the infection of host cells and consequently carcinogenesis.61
Bacteroides Fragilis
Bacteroides fragilis (B. fragilis) is a Gram-negative bacterium that belongs to the phylum Bacteroidetes and is known for its anti-inflammatory effects in the intestinal tract. In contrast, enterotoxigenic B. fragilis (ETBF), a specific subtype, produces an enterotoxin (BFT; B. fragilis toxin), leading to chronic infections without symptoms, DNA damage and the formation of colonic tumors.23 Meta-analysis reveals that most studies supported the notion of an association or causal role of ETBF in CRC.62
ETBF plays a role in the development and progression of CRC by affecting the mucosal immune response and causing changes in epithelial cells.63 A study was conducted to evaluate the prevalence of ETBF in stool samples from 60 CRC patients and 60 healthy individuals without a history of colorectal disease. The results of the screening using PCR targeting neu and bft marker genes reveal that the frequency of B. fragilis was higher in CRC cases (58.3%) compared to controls (26.6%). Moreover, the presence of the bft gene was significantly more common in CRC cases, particularly in stage III patients compared to stages I and II. Specifically, the enterotoxin isotype bft-2 was detected more frequently among CRC patients than in healthy controls. These findings suggest a correlation between fecal ETBF and CRC, indicating that ETBF detection could potentially serve as a marker for diagnosing CRC.64
A study in Québec, Canada analyzed fecal samples from healthy individuals (N = 62) and CRC patients (N = 94). Using PCR to detect pks island and bft marker genes, the results showed that 42% of healthy individuals and 46% of CRC patients were colonized by pks+ bacteria. The bft marker was found in 21% of healthy individuals and 32% of CRC patients, with both markers present in 8% of healthy controls and 13% of CRC patients. Early-onset CRC patients (under 50 years) had lower colonization rates of pks+ bacteria (20%) compared to late-onset patients (52%). The study suggests that high colonization rates by these bacteria may be a risk factor for developing CRC, and the lower prevalence in younger patients hints at an age-related component to bacterial colonization in CRC development.63
Specifically, ETBF forms a biofilm, produces toxins, and contributes to CRC development through chronic intestinal inflammation and tissue damage, whereas non-toxigenic B. fragilis (NTBF) does not produce such toxins.65 The abundance of B. fragilis was notably higher in tumor tissues compared to healthy ones, suggesting its potential role in CRC development through the modulation of cellular signaling pathways like WNT/β-catenin and the increased expression of CRC-related signaling pathway genes such as AXIN, CTNNB1, and BCL2.66 Studies conducted on colorectal cell lines have demonstrated that exposure to BFT leads to the disruption of E-cadherin, resulting in increased expression of the inflammatory cytokine IL-8.67
ETBF induces the expression of cyclooxygenase (COX)-2, which releases PGE2, initiating inflammation and controlling cell proliferation. This process involves the activation of signal transducers and activators of transcription (STAT)3, which consequently activates regulatory T-cells (Tregs), decreases interleukin (IL)-2 levels, increases IL-17 and IL-6 levels. This facilitates early intestinal inflammation and the promotion of cancer cell survival and proliferation.65 Furthermore, BFT degrades E-cadherin, altering signaling pathways that can upregulate spermine oxidase, affecting cell morphology, promoting carcinogenesis, and causing irreversible DNA damage.65
Other virulent factors linked with the pathogenicity of B. fragilis include Cytolethal distending toxins (CDT) which cause DNA damage, leading to genomic instability; FadA which is a surface protein that is involved in chemotaxis and motility; and Hemolysins (Holin, Zeta, and YoeB). These proteins contribute to the pathogenesis of B. fragilis infections by damaging host cells and tissues and promoting inflammation and immune evasion culminating in tumor initiation and progression.24–26
Helicobacter Pylori
Helicobacter pylori (H. pylori) is a Gram-negative bacterium. It belongs to the family Helicobacteraceae and is a spiral-shaped bacterium that colonizes the stomach and duodenum.68 Though primarily associated with gastric cancer, some studies indicate that chronic H. pylori infection may also be a risk factor for CRC, possibly due to its effects on systemic inflammation and gut microbiota.69 A study was conducted to investigate the link between H. pylori infection and the risk of developing CRC and adenomatous polyps. Fifty individuals diagnosed with colon cancer or adenomatous polyps were enrolled as the case group, while one hundred subjects without specific pathologies (such as polyps, neoplasms, or inflammatory diseases) constituted the control group. Blood samples were collected from all participants to assess the presence of anti-Helicobacter pylori antibodies. The serum levels of anti-Helicobacter pylori IgG and IgA antibodies were measured using an indirect enzyme-linked immunosorbent assay (ELISA). Among the participants, thirty-three were diagnosed with adenomatous polyps and 17 with colon cancer. The prevalence of H. pylori infection was significantly higher in individuals with colon cancer and adenomatous polyps compared to the healthy controls. These findings suggest that H. pylori infection may serve as a risk factor for both colon cancer and adenomatous polyps.70
Another study investigated the link between H. pylori infection and CRC using data from a nationwide population-based Chinese cohort. The study identified approximately 3936 individuals newly diagnosed with H. pylori infection and 15,744 age- and sex-matched patients without H. pylori infection from Taiwan’s National Health Insurance Research Database between 2000 and 2005. The cumulative incidence of CRC was found to be higher in the H. pylori-infected cohort compared to the comparison cohort. After adjusting for potential confounders, H. pylori infection was significantly associated with an increased risk of CRC.71
A systematic review and meta-analysis involving forty-seven studies with a total of 17,416 CRC cases and 55,811 controls was conducted by Zuo et al (2020). The analysis revealed an overall increased risk of CRC associated with H. pylori infection, although there was notable heterogeneity across the studies. Subgroup analysis indicated that the strength of this correlation might vary depending on the study design.69
Using H. pylori multiplex serologic assays, Butt et al (2019) analyzed serum samples from 4,063 incident CRC cases and 4,063 matched controls from ten prospective cohorts for antibody responses to thirteen H. pylori proteins, including VacA and CagA. Overall, 40% of controls and 41% of cases were H. pylori–seropositive, showing a marginal increase in CRC odds. Notably, H. pylori VacA-specific seropositivity was linked to an 11% rise in CRC odds, with a more pronounced effect observed among African Americans. Furthermore, CRC odds correlated with the level of VacA antibody in the entire cohort and specifically among African Americans. The analysis highlights the association between serologic responses to H. pylori VacA and an increased risk of CRC, particularly among African Americans.27
Enterococcus Faecalis
Enterococcus faecalis is a Gram-positive, facultative anaerobic bacterium belonging to the genus Enterococcus that has been associated with CRC development.72 In a recent study, a total of 80 formalin-fixed and paraffin-embedded (FFPE) colorectal tissue samples from CRC patients (n = 40) and healthy controls (n = 40) were analyzed for the presence and copy number of specific bacterial species using quantitative PCR. The relative quantification was calculated using the comparative cycle threshold method and expressed as a relative fold difference compared to a reference gene. The relative abundance and copy number of E. faecalis were significantly higher in CRC samples than in the control group. Additionally, the frequency of E. faecalis was significantly higher in the late stages (III/IV) of cancer compared to the early stages (I/II). This study demonstrated a higher concentration of E. faecalis in FFPE samples from CRC patients than from controls.73
Another study evaluated the mean copy number of E. faecalis in individuals with polyps and CRC compared to healthy controls. A total of 25 CRC patients and twenty-eight patients with intestinal polyps were selected, and stool specimens were collected. Additionally, twenty-four healthy individuals were selected as the control group. Bacterial DNA was extracted from the stool samples, and the number of E. faecalis was assessed using PCR for confirmation of the standard strain and absolute Real-Time PCR (qRT-PCR). The results showed that the mean copy number of E. faecalis was 11.2×109 per gram of stool in CRC patients, 9.4×108 per gram of stool in polyp patients, and 9×108 per gram of stool in healthy individuals. There was a significant difference in the copy numbers among the three groups. The presence of E. faecalis in the fecal flora of CRC patients was significantly higher than in those with polyps and healthy individuals, suggesting a potential role of this bacterium in inducing CRC.74
Zhang et al (2023) investigated the impact of specific metabolites produced by E. faecalis on CRC. The pro-tumor effects of E. faecalis were examined in colonic epithelial cells. It was observed that when cultured CRC cells were co-incubated with the conditioned medium of E. faecalis, their proliferation increased. Furthermore, liquid chromatography-mass spectrometry (LC-MS) screening identified biliverdin (BV) as the key metabolite produced by E. faecalis. BV enhanced colony formation, promoted cell proliferation, and prevented cell cycle arrest in cultured CRC cells. Additionally, BV significantly elevated the expression levels of IL-8 and VEGFA by modulating the PI3K/AKT/mTOR signaling pathway, thereby accelerating angiogenesis in CRC.13
The Fsr system is a quorum-sensing regulatory system in E. faecalis that involved is involved in the formation and the expression of virulence factors, including gelatinase and serine protease. This system plays a critical role in the pathogenicity of E. faecalis, which is known to be associated with various infections, including those related to CRC.75 The Fsr system allows E. faecalis to sense and respond to population density through the production and detection of signaling molecules. This system comprises the genes fsrA, fsrB, fsrC, and fsrD. The Fsr system regulates the expression of genes responsible for producing gelatinase (GelE) and serine protease (SprE), which are important for tissue invasion and degradation.28
Campylobacter Jejuni Subsp. Jejuni
Campylobacter jejuni subsp. jejuni, a Gram-negative bacterium, is the leading cause of bacterial gastroenteritis worldwide and has been associated with CRC. Studies examining tissue and fecal microbiomes have consistently shown an enrichment of Campylobacter in CRC samples.76
The prevalence of CRC has significantly risen in numerous Asian nations over the last 40 years. This swift shift in epidemiology implies that environmental factors play a role in the transformation of intestinal epithelial cells towards neoplasia. A diet resembling Western patterns (high in animal fats and proteins) is recognized as a risk factor. Furthermore, there is a strong association between dietary patterns and the microbial composition of the gut. These observations suggest that certain bacteria might contribute to the development of neoplasms in the colon and rectum. Campylobacter is prevalent in modern cattle and poultry farms and can adhere to colon epithelial cells, potentially causing genomic instability by releasing toxins that damage DNA. Consequently, Campylobacter is implicated in the advancement of colon adenocarcinoma.77
Campylobacter jejuni generates cytolethal distending toxin (CDT), a genotoxin with DNAse capabilities that induce DNA double-strand breaks, thus facilitating tumorigenesis.29 In a certain study, Germ-free (GF) ApcMin/+ mice were used in conjunction with a 1% dextran sulfate sodium diet to investigate the tumorigenic potential of CDT-producing C. jejuni. The findings revealed that GF ApcMin/+ mice colonized with the human clinical isolate C. jejuni 81–176 developed significantly more and larger tumors compared to uninfected mice. Moreover, C. jejuni with a mutated cdtB subunit (mutcdtB) attenuated tumorigenesis induced by C. jejuni in vivo and reduced the DNA damage response in cells and enteroids. C. jejuni infection triggered the expression of numerous colonic genes, with twenty-two genes being reliant on the presence of cdtB. Metatranscriptomic data indicated a distinct microbial gene expression profile between the C. jejuni-infected group and the mutcdtB group, along with differences in microbial communities observed through 16S rDNA sequencing. Notably, rapamycin could mitigate the tumorigenic potential of C. jejuni. In summary, the clinical isolate C. jejuni 81–176 fosters CRC development and leads to alterations in microbial composition and transcriptomic responses, a phenomenon dependent on CDT production.29
Other virulent factors facilitating C. jejuni infection include CadF (Campylobacter adhesion factor), and FlaB (Flagellar adhesin B).30
Clostridium Septicum
Clostridium septicum is a Gram-positive, toxin-producing, and spore-forming bacterium.78 From literature, the connection between Clostridium septicum infections and CRC patients has been well documented. These infections are present as bacteremia, septic embolism, cellulitis, myonecrosis, and gas gangrene of the limbs.79 There is a well-documented link between C. septicum bacteremia and a higher likelihood of a CRC diagnosis. A population-based cohort study was conducted in Southern Denmark and Region Zealand from 2007 to 2016, encompassing a population of about two million people, including 45,774 bacteremia episodes and 231,387 blood culture-negative cases. These bacteremia episodes were cross-referenced with the Danish central register for CRC. The study’s findings confirmed previous reports, demonstrating an increased risk of a CRC diagnosis following C. septicum bacteremia (20.8%).80
Abraham & Padam, (2023) report the clinical case involving an 81-year-old female whose primary presenting symptom of metastatic colon cancer was C. septicum bacteremia.81 Another case of colon adenocarcinoma linked with C. septicum endophthalmitis detected on 18F-FDG PET/CT has been reported.82 It is noteworthy that carcinogenesis typically spans several years and is nearly asymptomatic during its progression. Although C. septicum bacteremia is relatively uncommon, its occurrence is frequently linked to malignancies, particularly those of the lower gastrointestinal tract. This paraneoplastic symptom can often serve as the initial indication of an underlying malignant disease.79
There is a well-described association between bacteremia with C. septicum and an increased probability of a CRC diagnosis. A population-based cohort study in a population about two million people, including 45,774 bacteremia episodes and 231,387 blood culture-negative cases was performed in the Region of Southern Denmark and Region Zealand from 2007 to 2016. Episodes of bacteremia were combined with the Danish central register for CRC. The study results confirmed previous findings of an increased risk of a CRC diagnosis after bacteremia with C. septicum (20.8%).80
Virus
Several viruses have been associated with an increased risk of CRC through various mechanisms, including chronic inflammation, immune suppression, and direct oncogenic effects (Table 1).
