Back to Journals » OncoTargets and Therapy » Volume 13
Microarray Expression Profiles and Bioinformatics Analyses Reveal Aberrant Circular RNAs Expression in Bladder Cancer
Authors Yang J, Chen J, Wu S, Fei X, Wang X, Wang K ![]()
Received 7 July 2020
Accepted for publication 24 September 2020
Published 27 October 2020 Volume 2020:13 Pages 10889—10899
DOI https://doi.org/10.2147/OTT.S270747
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Jun Yang,1 Junwen Chen,2 Si Wu,3 Xiang Fei,2 Xia Wang,2 Kefeng Wang2
1Department of Gastroenterology, Shengjing Hospital of China Medical University, Shenyang 110004, People’s Republic of China; 2Department of Urology, Shengjing Hospital of China Medical University, Shenyang 110004, People’s Republic of China; 3Department of Biobank, Shengjing Hospital of China Medical University, Shenyang 110004, People’s Republic of China
Correspondence: Kefeng Wang
Department of Urology, Shengjing Hospital of China Medical University, 36# Sanhao Street, Heping District, Shenyang, Liaoning, People’s Republic of China
Tel +86 18940254849
Fax +86 24 31939077
Email [email protected]
Background: Increasing evidence shows that circular RNAs (circRNAs) are involved in many biological processes, functioning as microRNA (miRNA) sponges. The aim of this study is to identify differentially expressed circRNAs in bladder cancer (BCa).
Methods: The transcriptome of circRNAs in BCa was assayed by microarray. Quantitative real-time PCR was performed to verify the results. Then, potential miRNA response elements (MREs) between circRNAs and miRNAs were predicted. Pathway and ontology enrichment analyses were performed to identify mechanisms related to the gene regulation of differentially expressed circRNAs.
Results: Three hundred and eighty-six up-regulated and 394 down-regulated circRNAs were identified, and their potential MREs were predicted in BCa.
Conclusion: The differentially expressed circRNAs indicate that circRNAs could play important roles in the molecular pathogenesis of BCa.
Keywords: bladder cancer, microarray, noncoding RNAs, circular RNAs, bioinformatics analysis
Introduction
Bladder cancer (BCa) is one of the most prevalent malignant tumors worldwide. The morbidity and mortality of BCa ranks eleventh and fourteenth, respectively, of all tumor types.1 It is estimated that there were 81,400 new BCa cases and 17,980 deaths in the United States in 2020.2 Surgery, radiation therapy and chemotherapy only provide limited treatment for advanced BCa,3,4 which has a high recurrence rate.5 This high recurrence rate is partly due to our poor understanding of BCa pathogenesis. Therefore, it is necessary to search for new ways to improve the diagnosis and treatment of BCa.
Circular RNAs (circRNAs) are considered to represent splicing errors. Accumulating evidence has revealed that these low abundant but highly stable closed-loop structures could play vital roles in many biological processes,6,7 such as transcriptional regulation,8 RNA transport9 and proteins binding.10 Recently, many studies have revealed that circRNAs displayed vital roles in various tumor types.11 Although many circRNAs have been found by high-throughput sequencing, the detailed mechanisms of their functions in tumor onset and progression remain unclear.12–14
In this study, we detected expressed circRNA profiles in BCa and para-carcinoma normal tissues by microarray. Seven hundred and eighty circRNAs were significantly differentially expressed. Two circRNAs with the most significant differences were verified by quantitative real-time PCR (qRT-PCR). The results indicated that hsa_circRNA_103670 was up-regulated and hsa_circRNA_000367 was down-regulated in 30 pairs of BCa samples, consistent with our microarray assay results.
Materials and Methods
Clinical Tissues
Human BCa and para-carcinoma normal tissues were obtained from the Department of Urology, Shengjing Hospital of China Medical University (Shenyang, China) for microarray analysis and qRT-PCR. None of the patients received surgery or other oncotherapy prior to this study. The study was conducted in accordance with the Declaration of Helsinki, and all the samples were collected for experimental purposes. Informed consent forms were signed by all patients before the study. Ethics approval was authorized by the Medical Research and New Technology Ethics Committee of Shengjing Hospital of China Medical University with an approval ID No. 2016 PS449K.
RNA Extraction and Quality Control
Total RNA was isolated using Trizol reagent (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s instructions. The concentrations of the RNA samples were determined by OD260 using a NanoDrop ND-1000 (Thermo NanoDrop, Wilmington, DE, USA) instrument. The integrity of the RNA was assessed by electrophoresis on a denaturing agarose gel.
RNA Labeling and Hybridization
Total RNA from each sample was treated with RNase R (EpiCentre Inc., Madison, WI, USA) to enrich for circRNAs. The enriched circRNAs were then amplified and transcribed into fluorescent circRNAs utilizing random primers according to the Arraystar Super RNA Labeling protocol (Arraystar_Inc, Rockville, MD, USA). The labeled circRNAs were purified by the RNeasy Mini Kit (Qiagen, Dusseldorf, Germany). The concentration and specific activity of the labeled circRNAs (pmol Cy3/μg complementary RNA) were measured by NanoDrop ND-1000. Each labeled complementary RNA (1 μg) was fragmented by adding 5 μL 10 × Blocking Agent and 1 μL of 25 × Fragmentation Buffer, heated at 60°C for 30 min. Finally, 25 μL 2 × hybridization buffer was added to dilute the labeled circRNAs. Fifty μL of hybridization solution was dispensed into the gasket slide and assembled onto the circRNA expression microarray slide. The slides were incubated for 17 hours at 65°C in an Agilent Hybridization oven. The hybridized arrays were washed, fixed and scanned using the Agilent Scanner G2505C.
