Back to Journals » Cancer Management and Research » Volume 12
METTL14 Inhibits Hepatocellular Carcinoma Metastasis Through Regulating EGFR/PI3K/AKT Signaling Pathway in an m6A-Dependent Manner
Authors Shi Y, Zhuang Y, Zhang J, Chen M, Wu S
Received 3 November 2020
Accepted for publication 10 December 2020
Published 23 December 2020 Volume 2020:12 Pages 13173—13184
DOI https://doi.org/10.2147/CMAR.S286275
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Eileen O'Reilly
Yuntao Shi,1,* Yingying Zhuang,2,* Jialing Zhang,1 Mengxue Chen,1 Shangnong Wu1
1Department of Gastroenterology, The Affiliated Huai’an No. 1 People’s Hospital of Nanjing Medical University, Huai’an, People’s Republic of China; 2Department of Medical Imaging, The Affiliated Huai’an No. 1 People’s Hospital of Nanjing Medical University, Huai’an, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shangnong Wu
Department of Gastroenterology, The Affiliated Huai’an No. 1 People’s Hospital of Nanjing Medical University, No. 1, Huanghe West Road, Huai’an 223300, People’s Republic of China
Tel +86 517-80872251
Email [email protected]
Purpose: Hepatocellular carcinoma (HCC) ranks as the fourth leading cause of cancer-related deaths worldwide. N6-methyladenosine (m6A) RNA methylation is the most common modification of messenger RNAs (mRNAs). The prognosis of HCC patients with metastasis remains poor. Our study aimed to elucidate the regulatory role of m6A on HCC metastasis.
Patients and Methods: All HCC patients were enrolled from The Affiliated Huai’an No. 1 People’s Hospital of Nanjing Medical University. The expression levels of gene were tested by quantitative polymerase chain reaction (qPCR), Western blot, or immunohistochemistry (IHC) analysis. Wound healing assay, Transwell invasion assay, and lung metastasis model were implemented to investigate the migration and invasion ability of HCC cells. Candidate targets were selected by a comprehensive analysis of RNA-sequencing and m6A-sequencing of HepG2 cells.
Results: In this study, we demonstrated that METTL14 was significantly downregulated in HCC and significantly associated with the prognosis of HCC patients. METTL14 knockdown promoted the migration, invasion, and epithelial–mesenchymal transition (EMT) of HCC cells in vitro and in vivo. In addition, overlapping RNA-sequencing and m6A-sequencing data, we identified EGFR as a direct target of METTL14 in HCC. Mechanistically, METTL14 was found to inhibit HCC cell migration, invasion, and EMT through modulating EGFR/PI3K/AKT signaling pathway in an m6A-dependent manner.
Conclusion: Targeting METTL14/EGFR/PI3K/AKT signaling pathway may facilitate the development of a new treatment strategy against the metastasis of HCC.
Keywords: hepatocellular carcinoma, N6-methyladenosine, METTL14, EGFR
Introduction
Hepatocellular carcinoma (HCC), aggressive cancer, ranks as the fourth leading cause of cancer-related deaths worldwide.1 Despite advances in therapy and surgical methods, the overall survival (OS) of HCC patients with metastasis or recurrence remains poor.2 Therefore, it is critical to elucidate the molecular mechanisms of HCC cell metastasis.
N6-methyladenosine (m6A) RNA methylation is the most common modification of messenger RNAs (mRNAs).3 Generally, m6A modification exists in the thousands of transcripts in mammalian cells and prefers to occur at the consensus RRACH motif (R = G or A; H = A, C, or U).4 The m6A modification is catalyzed by a multicomponent methyltransferase complex (MTC) consisting of METTL3, METTL14, WTAP, KIAA1429, RBM15, and ZC3H13.5–9 Moreover, the m6A modification can also be removed by RNA demethylases, including FTO and ALKBH5.10,11 Interestingly, m6A-modified mRNAs may occur in different fates if they are bound by different RNA binding proteins of which the known ones are YTHDC1 and YTHDF1/2/3.12–14 METTL14, a core component of MTC, colocalizes with METTL3 in nuclear speckles, and catalyzes the m6A modification.15 The aberrant m6A modification caused by dysregulated METTL14 plays an important role in the progression of various cancers, such as colorectal cancer16 In HCC, METTL14 has been reported to suppress the metastasis of HCC cells by modulating m6A-dependent primary miR-126 processing.17 Considering m6A RNA methylation is the most common modification of mRNAs,3 we hypothesized that METTL14 may also inhibit the metastatic potential of HCC through regulating specific mRNAs in an m6A-dependent manner.
In this study, we revealed that METTL14 was significantly downregulated in HCC and significantly associated with the prognosis of HCC patients. Mechanistically, we demonstrated that METTL14 can destabilize EGFR mRNA in an m6A-dependent manner and inhibit HCC cell metastasis through EGFR/PI3K/AKT signaling pathway.
