Back to Journals » International Journal of Nanomedicine » Volume 11
Miconazole-loaded solid lipid nanoparticles: formulation and evaluation of a novel formula with high bioavailability and antifungal activity
Authors Aljaeid B, Ibrahim KMH ![]()
Received 16 November 2015
Accepted for publication 9 December 2015
Published 25 January 2016 Volume 2016:11 Pages 441—447
DOI https://doi.org/10.2147/IJN.S100625
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Bader Mubarak Aljaeid,1 Khaled Mohamed Hosny1,2
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Saudi Arabia; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Beni Suef University, Beni Suef, Egypt
Background and objective: Miconazole is a broad-spectrum antifungal drug that has poor aqueous solubility (<1 µg/mL); as a result, a reduction in its therapeutic efficacy has been reported. The aim of this study was to formulate and evaluate miconazole-loaded solid lipid nanoparticles (MN-SLNs) for oral administration to find an innovative way to alleviate the disadvantages associated with commercially available capsules.
Methods: MN-SLNs were prepared by hot homogenization/ultrasonication. The solubility of miconazole in different solid lipids was measured. The effect of process variables, such as surfactant types, homogenization and ultrasonication times, and the charge-inducing agent on the particle size, zeta potential, and encapsulation efficiency were determined. Furthermore, in vitro drug release, antifungal activity against Candida albicans, and in vivo pharmacokinetics were studied in rabbits.
Results: The MN-SLN, consisting of 1.5% miconazole, 2% Precirol ATO5, 2.5% Cremophor RH40, 0.5% Lecinol, and 0.1% Dicetylphosphate, had an average diameter of 23 nm with a 90.2% entrapment efficiency. Furthermore, the formulation of MN-SLNs enhanced the antifungal activity compared with miconazole capsules. An in vivo pharmacokinetic study revealed that the bioavailability was enhanced by >2.5-fold.
Conclusion: MN-SLN was more efficient in the treatment of candidiasis with enhanced oral bioavailability and could be a promising carrier for the oral delivery of miconazole.
Keywords: miconazole, Precirol ATO5, solid lipid nanoparticles, encapsulation, Cremophor RH40, antifungal activity
Introduction
Miconazole is a broad-spectrum antifungal drug that has an imidazole group.1,2 Miconazole is a weak base and shows poor aqueous solubility (<1 μg/mL); as a result, a reduction in its therapeutic efficacy has been reported.3,4 Miconazole has a dual mechanism of action: the inhibition of ergosterol biosynthesis and the inhibition of peroxidases, which causes the accumulation of peroxide within the cell, leading to cell death.5,6 This antifungal drug is available in different dosage forms, including topical, oral, and parenteral, depending on the type and severity of infection.7–9 Miconazole has been extensively used for the treatment of dermatophytosis, cutaneous mycosis, pityriasis, fungal vaginitis, and oropharyngeal candidiasis.10–13
Various approaches have been used to overcome its limitation of poor water solubility, such as the addition of surfactants, co-precipitation techniques, or by incorporating miconazole into colloidal carrier systems, including lipid nanoparticles.14–16 These systems have been shown to enhance the antifungal activity of miconazole.4 To date, few authors have attempted to enhance the oral bioavailability of miconazole, and the results are inconsistent.17,18
Solid lipid nanoparticles (SLNs) were developed at the beginning of the 1990s as an alternative particulate delivery system to liposomes, emulsions, microparticles, and polymeric nanoparticulate systems. SLNs combine the advantages of those systems, such as controlled release, biodegradability, biocompatibility, and the protection of the incorporated drug against chemical degradation.19 However, SLNs simultaneously overcome the drawbacks of other colloidal carrier systems.20,21 SLNs have drawn major attention as novel colloidal drug carriers for the administration of drugs by several routes, such as oral, topical, parenteral, ophthalmic, and rectal.22–25 SLNs can be prepared using various methods: high-pressure homogenization, ultrasonication/high-speed homogenization, solvent emulsification–diffusion, solvent evaporation, double emulsion, and microemulsion.26–28
The aim of this study was to prepare and evaluate miconazole-loaded SLN (MN-SLN) formulations for oral administration to provide an innovative way to enhance the antifungal activity of this drug. Furthermore, in vitro antifungal activity and in vivo pharmacokinetic studies were conducted using the optimal formulations.