Microarray Data Analysis
Scanned images were imported into Agilent Feature Extraction software for raw data extraction. Quantile normalization of raw data and subsequent data processing were performed using the R software package. When comparing two groups of profile differences (such as disease versus control), the “fold change” (ie the ratio of the group averages) between the two groups for each circRNA was computed. The statistical significance of the difference was estimated by t-test. circRNAs having fold changes ≥2 and P-values <0.05 were selected as significantly differentially expressed. A box plot was used to compare the distributions of the intensities from all the samples. A scatter plot was created to assess the circRNA expression variation between the two groups. Volcano plots and hierarchical clustering were used to show the differentially expressed circRNAs.
Quantitative Real-Time PCR Analysis
Hsa_circRNA_103670 and hsa_circRNA_000367 were analyzed with specific primers (for hsa_circRNA_103670: left primer CGGCTCTTCCAGCTACAAAC and right primer CATTGGCTACCTCCTCTGCT. For hsa_circRNA_000367: left primer ATCGAAGACTGGCGTGAAAC and right primer ATTGCACAGTGGATGGATCA). qRT-PCR was performed using the Bio-Rad CFX96 system with SYBR green to determine the expression level of each circRNA. The PCR reaction was conducted in a 15 μL volume, including 7.5 μL SYBR Mix, 6 μL cDNA, 0.75 μL forward primer (10 μM), and 0.75 μL reverse primer (10 μM). The qRT-PCR protocol was as follows: 50°C for 2 min, 95°C for 8 min 30 sec, followed by 45 cycles at 95°C for 15 sec, and 60°C for 1 min. Extension was performed at 95°C for 1 min, 55°C for 1 min and 55°C for 10 sec. GAPDH was used as a normalization control.
Statistics
Statistical analyses were performed by SPSS 17.0 (SPSS_Inc, Chicago, IL, USA). P <0.05 was considered to be statistically significant. Data differences between the two groups were analyzed by paired t-test. Data were expressed as mean ± SEM/SD from at least three independent experiments.
Results
Identification of Differentially Expressed circRNAs in BCa and Para-Carcinoma Normal Tissues
A microarray containing 13,617 human circRNA probes was conducted for three paired BCa and para-carcinoma normal tissues, following the Arraystar Human circRNA Array V2.0 protocol. Hierarchical clustering was performed to distinguish the circRNA expression profiles among the samples (Figure 1A). We compared the distributions of the expression intensity values from the six samples after normalization. The results showed that the normalized intensity values were similar across all six samples (Supplementary Fig. S1A). We then assessed the circRNA expression reproducibility between the two groups (Figure 1B). Volcano plots were constructed to exhibit the differentially expressed circRNAs between the two groups (Figure 1C).
 |
Figure 1 Differentially expressed circRNAs in BCa. (A) Hierarchical clustering shows distinguishable circRNA expression profiles among the samples. Each column represents a sample, including the normal control (first three columns) and BCa (last three columns). Each row represents a circRNA. Red band means relative high expression, while green one means relative low expression. (B) Scatter plot of circRNAs for assessing the expression reproducibility between the paired BCa and para-carcinoma normal tissues. The values plotted on X and Y axes are averaged normalized signal values of BCa and normal control (log2 scaled). Two green slashes represent 2.0 fold changes while the middle slash means no difference. CircRNAs above the upper green slash or below the lower green slash indicate a more than 2.0 fold change. (C) Volcano plots of circRNAs for exhibiting the differentially expressed circRNAs between the two groups. The vertical lines correspond to 2.0 fold up and down, respectively, and the horizontal line represents a P-value of 0.05. The red points in the plot represent the differentially expressed circRNAs with statistical significance. (D-E) The two pie charts display the sources of circRNAs. Most of the differentially expressed circRNAs derive from exons in up-regulated group (D) and down-regulated group (E). The other sources are intronic, sense overlapping, antisense and intergenic. (F) The bar diagram shows the locations of the circRNAs on the chromosomes. |
Statistical significance between the two groups was determined by a fold-change ≥2.0 and P <0.05. The results showed that a total of 780 circRNAs had different expressions between the two groups, containing 386 up-regulated and 394 down-regulated circRNAs (Supplementary data 1). Fifty-three circRNAs showed a more than five-fold increase, among which 18 revealed a more than ten-fold up-regulation. Thirty-four circRNAs displayed a more than five-fold decrease, among which 7 exhibited a more than ten-fold down-regulation. Most of the above 780 circRNAs were derived from exons, namely 82.90% (320/386) in the up-regulated group and 81.73% (322/394) in the down-regulated group. The other sources were introns, sense overlapping, antisense sequences and intergenic regions (Figure 1D and E). The locations of the circRNAs on the chromosomes are shown in Figure 1F. The top ten up-regulated and down-regulated circRNAs are shown in Table 1.