Patients and Methods
Clinical Samples
All clinical samples were collected from The Affiliated Huai’an No. 1 People’s Hospital of Nanjing Medical University. The inclusion criteria for the patients to be enrolled in the study were as follows: HCC patients whose histologic slides were identified by two pathologists independently; b HCC patients without any antitumor treatment before surgery, such as chemoradiotherapy. Otherwise, patients were excluded. The detailed clinicopathological characteristics of the HCC patients are listed in Table 1. The informed consent was obtained from each patient and the study was approved by the Ethics Committee of The Affiliated Huai’an No. 1 People’s Hospital of Nanjing Medical University.
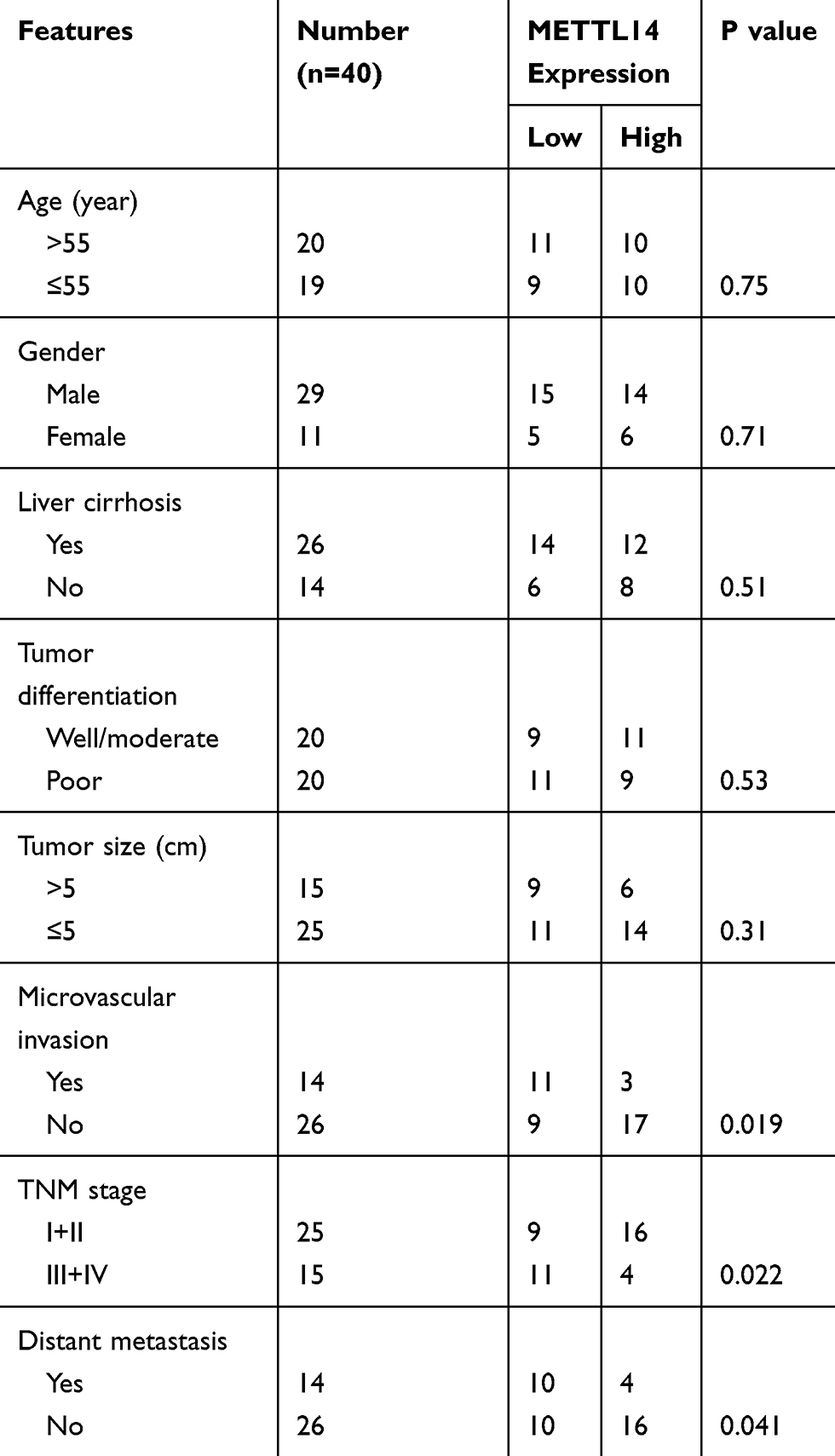 |
Table 1 The Correlations Between METTL14 Expression Levels and the Clinicopathological Features in HCC Patients |
Cell Culture
Normal human hepatocyte cells (LO2) and HCC cell lines (Hep3B, MHCC-LM3, YY8103, SMCC7721, HepG2) were purchased from Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences (Shanghai, China). All cells were routinely cultured with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Vienna, Austria) and cultured in a humidified atmosphere of 5% CO2 at 37°C.
