Back to Journals » Journal of Inflammation Research » Volume 18
METTL3/IGF2BP2-Mediated m6A RNA Methylation Drives Alveolar Macrophage-Dependent Neutrophil Recruitment in Cigarette Smoke-Induced COPD
Authors Lin A, Wang J, Wang L, Zhang Y
Received 30 April 2025
Accepted for publication 17 October 2025
Published 29 October 2025 Volume 2025:18 Pages 15033—15047
DOI https://doi.org/10.2147/JIR.S532958
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Cynthia Koziol-White
Andi Lin,1,* Jian Wang,2,* Lixing Wang,3 Yaping Zhang3,4
1The Second Clinical Medical College of Fujian Medical University, Quanzhou, Fujian, 362000, People’s Republic of China; 2Department of Pathology, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian, 362000, People’s Republic of China; 3Clinical Center for Molecular Diagnosis and Therapy, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian, 362000, People’s Republic of China; 4Fujian Key Laboratory of Lung Stem Cells, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian, 362000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yaping Zhang, Email [email protected]
Introduction: Chronic inflammatory infiltration caused by cigarette smoke is one of the primary characteristics that define chronic obstructive pulmonary disease (COPD), but the epigenetic mechanisms governing immune cell crosstalk remain poorly defined. This study aims to elucidate the critical role of m6A RNA methylation in modulating alveolar macrophage-neutrophil interactions during COPD progression.
Methods: A mouse COPD model was employed, combined with mechanistic studies that included pharmacological inhibition, RNA stability assay, RIP, m6A-qPCR, and cell chemotaxis assay in cells, to investigate the effect of cigarette smoke exposure on m6A regulatory molecules and immune cell interactions.
Results: Cigarette smoke exposure upregulated METTL3 and IGF2BP2 expression in alveolar macrophages. METTL3-mediated m6A modification promoted the stability of CXCL8 mRNA in an IGF2BP2-dependent manner, leading to enhanced CXCL8 secretion and neutrophil recruitment. Concurrently, METTL3-mediated m6A modification stabilized ICAM-1 mRNA in endothelial cells, facilitating neutrophil adhesion and transmigration. This dual-cell mechanism synergistically amplifies neutrophilic inflammation in COPD.
Discussion: Our results uncover a novel epitranscriptional pathway through which cigarette smoke promotes neutrophilic inflammation via m6A-dependent regulation of both CXCL8 in macrophages and ICAM-1 in endothelial cells. These findings position the METTL3/IGF2BP2/m6A axis as a central regulatory mechanism coordinating multicellular interactions in COPD pathogenesis and suggest its potential as a therapeutic target for modulating disease progression.
Keywords: cigarette, alveolar macrophages, METTL3, STM2457, COPD, CXCL8
Introduction
Chronic obstructive pulmonary disease (COPD) is a long-term respiratory disease caused by inflammation of the airways and lungs, resulting in persistent and progressive airflow obstruction.1 Presently, COPD ranks as the fourth leading cause of death globally, affecting approximately 400 million people and posing a global public health challenge.2 Although existing studies have revealed multiple pathogenic mechanisms, such as chronic inflammation and oxidative stress, effective treatment options are still limited, especially in the late stages of the disease.
The onset of COPD is primarily triggered by exposure to harmful environmental factors, notably cigarette smoke, and is pathologically characterized by chronic bronchial and pulmonary inflammation. A hallmark of this inflammation is the persistent infiltration of immune cells—particularly neutrophils and alveolar macrophages (AMs)—which collectively contribute to parenchymal lung damage.3 As the primary immune cells in the alveolar spaces, AMs play a central role in phagocytosing foreign particles and pathogens, as well as orchestrating inflammatory responses.4,5 In COPD, the number of AMs is significantly elevated.6 These activated macrophages secrete heightened levels of pro-inflammatory cytokines and chemokines, including interleukin-8 (CXCL8), CXCL1, and CXCL2, which are critical for recruiting neutrophils to sites of inflammation. Neutrophils represent major effector cells in COPD-related inflammation. Upon recruitment, they migrate from the circulation into lung tissue by adhering to activated vascular endothelial cells. Under inflammatory conditions, endothelial cells upregulate adhesion molecules such as ICAM-1 and VCAM-1, facilitating neutrophil rolling, firm adhesion, and transmigration into the alveolar spaces. Once activated, neutrophils release a range of damaging mediators—including proteases, reactive oxygen species, and additional cytokines—that perpetuate tissue destruction and amplify inflammatory responses across airways, parenchyma, and vasculature.7,8 Thus, the coordinated activation of AMs and subsequent neutrophil recruitment and infiltration form a vicious cycle that drives disease progression in COPD. Therapeutic strategies aimed at interrupting this cascade—particularly by targeting neutrophil migration or activation—hold significant promise for mitigating inflammation and tissue damage in COPD.9,10
N6-methyladenosine (m6A) is the most prevalent internal chemical modification on eukaryotic mRNA, playing a dynamic and pivotal role in post-transcriptional gene regulation. This modification is orchestrated by three classes of regulatory proteins: methyltransferases (“writers”), such as the METTL3-METTL14 complex, which catalyze m6A deposition; demethylases (“erasers”), including FTO and ALKBH5, which remove the mark; and binding proteins (“readers”), such as the YTHDF family, IGF2BPs, and HNRNPC, which interpret the m6A signal and dictate mRNA fate. Specifically, reader proteins can differentially influence mRNA stability—YTHDF2 often promotes decay, whereas IGF2BPs enhance stability and translation.11–14 Through these mechanisms, m6A modification extensively regulates RNA processing and metabolism, thereby influencing critical biological processes including cell differentiation, proliferation, and numerous physiological and pathological pathways.15,16 Growing evidence underscores the significance of m6A RNA methylation in disease mechanisms; for instance, in cervical cancer, METTL3 stabilizes PD-L1 and SOX2 mRNAs in an m6A-IGF2BP2-dependent manner,17 while the METTL3/YTHDF2 axis promotes degradation of circRNF13 to enhance radiosensitivity by modulating CXCL1 expression.18
While our previous research demonstrated that METTL3-mediated m6A modification drives epithelial-mesenchymal transition (EMT) in COPD airway epithelium via the SOCS3/STAT3/SNAI1 pathway,19 the role of m6A in immune dysregulation—particularly in alveolar macrophage-mediated neutrophil recruitment—remains largely unexplored in COPD. Given the central role of neutrophilic inflammation and macrophage activation in COPD pathogenesis, we hypothesize that m6A modification serves as a key regulatory layer in immune cell crosstalk. In this study, we aim to investigate the function and mechanism of m6A in regulating alveolar macrophage-dependent neutrophil recruitment, seeking to bridge this knowledge gap and potentially identify novel diagnostic or therapeutic targets for COPD.