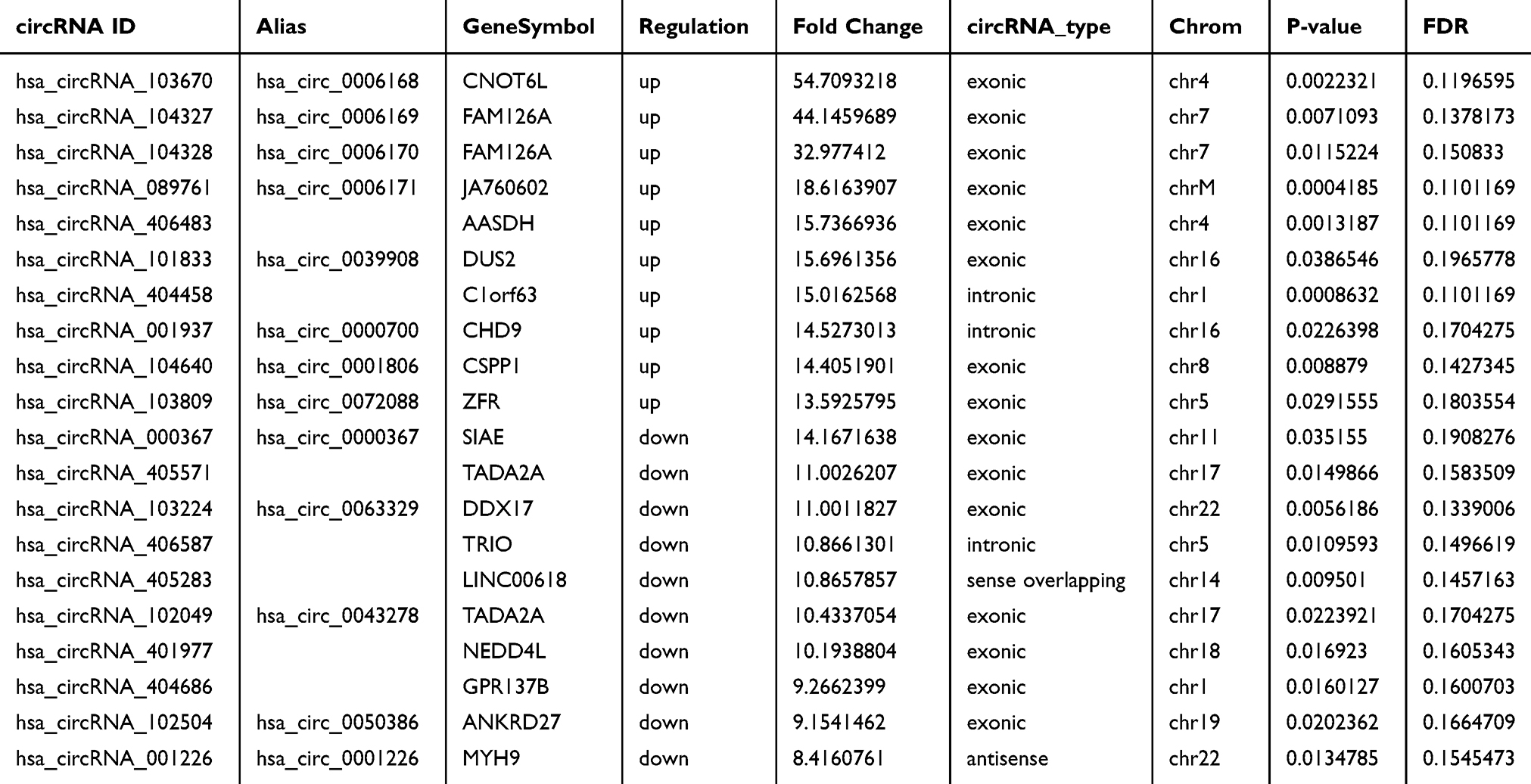 |
Table 1 The Top 10 Up- and Down-Regulated circRNAs Ranked by Fold Changes in Microarray Data |
Construction of circRNA-miRNA Interaction Network and miRNA Response Element (MRE) Analysis
Recent studies have demonstrated that circRNAs play important roles in fine-tuning miRNAs to regulate the expressions of genes by sequestering them.15,16 The interactions between circRNAs and their potential targeted miRNAs were predicted according to seed matching sequences using Arraystar miRNA target prediction software based on TargetScan17 and miRanda.18 The networks of the top 10 up-regulated and down-regulated circRNAs and their correlated miRNAs were delineated by Cytoscape (Figure 2). We listed the top five possible MREs of the highest up-regulated and down-regulated circRNAs. The potential MREs of hsa_circRNA_103670 included miR-570-5p, miR-592, miR-542-3p, miR-628-5p and miR-148b-5p. For hsa_circRNA_000367, the potential MREs included miR-331-3p, miR-4646-5p, miR-4797-5p, miR-3919 and miR-3190-3p. The results are shown in Figure 3A and B, which displays the 2D structures of seed sequence binding, nucleotide composition with regard to AU-richness, and MRE position.