Cell Transfection
The siRNAs were designed and synthesized by RiboBio (Guangzhou, China) and transfected into HepG2 and MHCC-LM3 cells using Lipofectamine 2000 (Invitrogen, USA). The sequences of siRNAs are listed in Table 2. Stable knockdown of METTL14 was achieved by subcloning siMETTL14 into pGLV3/H1/GFP/Puro vectors to construct shMETTL14 for animal studies (GeneChem, China). Then, HepG2 cells were infected with lentivirus at a multiplicity of infection (MOI) of 10, followed by selection with 2 μg/mL puromycin.
 |
Table 2 The RNA Sequences Used in This Study |
RNA Extraction and qPCR
Total RNA was isolated by TRIzol reagent (Invitrogen, China) and subsequent cDNA synthesis was performed with PrimeScript RT Reagent (Takara, Japan) according to the manufactures’ instructions. RNA expression was measured by qPCR by using the SYBR Premix Ex Taq™ (Takara, Japan). The relative expression of genes was calculated by using the 2−ΔΔCt method with the normalization to GAPDH. The primer sequences of genes are shown in Table 2.
Western Blot Analysis
HCC cells were washed and lysed in RIPA lysis buffer. After that, the extracts were boiled for 5 min, separated on SDS-PAGE, and transferred to a PVDF membrane (Millipore, Germany). Subsequently, the PVDF membrane was incubated with the corresponding primary antibody at 4°C overnight. Then, the membrane was washed five times and incubated with a secondary antibody. Finally, bands were visualized by using an imaging system (Bio-Rad, USA). Target genes were normalized to corresponding GAPDH using Image J software. The antibodies used in this study were as follows: METTL14 (Proteintech, 26158-1-AP); EGFR (Proteintech, 18986-1-AP); p-EGFR (CST, #3777); PI3K (Proteintech, 20584-1-AP); p-PI3K (CST, #17366); AKT (Abcam, ab8805); p-AKT (#4060); E-cadherin (Proteintech, 20874-1-AP); N-cadherin (Proteintech, 22018-1-AP); Vimentin (Proteintech, 10366-1-AP); GAPDH (Proteintech, 10494-1-AP).
Immunohistochemistry (IHC) Analysis
The paraffin sections were deparaffined and then treated in methanol with 3% H2O2 for 30 min through the steps of fractional ethanol series and rehydration of distilled water. Sections were rinsed twice with phosphate-buffered saline and incubated with goat serum. After washing, the sections were incubated with appropriate primary antibodies (anti-METTL14, Proteintech, 26158-1-AP) at 4°C overnight. Immunohistochemistry staining was performed with horseradish peroxidase (HRP) conjugates using Diaminobenzidine (DAB) detection. Images were taken with an Olympus FSX100 microscope (Olympus, Japan).
Lung Metastasis Model
For the lung metastasis model, stably transfected HepG2 cells (1 × 106/0.1 mL DMEM) were injected into each nude mouse through the tail vein. Five weeks later, mice were euthanized, and the lung tissues were collected. Then, the metastatic nodules were counted by using HE-staining. Our study was approved by the Animal Care Committee of Nanjing Medical College and mice were maintained under specific pathogen-free (SPF) conditions and manipulated according to the protocols of the Animal Research: Reporting in Vivo Experiments (ARRIVE) guideline and the American Veterinary Medical Association (AVMA) for euthanasia guideline.
Wound Healing Assay
HCC cells were transplanted into a 6-well culture plate and incubated for 24 h followed by being scraped three parallel lines with a sterile pipette tip, being washed twice with PBS, and being cultured in DMEM containing 10% FBS. Images were taken under a microscope at 0 and 24 h after scratching (Olympus, Japan).
Transwell Invasion Assay
Transwell invasion assays were conducted in transwell chambers (Corning, USA) which was coated with Matrigel mix (BD Biosciences, USA) according to the manufacturer’s protocol. After incubation for 24 h, non-invaded cells were scraped off, and the cells invaded on the bottom of the chamber were fixed with 4% paraformaldehyde for 10 min, then stained with crystal violet (0.1%) for 15 min. Thereafter, the stained cells were photographed and counted with an Olympus FSX100 microscope (Olympus, Japan).
Bioinformatics Analysis
GEO datasets were searched in the public GEO database (https://www.ncbi.nlm.nih.gov/geo/) by using keywords “METTL14 OR Methyltransferase-like protein 14” and “Hepatocellular carcinoma OR HCC”. Then, GSE54236 which contains 80 normal liver tissue and 81 HCC tissue samples was enrolled. The differentially expressed miRNAs were analyzed by using GEO2R based on the “limma” R package. The prognostic value of METTL14 in HCC was assessed by using the Kaplan–Meier plotter database (http://kmplot.com/analysis/index.php?p=background).