Materials and methods
Miconazole base was received as a kind gift from Pfizer, Inc. (New York, NY, USA). Precirol ATO5, Compritol 888 ATO, Gelucire, and Labrasol were obtained from Gattefosse (St-Priest, France). Cholesteryl stearate was purchased from Fluka Chemical Company (Buchs, Switzerland). Cremophor RH40 and Poloxamer 188 were purchased from Sigma-Aldrich (St Louis, MO, USA). Lecinol was a kind gift from Nikko chemicals Co., Ltd (Tokyo, Japan). All other reagents and chemicals were of analytical grade.
Methods
Measurement of the solubility of miconazole in lipids
The method depends on the measurement of the partitioning behavior of the miconazole base in various lipids (Precirol ATO5, Cholesteryl stearate, Nikkomulese, Tri-fat, and Compritol 888 ATO). Miconazole base (25 mg) was dispersed in a mixture of a melted lipid (500 mg) and 0.5 mL of hot distilled water with continuous shaking for 5 minutes in a hot water bath at 75°C. After cooling and centrifugation at 15,000 rpm and 10°C for 30 minutes, an aqueous phase was separated. Then, the samples were analyzed for their miconazole base content by measuring the absorbance using ultraviolet spectroscopy at 274 nm, for the determination of the amount dissolved in aqueous phase. The amount dissolved in lipid phase was determined indirectly by subtracting the amount dissolved in water from the total amount of miconazole used during the preparation. Then, the partition coefficient for miconazole in different mixtures was determined.
Preparation of MN-SLNs
MN-SLNs were prepared by a hot emulsification/ultrasonication method. According to the partitioning results, Precirol ATO5 was selected as a solid lipid for the formulation of MN-SLNs. Miconazole powder (1.5%), Precirol ATO5 (2%), and Lecinol (0.5%) were dissolved in a mixture of chloroform and methanol (1:1) (20 mL). The solvent was then completely removed using a rotary evaporator. The drug-embedded lipid layer was melted by heating at 75°C. An aqueous phase was then formulated by dissolving one of the surfactants (2.5%), such as Gelucire, Poloxamer, Labrasol, or Cremophor RH40, in double-distilled water and adding it to the molten lipid phase. This was followed by homogenization for 3 minutes. Coarse hot oil in a water emulsion was obtained, which was then ultrasonicated using a probe sonicator. Finally, the obtained hot nanoemulsion was allowed to cool to room temperature to prepare MN-SLNs. The impact of varying the critical parameter variables on the preparation of different formulations is shown in the following sections.
SLN preparation
SLNs were prepared by varying the surfactant types (Gelucire, Poloxamer 188, Labrasol, and Cremophor RH40) and ultrasonication time (3 and 7 minutes), while the surfactant concentrations remained constant at 2.5% and the homogenization time was 3 minutes.
Particle size and zeta potential measurements
The hydrodynamic diameter and zeta potential of the resulting dispersions were measured by dynamic light scattering using a Zetatrac machine (Microtrac, Inc., PA, USA).
Measurement of the entrapment efficiency
The SLN dispersion was centrifuged at 5,000 rpm for 30 minutes. The quantity of free drug in the dispersion medium was determined. The amount of miconazole in the aqueous phase was recorded at 274 nm.
Encapsulation efficiency was calculated as follows:
Encapsulation efficiency (%) + 100 – (Fs/Ts × 100) | (1) |
where Fs is the free soluble drug and Ts is the initial amount of drug added during the preparation of MN-SLNs.
Drug release study
A drug release study was carried out following the dialysis bag method.29 An MN-SLN suspension was placed in dialysis bags that had a 12 kDa molecular weight cutoff (Sigma-Aldrich) and was then immersed in 250 mL of 0.1 N HCl (pH =1.2) at 37°C as a dissolution apparatus with a paddle rotating at 50 rpm for 2 hours. Then, the pH was increased to 7.4 for the remaining 10 hours. An aliquot of the sample (5 mL) was taken from the dissolution medium at different time intervals (0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, and 12 hours) and analyzed for its miconazole content at 247 nm.