Human Papillomavirus (HPV)
Human Papillomavirus (HPV) is a non-enveloped DNA virus belonging to the Papillomaviridae family. It primarily infects epithelial cells of the skin and mucous membranes and has been associated with the development of CRC.83
Meta-analysis of data from nineteen studies indicated a statistically significant presence of HPV infection in CRC tumor tissue.84 Using immunohistochemistry (IHC), real-time PCR, and RNA sequencing, HPV infection in 50 CRC samples from various regions (excluding the anal area) was investigated in a study. HPV infection was identified in 20% of CRC cases from the right side (caecum, ascending, and transverse colon) and in 40% from the left side (descending colon and rectum). In all HPV-positive CRCs, there was no expression of tumor suppressor protein (p53) and the retinoblastoma protein (pRB), indicating HPV’s role in tumorigenesis. In terms of the tumor microenvironment, HPV-related cancers exhibited reduced immune surveillance but an enhanced cytotoxic response by lymphocytes. CRC tumors testing positive for HPV have a distinct gut microbiome compared to HPV-negative CRC tumors, with HPV-positive samples showing the absence of certain bacterial species commonly found in CRC. These findings support the carcinogenic role of HPV in CRC and suggest HPV’s influence in modulating the tumor immune microenvironment.85
Hafez et al (2022) investigated the relationship between P16 expression, as an indicator of HPV infection, and CRC in Egyptian patients, along with its association with histopathological characteristics. The study examined fifty-nine cases of colorectal carcinoma and thirty specimens of normal colonic mucosa. P16 (also known as p16INK4a), a tumor suppressor protein was detected in 22% (13 out of 59) of patients with CRC, while no P16 expression was found in all thirty cases of non-neoplastic colonic mucosa. P16 expression was more frequently observed in distal carcinomas. The study concluded that P16 protein is expressed in a significant percentage of CRC cases, suggesting a potential role of HPV in colorectal carcinogenesis. These findings underscore the importance of P16 protein expression in the pathogenesis of colorectal carcinoma, particularly in distal tumors.86
In a recent study, mutated genes that link HPV infection to CRC carcinogenesis have been identified. HPV positivity was observed in 15 (31.9%) cases of CRC patients, with HPV antigens expressed in tumor tissues rather than in adjacent noncancerous tissues. Confirmation tests showed that four differentially expressed genes MMP-7, MYC, WNT-5A, and AXIN2 were upregulated in cancerous tissues compared to adjacent non-cancerous tissues, with MYC, WNT-5A, and AXIN2 also upregulated in HPV-positive CRC tissues compared to HPV-negative tissues. The HPV genome may integrate into tumor genomes, influencing multiple-signaling pathways.87
HPVs are double-stranded DNA viruses without an envelope, capable of infecting epithelial cells in the skin and mucosa. The viral genome includes genes for early (E1-E7) and late (L1 and L2) proteins, essential for infection. HPV integrates its DNA into the host cell genome, disrupting the E2 gene, which normally regulates the oncoproteins E6 and E7. Loss of E2 regulation leads to uncontrolled E6 and E7 expression.88 E6 deactivates p53, disrupting cell cycle control and apoptosis, while E7 deactivates the pRB, increasing p16INK4A levels and inhibiting cyclin-dependent kinases. This leads to the entry into the S phase without undergoing G1 arrest. The combined inactivation of p53 by E6 and pRB by E7 leads to the disruption of various cellular processes, including DNA repair, angiogenesis, and apoptosis, leading to genomic instability, and colorectal cancer development.89,90
Eight studies reported HPV typing in 302 hPV-positive colorectal carcinomas. HPV type 18 was the most commonly identified virus in CRC cases from Asia and Europe, whereas HPV type 16 was more frequently found in colorectal tumors from South America.91 Ibragimova et al (2018) studied the impact of high-risk HPV type 16’s E6/E7 oncoproteins on two human primary normal colorectal mesenchymal cell lines (NCM1 and NCM5). The study found that E6/E7 oncoproteins increased the expression of D-type cyclins, cyclin E, and Id-1, promoting cell proliferation, transformation, and migration.84
Fernandes et al (2020) suggested that the E5 oncoprotein from high-risk HPVs might induce cellular changes and contribute to oncogenesis through interactions with EGF-R1, MAP kinase, PI3K-Akt, and pro-apoptotic proteins.31 Furthermore, the HPV16 E5 oncoprotein directly interferes with the host’s immune response, helping the virus remain undetected by the immune system. This invisibility is critical for the initiation and persistence of the infection and, consequently, for cell transformation and cancer progression. The HPV16 E5 oncoprotein executes significant anti-immune functions, such as downregulating MHC class I and II on the cell surface, affecting the COX-2 pathway, and inhibiting natural killer (NK) cell and interferon activities.32
Epstein–Barr Virus (EBV)
Epstein–Barr Virus (EBV), a double-stranded DNA gamma-herpes virus, is known primarily for causing infectious mononucleosis and being associated with several types of cancers. EBV is composed of two strains: B95-8 (type 1) and AG876 (type 2), also referred to as type A and type B, respectively.92,93 Several studies have established a causative link between EBV and colorectal carcinogenesis through its ability to promote cell proliferation and inhibit apoptosis. Using techniques such as in situ hybridization (ISH), immunohistochemistry (IHC), and PCR-based methods to detect the presence of viral EBV DNA in tumor samples.31
EBV displays a dual tropism, being capable of infecting both B and epithelial cells through the alteration of its envelope proteins.94 Therefore, EBV infection is commonly associated with B-cell lymphomas like Burkitt and Hodgkin lymphoma, as well as epithelial malignancies such as nasopharyngeal, gastric, and potentially rectal carcinomas.94
In a systematic review and meta-analysis by Maskouni et al (2023), the prevalence of EBV infection in CRC patients and any potential association between the two was examined. A total of twenty-three articles were included in the meta-analysis, comprising eight case/control studies and fifteen cross-sectional studies. The combined prevalence of EBV among 1,954 CRC patients was found to be 18%. Regionally, South America had the highest prevalence of EBV at 30%, while Africa had the lowest at 0%. This data suggests that EBV infection is linked to CRC and may be considered a potential risk factor for its development.95
A report by Qualtrough et al (2009), highlighted the involvement of the Fascin gene in the advancement of various cancers, including CRC, as Fascin tends to be overexpressed in numerous types of invasive cancers.96 The presence of EBV in CRC often coincides with heightened Fascin expression.97 On the contrary, Kim et al (2017) opined that colorectal and gastric cancer characterization showed the presence of EBV infection, alongside reports of loss of ARID1A protein expression linked to advanced grade and tumor differentiation.98
During lytic infection, genes encoded by EBV selectively replicate virion components, including viral DNA genomes and proteins. However, in latent infection cycles, EBV encodes viral genes such as the six nuclear antigens (EBNA-1, −2, −3A, −3B, −3C, and LP), three latent membrane proteins (LMP-1, −2A, and −2B), two small non-coding RNAs (EBER-1 and −2), and the BamHI-A rightward transcripts. The primary function of these EBV proteins is to maintain viral existence by evading immune surveillance mechanisms. Additionally, it is now well established that these genes can function as “oncogenes” in infected cells.31
EBV infection can trigger inflammatory pathways and cytokine/chemokine signaling, potentially altering the tumor immune microenvironment (TIM) in CRC.99 It exerts several immune-suppressive effects that facilitate carcinogenic transformation, including silencing the anti-EBV effect of interferon-gamma (IFN-γ) in B cells and modulating the production of antiviral cytokines like TNF-α, IL-1β, and IL-6. Additionally, EBV can mimic the characteristics of IL-10, allowing it to evade the host’s antiviral response. Overall, a compromised immune system and a chronic inflammatory microenvironment enhance the oncogenic properties of EBV.31
John Cunningham Virus (JCV)
JCV is a human polyomavirus, which is epitheliotropic and neurotropic and known to cause severe complications such as progressive multifocal leukoencephalopathy (PML) and has been linked to CRC.100 The strain most frequently detected in CRC tissues is Mad-1.101 Detected in CRC tissues, the viral T-antigen is known to be oncogenic as it can disrupt cell cycle regulation and DNA repair mechanisms, potentially leading to carcinogenesis.102 The JC virus (JCV) T-antigen, a viral oncoprotein, functions by interacting with and deactivating key tumor suppressor proteins such as p53 and pRB, thereby causing cell cycle disruption and cellular transformation.33
Research indicates that JCV infection can heighten the invasion and migration abilities of colorectal cancer cells, thereby enhancing their potential for metastasis. This influence is facilitated by the viral T-antigen and its interactions with host cell signaling pathways involved in cell motility and invasion. Furthermore, JCV T-antigen has been found to prompt chromosomal instability and aneuploidy, characteristic features of cancer that contribute to the accumulation of genetic changes.103 JCV might induce genetic instability by transiently engaging with p53 and β-catenin in colonic cells, resulting in a “hit-and-run” mechanism of carcinogenesis.104
A study was conducted to evaluate the association of JCV DNA in patients with adenocarcinoma of the colon. Among the test group, four out of forty samples (10%) and 10 out of 80 control samples (12.5%) were positive for JCV DNA. The prevalence of JCV DNA was 10% among patients with CRC and 12.5% in benign tumors. The expression of the T-Ag protein could explain the increased risk of CRC.105 In another study conducted in Khartoum State, Sudan, the association of JCV DNA in patients with colorectal cancer was evaluated. Using nested PCR to detect the Vp1/T Ag junction genome in the JCV genome, the results show that three out of 70 (4.2%) samples tested positive for JCV DNA. This finding also suggests that the expression of the T-Ag protein may explain the increased risk of CRC.106
Human Cytomegalovirus (HCMV)
HCMV is a double-stranded DNA virus belonging to the Herpesviridae family There is some evidence suggesting that HCMV infection may be associated with CRC through its effects on the immune system and its ability to induce chronic inflammation.107 HCMV encodes several proteins with oncogenic potential, including immediate-early (IE) and early (E) proteins, which can disrupt cell cycle regulation and apoptosis. These IE/E proteins have been detected in colorectal cancer tissues and may contribute to cancer development by interacting with host cell proteins like p53 and pRB. HCMV infection can lead to genomic instability, chromosomal abnormalities, and an accumulation of mutations in host cells, which could drive the initiation and progression of CRC.34
A recent study aimed to detect the HCMV UL55 gene and immediate early and early (IE/E) proteins in colorectal tumor tissues and adjacent non-neoplastic tissues (ANNT). It also aimed to correlate the presence of HCMV with clinicopathological features of CRC. The study enrolled 50 hCMV seropositive patients with resectable CRC. The study concluded that CRC tumor tissues are more infected by HCMV than ANNT, and there is a significant association of HCMV presence with higher CRC tumor stage and nodal involvement, particularly in an age-dependent manner, suggesting a potential oncomodulatory and disease progression role of HCMV.35