Statistical Analysis
All data analysis was performed by using SPSS 20.0 (SPSS, USA) and GraphPad Prism 7 (GraphPad, USA). The differences between the two groups were determined by a two-tailed t-test. The differences between multiple groups were determined by analysis of variance (ANOVA). The chi-square test was used to analyze the different distributions of clinicopathological variables. Overall survival was estimated with the Kaplan–Meier method, and the Log rank test was employed to evaluate the difference. p < 0.05 was considered to be statistically significant.
Results
METTL14 Was Significantly Downregulated in HCC and Significantly Associated the Prognosis of HCC Patients
Analysis of public data (TCGA and GSE54236) showed that METTL14 was significantly downregulated in HCC tissues compared with normal liver tissues (Figure 1A and B). Consistently, qPCR analysis of clinical samples and HCC cell lines verified that the expression of METTL14 significantly decreased in HCC tissues and cell lines, especially in distant metastasis tissues and MHCC-LM3 cells (Figure 1C and D). In addition, IHC analysis showed that the protein levels of METTL14 were also significantly downregulated in HCC tissues (Figure 1E). Chi-square analysis exhibited that low METTL14 expression was significantly correlated with advanced TNM stage, microvascular invasion, and distant metastasis (Table 1). Kaplan–Meier survival curves showed that HCC patients with low METTL14 expression underwent a poorer overall survival (Figure 1F). Moreover, the analysis of the Kaplan–Meier Plotter database of HCC exhibited that HCC patients with downregulated METTL14 trend to undergo poorer relapse-free survival (RFS), progression-free survival (PFS), and disease-specific survival (DSS) (Figure 1G).
 |
Figure 1 METTL14 was significantly downregulated in HCC and significantly associated the prognosis of HCC patients. (A and B) The expression value of METTL14 in HCC based on TCGA and GSE54236 analysis. (C and D) The relative expression of METTL14 in HCC tissues and cell lines compared to ANTs and LO2 cells. (E) IHC analysis of METTL14 expression in HCC tissues. (F) Kaplan–Meier survival curves of HCC patients. (G) Analysis of Kaplan-Meier Plotter database of HCC exhibited that HCC patients with downregulated METTL14 trend to undergo poor OS, RFS, PFS, and DSS. ANT, adjacent normal tissue; HCC, hepatocellular carcinoma; PCT, primary cancer tissue; DMT, distant metastasis tissue; *p<0.05; **p<0.01; ***p<0.001. |
METTL14 Knockdown Promoted the Migration, Invasion, and EMT of HCC Cells
To explore the regulatory role of METTL14 in HCC, we knocked down its expression using two small interference RNA (siM14-1 and siM14-2). qPCR and Western blot analysis showed that siM14 transfection markedly inhibited the expression of METTL14 at both mRNA and protein levels (Figure 2A and B). The wound healing assays showed that METTL14 downregulation significantly improved the migratory capabilities of HCC cells (Figure 2C). The Transwell Matrigel invasion assays showed that METTL14 downregulation significantly promoted the invasion ability of HCC cells (Figure 2D). Mounting evidence has verified that EMT is an essential step during metastasis of cancer cells.18,19 Western blot assay exhibited that METTL14 knockdown inhibited the expression of epithelial markers (E-cadherin) and increased the expression of mesenchymal markers (N-cadherin, Vimentin) (Figure 2E). To test whether METTL14 could suppress the metastasis of HCC cells in vivo, we established lung metastasis models through intravenously injecting METTL14-silenced HepG2 cells. Then, we observed that there were more metastatic nodules on the lung surfaces in the METTL14 stable knockdown group than that in the control group (Figure 2F).