Determination of the flow properties
The flow properties of pure miconazole and its SLN formulations were described by the following methods:
Bulk and tapped densities method
Two grams of tested powder was weighed and poured into a 10 mL graduated cylinder, and then, the cylinder was tapped three times at 3-second intervals from a height of 2.5 cm. The volume of the powder (bulk volume) was recorded, which was used to calculate the bulk density according to the following equation:
|
|
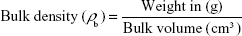
The tapped density of the drug was determined by tapping the cylindrical measure several times until a constant volume was achieved (tapped volume), which was used to calculate the tapped density of the drug according to the following equation:
|
|
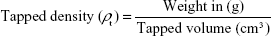
After calculating the bulk and tapped density, two parameters indicating the flowability were determined: the Hausner ratio and Carr’s index.
The Hausner ratio is the ratio between the tapped density and the bulk density, and Carr’s index can be estimated from the following equation:
|
|
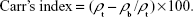
The obtained value gives an idea of the flow characteristic of powder particles.
Angle of repose method
By using a fixed-base funnel method, the angle of repose was measured as follows: The powder sample, either miconazole alone or MN-SLN, was allowed to fall through a funnel that was hung 2 cm above the surface until the apex of the pile reached the lower tip of the funnel. Then, the diameter and the height of the pile were measured and used to calculate the angle of repose according to the following equation:
Tan θ = h/r | (5) |
where θ is the angle of repose, h is the pile height, and r is the radius of the formed pile.
Triplicate samples were carried out for each experiment.
Assessment of antifungal activity
In vitro antifungal studies were conducted against Candida albicans in Sabouraud dextrose agar medium (SDA).
Inoculum preparation
Three to five colonies of standard strain C. albicans were suspended in 2 mL of sterile normal saline and simultaneously vortexed. McFarland standards (0.5 meq) were used to adjust the turbidity of the homogenous suspension. A sterile swab was dipped into the suspension and swabbed on dried plates containing SDA to obtain a lawn culture.
Disk diffusion method
Sterile filter paper disks (6 mm) were placed and inoculated on dried SDA plates. Then, 100 μL of the MN-SLN suspension in sterile water was placed on the disk. The plates were incubated at 25°±1°C. The zone of inhibition was observed around the disk after 1 and 2 days, respectively. Ethanol (99.9%) was used as a positive control. Data were analyzed using one-way analysis of variance (ANOVA). The level of significance was set at P<0.05.
In vivo pharmacokinetic study
An in vivo drug absorption study was conducted according to the institutional guidelines of the Animal Ethics Committee of the Faculty of Pharmacy, King Abdulaziz University, Jeddah, Saudi Arabia. Twelve albino male rabbits weighing between 2 and 2.5 kg were used in this study. Based on in vitro and antifungal activity results, negatively charged MN-SLN formulations were selected for in vivo evaluation. These formulations were composed of 150 mg of miconazole, 200 mg of Precirol ATO5 as a solid lipid, 250 mg of Cremophor RH40 as a surfactant, 50 mg of Lecithin, and 10 mg of Dicetylphosphate and were prepared by 3 minutes of homogenization followed by 7 minutes of an ultrasonication method.
The rabbits were kept in a fasting condition for 24 hours before the experiment was commenced. In this experiment, two groups of rabbits (six rabbits/group) were orally administered with either a miconazole capsule (as a control) or its SLN formulation (10 mg/kg). Suspensions of formulations in 5 mL of normal saline were orally administered via gastric intubation.
Blood samples (2 mL) were collected before and after drug administration at different time intervals (0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 9, 12, and 24 hours). The obtained serum samples were frozen at −20°C until the analysis of high-performance liquid chromatography was performed.