The relationship between HCMV infection and the expression of inflammatory enzymes like 5 lipoxygenase (5 LO) and cyclooxygenase 2 (COX 2), along with other molecular, genetic, and clinicopathological features of CRC was explored in a recent study. The study involved analyzing 146 individual paraffin-embedded CRC tissue microarray (TMA) cores previously characterized for TP53 and KRAS mutations, microsatellite instability (MSI) status, Ki 67 index, and EGFR through immunohistochemistry (IHC). Results showed that approximately 90% of the CRC cases tested positive for HCMV IE and pp65 proteins, which correlated with the expression of COX 2, 5 LO, and KI 67 but not with EGFR immunostaining, TP53 and KRAS mutations, or MSI status. In vitro experiments demonstrated that HCMV infection increased the expression of 5 LO and COX 2 transcripts and proteins in both Caco2 and LS 174T cells, leading to enhanced cell proliferation. Treatment with GCV and CCX significantly reduced the transcript levels of COX 2, 5 LO, HCMV IE, and pp65 in infected cells. The widespread presence of HCMV in CRC tissues suggests its potential role in promoting inflammation and presents itself as a novel target for CRC therapy.36
Chelbi et al (2021) conducted a study to investigate the potential associations between CRC progression and the presence of HCMV. Using Nested PCR to validate the association between HCMV and CRC, and enzyme-linked immunosorbent assay to detect HCMV-specific serum IgG and IgM antibodies. The study analyzed forty tumor and thirty-five peri-tumor tissues, along with blood samples from 100 CRC patients and 140 healthy subjects. Additionally, serum samples from 80 CRC patients and one hundred healthy individuals were examined. Results indicated a significant presence of active HCMV in 93% of CRC patients compared to 60% in the control group. CRC patients also showed significantly higher titers of IgG and IgM anti-CMV antibodies compared to healthy subjects. In conclusion, the study confirmed a relationship between HCMV infection and CRC development, suggesting that CRC cells may be more susceptible to HCMV infection.108
HCMV leverages multiple mechanisms to facilitate the initiation, progression, and metastasis of CRC by directly affecting tumor cell behavior and altering the tumor microenvironment.36 HCMV infection can exert onco-modulatory effects by influencing cellular signaling pathways, microenvironmental factors and transcription factors, thereby promoting malignant transformation and tumor advancement.109 The viral chemokine receptor US28, encoded by HCMV, has been implicated in enhancing tumor migration, invasion and growth in colorectal cancer and enhancing intestinal neoplasia. Additionally, HCMV infection can activate inflammatory pathways and cytokine/chemokine signaling (eg, IL-6, IL-17), modifying the tumor immune microenvironment and potentially aiding in tumor progression and metastasis.36
Four commonly used HCMV strains—AD169, Towne, TB40E, and VR1814—cause replication stress in host cells. This is evident from irregularities in DNA replication and the formation of DNA damage markers (53BP1 foci). This replication stress leads to a host DNA damage response and chromosomal instability in both cells that support viral replication (permissive) and those that do not (non-permissive). This is particularly significant for CRC development, as non-permissive cells can continue to survive and grow even after being infected by HCMV.110
Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV)
While primarily associated with liver cancer, chronic infections with HBV and HCV can lead to systemic inflammation and immune suppression, which may indirectly increase the risk of colorectal cancer. The presence of these viruses in colorectal tumors suggests a potential role in cancer development, although the exact mechanisms and extent of their involvement are still being studied.111
A population-based case-control study was conducted in Taiwan to investigate the potential link between chronic hepatitis B virus (HBV) infections and CRC risk. Using data from the National Health Insurance Research Database of Taiwan, 69,478 newly diagnosed CRC patients between 2005 and 2011 were identified. Additionally, 69,478 age- and gender-matched individuals without CRC were randomly selected as controls from the same database. Logistic regression analysis was employed to calculate odds ratios (ORs) and assess the association between chronic HBV infection and CRC risk. The findings revealed that HBV infection was indeed associated with an increased risk of CRC. This association remained consistent across both genders. Further analysis based on age groups indicated that the CRC risk attributed to HBV decreased with increasing age. In summary, the study suggests a significant association between chronic HBV infection and an elevated risk of CRC. It emphasizes the importance of monitoring CRC development risk, especially in young patients with HBV infection.112
The potential link between hepatitis B virus (HBV) and hepatitis C virus (HCV) infections with the risk of colorectal neoplasia (CRN) was investigated. A cross-sectional study was conducted on asymptomatic individuals who underwent both colonoscopy and serologic testing for HBV surface antigen (HBsAg) and HCV antibody (HCV Ab) from 2004 to 2015. Among 155,674 participants tested for HBsAg, 5476 (3.5%) were positive. The average age of the participants was 41.1 ± 9.1 years. CRN prevalence was higher in HBsAg-positive participants compared to negative ones (16.9% vs 15.6%). Even after adjusting for confounding factors, HBsAg positivity was associated with an increased CRN risk. Out of 155,180 participants tested for HCV Ab, only 240 (0.15%) were positive. CRN prevalence was higher in HCV Ab-positive participants (22.9% vs 15.6%), but this association disappeared after adjusting for confounders. In conclusion, HBV infection was independently linked to an elevated risk of CRN, suggesting a potential contribution to colorectal carcinogenesis.113
In another case-control study, the association between chronic HCV infection and the risk of CRC was examined. Newly diagnosed CRC cases (N = 67,670) between 2005 and 2011 were identified and 67,670 controls were randomly selected without HCV or CRC from the same database, matching them by age and sex of the cases. Results from logistic regression analysis showed that the adjusted odds ratio (aOR) for CRC was 1.16 in relation to chronic HCV infection. The findings indicate that individuals with chronic HCV infection are at a heightened risk of developing CRC.114
HBV proteins like HBx can dysregulate cellular signaling pathways involved in cell proliferation, survival, and oncogenesis, such as Wnt, p53, and PI3K/Akt pathways. This may contribute to the initiation and progression of CRC.37 Integration of HBV DNA into the host genome can cause instability of the genomic system, insertional mutagenesis of cancer-genes, and chromosomal abnormalities, which is the hallmarks of cancer.115
Chronic HBV infection leads to immune dysregulation and persistent inflammation, creating a tumor-promoting microenvironment conducive to colorectal carcinogenesis. HBV DNA and proteins have been detected in extrahepatic tissues like the colorectum, suggesting a potential direct role in colorectal carcinogenesis. On the other hand, the mechanisms by which HCV may contribute to CRC are less clear, but it has been shown that chronic HCV infection can lead to persistent inflammation, which may promote colorectal carcinogenesis.116
HCV RNA and proteins have been detected in extrahepatic tissues, including the colorectum, indicating a potential direct role in CRC development. However, the evidence for a direct association between HCV and CRC has not been shown, with some studies failing to find a significant link after adjusting for confounding factors. Hence, while the mechanisms are not fully elucidated, HBV appears to have a stronger association with increased CRC risk compared to HCV, potentially through direct and indirect effects on cellular processes, inflammation, genomic instability, and immune dysregulation.113
Others
In addition to bacteria and viruses’ other microbes like fungi and protozoa can potentially influence the risk of CRC through various mechanisms such as chronic inflammation, immune modulation, and direct tissue damage.117
A meta-analysis was conducted to explore the role of the fungal mycobiota in CRC. Researchers examined fecal metagenomic datasets from seven previous studies, supplemented by an in-house cohort, totaling 1329 metagenomes (454 with CRC, 350 with adenoma, and 525 healthy individuals). The multicohort analysis identified fungi linked to CRC or adenoma across multiple cohorts. Key CRC-associated fungi included six enriched species (Aspergillus rambellii, Cordyceps sp. RAO-2017, Erysiphe pulchra, Moniliophthora perniciosa, Sphaerulina musiva, and Phytophthora capsici) and one depleted species (Aspergillus kawachii). The interactions among these fungi were more pronounced in CRC compared to adenoma and healthy individuals. Additionally, there was a significant trans-kingdom interaction between Fusobacterium nucleatum and Aspergillus rambellii, which promoted CRC cell growth in vitro and tumor growth in xenograft mice. Combined fungal and bacterial biomarkers were found to be more accurate than those with only bacterial species in distinguishing CRC patients from healthy individuals.118
Candida albicans is not only a common commensal organism in the vaginal and gastrointestinal tract (GIT) of humans but also a significant cause of infections globally, earning it the classification of an opportunistic pathogen. C. albicans can lead to superficial infections as well as more severe, often life-threatening systemic infections. These severe infections typically occur when the microbiota is disrupted, and immune defenses are weakened, allowing the fungus to spread from commensal pools, especially in the GIT, to vital organs. Thus, gastrointestinal colonization by C. albicans is a predisposing factor for serious infections.119
A study involving fifty-two patients newly diagnosed with adenoma/CRC and fifty-two age-matched controls was conducted to explore the association between C. albicans cultured from rectal swabs and newly diagnosed CRC. A notable and significant overrepresentation of C. albicans was found in CRC cases. This evidence suggests that the presence of C. albicans in the gut may induce or facilitate the development of some sporadic CRC cases.120
Entamoeba histolytica infection is characterized by the adherence of trophozoites to colonic epithelial cells through a specific galactose-N-acetylgalactosamine lectin.121 This direct adherence results in cytolysis and apoptosis of the colonic epithelial cells, leading to the release of IL-1α and precursor IL-1β. Activated IL-1β subsequently activates NF-κB in distal cells, inducing the production of cytokines and other inflammatory mediators, including COX-2, IL-1, and IL-8.122 The chronic inflammation and immune response elicited by E. histolytica infection create a microenvironment that can contribute to CRC development. Persistent inflammation, characterized by the continuous presence of cytokines and inflammatory mediators, leads to DNA damage, mutations, and the dysregulation of oncogenes and tumor suppressor genes. The production of COX-2, IL-1, and IL-8, as well as the presence of TNF-α, are critical factors in this process, as they are known to be involved in carcinogenesis.123
Haghighi et al (2024) conducted a study to detect Eh-lectin (a specific lectin from the organism E. histolytica), miRNA-643, XIAP (a gene involved in inhibiting apoptosis), and microsatellite instability (MSI) in 150 CRC biopsy samples using immunohistochemistry, Multiplex PCR, RT-qPCR, and Real-Time PCR. The result showed that of the 150 CRC biopsy samples analyzed, 39 (28 MSI-H and 11 MSI-L) exhibited MSI, while the remaining 111 were MSI-negative. Notably, co-occurrence of MSI and E. histolytica antigen was observed in eleven samples, which also showed increased miRNA-643 expression and decreased XIAP expression. This strongly supports the hypothesis that this protozoan parasite plays a role in MSI development through its potential involvement in apoptosis.124
Mechanism of Action of a Microbial Antigen-Derived CRC Vaccine
A microbial antigen-derived CRC vaccine is one in which epitopes are selected from highly antigenic, immunodominant proteins sourced from microbes. These antigens are capable of triggering responses from both T cells and B cells to recognize and attack cancer cells to combat cancer cells.125 (Figure 4).