Methods
Analysis of Public Datasets from COPD Patients
To characterize cellular heterogeneity in the COPD lung at single-cell resolution, we analyzed the publicly available scRNA-seq dataset GSE173896 from the Gene Expression Omnibus (GEO), which includes lung tissue samples from 9 COPD patients, 4 non-COPD smokers (smoker controls), and 3 never-smokers (healthy controls). We utilized the preprocessed count matrices, principal component analysis (PCA) embeddings, and cell cluster annotations generated by the original authors using the Seurat package (v4.4.3).20 Subcluster analysis was specifically performed on macrophage and endothelial cell populations to compare transcriptional profiles across the three groups. In addition, we interrogated another independent dataset, GSE130928, which contains gene expression data from alveolar macrophages obtained via bronchoalveolar lavage fluid (BALF) of COPD patients, smokers, and non-smokers, thereby enabling further validation of alveolar macrophage-specific signatures.6
Establishment of Cigarette Smoke-Induced COPD Mouse Model
Male C57BL/6 mice (6–8 weeks old, weight 20 ± 2 g) were maintained under specific pathogen-free conditions with a 12-hour light/dark cycle at 22 ± 1°C and 50 ± 10% humidity. Animals were provided ad libitum access to autoclaved food and water. After one week of acclimatization, mice were randomly divided into two experimental groups (n = 15/group): Control group (Received ambient air exposure) and COPD group (Subjected to chronic cigarette smoke exposure). The COPD model was established via a standardized CS exposure protocol adapted from.21 Mice in the COPD group were placed in a 30-L plexiglass exposure chamber and exposed to mainstream smoke generated from 12 commercially filtered cigarettes (Shishi brand, China) twice daily. This regimen was performed 5 days/week for 24 consecutive weeks. Control mice underwent identical handling in a separate chamber without CS exposure. After 24 weeks, mice were anesthetized by intraperitoneal injection of pentobarbital sodium (150 mg/kg). Finally, lungs were perfused with PBS via right ventricular puncture, harvested, and divided for histopathology.
Pathological semi-quantitative analysis of airway inflammation: for the assessment of airway inflammation, H&E-stained lung tissue sections were evaluated using a double-blinded protocol. Two experienced pathologists, who were blinded to the experimental groups, independently scored each section to minimize subjective bias. For each specimen, five non-overlapping high-power fields (×400 magnification) containing bronchial structures were randomly selected using a systematic sampling approach. Airway inflammation was semi-quantitatively scored based on the degree of inflammatory cell infiltration around the bronchial walls, following the modified scoring system.22 The final score for each sample was determined by calculating the mean of the scores provided by the two pathologists. All measurements and scoring were performed in triplicate to ensure consistency.
Primary Alveolar Macrophage Isolation
Bronchoalveolar lavage fluid (BALF) was collected through three instillations of 0.8 mL PBS per mouse. BALF cells underwent adherence selection (2 h, 37°C in RPMI-1640/10% FBS), yielding alveolar macrophages, and were lysed directly by TRIzol™ Reagent (Invitrogen, #15596026) to obtain total RNA.
Preparation of Cigarette Smoke Extract (CSE)
Cigarette smoke extract (CSE) can effectively simulate the effects of cigarette smoke on cells and is widely used in in vitro cell models for studying COPD. CSE was prepared according to an established method23 with modifications. Specifically, mainstream smoke from an unfiltered commercial cigarette (Shishi brand, China) was drawn through a peristaltic pump and bubbled into 10 mL culture medium. The smoke-bubbled medium was then sterilized by filtration through a 0.22-μm PVDF membrane, designated as 100% CSE stock solution. Absorbance standardization was performed at λ=320 nm using a spectrophotometer to calibrate each batch of CSE.
Cell Culture and Pharmacological Treatment
Human monocytic THP-1 cells (Procell, China) and endothelial EA.hy926 cells (Procell, China) were maintained under standard conditions (37°C, 5% CO2, 95% humidity). THP-1 cells were maintained in RPMI-1640 medium (Gibco, #11875093) containing 10% fetal bovine serum (FBS; Gibco #10270106) and 1% penicillin/streptomycin (Gibco, #15140122). EA.hy926 cells were cultivated in DMEM (Gibco, #11965092) supplemented with 10% FBS and 1% penicillin/streptomycin. For differentiation into macrophages, THP-1 cells were treated with 100 ng/mL phorbol 12-myristate 13-acetate (PMA; Biosharp, #BL1127A) over a 48-hour period. 5 μM STM2457 (MCE, #HY-134836) was used to treat differentiated THP-1 cells for 24 hours to inhibit the activity of METTL3.
RNA Interference
Double-stranded siRNAs targeting human IGF2BP2 and a non-targeting scramble control were designed and synthesized by GenePharma (Shanghai, China). IGF2BP2-siRNA: 5’-CCGCAUCAUCACUCUUAUCTT-3’ / 5’-GAUAAGAGUGAUGAUGCGGTT-3’; Scramble siRNA: 5’-UUCUCCGAACGUGUCACGUTT-3’ / 5’-ACGUGACACGUUCGGAGAATT-3’. siRNAs were transfected into PMA-differentiated THP-1 cells using Thermo Fisher Scientific’s Lipofectamine 3000 following the manufacturer’s protocol.