Statistical analysis for pharmacokinetic data
Cmax and Tmax were calculated using the plasma concentration–time curve in the WinNonlin™ Nonlinear Estimation Program. ANOVA was employed to assess the significance of the difference between the Tmax, Cmax, and area under plasma concentration-time curve from time zero to infinity (AUC0–∞) data of the miconazole obtained from the control capsule and the MN-SLN formulations (P≤0.05) using the statistical product and service solutions program. The area under plasma concentration-time curve from time zero to 24 hrs (AUC0–t) was calculated using the linear trapezoidal rule. In addition, the AUC0–∞ and relative bioavailability (BAR) of the MN-SLNs were calculated using the following equation:30
|
|
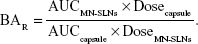
Results and discussion
Screening of lipid phase
The solubility of miconazole base in lipids was assessed by measuring the ratio of the amount of miconazole dissolved in the lipid phase to the amount of miconazole dissolved in the aqueous phase. The results indicated that the highest partition coefficient (14.6) was observed in Precirol ATO5 which was selected as a core lipid for the MN-SLN formulations, whereas it was 11.3, 8.6, 6.7, and 5.9 in cases of Cholesteryl stearate, Nikkomulese, Trifat, and Compritol 888 ATO, respectively. This could be attributed to the fact that Precirol ATO5 has a longer-chain fatty alcohol in its structure than the other lipids used, which results in the creation of a less ordered solid lipid matrix, leaving enough space to accommodate the drug molecule.31
Preparation and characterization of miconazole SLNs
MN-SLNs were successfully produced by hot homogenization followed by ultrasonication. Figures 1 and 2 indicate that the ultrasonication time has a significant impact on the particle size, while the incorporation of a charge-inducing agent during the preparation of the MN-SLN has a significant effect on the zeta potential. Moreover, the results indicated that the hydrodynamic diameter of the MN-SLNs containing Cremophor RH40 as a surfactant was 22 nm. The reduction in particle size during the production of the SLNs leads to an increase in the attraction force between the particles, which increases the surface tension at the interface, leading to physical instability.32 The incorporation of Cremophor RH40 as a surfactant in the formulation imparts a repulsion force between the nanoparticles and reduces not only the surface energy but also the interfacial tension of the system.33
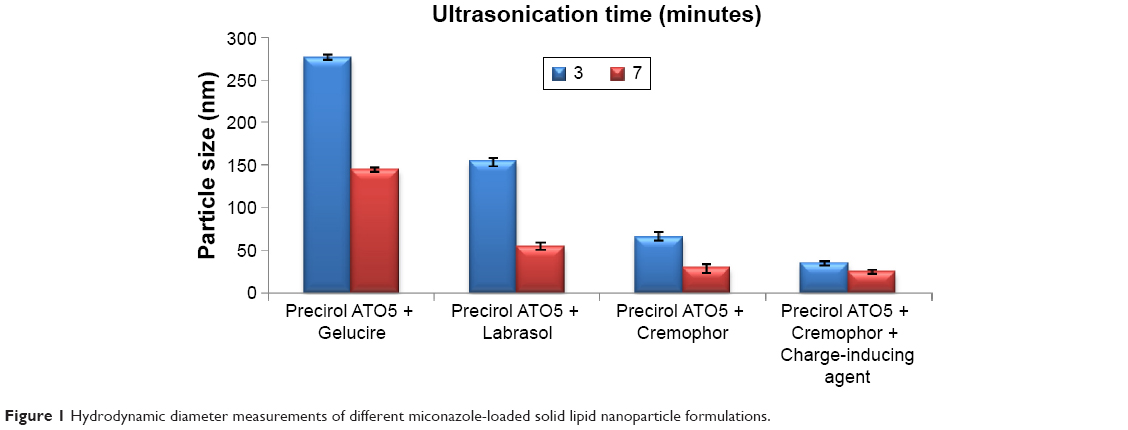 | Figure 1 Hydrodynamic diameter measurements of different miconazole-loaded solid lipid nanoparticle formulations. |
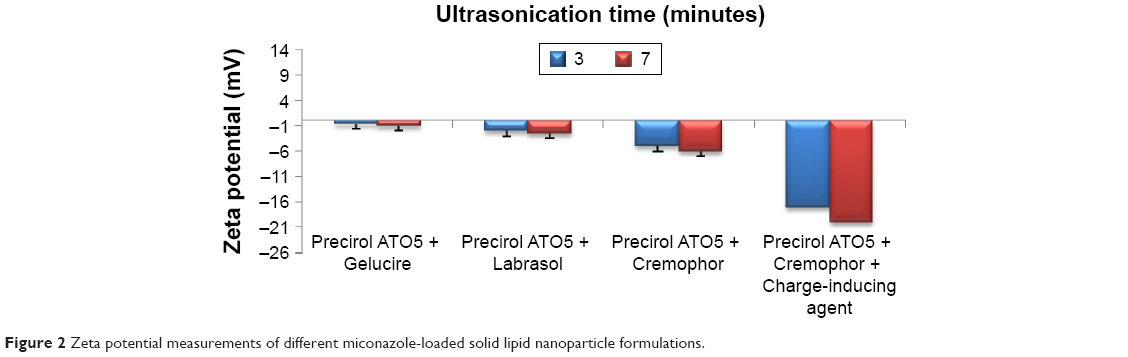 | Figure 2 Zeta potential measurements of different miconazole-loaded solid lipid nanoparticle formulations. |