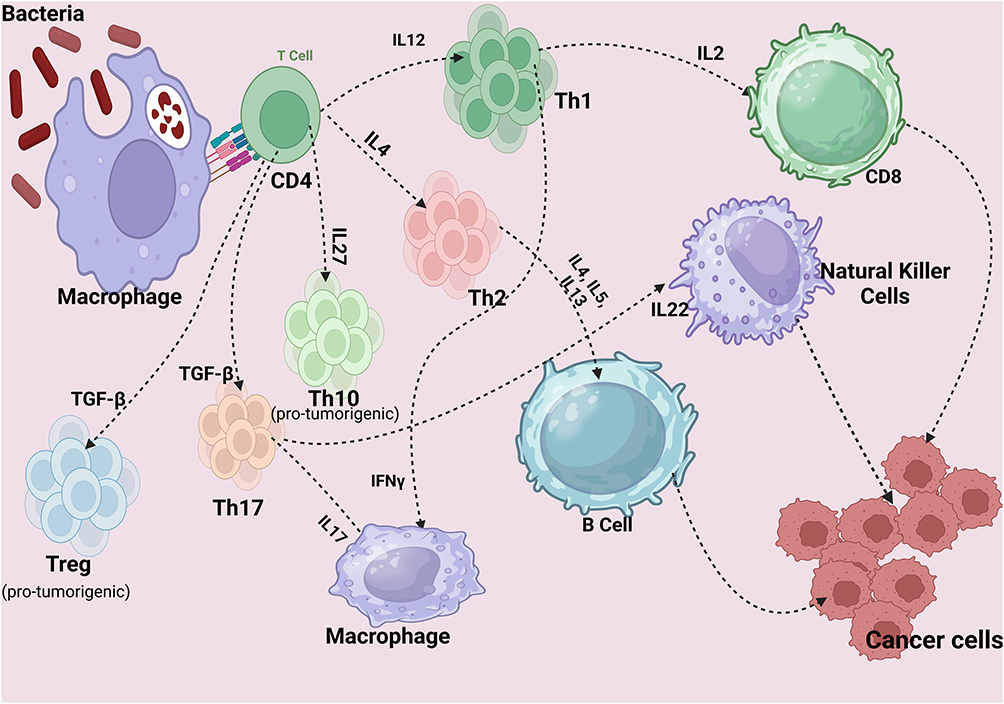 |
Figure 4 Antitumor Immunity. Created in BioRender. Rowaiye, A. (2024) https://BioRender.com/s21s679. |
APCs in peripheral tissues, such as the skin and mucosal layers, recognize pathogen-associated molecular patterns (PAMPs) on microbes through pattern recognition receptors (PRRs) like Toll-like receptors (TLRs). This engagement triggers the internalization of the antigen via endocytosis or phagocytosis. The APC process and display peptide fragments on their surfaces with major histocompatibility complex (MHC) molecules, effectively flagging the immune system to respond. Extracellular bacterial antigens primarily activate CD4+ T-cells through MHC class II presentation. These helper T-cells then release cytokines, which enhance immune responses by activating other immune cells, including B-cells and cytotoxic T-cells.11,126
When a naive CD4+ T-cell encounters an APC displaying an antigen on an MHC class II molecule that matches its T-cell receptor (TCR), it binds to this antigen-MHC complex. This initial binding, called “Signal 1”, is insufficient on its own to fully activate the CD4+ T-cell. Therefore, a second signal, known as the Co-stimulatory Signal or “Signal 2”, is required. Signal 2 is initiated when co-stimulatory molecules on the APC (such as CD80 or CD86) interact with specific receptors on the T-cell (like CD28). This co-stimulatory interaction provides confirmation that the antigen is foreign, not self, which helps prevent autoimmunity.127
Following T-cell activation, cytokine signaling is initiated. Cytokine signaling promotes T cell proliferation, differentiation into effector cells, and expression of activation markers. Inducer cytokines such as Interleukin-12 (IL-12) promotes differentiation into a Th1 cell, IL-4 promotes Th2 cell differentiation, and TGF-β promote Th17 and Treg cells. Upon full activation, the CD4+ T-cell transitions from a naive to an active helper T-cell, expressing genes that enable it to divide, secrete cytokines, and adopt a specialized helper role in the immune response. During clonal expansion, the activated CD4+ T-cell divides repeatedly, creating many identical daughter cells with the same TCR specific for the antigen.128,129
The differentiation pathway of the CD4+ T-cell (Th1, Th2, Th10, Th17, or Treg) depends on the cytokine environment. Each type of helper T-cell has distinct functions and cytokine profiles that tailor the immune response to specific pathogens or immune challenges. These effector cells in turn produce effector cytokines that can activate other immune cells, such as macrophages, B-cells, and cytotoxic T-cells. For example, Th1 cells produce interferon-gamma (IFN-γ) to enhance macrophage activity and IL-2, which play a central role in the activation and proliferation of CD8+ cytotoxic T-cells. Th2 cells produce IL-4, IL-5, and IL-13 to help B-cells produce antibodies. Th17 produces IL-17A, IL-17F and IL22 that activate macrophages and natural killer cells, and Treg cells produce IL-10. Th10 produce IL10 and have immunosuppressive activity like Treg.129,130
Of key importance in antitumor immunity are the cytotoxic CD8+ T and B-cells, which play complimentary roles each contributing to the detection, targeting, and elimination of cancer cells through specialized mechanisms.131,132
CD8+ T-cells, also known as cytotoxic T lymphocytes (CTLs), play an essential role in the body’s antitumor immunity by directly targeting and eliminating tumor cells. Their ability to identify, attack, and destroy cancerous cells relies on a series of well-coordinated steps that enhance their effectiveness in combating tumor growth and providing lasting immunity. The initial step in CD8+ T-cell antitumor activity is the recognition of tumor antigens. CD8+ T-cells are equipped to recognize specific antigens displayed by MHC class I molecules on the surface of tumor cells. These antigens may consist of mutated self-proteins, cancer-testis antigens, or viral proteins in cases where cancers are virus-induced. This recognition is critical because it enables CD8+ T-cells to distinguish tumor cells from normal cells, allowing them to selectively target and attack cells that express these specific tumor-associated antigens.131
Once a CD8+ T-cell recognizes a tumor antigen, it undergoes activation and clonal expansion. Activation requires two signals: the first is “Signal 1”, which involves antigen recognition, and the second is “Signal 2”, which is a co-stimulatory signal typically provided by professional APCs, such as dendritic cells. When both signals are received, CD8+ T-cells become fully activated and begin to proliferate, producing a large population of identical, tumor-specific T-cells. This clonal expansion ensures that the immune response is robust, with enough CD8+ T-cells available to tackle the tumor effectively.133
Following activation and expansion, CD8+ T-cells employ multiple cytotoxic mechanisms to eliminate tumor cells. One key mechanism is the perforin and granzyme pathway. CD8+ T-cells release perforin, a protein that forms pores in the tumor cell membrane, creating an entry point for granzymes. Granzymes are enzymes that penetrate through these pores and induce apoptosis, or programmed cell death, within the tumor cell.134 Another mechanism used by CD8+ T-cells is the Fas/FasL pathway, wherein the CD8+ T-cell expresses Fas ligand (FasL), which binds to Fas receptors on the tumor cell surface. This interaction also triggers apoptosis, leading to the death of the tumor cell.135 Additionally, CD8+ T-cells secrete IFN-γ, a cytokine with dual roles in antitumor immunity. IFN-γ can directly inhibit tumor cell growth, and it also enhances MHC class I expression on tumor cells, making them more visible and susceptible to CD8+ T-cell recognition and attack.136
An important feature of CD8+ T-cell-mediated immunity is the formation of memory cells after tumor clearance. Once the immediate tumor threat is eliminated, some CD8+ T-cells transition into memory T-cells. These memory cells persist in the body, providing long-lasting immunity. If the same tumor re-emerges, memory T-cells can mount a rapid and efficient response, often preventing tumor recurrence or significantly reducing its progression.137
The CD8+ T-cells serve as a critical component of the immune system’s response to tumors through a multifaceted approach that includes recognition of tumor antigens, activation and clonal expansion, direct cytotoxic mechanisms, and memory formation. This combination of targeted killing and lasting immunity makes CD8+ T-cells indispensable in the body’s natural defense against cancer, and understanding their function provides valuable insights for developing immunotherapies to harness and enhance these cells’ tumor-fighting abilities.131
B-cells play a vital role in antitumor immunity, primarily through producing tumor-specific antibodies and modulating immune responses. Their actions complement those of other immune cells, such as CD8+ T-cells, to generate a multifaceted and effective response against tumor cells. B-cells contribute to antitumor immunity through several mechanisms, including antibody production, antigen presentation, cytokine release, and the formation of tertiary lymphoid structures (TLS).132,138
The production of antibodies is one of the primary ways B-cells participate in antitumor immunity. When B-cells recognize tumor-associated antigens, they can differentiate into plasma cells, which produce antibodies specifically tailored to these antigens. These tumor-specific antibodies can then bind to antigens present on the surface of tumor cells, marking them for destruction.139
This process enables mechanisms such as antibody-dependent cellular cytotoxicity (ADCC), where natural killer (NK) cells recognize the Fc region of these antibodies through their Fc receptors. This recognition triggers ADCC, leading to the death of the tumor cell.140 Additionally, antibodies produced by B-cells can activate the complement system, a series of proteins in the blood that contribute to immune responses. When activated, the complement system forms a membrane attack complex on the surface of tumor cells, directly lysing and destroying them.141
Beyond antibody production, B-cells also act as APCs, an essential function in antitumor immunity. B-cells can capture tumor antigens and present them to CD4+ T-cells through MHC class II molecules. This antigen presentation is crucial for the activation of CD4+ helper T-cells, which play a supportive role in enhancing the functions of other immune cells, including CD8+ T-cells and additional B-cells. By presenting antigens and promoting helper T-cell activation, B-cells indirectly strengthen the immune response against tumor cells, making it more effective and sustained.139
B-cells further influence the immune response through cytokine production, which helps modulate the tumor microenvironment. B-cells secrete various cytokines that regulate the recruitment, survival, and function of different immune cells. By releasing pro-inflammatory cytokines, B-cells can enhance the immune response against tumors, increasing the presence and activity of cells that target tumor cells.142
In some tumors, B-cells aggregate to form structures resembling lymph nodes, known as tertiary lymphoid structures (TLS). TLS can serve as localized sites for B-cell activation, antigen presentation, and T-cell priming, creating a microenvironment within the tumor that strengthens the immune response. This concentrated activity of immune cells within TLS enhances the body’s ability to recognize and attack tumor cells at a local level, contributing to more robust and sustained antitumor immunity.143
The interplay between CD8+ T-cells and B-cells highlights the synergistic nature of the immune response in targeting tumors. B-cell-produced antibodies enhance CD8+ T-cell activity by increasing the visibility of tumor cells through ADCC and complement activation. In return, CD8+ T-cell-released IFN-γ can stimulate B-cells, encouraging them to produce more antibodies and further bolster the immune response. This collaboration between CD8+ T-cells and B-cells enables the immune system to attack tumors from multiple angles, leading to an effective and comprehensive defense against cancer.125
Future Perspectives and Conclusion on Microbiome-Based Vaccine Antigen Discovery in Colorectal Cancer
The field of microbiome research, particularly as it relates to CRC, is poised to benefit greatly from the continued advancement of metagenomic and bioinformatics technologies. Such tools will enable the precise and high-throughput identification of microbial antigens that may play a role in CRC. A major focus of future research will be on systematically cataloging and characterizing these antigens to identify those with the highest potential for stimulating immune responses that can combat or prevent tumor growth. In particular, the integration of single-cell sequencing and spatial transcriptomics could further elucidate the localization and activity of microbial antigens within CRC tissues, shedding light on which microbial profiles correlate most strongly with cancerous or pre-cancerous states.
