Back to Journals » International Journal of Nanomedicine » Volume 14
Metoclopramide nanoparticles modulate immune response in a diabetic rat model: association with regulatory T cells and proinflammatory cytokines
Authors Abd Ellah NH ![]() , Ahmed EA
, Ahmed EA ![]() , Abd-ellatief RB
, Abd-ellatief RB ![]() , Ali MF, Zahran AM
, Ali MF, Zahran AM ![]() , Hetta HF
, Hetta HF ![]()
Received 3 December 2018
Accepted for publication 8 February 2019
Published 3 April 2019 Volume 2019:14 Pages 2383—2395
DOI https://doi.org/10.2147/IJN.S196842
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Noura H Abd Ellah,1,2 Esraa A Ahmed,3 Rasha B Abd-ellatief,3 Marwa F Ali,4 Asmaa M Zahran,5 Helal F Hetta6,7
1Division of Pharmaceutical Sciences, James L Winkle College of Pharmacy, University of Cincinnati, Cincinnati, OH 45267, USA; 2Department of Pharmaceutics, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt; 3Department of Pharmacology, Faculty of Medicine, Assiut University, Assiut 71515, Egypt; 4Department of Pathology and Clinical Pathology, Faculty of Veterinary Medicine, Assiut University, Assiut 71515, Egypt; 5Department of Clinical Pathology, South Egypt Cancer Institute, Assiut, Egypt; 6Department of Internal Medicine, University of Cincinnati College of Medicine, Cincinnati, OH 45267-0595, USA; 7Department of Medical Microbiology and Immunology, Faculty of Medicine, Assiut University, Assiut 71526, Egypt
Background: The inflammatory basis of diabetes mellitus directed the researchers’ attention to the immune system for better management and prevention of complications. Metoclopramide (MCA; the only US Food and Drug Administration-approved for gastroparesis) has the ability to restore immune function through increasing prolactin secretion. This study aimed to test the effect of BSA/MCA nanoparticles (NPs) on modulating immune response.
Methods: BSA/MCA NPs were fabricated by desolvation and evaluated in vitro via measuring loading efficiency, particle size, and surface charge. The selected formula was further evaluated via differential scanning calorimetry and release behavior. Then, NPs were injected into rats (25 mg MCA/kg/week) for 3 weeks to be evaluated histopathologically and immunologically via measuring proinflammatory cytokines, such as IL1β, IL6, and TNFα, in addition to measuring regulatory T-cell frequency.
Results: MCA was successfully loaded on BSA, achieving high encapsulation efficiency reaching 63±2%, particles size of 120–130 nm with good polydispersity, and a negative surface charge indicating that entire positively charged drug was encapsulated inside NPs. Differential scanning calorimetry thermography of selected NPs showed an obvious interaction between components and cross-linking of BSA molecules using glutaraldehyde, resulting in sustained release of MCA (around 50% within 3 days). MCA NPs significantly restored the immune response via decreasing proinflammatory cytokines and increasing regulatory T-cell frequency when compared to control and free MCA (drug not loaded in NPs)-treated groups. Histopathological examination of this MCA NPs-treated group did not show the characteristic lesions of diabetes, and apoptosis nearly disappeared.
Conclusion: BSA/MCA NPs could be considered a new modality for treatment of gastroparesis, in addition to management of diabetes itself and preventing its complications via an MCA-immunomodulatory effect.
Keywords: metoclopramide, bovine serum albumin, diabetes, immunity, nanoparticles, gastroparesis
Introduction
Diabetes mellitus (DM) is a chronic disease, divided mainly into type 1 diabetes (T1D) and type 2 diabetes (T2D). Autoimmunity is recognized as the main cause of T1D, where the immune system attacks insulin-producing (β) cells, resulting in insufficient insulin. However, T2D is associated with inability of the body to use the produced insulin effectively. Autoimmunity has been reported as an overlapping issue between T1D and T2D, due to the presence of circulating islet autoantibodies, self-reactive T cells, and defects in regulatory T (Treg) cells of T2D patients.1,2
For a long time, it has been known that inflammation is involved in T1D pathogenesis, and currently there is strong evidence that inflammation also has a critical role in the pathogenesis of T2D.3–5 T2D has been suggested to compose the final result of an acute-phase reaction during which cytokines, including IL1β, IL6, TNFα, and IL17, are released in large amounts, sustaining inflammation.6–11 The uncontrolled inflammation results in pathogenesis of T2D through tissue destruction and activation of autoimmune response toward β-cells.1,12,13
Accordingly, extensive attention has been paid to the role of the immune system and inflammation in progression and complications of DM. In general, reduction in inflammation and restoring normal functions of the immune system should be kept in mind while controlling DM to decrease the risk of any further complications.14,15 Metoclopramide (MCA; dopamine D2-receptor antagonist), which is the only US Food and Drug Administration (FDA)-approved drug for diabetes-induced gastroparesis, has been used extensively as an immunomodulating agent, due to its ability to restore depressed cellular immune function through increasing prolactin secretion, which consequently affects cytokine release.16–19
In this study, we hypothesized that MCA use in DM could be beneficial in modulating immune response. To test this hypothesis, diabetic male rats were injected intraperitoneally with MCA. Serum levels of both proinflammatory cytokines (IL1β, IL6, and TNFα) and peripheral Treg-cell frequency were measured. Additionally, liver, lymph nodes, and spleen tissue were histopathologically examined for further support for this hypothesis.
An additional challenge in this study was combining the advantage of using MCA as an immunoenhancing agent and nanotechnology. MCA nanoparticles (NPs) were encapsulated into BSA, and outcomes of BSA/MCA NPs were compared with those of free MCA (MCA not loaded in NPs). The successful implementation of NPs in drug delivery depends on their ability to penetrate several anatomical barriers and sustain release of their contents, especially with drugs of short biological half-life (such as MCA). Dependently, using MCA NPs, it was possible to decrease frequency of administration to once a week (25 mg/kg/week). The use of albumin NPs has several advantages, including stability in blood, biocompatibility, biodegradability, and nonimmunogenicity.20–22
In this work, BSA/MCA NPs were fabricated and evaluated in vitro via measurement of drug-loading efficiency, particle size, surface charge, and transmission electron microscopy (TEM). Differential scanning calorimetry (DSC) and in vitro release also were studied to detect any interactions between NP components, achieving retarded drug release from these NPs. Then, the optimized NPs were injected into rats and evaluated histopathologically and immunologically.
Materials and methods
Materials
MCA hydrochloride and BSA were purchased from Sigma-Aldrich (St Louis, MO, USA). Glutaraldehyde was purchased from El-Gomhoria (Cairo, Egypt). Ethanol, sodium hydroxide, and saline (0.9%) were obtained from El-Nasr Chemical (Cairo, Egypt). An ELISA kit was purchased from RayBiotech (Peachtree Corners, GA, USA). Fluorescein isothiocyanate-conjugated antirat CD4 (Isotype rat IgG2b), PE-Cy5-conjugated antirat CD25 (Isotype rat IgG1) and PE-conjugated antirat FOXP3 (Isotype rat IgG2a) were purchased from eBioscience (San Diego, CA, USA).
Animals
Adult male Wistar rats weighing 200–250 g were obtained from the animal house at Assiut University. All animal experiments were done according to guidelines of the Council for International Organizations of Medical Sciences, which was founded under the auspices of the World Health Organization (WHO) and United Nations Educational, Scientific, and Cultural Organization. The study was approved by the ethical committee of the Faculty of Medicine, Assiut University, Assiut, Egypt. Rats were housed in clean capacious Macrolane cages under standard laboratory conditions, including a well-aerated room with suitable temperature (25°C±5°C) and maintained in good light.
Fabrication of metoclopramide nanoparticles
BSA powder was dissolved in distilled water. The pH of the solution was adjusted to be in the range of 8–9 using 0.1 M NaOH. An ethanol solution of MCA (10 mg/mL) was added dropwise under magnetic stirring into BSA solution. Excess desolvating agent (ethanol) was added dropwise under continuous stirring until the appearance of turbidity. After 10 minutes of continuous stirring, 100 μL (8% v:v) glutaraldehyde was added as a cross-linker. Overnight stirring at room temperature was done. MCA NPs were collected via centrifugation (14,000 rpm, 4°C for 30 minutes). The collected MCA particles were dried by solvent evaporation. The composition of different MCA NPs is shown in Table 1.