Drug release study
According to the particle size and entrapment efficiency, the release of the optimal MN-SLN formula containing 150 mg of miconazole, 200 mg of Precirol ATO5 as a solid lipid, 250 mg of Cremophor RH40 as a surfactant, 50 mg of lecithin, and 10 mg of dicetylphosphate, which was prepared by 3 minutes of homogenization followed by 7 minutes of ultrasonication, was compared with the release of pure drug. MN-SLNs displayed 90% release at the end of the test, whereas the percentage of miconazole released in the case of the plain drug powder was only 50%, as shown in Figure 3. The release of miconazole from the MN-SLN formulations displayed a biphasic drug release pattern with a burst release within 2 hours (~45% of miconazole were released) followed by a sustained release.34 The reason for the initial burst in the release profile may be due to the drug adsorbed on the surface of the SLNs. The slow release in the later stage was attributed to the fact that the solubilized or dispersed drug can only be released slowly from the lipid matrices through dissolution and diffusion mechanisms.
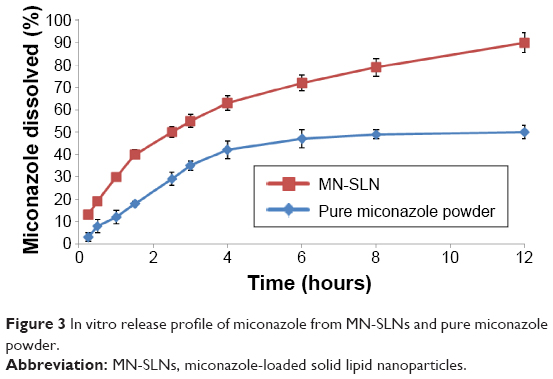 | Figure 3 In vitro release profile of miconazole from MN-SLNs and pure miconazole powder. |
Flow properties
The flow properties of the miconazole in its pure state and the MN-SLN formulations were confirmed by the values of the Hausner ratio, Carr’s index, and angle of repose.
According to United States Pharmacopeia 29-National Formulary 24 (USP 29-NF24), materials that have Hausner ratios ranging from 1.26 to 1.34, Carr’s index from 21 to 25, and angle of repose from 31° to 45° are likely to flow. This means that the miconazole powder has bad flowability as the Hausner ratio, Carr’s index, and angle of repose were 1.6°, 35°, and 55°, respectively. On the other hand, the flow parameters for the MN-SLNs were 1.021 for the Hausner ratio, 15.21% for Carr’s index, and 22.2° for angle of repose, which indicate an enhancement in all the flowability parameters. The SLNs have good flowability and can be incorporated during manufacturing processes without a need for the addition of a glidant, which has deleterious effects on the solubility and bioavailability of some drugs.
Assessment of antifungal activity
The antifungal efficacies of miconazole SLNs against C. albicans were investigated. As shown in Figure 4, marketed miconazole capsules showed an inhibition zone that was better than miconazole powder alone at 14 and 9 mm; however, the MN-SLNs showed the maximum inhibition zone of 22 mm. These results indicated that the formulation of miconazole as MN-SLNs enhanced the antifungal activity. This could be due to the small particle size (nanoscale) of the SLNs, which enhances the permeation across the cell membrane of C. albicans fungus.