Another essential direction will be to deepen our understanding of the dynamic interactions between host immune responses and microbial communities within the tumor microenvironment. Microbial antigens do not function in isolation; instead, their immunogenic potential is influenced by factors such as inflammation, immune evasion tactics of the tumor, and host microbiome variability. Investigating these complex host–microbe interactions at cellular and molecular levels, using models that replicate human immune responses, will be crucial. This knowledge will refine antigen selection, guiding researchers toward those microbial markers that are not only immunogenic but can also overcome the immunosuppressive environment characteristic of CRC.
Clinical development of microbiome-based vaccines will require a shift from preclinical findings to human trials. While animal models and organoid studies are essential first steps, the transition to rigorous, phased human trials will be necessary to validate the efficacy, safety, and long-term effects of these candidate vaccines. Additionally, clinical trials will need to assess how variations in the microbiome between individuals may impact vaccine efficacy, underscoring the importance of personalized approaches.
The concept of personalized CRC vaccines tailored to an individual’s unique microbiome composition is particularly promising. The potential to develop custom vaccine formulations that target microbial antigens specific to each patient’s CRC-associated microbiota could lead to highly targeted and effective prophylactic or therapeutic interventions. Advances in bioinformatics, such as machine learning algorithms trained to analyze individual microbiome data, could enable the selection of antigens that are both highly immunogenic and unique to the patient’s microbiome profile.
Put together, the future of CRC vaccine development lies in the convergence of advanced metagenomic technologies, sophisticated understanding of host–microbe interactions, and personalized immunotherapy approaches. Such progress could redefine CRC prevention and treatment, offering patients tailored, microbiome-specific immune interventions with the potential to transform CRC care on both individual and population-wide scales.
Given the role of microbial antigens in influencing the tumor microenvironment and immune response, CRC vaccine candidates would be developed from microbial peptides that are antigenic, non-toxic, non-allergenic, and capable of activating both T and B cell responses. To boost immunogenicity, linker and adjuvant sequences should be incorporated into these peptide constructs. This multi-antigenic vaccine design would then undergo extensive preclinical and clinical testing to confirm its efficacy and safety.
In conclusion, the identification of microbial antigens associated with colorectal cancer offers a novel and promising approach for vaccine development. The significant overrepresentation of specific bacteria and viruses in CRC tissues, coupled with their roles in promoting tumorigenesis through inflammation and immune modulation, positions these microbes as key targets for antigen selection. Future research should focus on the validation of these antigens through comprehensive metagenomic analyses, in vitro assays, and animal models to pave the way for the development of effective prophylactic or therapeutic CRC vaccines. Such advancements could revolutionize CRC prevention and treatment, leveraging the body’s immune system to combat one of the most prevalent cancers worldwide.
Abbreviations
18F-FDG PET/CT, 8F-fluorodeoxyglucose positron emission tomography-computed tomography; 5 LO, Arachidonate 5-lipoxygenase; AI-2, Autoinducer-2; AKT, protein kinase B; AMER, APC Membrane Recruitment Protein 1; ANNT, Adjacent non-neoplastic tissues; AP-PCR, Arbitrarily primed PCR; ARID1A, AT-rich interaction domain 1A; ATM, Ataxia-telangiectasia mutated; AUC, Area Under the Curve; BCL2, B cell leukemia/lymphoma 2; BFT, B. fragilis toxin; CadF, Campylobacter adhesion factor; CagA, Cytotoxin-associated gene A; CDT, Cytolethal distending toxin.; CIMP, CpG island methylator phenotype; CoPEC, colibactin-producing E. coli; COX-2, Cyclooxygenase; CTNNB1, Catenin Beta 1; DNA, Deoxyribonucleic acid; EBV, Epstein–Barr virus; EGFR, epidermal growth factor receptor; EHEC, Enterohemorrhagic Escherichia coli; ELISA, Enzyme-linked immunosorbent assay; EPEC, Enteropathogenic E. coli; ETBF, enterotoxigenic B. fragilis; FadA, Fusobacterium adhesin A; Fap2, Fibroblast Activation Protein-2; FFPE, formalin-fixed and paraffin-embedded; FlaB, Flagellar adhesin B; Fsr, Fosmidomycin resistance; GCV, Ganciclovir; GelE, gelatinase; GF, Germ-free; GIT, Gastrointestinal tract; HBsAg, HBV surface antigen; HBV, Hepatitis B virus; HBx, HBV-encoded oncogene X protein; HCMV, Human cytomegalovirus; HCV, Hepatitis C virus; HPV, Human Papillomavirus; IbeA, Invasion of brain endothelium protein A; IFN-γ, interferon-gamma; Ig, Immunoglobulin; IHC, immunohistochemistry; IL, Interleukin; ISH, In situ hybridization; JCV, John Cunningham Virus; LMP, Latent membrane proteins; miRNA, MicroRNA; MSI, Microsatellite instability; mTOR, mammalian target of rapamycin; MYD88, myeloid differentiation primary response gene 88; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; NTBF, non-toxigenic B. fragilis; OR, odds ratios; PCR, Polymerase Chain Reaction; PGE2, Prostaglandin E2; PI3K, Phosphoinositide 3-kinase; pks+, polyketide synthesis positive; Progressive multifocal leukoencephalopathy (PML); RAS/MAPK, rat sarcoma/mitogen-activated protein kinase; RNA, Ribonucleic acid; ROC, Receiver operating characteristic; rRNA, ribosomal RNA; RT-qPCR, reverse transcriptase-Quantitative-PCR; SGG, Streptococcus gallolyticus subspecies gallolyticus; SMO, Smoothened; SprE, serine protease; STAT, signal transducers and activators of transcription; STEC, Shiga toxin-producing E. coli; TGF-β pathway,; TGF-β, transforming growth factor-B; TIM, Tumor immune microenvironment; TLR, Toll-like receptor; TMA, Tissue microarray; TNF-α, Tumour Necrosis Factor alpha; TP53, Tumor protein p53; Tregs, regulatory T-cells.; US28, Human cytomegalovirus G protein-coupled receptor; VacA, Vacuolating cytotoxin A; VEGFA, Vascular endothelial growth factor A; Wnt, Wingless-related integration site; XIAP, X-Linked Inhibitor of Apoptosis.
Consent for Publication
The images featured were generated using BioRender.com under the terms of their licensing agreement.
Author Contributions
All authors played a significant role in the work presented, whether through conceptualization, study design, execution, data acquisition, analysis, interpretation, or a combination of these areas. They actively participated in drafting, revising, or critically reviewing the manuscript, granted final approval for the version submitted for publication, consented to the choice of journal, and accept responsibility for all aspects of the work.
Disclosure
The authors declare that there are no financial or non-financial competing interests.
References
1. Hashemi Goradel N, Heidarzadeh S, Jahangiri S. et al. Fusobacterium nucleatum and colorectal cancer: a mechanistic overview. J Cell Physiol. 2019;234(3):2337–2344. doi:10.1002/jcp.27250
2. Cancer trends: colorectal cancer statistics [Internet]. World Cancer Research Fund; 2023. Available from: https://www.wcrf.org/cancer-trends/colorectal-cancer-statistics/.