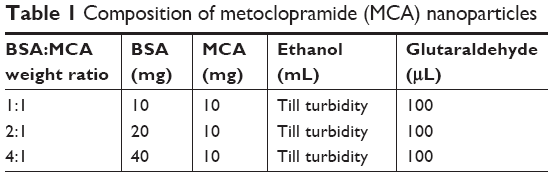 | Table 1 Composition of metoclopramide (MCA) nanoparticles |
Drug entrapment efficiency and loading capacity
The amount of MCA entrapped inside the BSA NPs was determined indirectly. NPs were centrifuged (14,000 rpm, 4°C for 30 minutes) and the supernatant diluted with distilled water and analyzed spectrophotometrically at 310 nm against blank prepared similarly (ultraviolet spectrophotometry; Jenway, Stone, UK). Drug entrapment efficiency (EE) was calculated according to Equation 1. Drug loading (DL) capacity was determined via measuring the MCA content of 1 mL formulation using the supernatant divided by the total weight of the 1 mL formulation using the dried pellet via solvent evaporation. DL% was calculated according to Equation 2.
|
|
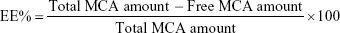
|
|
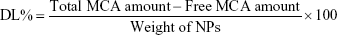
Particle-size and Zeta-potential measurements
Particle size, polydispersity index and Zeta-potential of different MCA NPs were measured using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). Pelleted NPs were diluted with distilled water for size and polydispersity measurements at 25°C and diluted with 0.05 M phosphate buffer pH 7.4 for Zeta-potential measurements.
Transmission electron microscopy (TEM)
BSA/MCA NP morphology was revealed by TEM (JEM-100CX II; Jeol, Tokyo, Japan). Selected NPs were negatively stained using 2% w:v uranyl acetate solution and deposited on 200-mesh carbon-coated copper grids. A clear image of deposited NPs was captured using digital microscopy and the image analyzed using imaging-viewer software.
Differential scanning calorimetry (DSC)
DSC analysis was performed for selected NPs and components to investigate the presence of any possible interactions (DSC50; Shimadzu, Kyoto, Japan). MCA, BSA, physical mixture, and selected BSA/MCA NPs were weighed in an aluminum pan (3–5 mg), then the temperature of all samples was elevated to 300°C at a constant rate (10°C/min).
In vitro release studies
In vitro MCA release from BSA NPs was carried out in a release tube capped with a standard semipermeable cellophane membrane at one side.23–25 A selected NP suspension (1 mL) was placed on the surface of the membrane. The tube was moved into a container containing 25 mL PBS pH 7.4, which was placed in a shaking water bath (50 strokes/minute and 37°C±0.2°C). Released MCA was calculated by measuring the absorbance of aliquots at 310 nm (ultraviolet spectrophotometry). Kinetic analysis of release data using the kinetic models zero- and first-order kinetics and Higuchi and Korsmeyer–Peppas models was done to determine the exact mechanism of MCA release from BSA NPs.
In vivo experimental protocol
Animals were divided into four groups (ten each group). The first group was control nondiabetic rats, the second was control diabetic rats, the third was MCA-treated diabetic rats, and the fourth was MCA NP-treated diabetic rats. Induction of diabetes was done in 1 week, then the third and forth groups were injected intraperitoneally with MCA (25 mg/kg/day)24 and MCA NPs (25 mg/kg/week), respectively, for 3 weeks. On the last day of the experiment, rats were sacrificed by decapitation. Blood samples were collected in sterile heparinized tubes and examined. The liver, lymph nodes, and spleen were dissected and fixed in buffered 10% formalin for tissue histopathology.
Diabetic induction
Diabetes was induced in the rats using streptozotocin (60 mg/kg intraperitoneally).26 Streptozotocin induces diabetes within 3 days through damaging β-cells. Diabetic animals were separated from nondiabetic animals and maintained under feeding and metabolism control. Blood samples were obtained from the tail vein to detect glucose levels in rats that exceeded 135 mg/dL.
Evaluation of proinflammatory cytokines in plasma
Proinflammatory cytokines (IL1β, IL6, and TNFα) were measured in all groups with the ELISA kit according to manufacturer protocols. Kit levels of sensitivity were 80 pg/mL for IL1, 30 pg/mL for IL6, and 25 pg/mL for TNFα.
Flow cytometric analysis of rat Treg cells
Treg cells were assessed in blood by staining blood samples for 20 minutes at 4°C in the dark with fluorescein isothiocyanate–conjugated antirat CD4 (Isotype rat IgG2b and PE-Cy5-conjugated antirat CD25 (Isotype rat IgG1). Following incubation for 15 minutes at 4°C, red blood cells lysis and washing with PBS were done. Then, fixation, permeabilization, and intracellular staining with PE-conjugated antirat FOXP3 (Isotype rat IgG2a) for 15 minutes were done. After washing with PBS, flow cytometric analysis was performed with a FACSCalibur with CellQuest software (BD Biosciences, San Jose, CA, USA). An isotype-matched negative control was used for each sample. A forward and side scatter histogram was used to define the lymphocyte population (R1). CD4+ cells were gated, and then CD25 and FOXP3 expression was assessed on CD4+ lymphocytes to detect Treg cells (CD4+CD25+Foxp3+Treg cells), as shown in Figure 1.
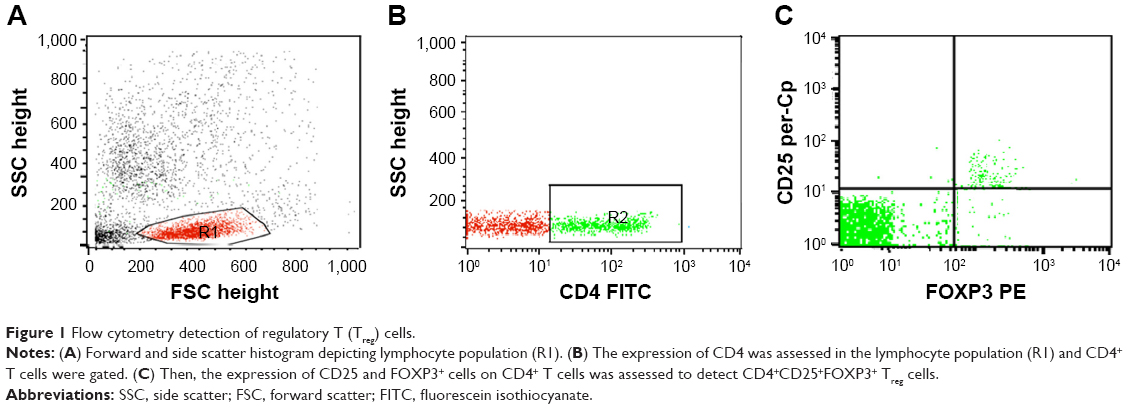 | Figure 1 Flow cytometry detection of regulatory T (Treg) cells. |
Histopathological examination
Fresh specimens from spleen, liver, and lymph nodes of rats of all experimental groups were collected and fixed in 10% neutral buffered formalin. Tissue samples were processed in routine fashion, sectioned at 4 μm thickness, and stained with hematoxylin and eosin (H&E) for histopathological examination by light microscopy (CX31; Olympus, Tokyo Japan) and photographed using a digital camera (Camedia C-506; Olympus).27
Statistical analysis
All experiments were done at least in triplicate, and results are reported as mean ± SD. Statistical differences among various treatment groups were assessed. P<0.05 was considered statistically significant (Prism 6.0; GraphPad Software, San Diego, CA, USA).
Results
Drug entrapment efficiency (EE%) and loading capacity (DL%)
MCA EE was increased with increasing BSA content, reaching 63%±2%. EE was significantly higher at BSA:MCA ratios of 2:1 and 4:1. DL was decreased with increasing BSA:MCA ratio. Both EE% and DL% are shown in Figure 2.