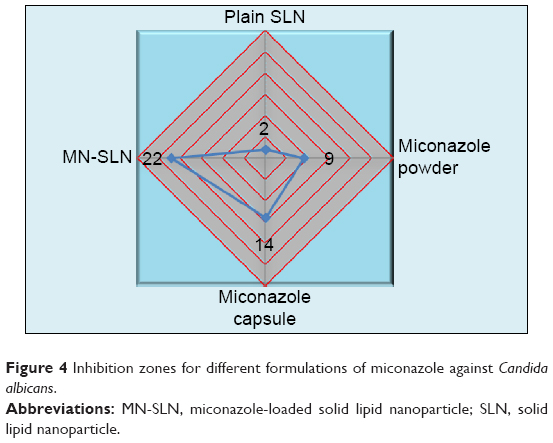 | Figure 4 Inhibition zones for different formulations of miconazole against Candida albicans. |
In vivo pharmacokinetic study
There was a statistically significant difference in the Tmax, Cmax, and AUC0–∞ data between the MN-SLNs and the marketed capsules (Table 1 and Figure 5) at P≤0.05. The mean AUC0–∞ for the marketed capsules was significantly different from the AUC0–∞ for the MN-SLNs. These results confirmed that the formulation of MN-SLNs increased the bioavailability of miconazole by >2.5-fold.
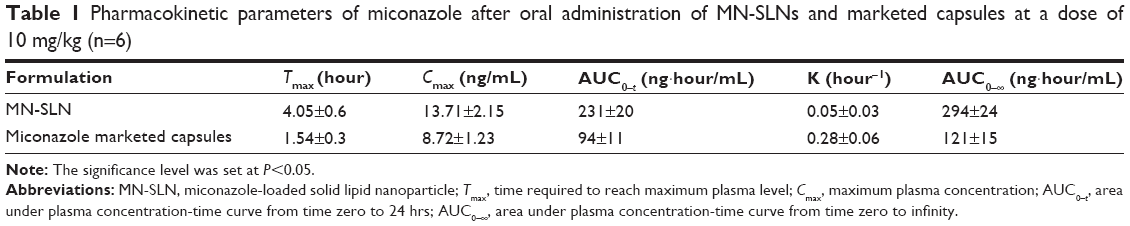 | Table 1 Pharmacokinetic parameters of miconazole after oral administration of MN-SLNs and marketed capsules at a dose of 10 mg/kg (n=6) |
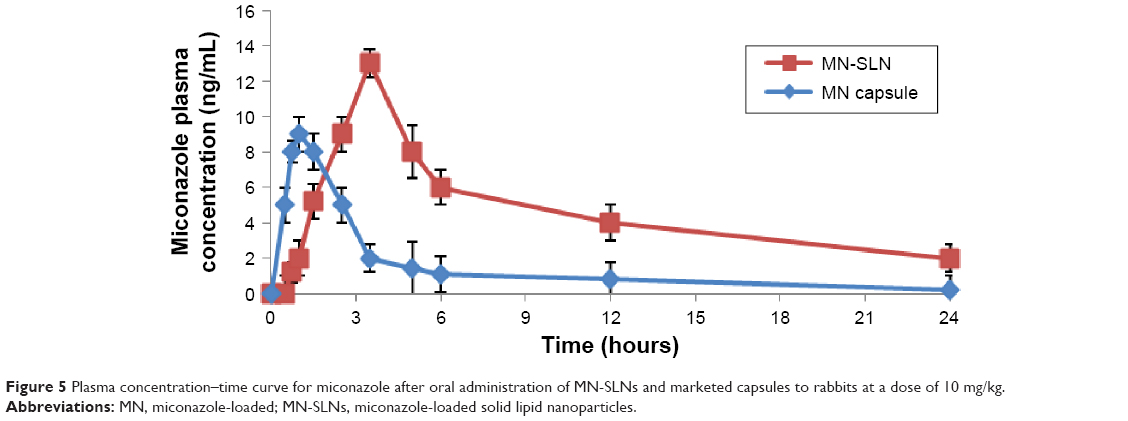 | Figure 5 Plasma concentration–time curve for miconazole after oral administration of MN-SLNs and marketed capsules to rabbits at a dose of 10 mg/kg. |
These results indicated that the MN-SLN formulation enhanced the miconazole absorption and permeation characteristics across the membrane of gastrointestinal tract. The results also revealed that MN-SLNs can significantly modify the pharmacokinetic profile and can increase the drug bioavailability by >2.5-fold in comparison with the marketed oral capsule formulation, which was because miconazole possesses poor aqueous solubility. The preparation of this drug as an SLN enhanced not only its solubility but also its tissue permeability. Furthermore, the pharmacokinetic characteristics of the drug upon delivery in an SLN formulation are dictated by the properties of the SLN rather than by the physicochemical characteristics of the drug molecule. Additionally, the presence of Cremophor as a surfactant in the SLN formula causes a steric hindrance that helps reduce the tissue uptake by evading the reticuloendothelial system, which will improve the residence time of SLNs in the blood circulation.22
Conclusion
The formulation of miconazole in oral SLNs, which is a novel drug delivery system, enhanced the bioavailability by >2.5-fold compared with the marketed capsules and was more efficient in the treatment of candidiasis. The optimum MN-SLN formula was composed of 1.5% miconazole, 2% Precirol ATO5, 0.5% Lecithin, 2.5% Cremophor RH40, 0.5% Lecinol, and 0.1% Dicetylphosphate. The usage of this formulation could eliminate the major drawbacks of the conventionally used capsules. MN-SLNs could be a promising carrier for the oral delivery of miconazole.
Acknowledgment
The authors gratefully acknowledge the Deanship of Scientific Research, King Abdulaziz University, Jeddah, Saudi Arabia (under Grant No D1435-869-166) for its technical and financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
Bensadoun R-J, Daoud J, El Gueddari B, et al. Comparison of the efficacy and safety of miconazole 50-mg mucoadhesive buccal tablets with miconazole 500-mg gel in the treatment of oropharyngeal candidiasis. Cancer. 2008;112(1):204–211. | ||