3. Hossain MS, Karuniawati H, Jairoun AA, et al. Colorectal cancer: a review of carcinogenesis, global epidemiology, current challenges, risk factors, preventive and treatment strategies. Cancers. 2022;14(7):1732. doi:10.3390/cancers14071732
4. Sharma VR, Singh M, Kumar V, et al. Microbiome dysbiosis in cancer: exploring therapeutic strategies to counter the disease. Semin Cancer Biol. 2021;70:61–70. doi:10.1016/j.semcancer.2020.07.006
5. Sánchez-Alcoholado L, Ramos-Molina B, Otero A, et al. The role of the gut microbiome in colorectal cancer development and therapy response. Cancers. 2018;12(6):1406. doi:10.3390/cancers12061406
6. Gagnière J, Raisch J, Veziant J, et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol. 2016; 22(2):501. doi:10.3748/wjg.v22.i2.501
7. Siddiqui R, Boghossian A, Alharbi AM, Alfahemi H, Khan NA. The pivotal role of the gut microbiome in colorectal cancer. Biology. 2022;11(11):1642. doi:10.3390/biology11111642
8. Lucas C, Barnich N, Nguyen HT. Microbiota, inflammation and colorectal cancer. Int J Mol Sci. 2017; 18(6):1310. doi:10.3390/ijms18061310
9. Veziant J, Villéger R, Barnich N, Bonnet M. Gut microbiota as potential biomarker and/or therapeutic target to improve the management of cancer: focus on colibactin-producing Escherichia coli in colorectal cancer. Cancers. 2021;13(9):2215. doi:10.3390/cancers13092215
10. Yu I, Wu R, Tokumaru Y, Terracina KP, Takabe K. The role of the microbiome on the pathogenesis and treatment of colorectal cancer. Cancers. 2022;14(22):5685. doi:10.3390/cancers14225685
11. Gaudino SJ, Kumar P. Cross-talk between antigen presenting cells and T cells impacts intestinal homeostasis, bacterial infections, and tumorigenesis. Front Immunol. 2019;10:360. doi:10.3389/fimmu.2019.00360
12. Li S, Liu J, Zheng X, et al. Tumorigenic bacteria in colorectal cancer: mechanisms and treatments. Cancer Biol Med. 2021;19(2):147–162. doi:10.20892/j.issn.2095-3941.2020.0651
13. Zhang L, Liu J, Deng M, et al. Enterococcus faecalis promotes the progression of colorectal cancer via its metabolite: biliverdin. J Transl Med. 2023;21(1):72. doi:10.1186/s12967-023-03929-7
14. Pang X, Tang YJ, Ren XH, Chen QM, Tang YL, Liang XH. Microbiota, epithelium, inflammation, and TGF-β signaling: an intricate interaction in oncogenesis. Front Microbiol. 2018;9:367461. doi:10.3389/fmicb.2018.01353
15. Guo P, Tian Z, Kong X, et al. FadA promotes DNA damage and progression of Fusobacterium nucleatum-induced colorectal cancer through up-regulation of chk2. J Exp Clin Cancer Res. 2020;39:1–13. doi:10.1186/s13046-020-01677-w
16. Wu J, Li K, Peng W, et al. Autoinducer-2 of Fusobacterium nucleatum promotes macrophage M1 polarization via TNFSF9/IL-1β signaling. Int Immunopharmacol. 2019;74:105724. doi:10.1016/j.intimp.2019.105724
17. Pignatelli P, Nuccio F, Piattelli A, Curia MC. The role of Fusobacterium nucleatum in oral and colorectal carcinogenesis. Microorganisms. 2023;11(9):2358. doi:10.3390/microorganisms11092358
18. Abed J, Emgård JE, Zamir G, et al. Fap2 mediates Fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed Gal-GalNAc. Cell Host Microbe. 2016;20(2):215–225. doi:10.1016/j.chom.2016.07.006
19. Nobbs AH, Jenkinson HF, Everett DB. Generic determinants of Streptococcus colonization and infection. Infect Genet Evol. 2015;33:361–370. doi:10.1016/j.meegid.2014.09.018
20. Pardo CP, Gonzalez RC, Pinto MES, Galiana A. Streptococcus gallolyticus and its implication in colorectal cancer. In: Colorectal Neoplasia and the Colorectal Microbiome. Academic Press; 2020:35–55.
21. Iyadorai T, Mariappan V, Vellasamy KM, et al. Prevalence and association of pks+ Escherichia coli with colorectal cancer in patients at the University Malaya Medical Centre, Malaysia. PLoS One. 2020;15(1):e0228217. doi:10.1371/journal.pone.0228217
22. Taieb F, Nougayrède JP, Watrin C, Samba‐Louaka A, Oswald E. Escherichia coli cyclomodulin Cif induces G2 arrest of the host cell cycle without activation of the DNA‐damage checkpoint‐signalling pathway. Cellular Microbiology. 2006;8(12):1910–1921. doi:10.1111/j.1462-5822.2006.00757.x
23. Matsumiya Y, Suenaga M, Ishikawa T, et al. Clinical significance of Bacteroides fragilis as a potential prognostic factor in colorectal cancer. Anaerobe. 2023;84:102784. doi:10.1016/j.anaerobe.2023.102784
24. Lai YR, Chang YF, Ma J, Chiu CH, Kuo ML, Lai CH. From DNA damage to cancer progression: potential effects of cytolethal distending toxin. Front Immunol. 2021;12:760451. doi:10.3389/fimmu.2021.760451
25. Silbergleit M, Vasquez AA, Miller CJ, Sun J, Kato I. Oral and intestinal bacterial exotoxins: potential linked to carcinogenesis. Prog Mol Biol Transl Sci. 2020;171:131–193. doi:10.1016/bs.pmbts.2020.02.004
26. Yekani M, Baghi HB, Naghili B, Vahed SZ, Sóki J, Memar MY. To resist and persist: important factors in the pathogenesis of Bacteroides fragilis. Microb Pathog. 2020;149:104506. doi:10.1016/j.micpath.2020.104506
27. Butt J, Varga MG, Blot WJ, et al. Serologic response to Helicobacter pylori proteins associated with risk of colorectal cancer among diverse populations in the United States. Gastroenterology. 2019;156(1):175–186. doi:10.1053/j.gastro.2018.09.054
28. Venkateswaran P, Lakshmanan PM, Muthukrishnan S, et al. Hidden agenda of Enterococcus faecalis lifestyle transition: planktonic to sessile state. Future Microbiol. 2022;17(13):1051–1069.
29. He Z, Gharaibeh RZ, Newsome RC, et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. 2019;68(2):289–300. doi:10.1136/gutjnl-2018-317200
30. Lopes GV, Ramires T, Kleinubing NR, Scheik LK, Fiorentini ÂM, da Silva WP. Virulence factors of foodborne pathogen Campylobacter jejuni. Microb Pathog. 2021;161:105265. doi:10.1016/j.micpath.2021.105265
31. Fernandes Q, Gupta I, Vranic S, Al Moustafa AE. Human papillomaviruses and Epstein–Barr virus interactions in colorectal cancer: a brief review. Pathogens. 2020;9(4):300. doi:10.3390/pathogens9040300
32. de Freitas AC, de Oliveira TH, Barros MR, Venuti A. hrHPV E5 oncoprotein: immune evasion and related immunotherapies. J Exp Clin Cancer Res. 2017;36:1–15. doi:10.1186/s13046-017-0541-1
33. Matalka I, Swedan S, Khabaz MN, Barahmeh M. JC virus in colorectal cancer: where do we stand? Future Virol. 2013;8(6):607–615. doi:10.2217/fvl.13.36
34. Yu C, He S, Zhu W, Ru P, Ge X, Govindasamy K. Human cytomegalovirus in cancer: the mechanism of HCMV-induced carcinogenesis and its therapeutic potential. Front Cell Infect Microbiol. 2023;13:1202138. doi:10.3389/fcimb.2023.1202138
35. Raouf M, Sabry AA, Ragab MA, Achy SE, Amer A. Detection of human cytomegalovirus UL55 gene and IE/E protein expression in colorectal cancer patients in Egypt. BMC Cancer. 2023;23(1):723. doi:10.1186/s12885-023-11200-x
36. Pantalone MR, Martin Almazan N, Lattanzio R, et al. Human cytomegalovirus infection enhances 5 lipoxygenase and cycloxygenase 2 expression in colorectal cancer. Int J Oncol. 2023;63(5):1–17. doi:10.3892/ijo.2023.5564
37. Liu T, Li W, Zhang Y, et al. Associations between hepatitis B virus infection and risk of colorectal cancer: a population-based prospective study. BMC Cancer. 2021;21(1):1–10. doi:10.1186/s12885-021-08846-w
38. Shang FM, Liu HL. Fusobacterium nucleatum and colorectal cancer: a review. World J Gastrointest Oncol. 2018;10(3):71. doi:10.4251/wjgo.v10.i3.71
39. Luo K, Zhang Y, Xv C, et al. Fusobacterium nucleatum, the communication with colorectal cancer. Biomed Pharmacother. 2019;116:108988. doi:10.1016/j.biopha.2019.108988
40. Lee DW, Han SW, Kang JK, et al. Association between Fusobacterium nucleatum, pathway mutation, and patient prognosis in colorectal cancer. Ann Surg Oncol. 2018;25:3389–3395. doi:10.1245/s10434-018-6681-5
41. Sun CH, Li BB, Wang B, et al. The role of Fusobacterium nucleatum in colorectal cancer: from carcinogenesis to clinical management. Chronic Dis Transl Med. 2019;5(3):178–187. doi:10.1016/j.cdtm.2019.09.001
42. Yamaoka Y, Suehiro Y, Hashimoto S, et al. Fusobacterium nucleatum as a prognostic marker of colorectal cancer in a Japanese population. J Gastroenterol. 2018;53:517–524. doi:10.1007/s00535-017-1382-6
43. Komiya Y, Shimomura Y, Higurashi T, et al. Patients with colorectal cancer have identical strains of Fusobacterium nucleatum in their colorectal cancer and oral cavity. Gut. 2019;68(7):1335–1337. doi:10.1136/gutjnl-2018-316661
44. De Carvalho AC, de Mattos Pereira L, Datorre JG, et al. Microbiota profile and impact of Fusobacterium nucleatum in colorectal cancer patients of Barretos Cancer Hospital. Front Oncol. 2019;9:813. doi:10.3389/fonc.2019.00813
45. Wu J, Li Q, Fu X. Fusobacterium nucleatum contributes to the carcinogenesis of colorectal cancer by inducing inflammation and suppressing host immunity. Transl Oncol. 2019;12(6):846–851. doi:10.1016/j.tranon.2019.03.003
46. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14(2):195–206. doi:10.1016/j.chom.2013.07.012
47. Pasquereau-Kotula E, Martins M, Aymeric L, Dramsi S. Significance of Streptococcus gallolyticus subsp. gallolyticus association with colorectal cancer. Front Microbiol. 2018;9:341818. doi:10.3389/fmicb.2018.00614
48. Sheikh AF, Masjedi Zadeh AR, Saki M, et al. Detection of Streptococcus gallolyticus in colorectal cancer and inflammatory bowel disease patients compared to control group in southwest of Iran. Mol Biol Rep. 2020;47(11):8361–8365. doi:10.1007/s11033-020-05807-7
49. Guven DC, Dizdar O, Alp A, et al. Analysis of Fusobacterium nucleatum and Streptococcus gallolyticus in saliva of colorectal cancer patients. Biomarker Med. 2019;13(9):725–735. doi:10.2217/bmm-2019-0020
50. Aymeric L, Donnadieu F, Mulet C, et al. Colorectal cancer specific conditions promote Streptococcus gallolyticus gut colonization. Proc Natl Acad Sci USA. 2018;115(2). doi:10.1073/pnas.1715112115
51. Zhang Y, Weng Y, Gan H, Zhao X, Zhi F. Streptococcus gallolyticus conspires myeloid cells to promote tumorigenesis of inflammatory bowel disease. Biochem Biophys Res Commun. 2018;506(4):907–911. doi:10.1016/j.bbrc.2018.10.136
52. Kumar R, Herold JL, Taylor J, Xu J, Xu Y. Variations among Streptococcus gallolyticus subsp. gallolyticus strains in connection with colorectal cancer. Sci Rep. 2018;8(1):1514. doi:10.1038/s41598-018-19941-7
53. Agnes A, Biondi A, Belia F, et al. Association between colorectal cancer and Streptococcus gallolyticus subsp. pasteuranus endocarditis: clinical relevance and cues for microbiota science. Eur Rev Med Pharmacol Sci. 2021;25(1):480–486. doi:10.26355/eurrev_202101_24417
54. Butt J, Blot WJ, Teras LR, et al. Antibody responses to Streptococcus Gallolyticus subspecies Gallolyticus proteins in a large prospective colorectal cancer cohort consortium. Cancer Epidemiol Biomarkers Prev. 2018;27(10):1186–1194. doi:10.1158/1055-9965.EPI-18-0249
55. Butt J, Jenab M, Willhauck-Fleckenstein M, et al. Prospective evaluation of antibody response to Streptococcus gallolyticus and risk of colorectal cancer. Int, J, Cancer. 2018;143(2):245–252. doi:10.1002/ijc.31283
56. Genua F, Butt J, Waterboer T, Hughes DJ. Association of antibody responses to Fusobacterium nucleatum and Streptococcus gallolyticus proteins with colorectal adenoma and colorectal cancer. Dig Dis Sci. 2023;68(8):3300–3311. doi:10.1007/s10620-023-08001-4
57. Basavaraju M, Gunashree BS. Escherichia coli: an overview of main characteristics. In: Escherichia Coli - Old and New Insights. IntechOpen; 2022.