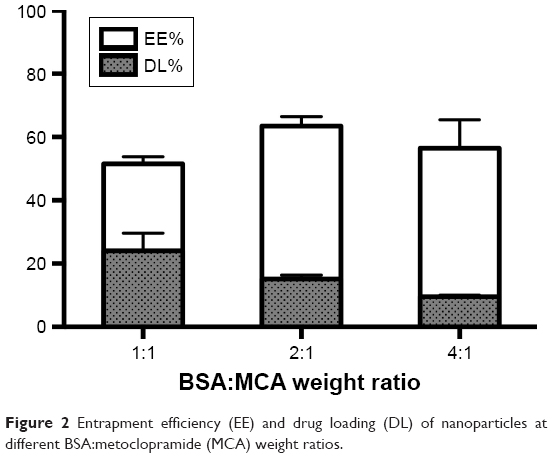 | Figure 2 Entrapment efficiency (EE) and drug loading (DL) of nanoparticles at different BSA:metoclopramide (MCA) weight ratios. |
Particle-size and Zeta-potential measurements
Figure 3 shows that particle sizes with the different BSA:MCA ratios were not significantly different. However, these different BSA:MCA ratios resulted in NPs with good polydispersity index, ranging from 0.13±0.02 to 0.19±0.01 (Figure 3). The NPs formed were negatively charged, and surface charge ranged from −31±9 to −40±2 mV.
 | Figure 3 Particle size, polydispersity index (PDI), and Zeta-potential of nanoparticles at different BSA:metoclopramide (MCA) weight ratios. |
Based on the aforementioned in vitro evaluations for the different formulated NPs, a BSA:MCA ratio of 2:1 was selected for further evaluation, due to its relatively higher EE and DL values.
Transmission electron microscopy (TEM)
TEM of selected BSA/MCA NPs (Figure 4A) showed spherical NPs and the particle size of these NPs using TEM was ≤100 nm.
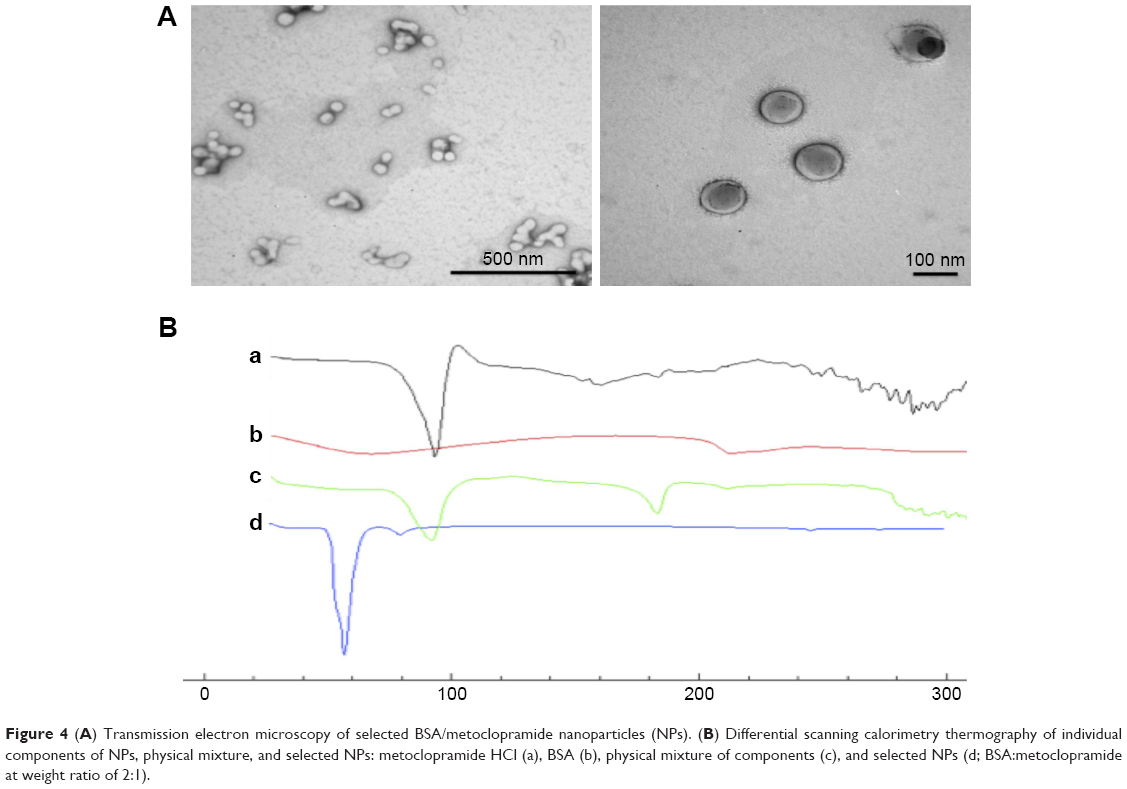 | Figure 4 (A) Transmission electron microscopy of selected BSA/metoclopramide nanoparticles (NPs). (B) Differential scanning calorimetry thermography of individual components of NPs, physical mixture, and selected NPs: metoclopramide HCl (a), BSA (b), physical mixture of components (c), and selected NPs (d; BSA:metoclopramide at weight ratio of 2:1). |
Differential scanning calorimetry (DSC)
Thermal analysis of MCA HCl, BSA, physical mixture, and selected NPs was performed to detect any possible interactions (Figure 4B). MCA HCl thermography showed characteristic endothermic peaks at 95°C, 158°C and 185°C. BSA thermography showed characteristic broad endothermic peaks at 69°C and 210°C. The thermal behavior of the physical mixture of BSA and MCA (1:1) in the absence of glutaraldehyde exhibited the characteristic peaks of the two components. However, thermography of developed NPs did not show endothermic BSA peaks. Furthermore, shifted endothermic peaks at 58°C and 80°C were observed.
In vitro release studies
In vitro MCA release from desolvated BSA NPs was studied and showed controlled and sustained release behavior. MCA was released from the NPs at 26%±5% after 24 hours, and only around 55% had released after 3 days (Figure 5A). However, the release pattern of free MCA revealed that around 100% of the drug had been released after 24 hours. Kinetic analysis of these data showed that the highest correlation was observed with the Higuchi model, indicating that MCA was released by diffusion (Figure 5B). The diffusion exponent n of the Korsmeyer–Peppas model was 0.489, which revealed that MCA release from BSA NPs followed non-Fickian diffusion.