RT-qPCR Analysis
Total RNA was extracted with TRIzol (Invitrogen, #15596026CN) and quantified via NanoDrop™. cDNA synthesis was performed with 1 μg RNA using PrimeScript™ RT Reagent Kit (Takara, #RR047A). Quantitative PCR utilized TB Green™ Premix Ex Taq™ II (Takara, #RR820A) on a QuantStudio™ 6 system (Applied Biosystems) with the Human GAPDH Endogenous Reference Genes Primers (Sangon, #B662104) and the following parameters: METTL3-F: 5′-GTGATCGTAGCTGAGGTTCGTTCC; METTL3-R: 5′-CTCAATCTTGCGAGTGCCAGGAG; IGF2BP2-F: 5′-GGCAAGACCGTGAACGAACTG; IGF2BP2-R: 5′-CCGATAATTCTGACGATCACTTCCTC. Melt curve analysis confirmed amplification specificity. Relative mRNA expression was calculated via the 2(-ΔΔCt) method, normalized to GAPDH.
Western Blot Analysis
Protein lysates (15 μg/sample) were resolved on 15% Tris-glycine SDS-PAGE gels and transferred to PVDF membranes (Millipore, #IPVH00010). Membranes were blocked with 5% dry milk in TBST for 2 h at room temperature. Primary antibody incubations were performed overnight at 4°C in antibody dilution buffer, respectively, including METTL3 (Diagbio, #db15992), IGF2BP2 (Diagbio,#db16009), ICAM-1 (Proteintech, #60299-1-Ig), and β-actin (Beyotime, #AF5003). After the 1-hour incubation with species-matched HRP-conjugated secondary antibodies (Beyotime, #A0208, #A0216), the immunoreactive signals were detected by employing a highly sensitive ECL luminescence reagent (Beyotime, #P0018AS) and captured using the ImageQuant LAS 4000 system. The intensity of Western blot bands was quantified with ImageJ software.
mRNA Stability Analysis
Cells (5 × 105) were subjected to exposure to actinomycin D (Sigma, #A1410) with a concentration of 5 μg/mL for 0, 1,3, or 5 h to inhibit the generation of new RNA. Following this, the total RNA of cells was harvested, and the mRNA level of genes was detected by RT-qPCR.13
RNA Immunoprecipitation (RIP) Assay
To detect direct binding between IGF2BP2 and CXCL8 mRNA, the assay was performed using the RNA Binding Protein Immunoprecipitation (RIP) Kit (Sangon, #B605109). 1×107 cells PMA-differentiated THP-1 cells were seeded and lysed in RIP Lysis Buffer for 30min on ice, supernatant was collected and divided into three portions: 150 μL (IP), 150 μL (IgG), and 150 μL (Input). 50 μL ProteinA/G Magnetic Beads were washed twice with RIP Wash Buffer. The IP beads and IG beads were separately incubated with 5 μg anti-IGF2BP2 (Abcam, #ab117809) or Normal Rabbit IgG (kit provided) in 100 μL RIP Wash Buffer (4°C, 2 h, rotation). Then, the beads were washed three times to remove unbound antibody. Conjugated beads were mixed with 900 μL RIP Buffer and 150 μL cell lysis, incubated at 4°C overnight with rotation. Beads were washed five times with RIP Wash Buffer to remove contaminants. Beads were added with 500 μLTRIzol to extract RNA. Finally, RT-qPCR was performed with the following parameters: CXCL8-F: 5′-CTCTTGGCAGCCTTCCTGATTTC; CXCL8-R: 5′-GGGGTGGAAAGGTTTGGAGTATG. Relative enrichment was calculated via 2(-ΔΔCt).
m6A-qPCR Analysis
The m6A qPCR was performed using an m6A MeRIP kit (A&D Technology, #A-P-9018). Briefly, 10 µg of total RNA was fragmented into fragments in the Immuno Capture Buffer. The fragmented RNA was incubated with anti-m6A antibody or control antibody (IgG antibody) conjugated to magnetic Beads. m6A capture antibodies containing m6A target fragments were pulled down. Then, the enriched RNA was released, purified, and eluted, and finally subjected to real-time qPCR.
Neutrophil Chemotaxis Assay
To isolate human neutrophils for use in the cell chemotaxis assay, leukocytes were isolated from fresh blood samples of healthy donors (n=5, aged 18–30 years) using red blood cell lysis buffer (Biosharp, #BL503A), with approval from the Ethics Committee of The Second Affiliated Hospital of Fujian Medical University (Approval No. #2016 [46]). A Transwell migration assay was performed using transwell chambers (Corning, #3421). Briefly, 100 μL of leukocyte suspension (1×105 cells) was added to the upper chamber, while the lower chamber contained chemotactic supernatant collected from THP-1 cells treated with CSE and/or inhibitor for 12 hours, followed by an additional 12 hours in fresh medium. After 2 hours of incubation at 37°C with 5% CO2, cells that migrated to the lower chamber were collected for analysis.
Migrated cells in the lower chamber were analyzed by flow cytometry to quantify neutrophils. Two complementary gating strategies—classical FSC/SSC properties and CD45⁺/SSC‑high characteristics—were applied to identify neutrophils and ensure accurate counting, with results showing good consistency between both methods. To minimize inter-batch variation in FSC/SSC signals, a reference gating standard was established for each experiment using an aliquot of leukocytes from the same donor that had not undergone migration. This reference sample was used to define the FSC/SSC threshold, which was then consistently applied to all test samples.
Analytical Statistics
Data analysis was conducted using GraphPad Prism 9 software. The determination of statistical significance relied on appropriate tests—including t-test, one-way ANOVA, or two-way ANOVA—with the specific choice depending on the experimental design characteristics and data distribution patterns. All data are reported as the mean ± standard deviation (SD), which were obtained from three independent experiments. P-value < 0.05 was regarded as indicating statistical significance.