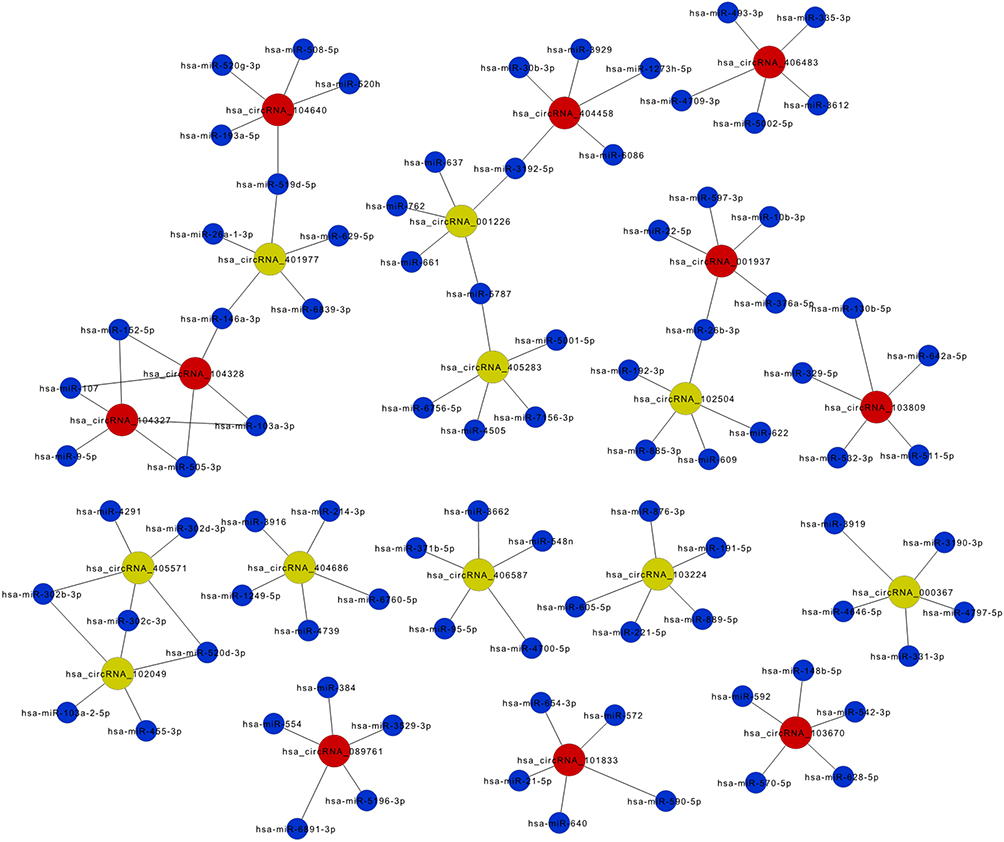 |
Figure 2 The circRNA-miRNA network analysis, which comprises the top 10 up- and down-regulated circRNAs and their targeted miRNAs. |
 |
Figure 3 The detailed annotation for circRNA-miRNA interactions. (A-B) The top five MREs of hsa_circRNA_103670 (A) and hsa_circRNA_000367 (B) are predicted. |
Analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO)
KEGG pathway analysis was conducted to investigate the impact of circRNAs in gene regulation. The results showed that 42 pathways were related to the up-regulated mRNAs (Supplementary data 2), while 41 pathways were associated with the down-regulated mRNAs (Supplementary data 3). The top ten KEGG pathways of the up-regulated and down-regulated mRNAs are illustrated in Figure 4A and B. Hypertrophic cardiomyopathy and the prolactin signaling pathway were the top pathways among the up-regulated and down-regulated transcripts, respectively.
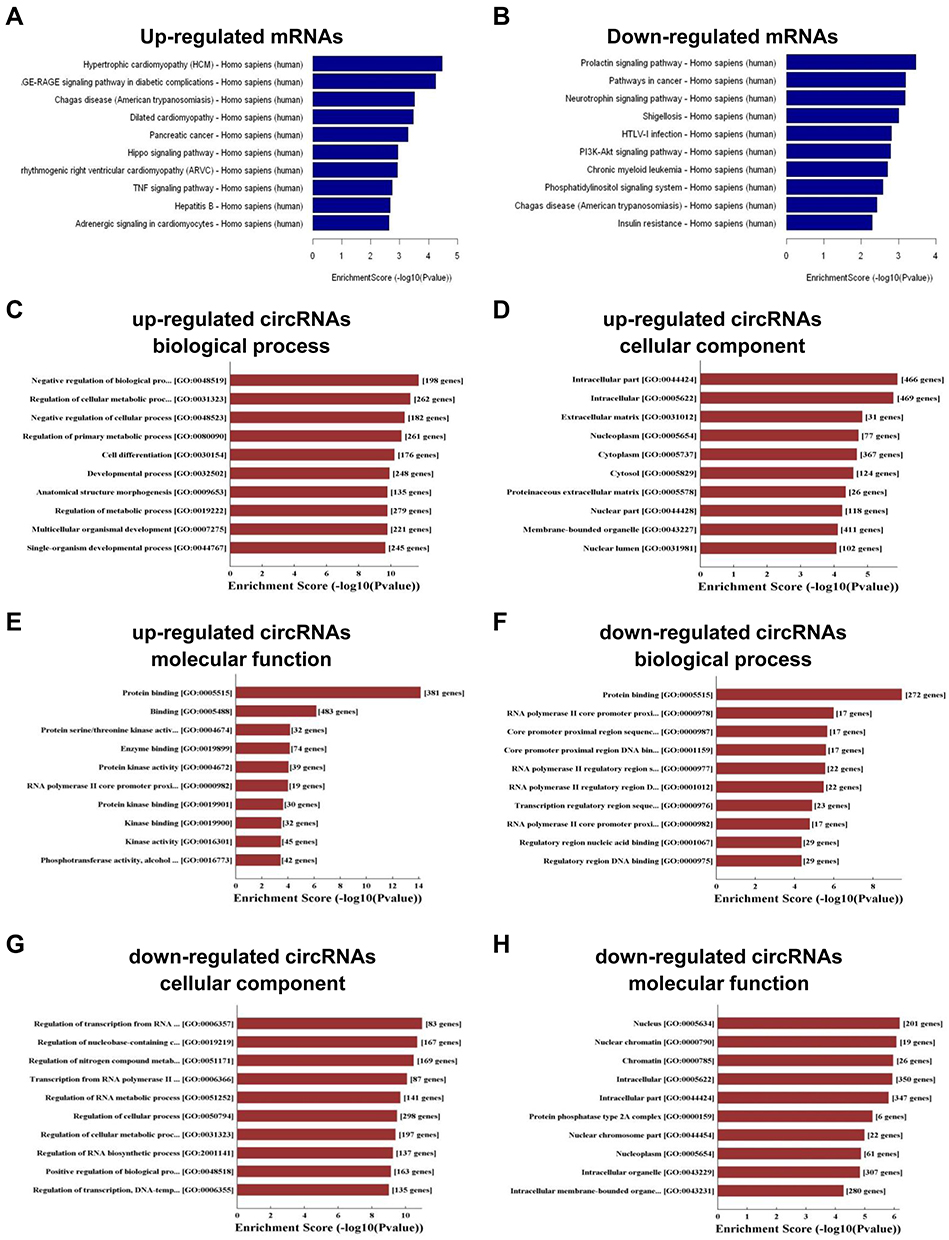 |
Figure 4 KEGG and GO analyses of the genes that produce up-regulated and down-regulated circRNAs. (A-B) KEGG pathway enrichment analyses of up-regulated mRNAs (A) and down-regulated mRNAs (B). (C-H) GO enrichment analyses of up-regulated circRNAs in biological process (C), cellular component (D) and molecular function (E) and down-regulated circRNAs in biological process (F), cellular component (G) and molecular function (H). |
Furthermore, GO analysis was also performed to parse the linear transcripts of the differentially expressed circRNAs. The results revealed that negative regulation of biological process, intracellular part, and protein binding were the top gene and gene product attributes in the biological process, cellular component, and molecular function, respectively, for the up-regulated transcripts (Figure 4C-E). For the down-regulated transcripts, the top GO terms were regulation of transcription from RNA polymerase II promoter, and nucleus and protein binding (Figure 4F-H). The above results revealed that these pathways may take part in the onset and progression of BCa.