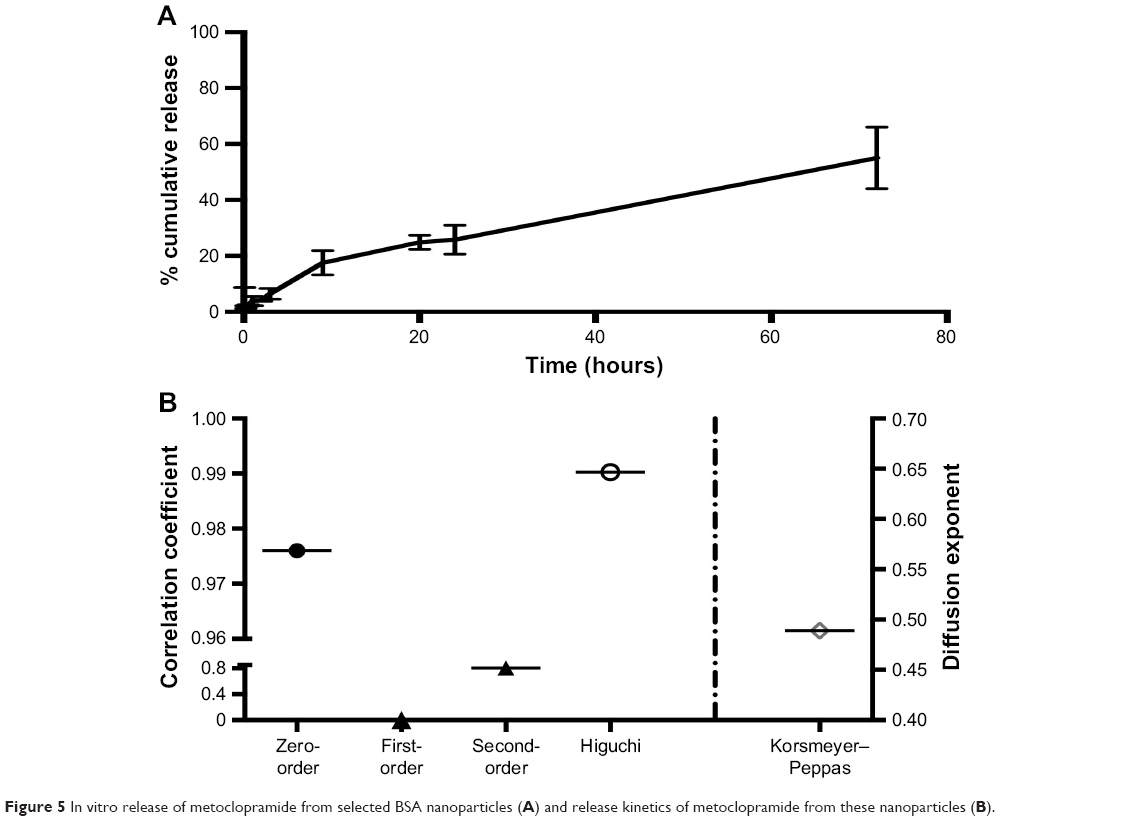 | Figure 5 In vitro release of metoclopramide from selected BSA nanoparticles (A) and release kinetics of metoclopramide from these nanoparticles (B). |
Effect of MCA and MCA NPs on serum levels of proinflammatory cytokines
Serum levels of IL1β, IL6, and TNFα were significantly higher among control diabetic rats than control nondiabetic rats (P<0.001 for all). Serum levels of IL1β, IL6, and TNFα were significantly decreased in diabetic rats treated with MCA when compared to untreated diabetic rats (P=0.01, P=0.04, and P=0.001, respectively). Moreover, serum levels of IL1β, IL6, and TNFα were significantly decreased in diabetic rats treated with MCA NPs when compared to untreated diabetic rats (P<0.001, P=0.01, and P=0.03, respectively). Also, there were significant decreases in serum levels of IL6 and TNFα in diabetic rats treated with MCA NPs when compared to free MCA-treated rats (P=0.04 and P=0.01, respectively), as shown in Table 2.
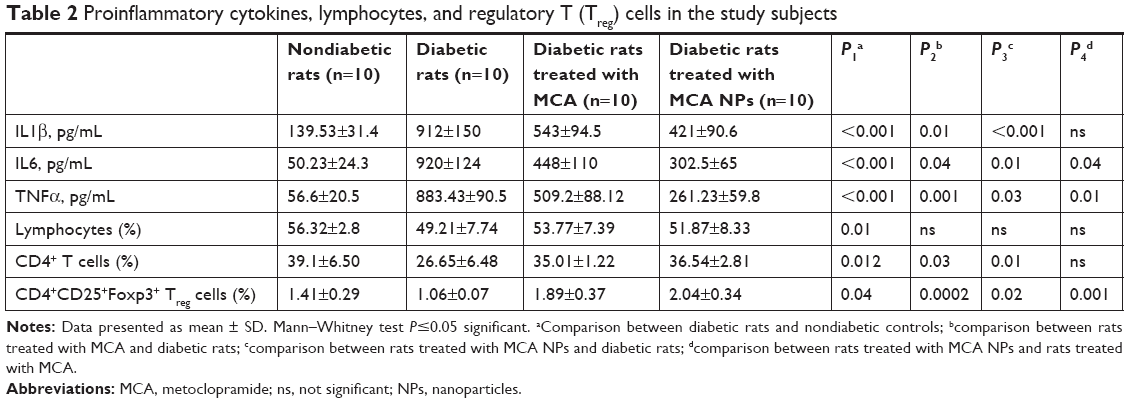 | Table 2 Proinflammatory cytokines, lymphocytes, and regulatory T (Treg) cells in the study subjects |
Effect of MCA and MCA NPs on frequency of Treg cells
The frequency of lymphocytes was significantly decreased in diabetic rats compared to nondiabetic rats (P=0.01). Specifically, the frequency of CD4+ T cells was significantly decreased in diabetic rats compared to nondiabetic rats (P=0.012). Additionally, the frequency of Treg cells was significantly lower in diabetic rats than nondiabetic rats (P=0.04).
Table 2 shows that the frequency of CD4+ T cells and Treg cells was significantly increased in diabetic rats treated with MCA when compared to the control diabetic rats (P=0.03 and P=0.0002), respectively. Moreover, the frequency of CD4+ T cells and frequency of Treg cells were significantly increased in diabetic rats treated with MCA NPs when compared to untreated diabetic rats (P=0.01 and P=0.02, respectively). However, MCA NPs showed significant increases in only Treg cells when compared to MCA-treated rats (P=0.001).
Histopathological examination
The diabetic rat group showed different lesions in different organs. Livers showed centrilobular hepatic necrosis and clear nonstaining vacuoles in hepatic cells, which may have contained intracytoplasmic material, such as lipids, Kupffer-cell proliferation in the portal area, and vascular changes characterized by dilatation and congestion of central veins and hepatic sinusoids (Figure 6A–D). Apoptosis of some lymphocytes characterized by cell shrinkage and fragmentation of cell cytoplasm appeared in cortical lymphoid follicles in lymph nodes, accompanied by subcapsular vascular congestion (Figure 6E and F). Histopathological examination of spleens showed collagen deposition in different areas of red pulp (Figure 6G and H).
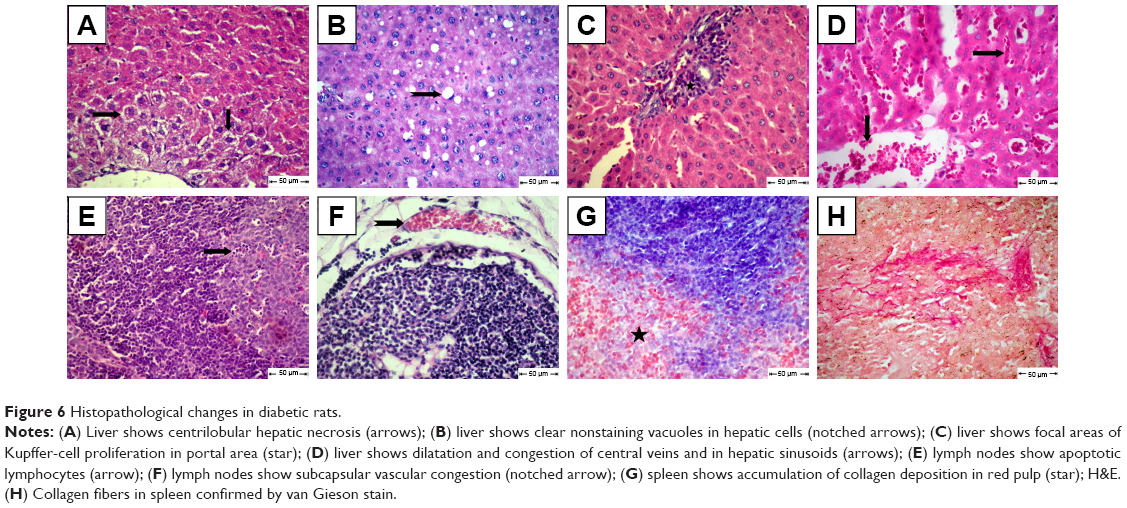 | Figure 6 Histopathological changes in diabetic rats. |
For the MCA-treated diabetic group, vacuolar degeneration in hepatocytes, which appeared as clear vacuoles, were detected in moderate distribution in liver tissue (Figure 7A). Also, Kupffer-cell proliferation was seen in the portal area (Figure 7B). Congestion of blood vessels associated with apoptosis in lymphocytes of lymphoid follicles was seen in most examined lymph nodes in this group (Figure 7C). Focal areas of hemorrhage accompanied by mild hemosiderosis were observed in red pulp of spleens (Figure 7D).