Results
Dysregulated Expression of Major m6A Enzymes and Chemokines in Alveolar Macrophages of COPD Patients
To understand the expression profiles of major m6A-modifying enzymes and chemokines within AMs associated with COPD, we examined single-cell RNA sequencing (scRNA seq) data of lung tissues from COPD patients, smokers, and healthy controls in the public database GSE173896. Unsupervised clustering of the integrated dataset identified 24 distinct cell clusters (Figure 1A), comprising 12 immune cell clusters, 5 epithelial cell clusters, 2 stromal cell clusters, and 5 clusters of unassigned/other cell types. Focusing specifically on macrophage populations derived from this analysis, we detected a marked increase in the expression of CXCL8 – a gene related to neutrophil chemokines – in AMs derived from COPD patients and smokers as opposed to non-smokers (Figure 1B), suggesting that smoking promotes the secretion of IL-8, and AMs are a significant source of this chemokine in the COPD lung microenvironment. Furthermore, the expression profile of key m6A regulatory enzymes was observed, indicating that the key regulatory enzymes are dysregulated in AMs of COPD patients. Specifically, abnormal expression patterns were detected for the major m6A “writer” enzyme METTL3 and the m6A “reader” protein IGF2BP2 (Figure 1C). These findings implicate dysregulated m6A RNA methylation as a potential regulatory mechanism operative in AMs during COPD pathogenesis. To validate this result, the public dataset GSE130928 database was used as the validation set to analyze the gene abundance data of alveolar macrophages isolated from BALF of patients with COPD, smokers, and non-smokers. The validation results demonstrated that the expression levels of METTL3 and IGF2BP2 in alveolar macrophages were notably elevated in the COPD group when compared to non-smokers (Figure 1D), consistent with the previous analysis results. Moreover, our analysis also identified a notable positive correlation between the abundance of IGF2BP2 and that of CXCL8 (Figure 1E), conjecturing that the reading protein IGF2BP2 may regulate the expression of CXCL8 in alveolar macrophages during COPD.
 |
Figure 1 The gene expression patterns of major m6A enzymes and CXCL8 in alveolar macrophages of COPD patients. (A) A Unified manifold approximation and projection (UMAP) map showing single-cell clustering of various cell types in the scRNA-seq dataset GSE173896. Different colors represent distinct cell populations of human lung cells. (B) Violin plots depicting the expression levels of CXCL8 in macrophage clusters from three groups: COPD, non-smoker, and smoker in GSE173896. (C) Violin plots illustrating the expression levels of METTL3 and IGF2BP2 in macrophage clusters from three groups: COPD, non-smoker, and smoker in GSE173896. (D) Bar graphs presenting the relative expression of METTL3 and IGF2BP2 in non–smokers (n=24), smokers (n=42), and COPD patients (n=22) in the GSE130928 database. Data are presented as mean ± standard deviation (SD). Statistical significance was determined by One-way ANOVA. ns for non - significant, ** for p < 0.01, *** for p < 0.001. (E) Scatter plot with a linear regression line showing the correlation between IGF2BP2 mRNA and CXCL8 mRNA expression in the GSE130928 database. The coefficient of determination (R2), p-value, and number of samples (n) are provided, indicating the strength and significance of the correlation. |
Abnormal Expression of METTL3 and IGF2BP2 in Alveolar Macrophages from in vivo and in vitro COPD Models
To check the abnormal expression of METTL3 and IGF2BP2 in alveolar macrophages of COPD, we efficiently established an in vivo mouse model of COPD through cigarette smoke exposure. Lung sections were processed for hematoxylin-eosin (HE) staining to facilitate subsequent observation. Compared with the control group, which presented a healthy pulmonary condition, the COPD model group demonstrated significant pathological changes. These changes included alveolar enlargement, destruction of alveolar septa, and increased inflammatory cell infiltration in the submucosa and around blood vessels (Figure 2A), highlighting the successful establishment of the COPD model. To demonstrate neutrophil infiltration in COPD, we performed immunohistochemical staining on lung tissue sections of mice using the specific marker Ly6g for neutrophils of mice, as shown in Figure 2B. A notable accumulation of Ly6g-positive neutrophils was observed in the COPD group, particularly in and around the airways and alveolar regions. Meanwhile, to assess airway inflammation in mice, a double-blind approach was used to evaluate H&E-stained lung tissue sections. The pathological semi-quantitative analysis of airway inflammation showed that the COPD group mice exhibited enhanced airway inflammation (Figure 2C). These results indicate that neutrophil infiltration is a prominent feature in the COPD lung.
 |
Figure 2 Abnormal expression of METTL3 and IGF2BP2 in alveolar macrophages from in vivo and in vitro COPD models. (A) Hematoxylin and eosin (H&E) staining of lung tissues from control and COPD mice. Scale bars: 200 μm (low magnification) and 100 μm (high magnification, insets). (B) Immunohistochemical staining of lung tissues from control and COPD subjects using an antibody against the mouse neutrophil marker Ly6g. Scale bars: 200 μm (low magnification) and 100 μm (high magnification, insets). (C) Quantification of airway inflammation scores in lung tissue from control and COPD mice. Data are expressed as mean ± standard deviation (SD), and statistical significance was assessed using the T-test. ****P < 0.0001. (D) Relative mRNA levels of METTL3 (left) and IGF2BP2 (right) in primary alveolar macrophages isolated from bronchoalveolar lavage fluid (BALF) of control and COPD subjects, determined by quantitative real-time PCR. Data are expressed as mean ± standard deviation (SD), and statistical significance was assessed using the T-test. **P < 0.01, ***P < 0.001. (E) The protein expression of METTL3 and IGF2BP2 in PMA-differentiated THP-1 cells (treated with 4% CSE or 0% CSE as the control) is shown via representative Western blot images (left) and the associated quantitative analysis (right). Representative Western blot images (left) and quantitative analysis (right) of METTL3 and IGF2BP2 protein expression in PMA-differentiated THP-1 cells treated with 4% CSE or 0% CSE (control). Data are expressed as mean ± standard deviation (SD), and statistical significance was assessed using Two-way ANOVA. **P < 0.01, ***P < 0.001. |
We further isolated primary alveolar macrophages from the BALF of COPD mice and analyzed the mRNA levels of METTL3 and IGF2BP2 using real-time qPCR analysis. Compared to the control group, the relative mRNA levels of METTL3 and IGF2BP2 in AMs from the COPD group were significantly increased (Figure 2D). This finding is in accordance with the previous results obtained from COPD patients, suggesting abnormal expression of METTL3 and IGF2BP2 in both the mouse model and human patients, and further validating the potential significance of these two factors in the pathogenesis of COPD.