Confirmation of Differentially Expressed circRNAs in Clinical Samples and Their Correlations with Clinicopathological Features
We chose hsa_circRNA_103670 and hsa_circRNA_000367 for further validation because they had the highest up- and down-regulated fold changes in the microarray assay. A qRT-PCR assay was performed in 30 paired of BCa and para-carcinoma normal tissues. The results were consistent with our microarray data and showed that the expression of hsa_circRNA_103670 was higher in the BCa group than the para-carcinoma normal tissues, while hsa_circRNA_000367 expression was lower in the BCa group than the para-carcinoma normal tissues (Figure 5A and B). The results of a clinicopathological analysis showed that the high expression of hsa_circRNA_103670 was related to the high histological grade, poor T stage. Early T stage had a positive correlation with the expression of hsa_circRNA_000367 (Table 2).
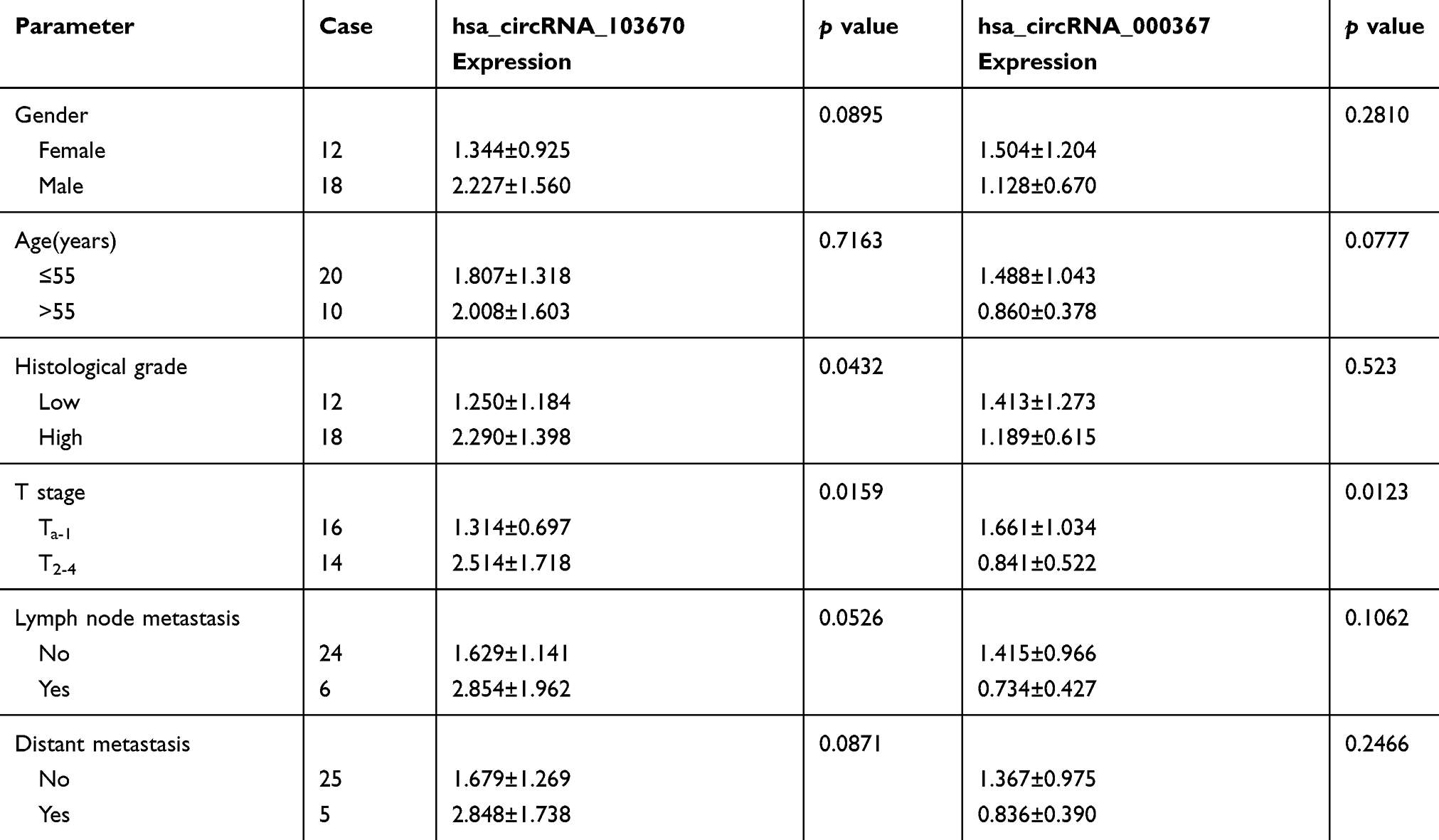 |
Table 2 Relationships Between hsa_circRNA_103670, hsa_circRNA_000367 Expression and Clinicopathologic Features of BCa |
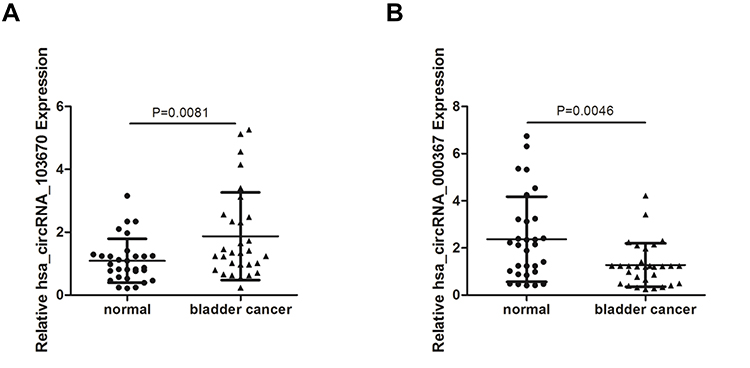 |
Figure 5 The verification of two differentially expressed circRNAs by qRT-PCR in 30 pairs BCa samples. (A and B) Hsa_circRNA_103670 (A) and hsa_circRNA_000367 (B) expression in 30 paired BCa samples. Each sample is run in triplicate and in multiple experiments for mean ± SEM. |
Discussion
CircRNAs were found in the 1970s as an abnormal splicing product by Sanger et al.19 Due to technological limitations, circRNAs were not highly studied in the following years.20–22 However, studies investigating circRNAs have become more common since two sequencing analyses were done by Jeck et al,23 and Memczak et al.24 With the improvement of high-throughput sequencing, circRNAs were found to exist in many tissues abundantly and stably.25 More and more studies have demonstrated that circRNAs have tissue or cell specificity, are relatively conserved, and have dynamic expression, which reflects their possible functions in eukaryotes.26–28 Many studies have shown that circRNAs function as miRNA sponges in various tumors.29,30 Our study shows evidence that some circRNAs are differentially expressed in BCa compared with para-carcinoma normal tissues.