 |
Figure 2 METTL14 knockdown promoted the migration and metastasis of HCC cells. (A and B) qPCR analysis and Western blot analysis showed that transfecting siMETTL14 significantly inhibited METTL14 expression in HCC cells. (C) Wound healing assay showed that METTL14 downregulation improved the migration ability of HCC cells. (D) Transwell invasion assay showed that METTL14 downregulation promoted the metastasis of HepG2 and MHCC-LM3 cells. (E) The effect of METTL14 knockdown on EMT of HCC cells. (F) Lung metastasis models showed that METTL14 knockdown increased the lung metastatic nodules. *p<0.05; **p<0.01; ***p<0.001. |
Identifying EGFR mRNA as the Target of METTL14 in HCC
To identify the targets of METTL14 in HCC, we analyzed the public RNA-sequencing profiling and m6A-sequencing profiling of HepG2 cells with METTL14 knockdown based on GSE90642. RNA sequencing data showed that 329 genes were significantly downregulated, but 530 genes were overexpressed after METTL14 knockdown in HepG2 cells (Figure 3A). In addition, METTL14 knockdown resulted in 705 transcripts with downregulated m6A modification (Figure 3A). Thereafter, we overlapped the RNA-sequencing data and m6A-sequencing data and selected these dysregulated mRNAs with downregulated m6A modification as the potential targets of METTL14 in HCC. KEGG analysis showed that these potential targets of METTL14 were significantly associated with 20 signaling pathways, such as phosphatidylinositol phosphorylation and regulation of phosphatidylinositol 3-kinase (PI3K) signaling (Figure 3B). Next, we conducted a protein-protein-interaction (PPI) analysis and selected out the top 10 hub genes among these potential targets of METTL14 (Figure 3C). Interestingly, EGFR ranked at the core position of these hub genes (Figure 3C). Therefore, we selected EGFR as the target of METTL14 in HCC. Analysis of GSE90642 showed that METTL14 knockdown resulted in downregulated m6A levels but upregulated mRNA levels of EGFR (Figure 3D). MeT-DB V2.0 database revealed that the m6A-modified peak in EGFR mRNA was located in the 3ʹuntranscriptional region (3ʹUTR) (Figure 3E). qPCR analysis and Western blot analysis verified that METTL14 downregulation significantly increased EGFR expression at mRNA and protein levels (Figure 3F and G). Recently, studies have reported that YTHDF2, an m6A reader, could target thousands of mRNA and destabilize mRNA via recognizing m6A motif.20 Therefore, we explored whether YTHDF2 affects EGFR expression. Two specific siRNAs targeting YTHDF2 were employed and Western blot was used to confirm the knockdown efficiency (Figure 3H). Similar to the results of METTL14 downregulation, we found that YTHDF2 knockdown strongly augmented EGFR expression (Figure 3H).
 |
Figure 3 Identifying EGFR as the target of METTL14 in HCC. (A) Analysis of RNA-sequencing and m6A-sequencing data of METTL14 knockdown in HepG2 cells deposited in GSE90642. (B) KEGG analysis of the potential targets of METTL14 in HCC. (C) PPI analysis and the top 10 hub genes of the potential targets of METTL14. (D) METTL14 knockdown resulted in downregulated m6A levels but upregulated mRNA levels of EGFR in GSE90642. (E) The m6A peak in EGFR mRNA. (F and G) qPCR and Western blot analysis showed that METTL14 knockdown increased EGFR expression. (H) Western blot analysis showed that YTHDF2 knockdown increased EGFR expression. **p<0.01; ***p<0.001. |
EGFR Knockdown Effectively Reversed the Effect of METTL14 Downregulation on HCC Cells
To explore the role of EGFR in HCC, we suppressed its expression using small interfering RNA (siEGFR). Western blot analysis showed that siEGFR transfection significantly inhibited the expression of EGFR at both mRNA and protein levels (Figure 4A). The wound healing assays showed that EGFR downregulation significantly inhibited the migratory capabilities of HCC cells (Figure 4B). The Transwell Matrigel invasion assays showed that EGFR downregulation also significantly inhibited the invasion of HCC cells (Figure 4C). Next, we co-transfected siEGFR and siMETTL14 into HCC cells to investigated whether suppressing EGFR can reverse the effect of METTL14 knockdown on HCC cells. The wound healing assays showed that METTL14 knockdown promoted cell migration, whereas EGFR downregulation reversed such effect (Figure 4B). Consistently, the Transwell Matrigel invasion assays showed that the improved invasive ability resulted from METTL14 knockdown was re-established after inhibiting EGFR expression in HCC cells (Figure 4C).