Mendes AI, Silva AC, Catita JA, et al. Miconazole-loaded nanostructured lipid carriers (NLC) for local delivery to the oral mucosa: improving antifungal activity. Colloids Surf B Biointerfaces. 2013;111:755–763. | ||
Pedersen M, Edelsten M, Nielsen VF, et al. Formation and antimycotic effect of cyclodextrin inclusion complexes of econazole and miconazole. Int J Pharm. 1993;90(3):247–254. | ||
Jain S, Jain S, Khare P, et al. Design and development of solid lipid nanoparticles for topical delivery of an anti-fungal agent. Drug Deliv. 2010;17(6):443–451. | ||
Fothergill AW. Miconazole: a historical perspective. Expert Rev Anti Infect Ther. 2006;4(2):171–175. | ||
van den Bossche H. Biochemical effects of miconazole on fungi – I: effects on the uptake and/or utilization of purines, pyrimidines, nucleosides, amino acids and glucose by Candida albicans. Biochem Pharmacol. 1974;23(4):887–899. | ||
Nafee NA, Ismail FA, Boraie NA, et al. Mucoadhesive buccal patches of miconazole nitrate: in vitro/in vivo performance and effect of ageing. Int J Pharm. 2003;264(12):1–14. | ||
Tsutsumi S, Iida M, Tada N, et al. Characterization and evaluation of miconazole salts and cocrystals for improved physicochemical properties. Int J Pharm. 2011;421(2):230–236. | ||
Shahzadi I, Masood MI, Chowdhary F, et al. Microemulsion formulation for topical delivery of miconazole nitrate. Int J Pharm Sci Rev Res. 2014;24(6):30–36. | ||
Ellepola AN, Samaranayake LP. Antimycotic agents in oral candidosis: an overview: 2. Treatment of oral candidosis. Dent Update. 2000;27(4): 165–174. | ||
Peira E, Carlotti ME, Trotta C, et al. Positively charged microemulsions for topical application. Int J Pharm. 2008;346(1–2):119–123. | ||
Sabri LA, Sulayman HT, Khalil YI. An investigation release and rheological properties of miconazole nitrate from Emulgel. Iraqi J Pharm Sci. 2009;18(2):26–31. | ||
Cerdeira AM, Mazzotti M, Gander B. Miconazole nanosuspensions: influence of formulation variables on particle size reduction and physical stability. Int J Pharm. 2010;396:210–218. | ||
Jacobsen J, Bjerregaard S, Pedersen M. Cyclodextrin inclusion complexes of antimycotics intended to act in the oral cavity-drug supersaturation, toxicity on TR146 cells and release from a delivery system. Eur J Pharm Biopharm. 1999;48(3):217–224. | ||
Levy MY, Polacheck I, Barenholz Y, et al. Efficacy evaluation of a novel submicron miconazole emulsion in a murine cryptococcosis model. Pharm Res. 1995;12(2):223–230. | ||
Müller RH, Shegokar R, Keck CM. 20 years of lipid nanoparticles (SLN & NLC): present state of development & industrial applications. Curr Drug Discov Technol. 2011;8(3):207–227. | ||
Tenjarla S, Puranajoti P, Kasina R, et al. Preparation, characterization, and evaluation of miconazole–cyclodextrin complexes for improved oral and topical delivery. J Pharm Sci. 1998;87(4):425–429. | ||