Microarray assays have been previously used to detect circRNAs, lncRNAs, miRNAs, or mRNAs in various tumors,31–33 including in BCa samples.34–37 Li et al identified 16,535 circRNAs and found that 524 circRNAs were significantly down-regulated, while 47 circRNAs were up-regulated in BCa compared to para-carcinoma normal tissues.35 Yang et al also performed next-generation sequencing in 5 BCa patients and revealed that 42 circRNAs were down-regulated and 14 circRNAs were up-regulated in BCa compared to non-cancerous tissues.36 The results of Zhong et al showed that 469 circRNAs were significantly altered based on microarray data, including 184 down-regulated circRNAs and 285 up-regulated circRNAs.37 In our study, we determined the profiles of circRNAs in three paired BCa and para-carcinoma normal tissues by microarray sequencing. The results showed that 386 circRNAs were significantly up-regulated and 394 circRNAs were down-regulated between the two groups. Moreover, 18 up-regulated and 7 down-regulated circRNAs exhibited a more than 10-fold change, which implied the possibility of these circRNAs functioning as oncogenes or tumor suppressors in BCa. Comparing the top 20 up- and down-regulated circRNAs of Zhong et al’s microarray data, we find that only hsa_circRNA_0072088, hsa_circRNA_0000520, hsa_circRNA_0003528, hsa_circRNA_0028173, hsa_circRNA_0082582, and hsa_circRNA_0005265 are consistent with our microarray results. The two results do not fit very well due to some reasons. One possible reason may be that the samples of each microarray are limited and insufficient to assess the differential expression of circRNAs, for which the heterogeneity among the patients is usually noticeable. Another reason may be that the clinical samples are derived from different regions and population ancestries. Research in large samples and different regions and population are needed to reduce the bias.