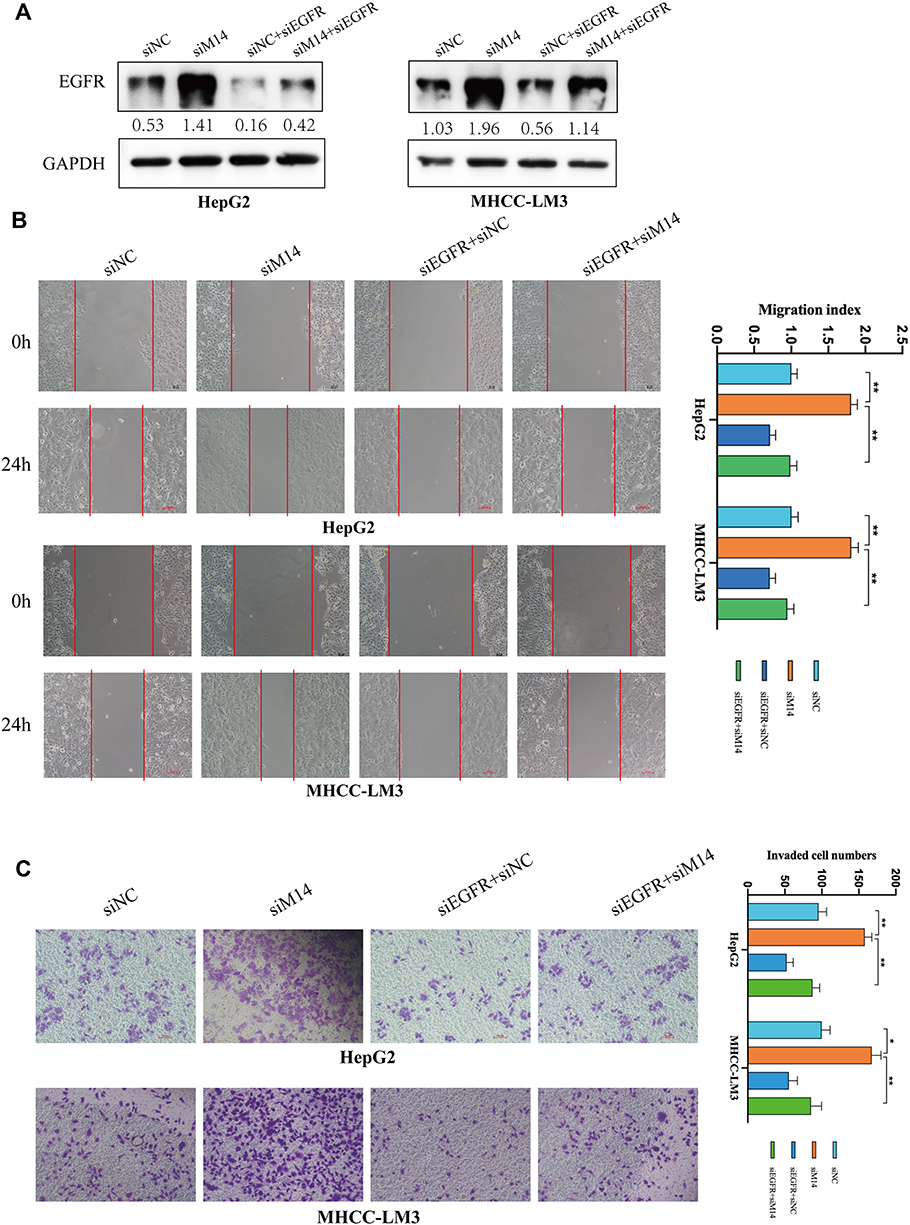 |
Figure 4 EGFR knockdown effectively reversed the effect of METTL14 downregulation on HCC cells. (A) The transfection efficiency was tested by Western blot. (B) The wound healing assays showed that METTL14 knockdown promoted cell migration, whereas EGFR downregulation reversed such effect. (C) The Transwell Matrigel invasion assays showed that the improved invasive ability resulted by METTL14 knockdown was re-established after inhibiting EGFR expression in HCC cells. *p<0.05; **p<0.01. |
METTL14 Inhibited the Metastatic Potential of HCC Cells Through Regulating EGFR/PI3K/AKT Signaling Pathway
It has been reported that EGFR can promote HCC metastasis through the activation of EGFR phosphorylation and subsequent PI3K-AKT signaling pathway.21 In addition, PI3K-AKT signaling pathway is significantly associated with EMT of cancer cells.22,23 KEGG analysis revealed the potential targets of METTL14 were significantly enriched in the regulation of PI3K signaling (Figure 3B). Therefore, we hypothesized that METTL14 may function through the EGFR-PI3K-AKT signaling pathway in HCC. Western blot analysis showed that the effect of METTL14 knockdown on the expression of epithelial markers (E-cadherin) and mesenchymal markers (N-cadherin, Vimentin) was reversed by EGFR inhibition (Figure 5A). Besides, METTL14 knockdown increased the expression of phosphorylated EGFR (p-EGFR), p-PI3K, and p-AKT (Figure 5B). On the contrary, suppressing EGFR decreased the expression of p-EGFR, p-PI3K, and p-AKT (Figure 5B). Moreover, EGFR knockdown could reverse the effect of METTL14 inhibition on the activation of EGFR, PI3K, and AKT (Figure 5B).