Subsequently, THP-1 cells were induced to differentiate into macrophages using PMA. These differentiated macrophages were then exposed to CSE to establish an in vitro model of COPD. Western blot analysis was performed, and the result revealed markedly higher protein levels of METTL3 and IGF2BP2 in the CSE-exposed group (Figure 2E). The above findings confirmed the abnormal expression of METTL3 and IGF2BP2 in AMs and implied the necessity for further in-depth investigations into the regulatory functions.
METTL3 Regulates CSE-Induced Recruitment of Neutrophils and Secretion of CXCL8 in Macrophages
To investigate the regulatory role of m6A modification in macrophage-mediated neutrophil recruitment, we inhibited m6A methylation using the METTL3 inhibitor STM2457 and evaluated neutrophil chemotaxis via a transwell assay. Compared with the control, supernatant from CSE-treated PMA differentiated macrophages significantly enhanced neutrophil migration (Figure 3A), indicating that CSE stimulation promotes the secretion of chemotactic factors by macrophages. However, treatment with STM2457 markedly attenuated this chemotactic response, as shown by a reduction in the number of neutrophils migrating to the lower chamber (Figure 3A). These results offer crucial insights that m6A methylation plays a critical role in regulating the ability of macrophages to recruit neutrophils under CSE stimulation.
 |
Figure 3 METTL3 modulates neutrophil recruitment and CXCL8 expression in response to cigarette smoke extract via m6A modification. (A) Representative microscopy images (top, scale bar = 100 μm) and flow cytometry plots (middle and bottom) showing neutrophil counts in different treatment groups. Neutrophils were gated based on classical FSC/SSC properties and CD45⁺/SSC‑high properties. Quantification of neutrophil counts is shown in the bar graph below. Data are presented as mean ± SD; Statistical significance was determined by Two-way ANOVA. *P < 0.05, ****P < 0.0001. (B) ELISA quantification of interleukin-8 - 8 (IL-8) levels in THP-1-derived macrophages supernatants from different treatment groups. Data are mean ± SD; Statistical significance was determined by Two-way ANOVA. ****P < 0.0001. (C) Analysis of CXCL8 mRNA stability following STM2457 treatment in THP-1-derived macrophages by RT-qPCR. Data are presented as mean ± SD; **P < 0.01, ****P < 0.0001. (D) The m6A peak abundance of CXCL8 mRNA transcript in cells transfected with siRNA-control or siRNA-METTL3 was detected by MeRIP-Seq and visualized in Integrative Genomics Viewer (IGV). The red peaks represent immunoprecipitation (IP), and the blue peaks represent Input. (E) m6A modification enriches CXCL8 mRNA in PMA-differentiated THP-1 cells. RNA immunoprecipitation (RIP) assay was conducted using IgG (negative control) or an anti-m6A antibody (m6A-IP). Relative enrichment of CXCL8 mRNA was measured through qRT-PCR analysis. Data were shown as mean ± SD, while statistical significance was determined using the T-test. *P < 0.05. |
To further investigate whether m6A modification modulates macrophage-dependent neutrophil recruitment through chemokine signaling, we focused on IL-8 (CXCL8), a key chemokine released by alveolar macrophages that promotes neutrophil migration via CXCR1/CXCR2 binding. Using ELISA, we quantified IL-8 secretion under different treatment conditions. We found that CSE stimulation significantly increased IL-8 production by macrophages compared to controls. In contrast, inhibition of METTL3 with STM2457 markedly reduced CSE-induced IL-8 secretion (Figure 3B). These results suggest that m6A methylation contributes to enhancing the expression of IL-8 in macrophages.
To further study the role of m6A in CXCL8 post-transcriptional regulation, we first evaluated whether this modification affects the stability of mRNA. We used actinomycin D to inhibit transcription and monitored the decline of CXCL8 mRNA over time. Compared with the control group (DMSO), treatment with METTL3 inhibitor STM2457 significantly accelerated the degradation of CXCL8 mRNA (Figure 3C), indicating that m6A methylation enhanced its stability. This result is consistent with our previous MeRIP seq screening after knocking out METTL3.24 This screening identified CXCL8 as a METTL3 targeted gene, and inhibition of METTL3 significantly reduced m6A modification on CXCL8 mRNA (Figure 3D), confirming that METTL3 is the main methyltransferase responsible for its m6A methylation. Furthermore, m6A qPCR analysis confirmed significant enrichment of m6A-modified CXCL8 transcripts in macrophages relative to IgG control immunoprecipitations (Figure 3E). Together, these results demonstrate that CSE promotes METTL3-mediated m6A deposition on CXCL8 mRNA, increasing its stability and ultimately enhancing secretion of the neutrophil-attracting chemokine IL-8.
IGF2BP2 Regulates CSE-Induced Recruitment of Neutrophils and Secretion of CXCL8 in Macrophages
Based on our previous analysis revealing a significant positive correlation between IGF2BP2 and CXCL8 expression, we hypothesized that IGF2BP2 recognizes m6A sites on CXCL8 mRNA and regulates its stability. To test this, we depleted IGF2BP2 using siRNA in THP-1-derived macrophages. Chemotaxis assays showed that IGF2BP2 silencing markedly reduced the number of neutrophils attracted by these macrophages compared to the siRNA control (Figure 4A). Furthermore, IGF2BP2 silencing decreased CXCL8 secretion (Figure 4B) and reduced CXCL8 mRNA stability (Figure 4C). To further verify the direct binding interaction, we performed an RNA immunoprecipitation (RIP) assay using an anti-IGF2BP2 antibody. The results showed a significant enrichment of CXCL8 mRNA in the IGF2BP2 immunoprecipitates relative to the IgG control group (Figure 4D), indicating a specific association between IGF2BP2 and CXCL8 transcripts. Together, these findings support that IGF2BP2 acts as an m6A reader protein that directly binds to and stabilizes CXCL8 mRNA, thereby enhancing its expression.