Consistent with our microarray data, the results of the qRT-PCR assay confirmed that hsa_circRNA_103670 and hsa_circRNA_000367 were up-regulated and down-regulated, respectively, in 30 paired BCa and para-carcinoma normal tissues. Hsa_circRNA_103670 was spliced from the CCR4-NOT transcription complex subunit 6 like (CNOT6L or CCR4b) gene. Zhou et al found that miR-146a-Cnot6L affected the stability of Zeb1 mRNA and induced epithelial-mesenchymal transformation.38 Hsa_circRNA_000367 was generated from the sialic acid acetylesterase (SIAE) gene and had been shown to participate in immune system disease.39,40 Due to the inseparable relationships between tumor formation and the immune system, we believe that SIAE may be correlated with tumorigenesis.
Patients with high histological grade and poor T stage had a high expression of hsa_circRNA_103670. Conversely, patients with poor T stage had a low expression of hsa_circRNA_000367. These data showed that circRNAs might play vital roles in carcinogenesis and the progression of BCa.
CircRNAs are known to act as miRNA sponges to regulate protein expression by sequestering miRNAs. Zhong et al found that the circRNA MYLK could promote BCa progression as a competing endogenous RNA through modulating VEGFA/VEGF2 signals.37 Yang et al showed that the circRNA ITCH could inhibit BCa progression through sponging miR-17/miR-224 and regulating p21, and PTEN expression.41 Our study predicted circRNA-miRNA interactions, which could provide information for further research. We also constructed a network of the top 10 up- and down-regulated circRNAs and their correlated miRNAs.
Furthermore, we detected the top five possible MREs of the highest up- and down-regulated circRNAs in BCa. The potential MREs of hsa_circRNA_103670 included miR-570-5p, miR-592, miR-542-3p, miR-628-5p, and miR-148b-5p. For hsa_circRNA_000367, the potential MREs included miR-331-3p, miR-4646-5p, miR-4797-5p, miR-3919, and miR-3190-3p. The results exhibited the 2D structure of seed sequence binding, nucleotide composition with regard to AU-richness, and MRE position.
Although we identified hundreds of differentially expressed circRNAs by microarray assay and predicted the potential interactions between circRNAs and their target miRNAs or mRNAs, additional in vitro cellular functional assays are needed to verify the effects of these circRNAs.
In summary, we showed that circRNAs may be dysregulated in BCa compared with para-carcinoma normal tissues. Confirmed by 30 paired clinical samples, the expression of hsa_circRNA_103670 was up-regulated and hsa_circRNA_000367 was down-regulated in BCa. Our study provides new data on circRNAs in the tumorigenesis of BCa.
Abbreviations
circRNAs, circular RNAs; BCa, bladder cancer; MREs, miRNA response elements; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology; SIAE, sialic acid acetylesterase.
Ethical Conduct of Research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Acknowledgments
We thank Yun Cui from the Department of Urology, Peking University Third Hospital and Institute of Urology for helping us preparing the manuscript.
Funding
This work was supported by the Key Research and Development Joint Program of Liaoning Province (Grant No. 2020JH 2/10300139) to K Wang, the Natural Science Foundation of Liaoning Province (Grant No. 2019-MS-360) to K Wang, Shenyang Science and Technology Bureau Plan Projects (Grant No. 20-205-4-076) to K Wang, the 345 Talent Project of Shengjing Hospital of China Medical University to K Wang and a natural scientific grant funded by Liaoning Province (Grant No.20170541054) to X Fei.
Disclosure
The authors declare that they have no competing interests.
References
1. Mahdavifar N, Ghoncheh M, Pakzad R, et al. Epidemiology, incidence and mortality of bladder cancer and their relationship with the development index in the world. Asian Pac J Cancer Prev. 2016;17(1):381–386. doi:10.7314/apjcp.2016.17.1.381
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
3. Jordan EJ, Iyer G. Targeted therapy in advanced bladder cancer: what have we learned? Urol Clin North Am. 2015;42(2):253–62, ix. doi:10.1016/j.ucl.2015.01.006
4. Chen CL, Liu CY, Cha TL, et al. Does radical cystectomy outperform other bladder preservative treatments in elderly patients with advanced bladder cancer? J Chin Med Assoc. 2015;78(8):469–474. doi:10.1016/j.jcma.2015.05.003
5. Dy GW, Gore JL, Forouzanfar MH, et al. Global burden of urologic cancers, 19902013. Eur Urol. 2017;71(3):437–446. doi:10.1016/j.eururo.2016.10.008
6. Salzman J. Circular RNA expression: its potential regulation and function. Trends Genet. 2016;32(5):309–316. doi:10.1016/j.tig.2016.03.002
7. Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17(4):205–211. doi:10.1038/nrm.2015.32
8. Guil S, Esteller M. RNA-RNA interactions in gene regulation: the coding and noncoding players. Trends Biochem Sci. 2015;40(5):248–256. doi:10.1016/j.tibs.2015.03.001
9. Ashwal-Fluss R, Meyer M, Pamudurti NR, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56(1):55–66. doi:10.1016/j.molcel.2014.08.019
10. Wilusz JE, Sharp PA. Molecular biology. A circuitous route to noncoding RNA. Science. 2013;340(6131):440–441. doi:10.1126/science.1238522
11. Li J, Yang J, Zhou P, et al. Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am J Cancer Res. 2015;5(2):472–80. eCollection 2015.