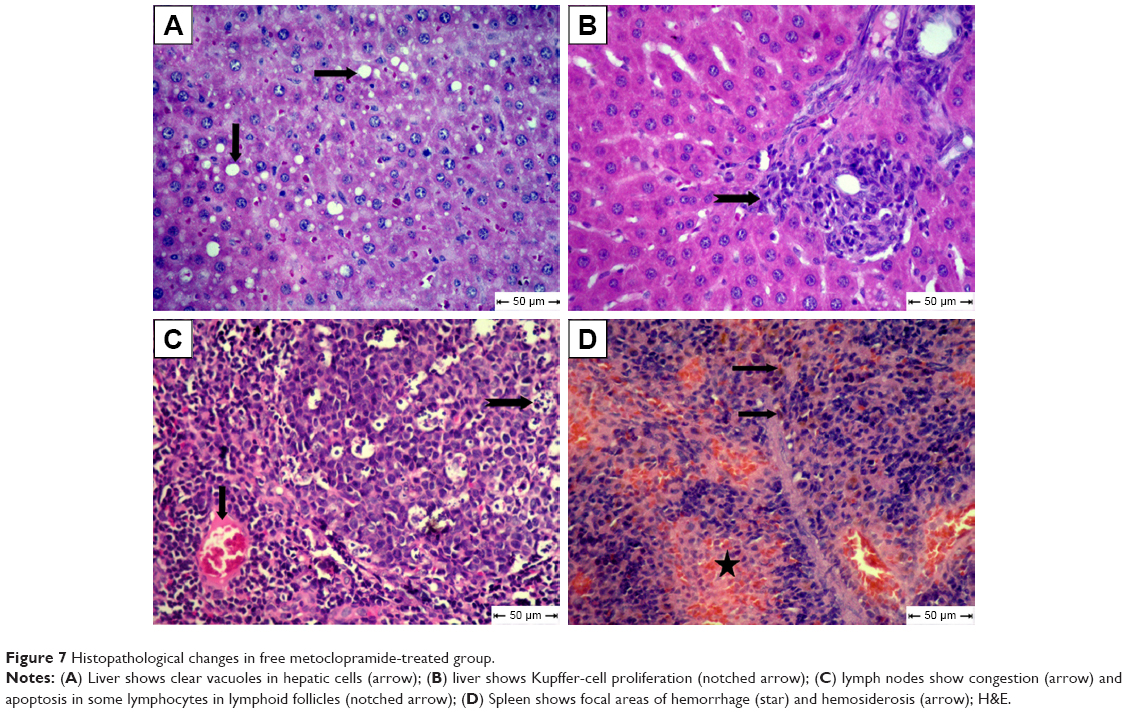 | Figure 7 Histopathological changes in free metoclopramide-treated group. |
The MCA NPs-treated group showed normal radiated hepatic cords (Figure 8A). Examination of lymph nodes showed follicular hyperplasia with variably sized reactive germinal centers (Figure 8B). Lymphoid follicles were heavily populated with small lymphocytes (Figure 8C). Extramedullary formation of hematopoietic cells accompanied with hemosiderosis was very obvious in red pulp of spleen (Figure 8D).
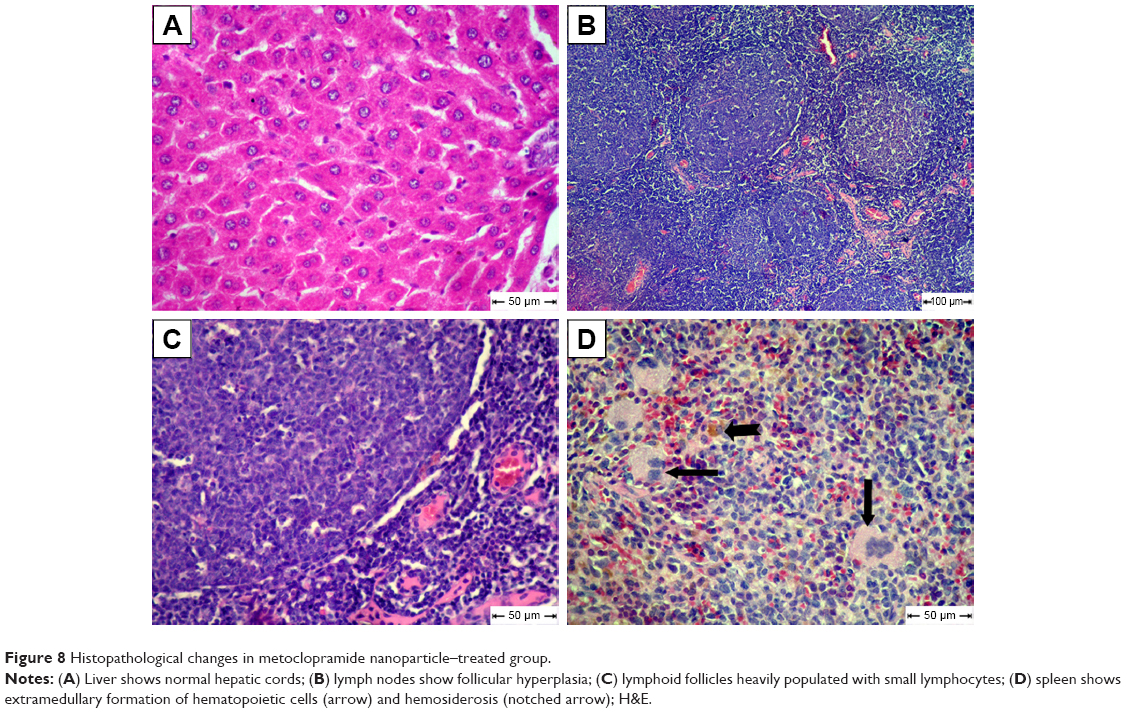 | Figure 8 Histopathological changes in metoclopramide nanoparticle–treated group. |
Discussion
In 2016, DM was classified by the WHO as the seventh-leading cause of death. Diabetes and its consequences can be managed with proper medical and physical intervention. Current antidiabetic medications might be able to ameliorate disease-induced inflammation via its hypoglycemic effect; however, this antidiabetic effect is inconsistent.5 Therefore, targeting the uncontrolled inflammation and improving the immune response of patients open new venues for better control of this disease and preventing its progression. Due to the reported immunomodulatory effect of MCA,16–19 this study aimed to test the effects of free MCA and BSA/MCA NPs on the immune system of diabetic rats via measuring the levels of IL1β, IL6, TNFα, and Treg cells frequency and additionally, via detection of histopathological changes on immunity-related organs (such as the liver, spleen, and lymph nodes).
BSA, which is homologous to human serum albumin, is a common carrier for many drugs in the form of microparticles and NPs, due to in vivo stability, low toxicity, nonimmunogenicity, and biodegradability.28 Despite the water-solubility of MCA, we successfully loaded it on BSA by desolvation followed by cross-linking with glutaraldehyde.29 Increasing BSA content showed a positive effect on EE and negative effect on DL (Figure 2). The positive effect on EE may be attributed to the well-defined structure of BSA. With fabrication pH of 8–9, BSA has negatively charged amino groups, which could electrostatically bind the positively charged aliphatic amines on MCA. Negative charges (−31±9 to −40±2 mV) on NPs revealed complete drug entrapment (Figure 3). Additionally, entrapping MCA inside the BSA matrix cross-linked using glutaraldehyde prevented MCA escape. This interaction between NP components was also revealed on DSC thermography (Figure 4B). The thermal behavior of the physical mixture of BSA and MCA (1:1) in the absence of glutaraldehyde exhibited characteristic endothermic peaks of both MCA and BSA, confirming absence of any interaction (Figure 4B). These characteristic endothermic peaks of MCA (at 95°C, 158°C, and 185°C) correspond to melting of the hydrated form, recrystallization of an anhydrous form, and final melting, respectively,30–32 and the characteristic endothermic peaks of BSA (69°C and 210°C) correspond to its melting (Figure 4B).25,33 However, thermography of BSA/MCA NPs did not show characteristic endothermic peaks of BSA or MCA, but new peaks appeared, indicating the interaction between NP components and cross-linking of BSA molecules using glutaraldehyde (cross-linker), where it is known that glutaraldehyde changes the thermal properties and crystallization temperatures of compounds.34 As a consequence of the aforementioned interactions between NP components, controlled and sustained release of MCA was observed (Figure 5A) over the faster release rate of the free MCA (100% release within 24 hours). The selected ratio (BSA:MCA 2:1) released only half the MCA content within 3 days, due to slow diffusion from the NP matrix (non-Fickian diffusion). MCA is considered a suitable candidate for sustained drug delivery due to its short half-life and has been incorporated in solid-lipid NPs, which controlled its release to around 15%–50% over 12 hours.24 Despite high EE of these solid-lipid NPs, our BSA NPs were characterized with fabrication simplicity, slower release pattern, and higher DL efficiency (10%–24% versus 5%–8%).