58. Wassenaar TME. coli and colorectal cancer: a complex relationship that deserves a critical mindset. Crit Rev Microbiol. 2018;44(5):619–632. doi:10.1080/1040841X.2018.1481013
59. Pleguezuelos-Manzano C, Puschhof J, Rosendahl huber A, et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature. 2020;580(7802):269–273. doi:10.1038/s41586-020-2080-8
60. Lopès A, Billard E, Casse AH, et al. Bonnet, Colibactin-positive Escherichia coli induce a procarcinogenic immune environment leading to immunotherapy resistance in colorectal cancer. Int, J, Cancer. 2020;146(11):3147–3159. doi:10.1002/ijc.32920
61. Leibiger K, Schweers JM, Schütz M. Biogenesis and function of the autotransporter adhesins YadA, intimin and invasin. Int J Med Microbiol. 2019;309(5):331–337. doi:10.1016/j.ijmm.2019.05.009
62. Scott N, Whittle E, Jeraldo P, Chia N. A systemic review of the role of enterotoxic Bacteroides fragilis in colorectal cancer. Neoplasia. 2022;29:100797. doi:10.1016/j.neo.2022.100797
63. Oliero M, Hajjar R, Cuisiniere T, et al. Prevalence of pks+ bacteria and enterotoxigenic Bacteroides fragilis in patients with colorectal cancer. Gut Pathog. 2022;14(1):51. doi:10.1186/s13099-022-00523-y
64. Haghi F, Goli E, Mirzaei B, Zeighami H. The association between fecal enterotoxigenic B. fragilis with colorectal cancer. BMC Cancer. 2019;19:1–4. doi:10.1186/s12885-019-6115-1
65. Cheng WT, Kantilal HK, Davamani F. The mechanism of Bacteroides fragilis toxin contributes to colon cancer formation. Malaysian J Med Sci. 2020;27(4):9. doi:10.21315/mjms2020.27.4.2
66. Dadgar-Zankbar L, Shariati A, Bostanghadiri N, et al. Evaluation of enterotoxigenic Bacteroides fragilis correlation with the expression of cellular signaling pathway genes in Iranian patients with colorectal cancer. Infect Agent Cancer. 2023;18(1):48. doi:10.1186/s13027-023-00523-w
67. Purcell RV, Permain J, Keenan JI. Enterotoxigenic Bacteroides fragilis activates IL-8 expression through Stat3 in colorectal cancer cells. Gut Pathog. 2022;14(1):16. doi:10.1186/s13099-022-00489-x
68. Mao LQ, Zhou YL, Wang SS, et al. Impact of Helicobacter pylori eradication on the gastric microbiome. Gut Pathog. 2021;13:1–13. doi:10.1186/s13099-021-00460-2
69. Zuo Y, Jing Z, Bie M, Xu C, Hao X, Wang B. Association between Helicobacter pylori infection and the risk of colorectal cancer: a systematic review and meta-analysis. Medicine. 2020;99(37):e21832. doi:10.1097/MD.0000000000021832
70. Teimoorian F, Ranaei M, Tilaki KH, Shirvani JS, Vosough Z. Association of Helicobacter pylori infection with colon cancer and adenomatous polyps. Iran J Pathol. 2018;13(3):325.
71. Liang CM, Hsu CH, Chung CH, et al. Risk for irritable bowel syndrome in patients with Helicobacter pylori infection: a nationwide population-based cohort study in Taiwan. Int J Environ Res Public Health. 2020;17(10):3737. doi:10.3390/ijerph17103737
72. de Almeida CV, Taddei A, Amedei A. The controversial role of Enterococcus faecalis in colorectal cancer. Ther Adv Gastroenterol. 2018;11:1756284818783606. doi:10.1177/1756284818783606
73. Khodaverdi N, Zeighami H, Jalilvand A, Haghi F, Hesami N. High frequency of enterotoxigenic Bacteroides fragilis and Enterococcus faecalis in the paraffin-embedded tissues of Iranian colorectal cancer patients. BMC Cancer. 2021;21(1):1353. doi:10.1186/s12885-021-09110-x
74. Geravand M, Fallah P, Yaghoobi MH, et al. Investigation of Enterococcus faecalis population in patients with polyp and colorectal cancer in comparison of healthy individuals. Arq Gastroenterol. 2019;56:141–145. doi:10.1590/s0004-2803.201900000-28
75. Dicks LM. How does quorum sensing of intestinal bacteria affect our health and mental status? Microorganisms. 2022;10(10):1969. doi:10.3390/microorganisms10101969
76. Kato I, Minkevitch J, Sun J. Oncogenic potential of Campylobacter infection in the gastrointestinal tract: narrative review. Scand J Gastroenterol. 2023;58(12):1453–1465. doi:10.1080/00365521.2023.2228954
77. Gervaz PA, De Campos Á, Caeiro A. Campylobacter jejuni causes colorectal cancer. World J Colorectal Surg. 2022;11(1):4–7. doi:10.4103/wjcs.wjcs_19_22
78. Thomas P, Abdel-Glil MY, Subbaiyan A, et al. First comparative analysis of Clostridium septicum genomes provides insights into the taxonomy, species genetic diversity, and virulence related to gas gangrene. Front Microbiol. 2021;12:771945. doi:10.3389/fmicb.2021.771945
79. Slezák M, Smolár M, Drobna Saniova B, Hosala M, Miklusica J. Clostridium septicum foot gangrene associated with colorectal cancer. Neuro Endocrinol Lett. 2022;43(2):57–64.
80. Justesen US, Nielsen SL, Jensen TG, et al. Bacteremia with anaerobic bacteria and association with colorectal cancer: a population-based cohort study. Clin Infect Dis. 2022;75(10):1747–1753. doi:10.1093/cid/ciac259
81. Abraham AT, Padam S. Clostridium septicum Bacteremia as the Presenting Sign of Colon Cancer. Cureus. 2023;15(9):1.
82. Plouznikoff N, Artigas C, Woff E, Ene D, Flamen P. Colon adenocarcinoma associated with Clostridium septicum endophthalmitis detected on 18F-FDG PET/CT. Clin Nucl Med. 2020;45(6):459–460. doi:10.1097/RLU.0000000000003025
83. Pandeya A, Khalko RK, Kotarya B, et al. Human Papillomavirus (HPV): molecular Epidemiology of Infection and its Associated Diseases. In: Biomedical Research, Medicine, and Disease. CRC Press; 2023:337–361.
84. Ibragimova MK, Tsyganov MM, Litviakov NV. Human papillomavirus and colorectal cancer. Med Oncol. 2018;35(11):1–6. doi:10.1007/s12032-018-1201-9
85. Ambrosio MR, Niccolai E, Petrelli F, et al. Immune landscape and oncobiota in HPV-associated colorectal cancer: an explorative study. Clin Exp Med. 2023;23(8):5101–5112. doi:10.1007/s10238-023-01165-3
86. Hafez FS, Meckawy GR, Alorabi M, Shakweer MM. Interpretation of P16 expression as a marker of HPV in colorectal cancer. Histol Histopathol. 2022;37:475–481. doi:10.14670/HH-18-439
87. Qiu Q, Li Y, Fan Z, et al. Gene expression analysis of human papillomavirus‐associated colorectal carcinoma. Biomed Res Int. 2020;2020:5201587. doi:10.1155/2020/5201587
88. Singh S, Ahmad S, Srivastava AN, Misra JS. A review on role of human papilloma virus (HPV) in health‐related diseases. Adv Med Dent Health Sci. 2020;3(3):34–40. doi:10.5530/amdhs.2020.3.9
89. Wellman TK Epigenetic alterations induced by human papillomavirus E6 and E7 oncoproteins [dissertation]. Cambridge, MA: Harvard University; 2018.
90. Szymonowicz KA, Chen J. Biological and clinical aspects of HPV-related cancers. Cancer Biol Med. 2020;17(4):864. doi:10.20892/j.issn.2095-3941.2020.0370
91. Damin DC, Ziegelmann PK, Damin AP. Human papillomavirus infection and colorectal cancer risk: a meta‐analysis. Colorectal Dis. 2013;15(8):e420–8. doi:10.1111/codi.12257
92. Cui X, Snapper CM. Epstein Barr virus: development of vaccines and immune cell therapy for EBV-associated diseases. Front Immunol. 2021;12:734471. doi:10.3389/fimmu.2021.734471
93. Gupta I, Al Farsi H, Jabeen A, et al. High-risk human papillomaviruses and Epstein–Barr virus in colorectal cancer and their association with clinicopathological status. Pathogens. 2020;9(6):452. doi:10.3390/pathogens9060452
94. Tsang CM, Tsao SW. The role of Epstein-Barr virus infection in the pathogenesis of nasopharyngeal carcinoma. Virol Sin. 2015;30(2):107–121. doi:10.1007/s12250-015-3592-5
95. Maskouni EJ, Jamalvandi T, Tabatabaei F, et al. Association between Epstein-Bar virus and colorectal cancer: a systematic review and meta-analysis. Microb Pathog. 2023;179:106087. doi:10.1016/j.micpath.2023.106087
96. Qualtrough D, Singh K, Banu N, Paraskeva C, Pignatelli M. The actin-bundling protein fascin is overexpressed in colorectal adenomas and promotes motility in adenoma cells in vitro. Br J Cancer. 2009;101(7):1124–1129. doi:10.1038/sj.bjc.6605286
97. Al-Antary N, Farghaly H, Aboulkassim T, Yasmeen A, Akil N, Al Moustafa AE. Epstein–Barr virus and its association with Fascin expression in colorectal cancers in the Syrian population: a tissue microarray study. Hum Vaccin Immunother. 2017;13(7):1573–1578. doi:10.1080/21645515.2017.1302046
98. Kim YS, Jeong H, Choi JW, Oh HE, Lee JH. Unique characteristics of ARID1A mutation and protein level in gastric and colorectal cancer: a meta-analysis. Saudi J Gastroenterol. 2017;23(5):268–274. doi:10.4103/sjg.SJG_184_17
99. Oleynikova NA, Danilova NV, Grimuta MO, Malkov PG. Epstein–Barr Virus in the Development of Colorectal Cancer. Sovremennye Tekhnologii v Meditsine. 2021;13(4 (eng)):82–91. doi:10.17691/stm2021.13.4.09
100. Niya MHK, Keshavarz M, Tameshkel FS, et al. Investigation of JC polyomavirus (JCV) genome in colorectal cancer patients from Iran. Iran J Public Health. 2020;49(3):1.