12. Sand M, Bechara FG, Sand D, et al. Circular RNA expression in basal cell carcinoma. Epigenomics. 2016;8(5):619–632. doi:10.2217/epi-2015-0019
13. Su H, Lin F, Deng X, et al. Profiling and bioinformatics analyses reveal differential circular RNA expression in radioresistant esophageal cancer cells. J Transl Med. 2016;14(1):225. doi:10.1186/s12967-016-0977-7
14. Zhong Z, Lv M, Chen J. Screening differential circular RNA expression profiles reveals the regulatory role of circTCF25-miR-103a-3p/miR-107-CDK6 pathway in bladder carcinoma. Sci Rep. 2016;6:30919. doi:10.1038/srep30919
15. Jost I, Shalamova LA, Gerresheim GK, et al. Functional sequestration of microRNA-122 from Hepatitis C Virus by circular RNA sponges. RNA Biol. 2018;15(8):1032–1039. doi:10.1080/15476286.2018.1435248
16. Bartsch D, Zirkel A, Kurian L. Characterization of circular RNAs (circRNA) associated with the translation machinery. Methods Mol Biol. 2018;1724:159–166. doi:10.1007/978-1-4939-7562-4_13
17. Enright AJ, John B, Gaul U, et al. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1. doi:10.1186/gb-2003-5-1-r1
18. Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13(4):271–282. doi:10.1038/nrg3162
19. Sanger HL, Klotz G, Riesner D, et al. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci USA. 1976;73(11):3852–3856. doi:10.1073/pnas.73.11.3852
20. Capel B, Swain A, Nicolis S, et al. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell. 1993;73(5):1019–1030. doi:10.1016/0092-8674(93)90279-y
21. Cocquerelle C, Daubersies P, Majerus MA, et al. Splicing with inverted order of exons occurs proximal to large introns. EMBO J. 1992;11(3):1095–1098. doi:10.1002/j.1460-2075.1992.tb05148.x
22. Burd CE, Jeck WR, Liu Y, et al. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6(12):e1001233. doi:10.1371/journal.pgen.1001233
23. Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–157. doi:10.1261/rna.035667.112
24. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338. doi:10.1038/nature11928
25. Dixon RJ, Eperon IC, Hall L, et al. A genome-wide survey demonstrates widespread non-linear mRNA in expressed sequences from multiple species. Nucleic Acids Res. 2005;33(18):5904–5913. doi:10.1093/nar/gki893
26. Rybak-Wolf A, Stottmeister C, Glazar P, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870–885. doi:10.1016/j.molcel.2015.03.027
27. Veno MT, Hansen TB, Veno ST, et al. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015;16:245. doi:10.1186/s13059-015-0801-3
28. Szabo L, Morey R, Palpant NJ, et al. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015;16(1):126. doi:10.1186/s13059-015-0690-5
29. Han D, Li J, Wang H, et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology. 2017;66(4):1151–1164. doi:10.1002/hep.29270
30. Chen L, Zhang S, Wu J, et al. circRNA_100290 plays a role in oral cancer by functioning as a sponge of the miR-29 family. Oncogene. 2017;36(32):4551–4561. doi:10.1038/onc.2017.89
31. Wang Y, Chen W, Chen J, et al. LncRNA expression profiles of EGFR exon 19 deletions in lung adenocarcinoma ascertained by using microarray analysis. Med Oncol. 2014;31(9):137. doi:10.1007/s12032-014-0137-y
32. Zhou J, Li W, Jin T, et al. Gene microarray analysis of lncRNA and mRNA expression profiles in patients with hypopharyngeal squamous cell carcinoma. Int J Clin Exp Med. 2015;8(4):4862–82. eCollection 2015.
33. Miska EA, Alvarez-Saavedra E, Townsend M, et al. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004;5(9):R68. doi:10.1186/gb-2004-5-9-r68
34. Luo H, Zhao X, Wan X, et al. Gene microarray analysis of the lncRNA expression profile in human urothelial carcinoma of the bladder. Int J Clin Exp Med. 2014;7(5):1244–54. eCollection 2014.
35. Li Y, Zheng F, Xiao X, et al. CircHIPK3 sponges miR-558 to suppress heparanase expression in bladder cancer cells. EMBO Rep. 2017;18(9):1646–1659. doi:10.15252/embr.201643581
36. Yang X, Yuan W, Tao J, et al. Identification of circular RNA signature in bladder cancer. J Cancer. 2017;8(17):3456–3463. doi:10.7150/jca.19345
37. Zhong Z, Huang M, Lv M, et al. Circular RNA MYLK as a competing endogenous RNA promotes bladder cancer progression through modulating VEGFA/VEGFR2 signaling pathway. Cancer Lett. 2017;403:305–317. doi:10.1016/j.canlet.2017.06.027
38. Zhou W, Thiery JP. Loss of Git2 induces epithelial-mesenchymal transition by miR146a-Cnot6L-controlled expression of Zeb1. J Cell Sci. 2013;126(Pt12):2740–2746. doi:10.1242/jcs.126367
39. Yamamoto M, Iguchi G, Bando H, et al. A missense single-nucleotide polymorphism in the sialic acid acetylesterase (SIAE) gene is associated with anti-PIT-1 antibody syndrome. Endocr J. 2014;61(6):641–644. doi:10.1507/endocrj.ej13-0539
40. Gan EH, MacArthur K, Mitchell AL, et al. The role of functionally defective rare germline variants of sialic acid acetylesterase in autoimmune Addison’s disease. Eur J Endocrinol. 2012;167(6):825–828. doi:10.1530/EJE-12-0579
41. Yang C, Yuan W, Yang X, et al. Circular RNA circ-ITCH inhibits bladder cancer progression by sponging miR-17/miR-224 and regulating p21, PTEN expression. Mol Cancer. 2018;17(1):19. doi:10.1186/s12943-018-0771-7
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.