Hostetler JS, Hanson LH, Stevens DA. Effect of cyclodextrin on the pharmacology of antifungal oral azoles. Antimicrob Agents Chemother. 1992;36(2):477–480. | ||
Muller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | ||
Boltri L, Canal T, Esposito P, et al. Lipid nanoparticles: evaluation of some critical formulation parameters. Proc Int Symp Control Release Bioact Mater. 1993;20(20):346–347. | ||
Yadav P, Soni G, Mahor A, et al. Solid lipid nanoparticles: an effective and promising drug delivery system-A review. Int J Pharm Sci Res. 2014;5(4):1152. | ||
Hosny KM, Aljaeid BM. Sildenafil citrate as oral solid lipid nanoparticles: a novel formula with higher bioavailability and sustained action for treatment of erectile dysfunction. Expert Opin Drug Deliv. 2014;11(7):1015–1022. | ||
Munster U, Nakamura C, Haberland A, et al. RU 58841-myristate-prodrug development for topical treatment of acne and androgenetic alopecia. Pharmazie. 2005;60(1):8–12. | ||
Wissing SA, Kayser O, Müller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev. 2004;56(9):1257–1272. | ||
Cavalli R, Gasco MR, Chetoni P, et al. Solid lipid nanoparticles (SLN) as ocular delivery system for tobramycin. Int J Pharm. 2002;238(1–2):241–245. | ||
Sinha VR, Srivastava S, Goel H, et al. Solid lipid nanoparticles (SLN’S)-trends and implications in drug targeting. Int J Adv Pharm Sci. 2010;1(3):212–238. | ||
Ekambaram P, Sathali AAH, Priyanka K. Solid lipid nanoparticles: a review. Sci Rev Chem Commun. 2012;2(1):80–102. | ||
Kamble MS, Vaidya KK, Bhosale AV, et al. Solid lipid nanoparticles and nanostructured lipid carriers-an overview. Int J Pharm Chem Biol Sci. 2012;2(4):681–691. | ||
Elshafeey A, Bendas E, Mohamed O. Intranasal microemulsion of sildenafil citrate: in vitro evaluation and in vivo pharmacokinetic study in rabbits. AAPS PharmSciTech. 2009;10(2):361–367. | ||
Yang Z, Gao S, Wang J, et al. Enhancement of oral bioavailability of 20(S)-ginsenoside Rh2 through improved understanding of its absorption and efflux mechanisms. Drug Metab Dispos. 2011;39(10):1866–1872. | ||
Sanna V, Caria G, Mariani A. Effect of lipid nanoparticles containing fatty alcohols having different chain length on the ex vivo skin permeability of econazole nitrate. Powder Technol. 2010;201(1):32–36. | ||
Tanvir S, Qiao L. Surface tension of nanofluid-type fuels containing suspended nanomaterials. Nanoscale Res Lett. 2012;7(1):226. | ||
Uner M, Wissing SA, Yener G, et al. Influence of surfactants on the physical stability of solid lipid nanoparticle (SLN) formulations. Pharmazie. 2004;59(4):331–332. | ||
Doijad RC, Manvi FV, Godhwani DM, et al. Formulation and targeting efficiency of cisplatin engineered solid lipid nanoparticles. Indian J Pharm Sci. 2008;70(2):203–207. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.