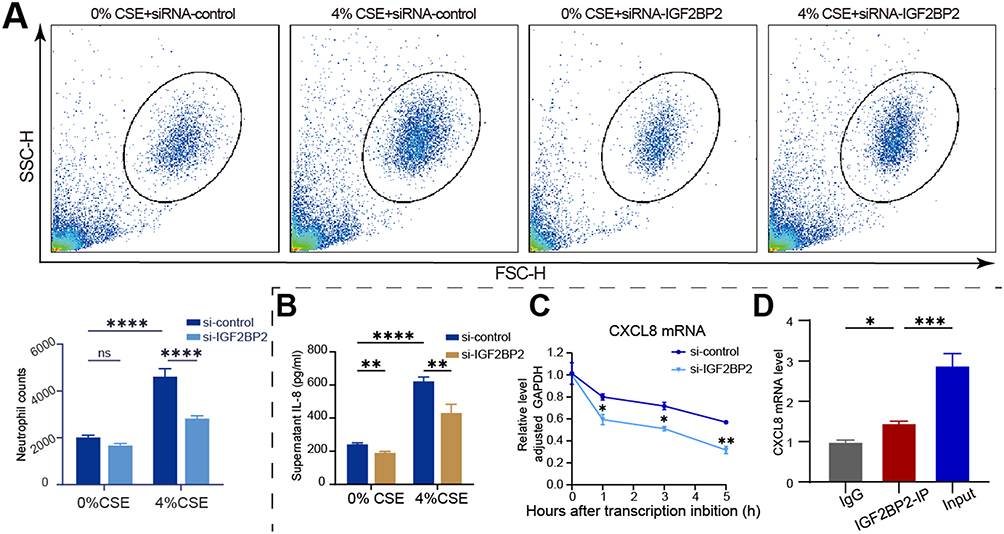 |
Figure 4 IGF2BP2 regulates recruitment of neutrophils and CXCL8 mRNA stability in macrophages in response to CSE. (A) Flow cytometry plots (with FSC-H and SSC-H) and quantification of neutrophil counts in the lower chamber of the transwell well to evaluate the effect of IGF2BP2 downregulation on THP-1-derived macrophage recruitment of neutrophils in response to CSE. Data are mean ± SD; Statistical significance was determined by Two-way ANOVA. ****P < 0.0001. (B) ELISA quantification of IL-8 levels in THP-1-derived macrophages’ supernatants from different treatment groups. Data are mean ± SD; Statistical significance was determined by Two-way ANOVA. **P < 0.01, ****P < 0.0001. (C) Analysis of CXCL8 mRNA stability following IGF2BP2 knockdown in THP-1-derived macrophages by RT-qPCR. Data are presented as mean ± SD; *P < 0.05, **P < 0.01. (D) RIP qPCR analysis of CXCL8 mRNA enrichment with IgG, IGF2BP2 – IP, and Input samples from THP-1-derived macrophages. Data are mean ± SD; One-way ANOVA was employed to determine statistical significance. *P < 0.05, ***P < 0.001. |
METTL3 Regulates ICAM-1 Expression in Vascular Endothelial Cells
Under inflammatory conditions, vascular endothelial cells become activated and upregulate the expression of adhesion molecules, including ICAM-1, which facilitates neutrophil binding, adhesion, rolling, and eventual transmigration into extravascular tissues. Analysis of the public dataset (GSE173896) revealed a marked increase in ICAM-1 expression and a moderate elevation of METTL3 in endothelial cells from COPD patients compared to non-smokers (Figure 5A). To model this response in vitro, we treated EA.hy926 endothelial cells with CSE and observed a significant upregulation of ICAM-1 protein (Figure 5B), consistent with the above observations. Importantly, the treatment with the METTL3 inhibitor STM2457 substantially attenuated ICAM-1 expression (Figure 5B). These results indicate that m6A methylation mediated by METTL3 contributes to the dysregulation of ICAM-1 in endothelial cells during COPD pathogenesis, potentially influencing neutrophil recruitment and disease progression. To investigate the regulatory role of m6A modification on ICAM-1 expression, we assessed the stability of ICAM-1 mRNA following METTL3 inhibition. Treatment with STM2457 significantly reduced the half-life of ICAM-1 transcripts (Figure 5C), indicating that m6A methylation enhances ICAM-1 mRNA stability. Consistent with this, analysis of our earlier MeRIP-seq data revealed the presence of m6A modification peaks on ICAM-1 mRNA, which were markedly reduced upon METTL3 knockdown (Figure 5D). Furthermore, m6A-specific RNA immunoprecipitation (RIP) followed by qPCR confirmed significant enrichment of ICAM-1 mRNA in the m6A antibody-precipitated fraction compared to the IgG control (Figure 5E). Collectively, these findings preliminarily demonstrate that METTL3-mediated m6A modification stabilizes ICAM-1 mRNA and upregulates its expression, revealing a functional epitranscriptional mechanism underlying ICAM-1 dysregulation in endothelial cells during COPD.
 |
Figure 5 METTL3 regulates ICAM-1 expression in vascular endothelial cells. (A) Violin plots depicting the expression levels of ICAM-1and METTL3 in endothelial cell clusters from COPD, non-smoker, and smoker in the GSE173896 database. (B) Representative Western blot images (left) and quantitative analysis (right) of ICAM-1 protein expression in EA.hy926 cells exposed to CSE or STM2457. Data are presented as mean ± SD; Statistical significance was determined by One-way ANOVA. *P < 0.05, ****P < 0.0001. (C) Analysis of CXCL8 mRNA stability following STM2457 treatment in EA.hy926 cells by RT-qPCR. Data are presented as mean ± SD; *P < 0.05, **P < 0.01. (D) The m6A peak abundance of ICAM-1 mRNA transcript in cells transfected with siRNA-control or siRNA-METTL3 was detected by MeRIP-Seq and visualized in Integrative Genomics Viewer (IGV). The red peaks represent immunoprecipitation (IP), and the blue peaks represent Input. (E) m6A modification enriches ICAM-1 mRNA in EA.hy926 cells exposed to CSE. RNA immunoprecipitation (RIP) assay was conducted using IgG (negative control) or an anti-m6A antibody (m6A-IP). The relative enrichment of ICAM-1 mRNA was quantified via qRT-PCR. Data are expressed as mean ± standard deviation (SD), and statistical significance was assessed using the T-test. *P < 0.05. |
Discussion
Chronic obstructive pulmonary disease is a respiratory disease with a high incidence rate and mortality, which is driven by a cycle of chronic inflammation and tissue destruction caused by infiltration of inflammatory cells.3 However, the molecular mechanism of m6A modification regulation in this pathological cascade is still not fully understood. In this study, we elucidate a novel epitranscriptional mechanism by which cigarette smoke extract promotes neutrophil recruitment in the pathogenesis of COPD, through METTL3-mediated m6A modification of key chemokine and adhesion molecules in both macrophages and endothelial cells. Our findings underscore the critical role of the m6A methylation machinery as a central regulator of neutrophilic inflammation.