101. Fang CY, Chen SY, Hsiao BX, et al. Unusually high incidence of polyomavirus JC infection in the higher grade of colorectal cancer tissues in Taiwan. Eur J Med Res. 2022;27(1):127. doi:10.1186/s40001-022-00756-2
102. Kimla LJ, Clark TG, Banerjee S, Campino S, Galli A. JC Polyomavirus T-antigen protein expression and the risk of colorectal cancer: systematic review and meta-analysis of case-control studies. PLoS One. 2023. 18(3):e0283642. doi:10.1371/journal.pone.0283642
103. Link A, Shin SK, Nagasaka T, et al. JC virus mediates invasion and migration in colorectal metastasis. PLoS One. 2009;4(12):e8146. doi:10.1371/journal.pone.0008146
104. Coelho TR, Almeida L, Lazo PA. JC virus in the pathogenesis of colorectal cancer, an etiological agent or another component in a multistep process? Virol J. 2010;7:1–8. doi:10.1186/1743-422X-7-42
105. Navand AH, Teimoori A, Makvandi M, et al. Study on JV virus in patients with colon cancer type adenocarcinoma. Asian Pac J Cancer Prev. 2019;20(4):1147. doi:10.31557/APJCP.2019.20.4.1147
106. Mohamed EI, El Hussein ARM, Elkhidir IM, Enan KA. Molecular detection of John Cunningham virus (JCV) in patients with colorectal cancer in Khartoum, Sudan. Drugs. 2019;29:30.
107. Herbein G. The human cytomegalovirus, from oncomodulation to oncogenesis. Viruses. 2018;10(8):408. doi:10.3390/v10080408
108. Chelbi H, Jelassi R, Belfkih S, et al. Association of CCR5Δ32 deletion and human cytomegalovirus infection with colorectal cancer in Tunisia. Front Genetics. 2021. 12:598635. doi:10.3389/fgene.2021.598635
109. Chen HP, Chan YJ. The oncomodulatory role of human cytomegalovirus in colorectal cancer: implications for clinical trials. 2014. Front Oncol. ;4:314. doi:10.3389/fonc.2014.00314
110. Merchut-Maya JM, Bartek JJ, Bartkova J, et al. Human cytomegalovirus hijacks host stress response fueling replication stress and genome instability. Cell Death Differ. 2022;29(8):1639–1653. doi:10.1038/s41418-022-00953-w
111. Zhou J, Guo X, Huang P, et al. HBV infection status indicates different risks of synchronous and metachronous liver metastasis in colorectal cancer: a retrospective study of 3132 patients with a 5-year follow-up. Cancer Manag Res. 2022:1581–1594. doi:10.2147/CMAR.S350276
112. Su F-H, Le TN, Muo C-H, Te SA, Sung F-C, Yeh -C-C. Chronic hepatitis B virus infection associated with increased colorectal cancer risk in Taiwanese population. Viruses. 2020;12(1):97. doi:10.3390/v12010097
113. Jung YS, Kim NH, Park JH, Park DI, Sohn CI. Correlation between hepatitis B virus infection and colorectal neoplasia. J Clin Med. 2019;8(12):2085. doi:10.3390/jcm8122085
114. Su FH, Bai CH, Le TN, et al. Patients with chronic hepatitis C virus infection are at an increased risk of colorectal cancer: a nationwide population-based case-control study in Taiwan. Front Oncol. 2021;10:2020. doi:10.3389/fonc.2020.561420
115. Min Y, Wei X, Xia X, et al. Hepatitis B virus infection: an insight into the clinical connection and molecular interaction between hepatitis B virus and host extrahepatic cancer risk. Front Immunol. 2023;14(March):1–20. doi:10.3389/fimmu.2023.1141956
116. Shen C, Jiang X, Li M, Luo Y. Hepatitis virus and hepatocellular carcinoma: recent advances. Cancers. 2023;15(2):533. doi:10.3390/cancers15020533
117. Saeed M, Shoaib A, Kandimalla R, et al. Microbe-based therapies for colorectal cancer: advantages and limitations. Semin Cancer Biol. 2022;86:652–665. doi:10.1016/j.semcancer.2021.05.018
118. Lin Y, Lau HC, Liu Y, et al. Altered mycobiota signatures and enriched pathogenic Aspergillus rambellii are associated with colorectal cancer based on multicohort fecal metagenomic analyses. Gastroenterology. 2022;163(4):908–921. doi:10.1053/j.gastro.2022.06.038
119. Alonso-Monge R, Gresnigt MS, Román E, Hube B, Pla J. Candida albicans colonization of the gastrointestinal tract: a double-edged sword. PLoS Pathog. 2021;17(7):e1009710. doi:10.1371/journal.ppat.1009710
120. Starý L, Mezerová K, Vysloužil K, et al. Candida albicans culture from a rectal swab can be associated with newly diagnosed colorectal cancer. Folia Microbiol. 2020;65(6):989–994. doi:10.1007/s12223-020-00807-3
121. Leroy A, Mareel M, De Bruyne G, Bailey G, Nelis H. Metastasis of Entamoeba histolytica compared to colon cancer: one more step in invasion. Invasion Metastasis. 1994–1995;14(1–6):177–191.
122. Chou A, Austin RL. Entamoeba histolytica infection. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557718/.
123. Ghosh S, Padalia J, Moonah S. Tissue destruction caused by Entamoeba histolytica parasite: cell death, inflammation, invasion, and the gut microbiome. Curr Clin Microbiol Rep. 2019;6(1):51–57. doi:10.1007/s40588-019-0113-6
124. Haghighi L, Dalimi A, Pirestani M, Ghaffarifar F. The effect of Entamoeba histolytica Lectin Antigen and MicroRNA-643 on the development of microsatellite instability (MSI) in colorectal adenocarcinoma. ResearchSquare. 2024;2024:1.
125. Mir MA, Hamdani SS, Mehraj U. Antigens and immunogens. In: Basics Fundam Immunol. Nova Science Publishers Inc.; 2020:77.
126. Suresh R, Mosser DM. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv Physiol Educ. 2013;37(4):284–291. doi:10.1152/advan.00058.2013
127. Azuma M. Co-signal molecules in T-cell activation: historical overview and perspective. Co-signal molecules in T Cell activation: immune regulation in health and disease. Advances in Experimental Medicine and Biology. 2019;1189:3–23. doi:10.1007/978-981-32-9717-3_1
128. Dong C. Cytokine regulation and function in T cells. Annu Rev Immunol. 2021;39(1):51–76. doi:10.1146/annurev-immunol-061020-053702
129. Lee V, Rodriguez DM, Ganci NK, et al. The endogenous repertoire harbors self-reactive CD4+ T cell clones that adopt a follicular helper T cell-like phenotype at steady state. Nat Immunol. 2023;24(3):487–500. doi:10.1038/s41590-023-01425-0
130. Basu A, Ramamoorthi G, Albert G, et al. Differentiation and regulation of TH cells: a balancing act for cancer immunotherapy. Front Immunol. 2021;12:669474. doi:10.3389/fimmu.2021.669474
131. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–668. doi:10.1038/s41577-020-0306-5
132. Kinker GS, Vitiello GA, Ferreira WA, Chaves AS, Cordeiro de Lima VC, Medina TD. B cell orchestration of anti-tumor immune responses: a matter of cell localization and communication. Front Cell Dev Biol. 2021;9:678127. doi:10.3389/fcell.2021.678127
133. Adams NM, Grassmann S, Sun JC. Clonal expansion of innate and adaptive lymphocytes. Nat Rev Immunol. 2020;20(11):694–707. doi:10.1038/s41577-020-0307-4
134. Zhu X, Shi Z, Mao Y, Lächelt U, Huang R. Cell Membrane Perforation: patterns, Mechanisms and Functions. Small. 2024;12:2310605. doi:10.1002/smll.202310605
135. Hua S, Gu X, Jin H, Zhang X, Liu Q, Yang J. Tumor-infiltrating T lymphocytes: a promising immunotherapeutic target for preventing immune escape in cholangiocarcinoma. Biomed Pharmacother. 2024;177:117080. doi:10.1016/j.biopha.2024.117080
136. Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-γ in tumor progression and regression: a review. Biomark Res. 2020;8:1–6. doi:10.1186/s40364-020-00228-x
137. Reina-Campos M, Scharping NE, Goldrath AW. CD8+ T cell metabolism in infection and cancer. Nat Rev Immunol. 2021;21(11):718–738. doi:10.1038/s41577-021-00537-8
138. Laumont CM, Banville AC, Gilardi M, Hollern DP, Nelson BH. Tumour-infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat Rev Cancer. 2022;22(7):414–430. doi:10.1038/s41568-022-00466-1
139. Yang H, Zhang Z, Li J, Wang K, Zhu W, Zeng Y. The Dual Role of B Cells in the Tumor Microenvironment: implications for Cancer Immunology and Therapy. Int J Mol Sci. 2024;25(21):11825. doi:10.3390/ijms252111825
140. Chin DS, Lim CS, Nordin F, Arifin N, Jun TG. Antibody-dependent cell-mediated cytotoxicity through Natural Killer (NK) cells: unlocking NK cells for future immunotherapy. Curr Pharm Biotechnol. 2022; 23(4):552–578. doi:10.2174/1389201022666210820093608
141. Golay J, Taylor RP. The role of complement in the mechanism of action of therapeutic anti-cancer mAbs. Antibodies. 2020. 9(4):58. doi:10.3390/antib9040058
142. Xue D, Hu S, Zheng R, Luo H, Ren X. Tumor-infiltrating B cells: their dual mechanistic roles in the tumor microenvironment. Biomed Pharmacother. 2024;179:117436. doi:10.1016/j.biopha.2024.117436
143. Fridman WH, Meylan M, Petitprez F, Sun CM, Italiano A, Sautès-Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol. 2022;19(7):441–457. doi:10.1038/s41571-022-00619-z
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.