 |
Figure 5 METTL14 inhibited the metastatic potential of HCC cells through regulating EGFR/PI3K/AKT signaling pathway. (A) Western blot analysis of the expression of EMT markers after siMETTL14 and siEGFR transfection. (B) Western blot analysis of the expression of p-EGFR, p-PI3K, and p-AKT after siMETTL14 and siEGFR transfection. |
Discussion
Although the surgical and medical treatment strategies have witnessed several advancements, the prognosis of HCC patients is still not so efficient because of frequent metastasis and recurrence.24 As the most abundant modification occurring in eukaryotic mRNAs,25 m6A modification has been widely reported to be associated with the metastasis of various cancers, including HCC.26–28 M6A modification is reversibly regulated by multicomponent methyltransferase complex and demethylases which are often dysregulated in cancers.5,10 For example, RNA N6-methyladenosine demethylase FTO is considerably upregulated in breast cancer and promotes breast tumor progression through N6-methyladenosine-dependent post-transcriptional degradation of BNIP3.29 METTL14 is a core component of MTC and colocalizes with METTL3 in nuclear speckles to catalyze the m6A modification.15 Gu et al discovered that METTL14 is lowly expressed in bladder cancer and METTL14 knockout promotes the proliferation, self-renewal, metastasis, and tumor-initiating capacity of bladder cancer.30 In HCC, METTL14 was reported to suppress the metastasis of HCC cells by modulating m6A-dependent primary miR-126 processing.17 In the present study, we verified the downregulated expression of METTL14 in HCC and further demonstrated that METTL14 can inhibit HCC metastasis through posttranscriptional destabilization of EGFR mRNA in an m6A-dependent manner.
EGFR is an important oncogene and presents at the majority of solid cancers, such as pancreatic cancer,31 breast cancer,32 prostate cancer,33 and colorectal cancer.34 In non–small cell lung cancer (NSCLC), there are several reports of EGFR overexpression which can lead to aggressive and clinically invasive characteristics.35,36 In HCC, the EGFR signaling pathway regulates the invasion and metastasis ability of cells.37 EGFR is significantly associated with the activation of PI3K-AKT signaling in HCC. Zheng and colleagues revealed that Tropomodulin3 modulates EGFR-PI3K-AKT signaling to drive HCC metastasis.21 In addition, CD133 confers cancer stem-like cell properties by stabilizing EGFR-AKT signaling in HCC.38 This preclinical evidence supports the hypothesis that targeting EGFR could be potentially promising for HCC prevention and treatment. In this study, we demonstrated that METTL14 can inhibit HCC cell migration, invasion and EMT through modulating EGFR/PI3K/AKT signaling pathway. Contrary to METTL14, METTL3, another key component of MTC, can facilitate tumor progression through post-transcriptional silencing of SOCS2 in HCC.39 Moreover, one study showed that both METTL3 and METTL14 have a methyltransferase domain to methylate RNA and the m6A methyltransferase activity is much higher in the METTL3/METTL14 complex than that in either subunit alone.5 However, the crystal structure and biochemical evidence indicated that METTL3, rather than METTL14, is the unique catalytic subunit, and METTL14 functions in structural stabilization and RNA substrate recognition.40 Therefore, further clarify the characterization of METTL3 and METTL14 in HCC is urgently needed.
Conclusions
In summary, our study convincingly demonstrates that the identified METTL14/EGFR/PI3K/AKT signaling pathway can regulate the migration, invasion and EMT of HCC cells, thereby facilitating the development of new treatment strategy against the metastasis of HCC.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.21492
2. Gyoeri GP, Pereyra D, Braunwarth E, et al. The 3-60 criteria challenge established predictors of postoperative mortality and enable timely therapeutic intervention after liver resection. Hepatobiliary Surg Nutr. 2019;8:111–124. doi:10.21037/hbsn.2019.02.01
3. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–3975. doi:10.1073/pnas.71.10.3971
4. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi:10.1038/nature11112
5. Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi:10.1038/nchembio.1432
6. Ping XL, Sun BF, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–189. doi:10.1038/cr.2014.3
7. Lan T, Li H, Zhang D, et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol Cancer. 2019;18:186. doi:10.1186/s12943-019-1106-z
8. Patil DP, Chen CK, Pickering BF, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–373. doi:10.1038/nature19342
9. Knuckles P, Lence T, Haussmann IU, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018;32:415–429. doi:10.1101/gad.309146.117
10. Mathiyalagan P, Adamiak M, Mayourian J, et al. FTO-dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation. 2019;139:518–532. doi:10.1161/CIRCULATIONAHA.118.033794
11. Li XC, Jin F, Wang BY, et al. The m6A demethylase ALKBH5 controls trophoblast invasion at the maternal-fetal interface by regulating the stability of CYR61 mRNA. Theranostics. 2019;9:3853–3865. doi:10.7150/thno.31868