We first demonstrated that CSE-stimulated macrophages exhibit an enhanced capacity to recruit neutrophils, and this effect is critically dependent on METTL3 activity. Inhibition of METTL3 with STM2457 effectively abolished this chemotactic response. We identified the neutrophil-attracting chemokine CXCL8 as a key downstream target. Our data indicate that METTL3 mediates m6A modification on CXCL8 mRNA, which in turn is recognized by the reader protein IGF2BP2. This recognition event significantly enhances the stability of CXCL8 mRNA, leading to increased CXCL8 protein synthesis and secretion. This axis represents a precise regulatory mechanism that amplifies the inflammatory response at the post-transcriptional level. Furthermore, we extended the regulatory scope of m6A beyond the immune cells to the vascular endothelium. We found that ICAM-1, a critical adhesion molecule for neutrophil transmigration, is also under m6A-dependent control. Similar to CXCL8, METTL3 inhibition reduced the stability of ICAM-1 mRNA and attenuated its CSE-induced protein expression. This parallel mechanism in endothelial cells suggests a coordinated epitranscriptional program that facilitates neutrophil recruitment from both the vascular and alveolar compartments.
The significance of our study is multifaceted. First, to our knowledge, we provide the first evidence that m6A modification directly regulates the expression of both CXCL8 and ICAM-1 within the context of COPD. While m6A has been previously implicated in various lung diseases, its specific role in governing neutrophil-driven inflammation in COPD has remained largely unexplored. Second, our research moves beyond mere correlation by delineating a clear mechanistic pathway: from METTL3 catalytic activity, through mRNA stabilization mediated by IGF2BP2, to enhanced protein output and functional outcomes, including neutrophil chemotaxis and adhesion. Importantly, our work establishes m6A modification as a central regulatory factor within the macrophage/CXCL8/neutrophil axis. We have demonstrated that METTL3 directly methylates CXCL8 mRNA, rather than exerting indirect effects through intermediate transcripts such as NF-κB p65 regulatory factors,25 a distinction that sets our findings apart from prior assumptions. Finally, by simultaneously examining macrophages and endothelial cells, we provide a more integrated perspective of the COPD inflammatory microenvironment, suggesting that the m6A pathway participates in multiple parallel mechanisms of neutrophil infiltration. Collectively, our study reveals a key role for m6A RNA methylation in alveolar macrophage-mediated neutrophil recruitment.
However, our study is subject to several limitations. Firstly, the findings are predominantly derived from in vitro cell line models and animal studies. Although these data provide compelling evidence supporting the role of m6A modification in COPD, further validation using primary human cells and clinical specimens is essential to confirm the translational relevance of our results. Secondly, the precise functional m6A sites on CXCL8 mRNA remain uncharacterized. We note that the CXCL8 gene region (chr4:73375020–7375200) contains multiple CA-repeat motifs, which may represent potential binding sites for IGF2BP2—a hypothesis that warrants further investigation. At the same time, the regulatory mechanism of ICAM-1 has not been deeply explored. Thirdly, while we established the significance of the METTL3–IGF2BP2–CXCL8 axis, it remains unclear whether m6A-dependent regulation also influences other inflammatory mediators or molecular pathways contributing to COPD pathogenesis. Finally, due to the presence of many targeted genes in METTL3, the potential systemic effects of METTL3 inhibition pose a challenge for clinical applications. The use of METTL3 inhibitors, such as STM2457, may lead to widespread off-target effects; Therefore, developing targeted interventions such as macrophage-specific METTL3 knockout or nanoparticle-based targeted delivery systems may help alleviate the risk of systemic immune suppression and improve treatment specificity.
In conclusion, our work establishes METTL3-mediated m6A modification as a pivotal upstream regulator of neutrophilic inflammation in COPD. By stabilizing the mRNAs of CXCL8 and ICAM-1, the m6A-IGF2BP2 axis powerfully augments the recruitment and retention of neutrophils in the airways, revealing a key function of m6A modification in coordinating the interaction between alveolar macrophages, endothelial cells, and neutrophils in COPD. Our findings not only advance our insight into the pathogenesis associated with COPD but also the METTL3-m6A pathway as a promising therapeutic target for curbing excessive neutrophil influx and ameliorating disease progression in COPD.
Conclusion
In summary, our study unveils a critical epitranscriptional pathway driving neutrophilic inflammation in COPD. We demonstrate that cigarette smoke exposure promotes METTL3-mediated m6A modification of CXCL8 and ICAM-1 mRNAs in alveolar macrophages and endothelial cells, respectively. This enhances mRNA stability and amplifies the expression of these key neutrophil-recruiting factors (Figure 6). This coordinated mechanism significantly contributes to the sustained neutrophil infiltration of COPD pathogenesis. Our findings position the METTL3-m6A-IGF2BP2 axis as a central regulator of inflammation, offer the evidence that smoking-induced epitranscriptomic dysregulation exacerbates inflammation in COPD, and provide a novel conceptual framework for understanding disease pathogenesis.
 |
Figure 6 Schematic model of m6A-dependent regulation of neutrophil recruitment in COPD. Cigarette smoke exposure upregulates METTL3 in alveolar macrophages, leading to m6A methylation of CXCL8 mRNA. The m6A reader IGF2BP2 recognizes and binds these modifications, enhancing CXCL8 mRNA stability and promoting CXCL8 protein synthesis. Secreted CXCL8 recruits neutrophils into the airways. Concurrently, METTL3-mediated m6A modification stabilizes ICAM-1 mRNA in endothelial cells, increasing ICAM-1 expression and further facilitating neutrophil adhesion and transmigration. This m6A-dependent synergistic mechanism sustains neutrophilic inflammation and contributes to COPD pathogenesis. |
Abbreviations
METTL3, Methyltransferase like 3; COPD, chronic obstructive pulmonary disease; CSE, cigarette smoke extract; BALF, Bronchoalveolar Lavage Fluid.