Sustained MCA release from NPs in small concentrations resulted in avoidance of most MCA side effects, such as extrapyramidal symptoms.35 The varied particle content of BSA had a slight effect on particle size (120–130 nm, Figure 3), similarly to previously reported.36,37 TEM shed light on particle morphology (Figure 4A), showing spherical particles smaller than obtained using light scattering in solution (120±3 nm), which has been attributed to determination of particle size in the dry form.38 The negative surface charge resulted in electrostatic repulsion among the formed NPs, preventing coagulation (polydispersity index <0.2, Figure 3).39,40 Protein–water interactions, which occur at any pH away from the isoelectric point of BSA (around 4.9), also decreased the coagulation of formed particles, resulting in more stabilized NPs in our preparation vehicles (pH 8–9).39,41 Small particles (120–130 nm) are beneficial in decreasing elimination from the body and increasing circulation time.32 Additionally, this size enables good penetration of biological barriers and interaction with biomolecules, which present in/on the cells, affecting the cellular responses.42 These promising in vitro findings of high EE, small particles and sustained release encourage us to use BSA/MCA NPs for further in vivo evaluation.
Currently, diabetes management is focusing on either optimizing available treatment or developing new antidiabetic drugs. MCA, which is the FDA-approved drug for diabetes-induced gastroparesis, is suggested in this study to be an adjunct for improving diabetes and decreasing its complications via working on the immune system. The principle behind this suggestion is the reported ability of MCA to improve inflammation and immune system dysfunction.16–19 This suggestion was supported with our findings that diabetic rats had significantly higher levels of TNFα, IL6, and IL1β but decreased percentage of peripheral CD4+CD25+Foxp3+ Treg when compared to nondiabetic rats (Table 2). T2D is currently regarded as a chronic inflammatory disease due to the implication of the inflammatory system in the pathogenesis of diabetes.43 Cytokines, a group of proteins that are expressed by several cell types, act as immune mediators and regulators. Treg cells and cytokines play important roles in the development of T2D. Moreover, research has demonstrated that the changes in the number and functions of Treg in the body are closely associated with the pathogenesis and development of various diabetic complications.44 Studies have reported that insulin resistance was associated with abnormal secretion of proinflammatory cytokines, such as TNFα, IL6 and IL1β, and decreased production of anti-inflammatory mediators, such as IL4 and IL10.45–47
When the diabetic rats were injected with free MCA (25 mg/kg/day) for 3 weeks, serum levels of proinflammatory cytokines (IL1β, IL6, and TNFα) were significantly decreased and the frequency of CD4+ T cell and Treg cells was significantly increased when compared to untreated diabetic rats (Table 2). The obvious MCA-lowering effect on proinflammatory cytokines is indirect via increased secretion of prolactin (immunoenhancing hormone).19,48–51 MCA (dopamine antagonist) increases prolactin secretion through blocking D2-receptors, preventing the inhibitory effect of dopamine (neurotransmitter) on prolactin release.19,52 Prolactin, which is responsible for many physiological functions, plays an important role in regulation of the immune system, where it can downregulate the expression of inflammatory cytokines.19,53
Treg cells, a small subset of T lymphocytes constituting only 5%–20% of the CD4+ compartment, are thought to be important to prevent excessive inflammatory responses and limit tissue impairment.54–58 In T2D, Treg cells can inhibit the inflammatory response by various pathways, such as surpassing cytokine secretion, modulating the microenvironment, and changing the expression of surface receptors.59
Nanomedicine, which is the integration of nanotechnology with medicine, has been extensively used to give an entirely new outlook to current medications. Advantages of NPs, such as sustained drug release, decreasing frequency of administration, decreasing side effects, and enabling cellular/molecular interactions due to smallness, make NPs an eye-catching tool for better management of diabetes. When diabetic rats were injected with BSA/MCA NPs (25 mg/kg/week) for 3 weeks, significant results were obtained. It was found that serum levels of IL1β, IL6, and TNFα were significantly decreased when compared to untreated diabetic rats, and when compared to free MCA-treated rats, both IL6 and TNFα showed a significant decrease (Table 2). Moreover, the frequency of CD4+ T cells and Treg cells was significantly increased when compared to untreated diabetic rats, and these NPs showed significant increases in Treg cells when compared to free MCA-treated rats. This significant difference produced with NPs, administered weekly, as opposed to the free drug, which was administered daily, could be attributed to the different benefits of nanotechnology. It was expected that albumin, which has a long half-life in circulation and has the ability to form NPs with sustained release (Figure 5A), would increase the circulation time of MCA.60 Long-circulating BSA NPs and release of small amounts of MCA guarantee that MCA in small concentrations will be available to work both peripherally and centrally through crossing the blood–brain barrier and exerting its effect through blocking the D2-receptors.61,62 Despite MCA readily crossing the blood–brain barrier, NPs are considered an efficient delivery carrier to the brain, where MCA releases gradually from NPs.60 Body exposure to small concentrations of MCA and consequently decreasing side effects is another advantage of using NPs over free drug.
Parallel to these serum-positive findings using MCA NPs, histopathological examination of the different animal groups confirmed the effect of MCA (Figures 6–8). Histopathological examination of diabetic rats showed hepatic lesions characterized by centrilobular hepatic necrosis, clear vacuoles in hepatic cells and cytoplasm that may have contained fat, Kupffer-cell proliferation in the portal area, and vascular changes, similar to typical hepatic lesions found in some studies.63,64 The liver plays a vital role in regulating glucose levels in pathological states of diabetes. Inflammation, and proliferation of Kupffer cells related to the role of liver in controlling glucose homeostasis by modifying the expression of hepatokines. Fetuin is one of the hepatokines that is responsible for increased inflammation and insulin resistance via inhibiting the insulin-receptor tyrosine kinase in hepatocytes.65 Also, accumulation of fat in hepatic cell cytoplasm is a characteristic lesion in diabetic rats. In diabetes, the liver usually stops the process of oxidizing fatty acids and uses them instead to synthesize triglycerides, which then accumulate abnormally in the liver. Additionally, in DM, insulin deficiency upregulates hormone-sensitive lipase in adipose tissue, subsequently leading to increased lipolysis and the circulation of free fatty acids, which then accumulate in the liver. The hepatic uptake of very-low-density lipoproteins increases due to these processes.66,67
The effect of diabetes on the immune system of rats in our experiment was demonstrated pathologically by the appearance of apoptotic lymphocytes in large number in lymphoid follicles accompanied by subcapsular vascular congestion in lymph nodes. Collagen deposition was observed in different areas in spleen red pulp, which is similar to previous findings.68 Diabetes decreases immune response, affecting immune cell function and the structure of immune organs, causing atrophy.69 In diabetes, increasing free-radical generation stimulates the release of proinflammatory cytokines, causing stressed lymphocytes and programmed cell death. TNF causes stimulation of death domains, such as Fas, which leads to activation of a signal transduction pathway inducing apoptosis.69,70
In this study, no significant improvement was observed on histopathological examination of livers or lymphatic tissue in free MCA-treated diabetic rats, while livers of MCA NPs-treated rats showed normally radiated hepatic cords without characteristic lesions of diabetes. There was hyperplasia of heavily populated lymphoid follicles with variably sized reactive germinal centers associated with extramedullary formation of hematopoietic cells. Apoptosis nearly disappeared in this group. These results are in line with previous studies, which showed that MCA treatment enhanced the depressed peritoneal and splenic macrophage function. The treatment of immunosuppressive effects of diabetes depends upon controlling apoptosis of lymphocytes in lymphatic tissue.71 MCA does not encourage apoptosis or influence the activation of caspases that complete apoptosis.72 MCA modifies splenocyte apoptosis and cytokine release in a murine model of sepsis.18 These encouraging histopathology results could be attributed to the aforementioned ability of MCA NPs to improve the immune function via decreasing proinflammatory cytokines and increasing the level of Treg cells (Table 2).