12. Roundtree IA, He C. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Trends Genet. 2016;32:320–321. doi:10.1016/j.tig.2016.03.006
13. Shi H, Wang X, Lu Z, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27:315–328. doi:10.1038/cr.2017.15
14. Du H, Zhao Y, He J, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626. doi:10.1038/ncomms12626
15. Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63:306–317. doi:10.1016/j.molcel.2016.05.041
16. Yang X, Zhang S, He C, et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol Cancer. 2020;19:46. doi:10.1186/s12943-020-1146-4
17. Ma JZ, Yang F, Zhou CC, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529–543. doi:10.1002/hep.28885
18. Chen Y, LeBleu VS, Carstens JL, et al. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol Med. 2018;10. doi:10.15252/emmm.201809085
19. Siddiqui MA, Gollavilli PN, Ramesh V, et al. Thymidylate synthase drives the phenotypes of epithelial-to-mesenchymal transition in non-small cell lung cancer. Br J Cancer. 2020. doi:10.1038/s41416-020-01095-x
20. Li M, Zhao X, Wang W, et al. Ythdf2-mediated m(6)A mRNA clearance modulates neural development in mice. Genome Biol. 2018;19:69. doi:10.1186/s13059-018-1436-y
21. Zheng H, Yang Y, Hong YG, et al. Tropomodulin 3 modulates EGFR-PI3K-AKT signaling to drive hepatocellular carcinoma metastasis. Mol Carcinog. 2019;58:1897–1907. doi:10.1002/mc.23083
22. Lee S, Choi EJ, Cho EJ, et al. Inhibition of PI3K/Akt signaling suppresses epithelial-to-mesenchymal transition in hepatocellular carcino- ma through the Snail/GSK-3/beta-catenin pathway. Clin Mol Hepatol. 2020;26(4):529–539. doi:10.3350/cmh.2019.0056n
23. Xue F, Jia Y, Zhao J. Overexpression of FYN suppresses the epithelial-to-mesenchymal transition through down-regulating PI3K/AKT pathway in lung adenocarcinoma. Surg Oncol. 2020;33:108–117. doi:10.1016/j.suronc.2020.02.002
24. Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012;62:394–399. doi:10.3322/caac.21161
25. Wu R, Jiang D, Wang Y, et al. N (6)-Methyladenosine (m(6)A) methylation in mRNA with A dynamic and reversible epigenetic modification. Mol Biotechnol. 2016;58:450–459. doi:10.1007/s12033-016-9947-9
26. Yue B, Song C, Yang L, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18:142. doi:10.1186/s12943-019-1065-4
27. Chen RX, Chen X, Xia LP, et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. 2019;10:4695. doi:10.1038/s41467-019-12651-2
28. Li J, You S, Zhang S, et al. Elevated N-methyltransferase expression induced by hepatic stellate cells contributes to the metastasis of hepatocellular carcinoma via regulation of the CD44v3 isoform. Mol Oncol. 2019;13:1993–2009. doi:10.1002/1878-0261.12544
29. Niu Y, Lin Z, Wan A, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18:46. doi:10.1186/s12943-019-1004-4
30. Gu C, Wang Z, Zhou N, et al. Mettl14 inhibits bladder TIC self-renewal and bladder tumorigenesis through N(6)-methyladenosine of Notch1. Mol Cancer. 2019;18:168. doi:10.1186/s12943-019-1084-1
31. Urtasun N, Vidal-Pla A, Pérez-Torras S, et al. Human pancreatic cancer stem cells are sensitive to dual inhibition of IGF-IR and ErbB receptors. BMC Cancer. 2015;15:223. doi:10.1186/s12885-015-1249-2
32. Issa A, Gill JW, Heideman MR, et al. Combinatorial targeting of FGF and ErbB receptors blocks growth and metastatic spread of breast cancer models. Breast Cancer Res. 2013;15:R8. doi:10.1186/bcr3379
33. Brizzolara A, Benelli R, Venè R, et al. The ErbB family and androgen receptor signaling are targets of Celecoxib in prostate cancer. Cancer Lett. 2017;400:9–17. doi:10.1016/j.canlet.2017.04.025
34. Sartore-Bianchi A, Amatu A, Porcu L, et al. HER2 positivity predicts unresponsiveness to EGFR-targeted treatment in metastatic colorectal cancer. Oncologist. 2019;24:1395–1402. doi:10.1634/theoncologist.2018-0785
35. Hastings K, Yu HA, Wei W, et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small-cell lung cancer. Ann Oncol. 2019;30:1311–1320. doi:10.1093/annonc/mdz141
36. Wang S, Ma P, Ma G, et al. Value of serum tumor markers for predicting EGFR mutations and positive ALK expression in 1089 Chinese non-small-cell lung cancer patients: a retrospective analysis. Eur J Cancer. 2020;124:1–14. doi:10.1016/j.ejca.2019.10.005
37. Wu L, Chen P, Ying J, et al. MAT2B mediates invasion and metastasis by regulating EGFR signaling pathway in hepatocellular carcinoma. Clin Exp Med. 2019;19:535–546. doi:10.1007/s10238-019-00579-2
38. Jang JW, Song Y, Kim SH, et al. CD133 confers cancer stem-like cell properties by stabilizing EGFR-AKT signaling in hepatocellular carcinoma. Cancer Lett. 2017;389:1–10. doi:10.1016/j.canlet.2016.12.023
39. Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–2270.
40. Zhou KI, Pan T. Structures of the m(6)A methyltransferase complex: two subunits with distinct but coordinated roles. Mol Cell. 2016;63:183–185. doi:10.1016/j.molcel.2016.07.005
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.