Data Sharing Statement
The dataset used in this investigation was derived from public repositories, including the GSE130928 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130928) and the GSE173896 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE173896). All data supporting the findings of this study are available within the paper.
Institutional Review Board Statement
The animal study protocol was approved by the Medical Ethics Committee of the Second Affiliated Hospital of Fujian Medical University, Ethics approval number: No. 193 [2024].
Acknowledgments
We thank the Natural Science Foundation of Fujian for the funding support.
Funding
This study was supported by the Natural Science Foundation of Fujian province (2024J01676) and the Joint Funds for the Innovation of Science and Technology, Fujian province (2023Y9240).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Priya V. GOLD COPD report: 2023 update. Lancet Respir Med. 2023;11(1):18. doi:10.1016/S2213-2600(22)00494-5
2. Chen W, Jianying X, Lan Y, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross-sectional study. Lancet. 2018;391(10131):1706–1717. doi:10.1016/S0140-6736(18)30841-9
3. Hogg James C, Fanny C, Soraya U, et al. The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease. N Engl J Med. 2004;350(26):2645–2653. doi:10.1056/NEJMoa032158
4. Shaykhiev R, Crystal RG. Early events in the pathogenesis of chronic obstructive pulmonary disease. Smoking-induced reprogramming of airway epithelial basal progenitor cells. Ann Am Thorac Soc. 2014;11(Suppl 5):S252–258. doi:10.1513/AnnalsATS.201402-049AW
5. Westphalen K, Gusarova GA, Islam MN, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. 2014;506(7489):503–506. doi:10.1038/nature12902
6. O’beirne SL, Kikkers SA, Oromendia C, et al. Alveolar Macrophage Immunometabolism and Lung Function Impairment in Smoking and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2020;201(6):735–739. doi:10.1164/rccm.201908-1683LE
7. Shapiro SD, Goldstein NM, Houghton AM, et al. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. 2003;163(6):2329–2335. doi:10.1016/s0002-9440(10)63589-4
8. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
9. Planagumà A, Domènech T, Pont M, et al. Combined anti CXC receptors 1 and 2 therapy is a promising anti-inflammatory treatment for respiratory diseases by reducing neutrophil migration and activation. Pulm Pharmacol Ther. 2015;34(37–45). doi:10.1016/j.pupt.2015.08.002
10. Tregay N, Begg M, Cahn A, et al. Use of autologous (99m)Technetium-labelled neutrophils to quantify lung neutrophil clearance in COPD. Thorax. 2019;74(7):659–666. doi:10.1136/thoraxjnl-2018-212509
11. Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–95. doi:10.1038/nchembio.1432
12. Zheng G, Dahl JA, Niu Y, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29. doi:10.1016/j.molcel.2012.10.015
13. Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–120. doi:10.1038/nature12730
14. Huang H, Weng H, Sun W, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–295. doi:10.1038/s41556-018-0045-z
15. Jiang X, Liu B, Nie Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6(1):74. doi:10.1038/s41392-020-00450-x
16. Roundtree IA, Evans ME, Pan T, et al. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169(7):1187–1200. doi:10.1016/j.cell.2017.05.045
17. Li T, Hu PS, Zuo Z, et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18(1):112. doi:10.1186/s12943-019-1038-7
18. Shi J, Rui X, Han C, et al. circRNF13, a novel N(6)-methyladenosine-modified circular RNA, enhances radioresistance in cervical cancer by increasing CXCL1 mRNA stability. Cell Death Discov. 2023;9(1):253. doi:10.1038/s41420-023-01557-0
19. Zhang Y, Wang L, Yan F, et al. Mettl3 Mediated m6A Methylation Involved in Epithelial-Mesenchymal Transition by Targeting SOCS3/STAT3/SNAI1 in Cigarette Smoking-Induced COPD.Int. J Chron Obstruct Pulmon Dis. 2023;18(1007–1017). doi:10.2147/copd.s398289
20. Watanabe N, Fujita Y, Nakayama J, et al. Anomalous Epithelial Variations and Ectopic Inflammatory Response in Chronic Obstructive Pulmonary Disease. Am J Respir Cell Mol Biol. 2022;67(6):708–719. doi:10.1165/rcmb.2021-0555OC
21. Su X, Chen J, Lin X, et al. FERMT3 mediates cigarette smoke-induced epithelial-mesenchymal transition through Wnt/β-catenin signaling. Respir Res. 2021;22(1):286. doi:10.1186/s12931-021-01881-y
22. Kang JY, Lee SY, Rhee CK, et al. Effect of aging on airway remodeling and muscarinic receptors in a murine acute asthma model. Clin Interv Aging. 2013;8(1393–1403). doi:10.2147/cia.s50496
23. Schamberger AC, Mise N, Jia J, et al. Cigarette smoke-induced disruption of bronchial epithelial tight junctions is prevented by transforming growth factor-β. Am J Respir Cell Ml Biol. 2014;50(6):1040–1052. doi:10.1165/rcmb.2013-0090OC
24. Yaping Z, Lixing W, Yifei L, et al. Mettl3-mediated transcriptome-wide m6A methylation induced by cigarette smoking in human bronchial epithelial cells. Toxicol in vitro. 2023;89(105584). doi:10.1016/j.tiv.2023.105584
25. He J, Zhou M, Yin J, et al. METTL3 restrains papillary thyroid cancer progression via m6A/c-Rel/IL-8-mediated neutrophil infiltration. Mol Ther. 2021;29(5):1821–1837. doi:10.1016/j.ymthe.2021.01.019
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.