Conclusion
NPs have been considered promising drug delivery systems for the treatment of many diseases. As DM prevalence has been increasing more rapidly, nanotechnology may provide better modalities for management of diabetes and prevention of its complications through reducing inflammation and restoring normal functions of the immune system. BSA/MCA NPs were designed and developed to have high DL efficiency, be small, and sustain the release of MCA from NPs (only 50% MCA content within 3 days).
MCA (dopamine D2-receptor antagonist) stimulates the release of prolactin, which in turns downregulates inflammatory cytokines and additionally increases the frequency of Treg cells. BSA/MCA NPs (25 mg/kg/week) injected into diabetic rats for 3 weeks showed significant reductions in serum levels of proinflammatory cytokines when compared to both control diabetic rats and free MCA-treated rats. Furthermore, these albumin NPs showed significant increases in frequency of Treg cells when compared to control diabetic rats and free MCA-treated rats. Based on the inflammatory basis of diabetes, immunomodulatory effects of MCA NPs on diabetic rats could help better control of the disease and prevent progression. Depending on these findings, implementation of immunotherapy should be considered in the management of diabetes.
Disclosure
The authors report no conflicts of interest in this work.
References
Itariu BK, Stulnig TM. Autoimmune aspects of type 2 diabetes mellitus – a mini-review. Gerontology. 2014;60(3):189–196. | ||
Wentworth JM, Fourlanos S, Harrison LC. Reappraising the stereotypes of diabetes in the modern diabetogenic environment. Nat Rev Endocrinol. 2009;5(9):483–489. | ||
Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol Med Microbiol. 1999;26(3–4):259–265. | ||
Peleg AY, Weerarathna T, McCarthy JS, Davis TM. Common infections in diabetes: pathogenesis, management and relationship to glycaemic control. Diabetes Metab Res Rev. 2007;23(1):3–13. | ||
Pollack RM, Donath MY, Leroith D, Leibowitz G. Anti-inflammatory agents in the treatment of diabetes and its vascular complications. Diabetes Care. 2016;39(Suppl 2):S244–S252. | ||
Koh KK, Han SH, Quon MJ. Inflammatory markers and the metabolic syndrome: insights from therapeutic interventions. J Am Coll Cardiol. 2005;46(11):1978–1985. | ||
Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation. 2005;111(11):1448–1454. | ||
Ruan H, Lodish HF. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev. 2003;14(5):447–455. | ||
El-Deeb TS, Bakkar SM, Eltoony L, et al. The adipokine chemerin and fetuin-A serum levels in type 2 diabetes mellitus: relation to obesity and inflammatory markers. Egypt J Immunol. 2018;25(1):191–202. | ||
Hetta HF, Elkady A, Morsy KH, Mohamed IS, Ibrahim MA. Serum level of IL17A among cirrhotic hepatitis C virus infected patients with incidence of diabetes mellitus. Egypt J Immunol. 2017;24(1):79–88. | ||
Hetta HF, Ez-Eldeen ME, Mohamed GA, et al. Visfatin serum levels in obese type 2 diabetic patients: relation to proinflammatory cytokines and insulin resistance. Egypt J Immunol. 2018;25(2):141–151. | ||
Hetta HF, Khairy HM, Ismail SM, et al. Circulating IL17A and IFN-gamma serum levels in cirrhotic hepatitis C virus infected patients with autoimmune thyroiditis. Int J Curr Microbiol Appl Sci. 2017;6(3):1972–1983. | ||
Hetta HF, Elkady A, Meshaal AK. TH17/TH1 role in endocrine disorders among chronic HCV infected patients. Int J Curr Microbiol App Sci. 2017;6(8):2542–2551. | ||
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. | ||
Maedler K, Sergeev P, Ris F, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110(6):851–860. | ||
Knöferl MW, Angele MK, Ayala A, Cioffi WG, Bland KI, Chaudry IH. Insight into the mechanism by which metoclopramide improves immune functions after trauma-hemorrhage. Am J Physiol Cell Physiol. 2000;279(1):C72–C80. | ||
Zellweger R, Wichmann MW, Ayala A, Chaudry IH. Metoclopramide: a novel and safe immunomodulating agent for restoring the depressed macrophage immune function after hemorrhage. J Trauma Acute Care Surg. 1998;44(1):70–77. | ||
Oberbeck R, Schmitz D, Wilsenack K, et al. Metoclopramide and cellular immune functions during polymicrobial sepsis. Eur Surg Res. 2004;36(2):116–122. | ||
Jarrar D, Wang P, Song GY, et al. Metoclopramide: a novel adjunct for improving cardiac and hepatocellular functions after trauma-hemorrhage. Am J Physiol Endocrinol Metab. 2000;278(1):E90–E95. | ||
Sebak S, Mirzaei M, Malhotra M, Kulamarva A, Prakash S. Human serum albumin nanoparticles as an efficient noscapine drug delivery system for potential use in breast cancer: preparation and in vitro analysis. Int J Nanomedicine. 2010;5:525–532. | ||
Lomis N, Westfall S, Farahdel L, Malhotra M, Shum-Tim D, Prakash S. Human serum albumin nanoparticles for use in cancer drug delivery: process optimization and in vitro characterization. Nanomaterials. 2016;6(6):E116. | ||
Elzoghby AO, Samy WM, Elgindy NA. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157(2):168–182. | ||
Ibrahim E-SA, Ismail S, Fetih G, Shaaban O, Hassanein K, Abdellah NH. Development and characterization of thermosensitive pluronic-based metronidazole in situ gelling formulations for vaginal application. Acta Pharm. 2012;62(1):59–70. | ||
Mohamed RA, Abass HA, Attia MA, Heikal OA. Formulation and evaluation of metoclopramide solid lipid nanoparticles for rectal suppository. J Pharm Pharmacol. 2013;65(11):1607–1621. | ||
Jenita JL, Chocalingam V, Wilson B. Albumin nanoparticles coated with polysorbate 80 as a novel drug carrier for the delivery of antiretroviral drug-Efavirenz. Int J Pharm Investig. 2014;4(3):142. | ||
Akbarzadeh A, Mehrabi MR, Mehrabi MR, et al. Induction of diabetes by streptozotocin in rats. Indian J Clin Biochem. 2007;22(2):60–64. | ||
Bancroft JD. Theory and Practice of Histological Techniques. NY: Churchill Livingstone; 1982. | ||
Lin W, Coombes AG, Garnett MC, et al. Preparation of sterically stabilized human serum albumin nanospheres using a novel Dextranox-MPEG crosslinking agent. Pharm Res. 1994;11(11):1588–1592. | ||
Xu R, Fisher M, Juliano RL. Targeted albumin-based nanoparticles for delivery of amphipathic drugs. Bioconjug Chem. 2011;22(5):870–878. | ||
Gattani SG, Khabiya SS, Amrutkar JR, Kushare SS. Formulation and evaluation of bilayer tablets of metoclopramide hydrochloride and diclofenac sodium. PDA J Pharm Sci Technol. 2012;66(2):151–160. | ||
Hemalatha K. Formulation and evaluation of metoclopramide hydrochloride microbeads by ionotropic gelation method. Int J Pharm Biol Arch. 2011;2(3). | ||
Mitchell AG. Polymorphism in metoclopramide hydrochloride and metoclopramide. J Pharm Pharmacol. 1985;37(9):601–604. | ||
Michnik A. Thermal stability of bovine serum albumin DSC study. J Therm Anal Calorim. 2003;71(2):509–519. | ||
Sathiyanarayanana P, Karunakarana RJ, Gomathib T, Sudhab P. Synthesis and characterization of carboxymethyl cellulose/polyethylene glycol/montmorillonite clay blends. Int J Novel Trends Pharm Sci. 2015;5(2):36–41. | ||
Yadav Deepak R, Ayyappan T, Shanmugam S, Sundaramoorthy K, Vetrichelvan T. Development and in-vitro evaluation of buccoadhesive metoclopramide hydrochloride tablet formulations. Development. 2011;3(1):516–525. | ||
Sebak S, Mirzaei M, Malhotra M, Kulamarvaa, Prakash S. Human serum albumin nanoparticles as an efficient noscapine drug delivery system for potential use in breast cancer: preparation and in vitro analysis. Int J Nanomed. 2010;5:525. | ||
Langer K, Balthasar S, Vogel V, Dinauer N, von Briesen H, Schubert D. Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int J Pharm. 2003;257(1–2):169–180. | ||
Radovic-Moreno AF, Lu TK, Puscasu VA, Yoon CJ, Langer R, Farokhzad OC. Surface charge-switching polymeric nanoparticles for bacterial cell wall-targeted delivery of antibiotics. ACS Nano. 2012;6(5):4279–4287. | ||
Jun JY, Nguyen HH, Chun HS, Kang BC, Ko S. Preparation of size-controlled bovine serum albumin (BSA) nanoparticles by a modified desolvation method. Food Chem. 2011;127(4):1892–1898. | ||
Rahimnejad M, Jahanshahi M, Najafpour G. Production of biological nanoparticles from bovine serum albumin for drug delivery. Afr J Biotechnol. 2006;5(20). | ||
Hoffmann MA, van Mil PJ. Heat-induced aggregation of beta-lactoglobulin as a function of pH. J Agric Food Chem. 1999;47(5):1898–1905. | ||
Yan S, Zhang H, Piao J, et al. Studies on the preparation, characterization and intracellular kinetics of JD27-loaded human serum albumin nanoparticles. Procedia Eng. 2015;102:590–601. | ||
Xiao J, Li J, Cai L, Chakrabarti S, Li X. Cytokines and diabetes research. J Diabetes Res. 2014;2014:920613. | ||
Yang TT, Song SJ, Xue HB, Shi DF, Liu CM, Liu H. Regulatory T cells in the pathogenesis of type 2 diabetes mellitus retinopathy by miR-155. Eur Rev Med Pharmacol Sci. 2015;19(11):2010–2015. | ||
Fernández-Real JM, Broch M, Ricart W, et al. Plasma levels of the soluble fraction of tumor necrosis factor receptor 2 and insulin resistance. Diabetes. 1998;47(11):1757–1762. | ||
de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582(1):97–105. | ||
Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. 2007;148(1):241–251. | ||
Mccallum RW, Sowers JR, Hershman JM, Sturdevant RA. Metoclopramide stimulates prolactin secretion in man. J Clin Endocrinol Metab. 1976;42(6):1148–1152. | ||
McNeilly A, Thorner M, Volans G, Besser G. Metaclopramide and prolactin. Br Med J. 1974;2(5921):729. | ||
Healy DL, Burger HG. Increased prolactin and thyrotrophin secretion following oral metoclopramide: dose-response relationships. Clin Endocrinol. 1977;7(3):195–201. | ||
Falaschi P, Frajese G, Sciarra F, Rocco A, Conti C. Influence of hyperprolactinaemia due to metoclopramide on gonadal function in men. Clin Endocrinol. 1978;8(5):427–433. | ||
Van den Berghe G, de Zegher F. Anterior pituitary function during critical illness and dopamine treatment. Crit Care Med. 1996;24(9):1580–1590. | ||
Zhu XH, Zellweger R, Ayala A, Chaudry IH. Prolactin inhibits the increased cytokine gene expression in Kupffer cells following haemorrhage. Cytokine. 1996;8(2):134–140. | ||
Zheng Y, Rudensky AY. FOXP3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8(5):457–462. | ||
Hetta HF, Mekky MA, Khalil NK, et al. Association of colonic regulatory T cells with hepatitis C virus pathogenesis and liver pathology. J Gastroenterol Hepatol. 2015;30(10):1543–1551. | ||
Mehta M, Hetta HF, Abdel-Hameed EA, et al. Association between IL28b rs12979860 single nucleotide polymorphism and the frequency of colonic Treg in chronically HCV-infected patients. Arch Virol. 2016;161(11):3161–3169. | ||
Zahran AM, Mohammed Saleh MF, Sayed MM, Rayan A, Ali AM, Hetta HF. Up-regulation of regulatory T cells, CD200 and Tim3 expression in cytogenetically normal acute myeloid leukemia. Cancer Biomark. 2018;22(3):587–595. | ||
Zahran AM, Zharan KM, Hetta HF. Significant correlation between regulatory T cells and vitamin D status in term and preterm labor. J Reprod Immunol. 2018;129:15–22. | ||
Guzmán-Flores JM, Portales-Pérez DP. Mecanismos de supresión de las células T reguladoras (Treg) [Mechanisms of suppression of regulatory T-cells (Treg)]. Gac Med Mex. 2013;149(6):630–638. Spanish. | ||
Bergonzi MC, Guccione C, Grossi C, et al. Albumin nanoparticles for brain delivery: a comparison of chemical versus thermal methods and in vivo behavior. ChemMedChem. 2016;11(16):1840–1849. | ||
Shilo M, Sharon A, Baranes K, Motiei M, Lellouche JPM, Popovtzer R. The effect of nanoparticle size on the probability to cross the blood-brain barrier: an in-vitro endothelial cell model. J Nanobiotechnol. 2015;13(1):19. | ||
Mariam J, Sivakami S, Dongre PM. Albumin corona on nanoparticles-a strategic approach in drug delivery. Drug Deliv. 2016;23(8):2668–2676. | ||
Mohamed J, Nazratun Nafizah AH, Zariyantey AH, Budin SB. Mechanisms of diabetes-induced liver damage: the role of oxidative stress and inflammation. Sultan Qaboos Univ Med J. 2016;16(2):e132–e141. | ||
Regnell SE, Lernmark Å. Hepatic steatosis in type 1 diabetes. Rev Diabet Stud. 2011;8(4):454. | ||
Stefan N, Sun Q, Fritsche A, et al. Impact of the adipokine adiponectin and the hepatokine fetuin-A on the development of type 2 diabetes: prospective cohort- and cross-sectional phenotyping studies. PLoS One. 2014;9(3):e92238. | ||
Longo D, Kasper D, Kauser S, Loscalzo J, Fauci A, Jameson J. Harrison’s Principle of Internal Medicine. Edisi ke-18. New York, McGraw-Hill; 2011. | ||
Al-Hussaini AA, Sulaiman NM, Alzahrani MD, Alenizi AS, Khan M. Prevalence of hepatopathy in type 1 diabetic children. BMC Pediatr. 2012;12(1):160. | ||
Ebaid H, Al-Tamimi J, Metwalli A, et al. Effect of STZ-induced diabetes on spleen of rats: improvement by camel whey proteins. Pakistan J Zool. 2015;47(4):1109–1116. | ||
Ebaid H. Promotion of immune and glycaemic functions in streptozotocin-induced diabetic rats treated with un-denatured camel milk whey proteins. Nutr Metab. 2014;11(1):31. | ||
Park GB, Kim YS, Lee HK, Cho DH, Kim D, Hur DY. CD80 (B7.1) and CD86 (B7.2) induce EBV-transformed B cell apoptosis through the Fas/FasL pathway. Int J Oncol. 2013;43(5):1531–1540. | ||
Wang N, Sun H, Shen Y, et al. Augmenter of liver regeneration inhibits apoptosis of activated human peripheral blood lymphocytes in vitro. Immuno Immunotoxicol. 2013;35(2):257–263. | ||
Jaber BM, Petroianu GA, Rizvi SA, et al. Protective effect of metoclopramide against organophosphate-induced apoptosis in the murine skin fibroblast L929. J Appl Toxicol. 2018;38(3):329–340. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.