Back to Journals » OncoTargets and Therapy » Volume 12
Methyloleanolate Induces Apoptotic And Autophagic Cell Death Via Reactive Oxygen Species Generation And c-Jun N-terminal Kinase Phosphorylation
Authors Jeong MS, Jung JH ![]() , Lee H
, Lee H ![]() , Kim CG, Kim SH
, Kim CG, Kim SH ![]()
Received 11 April 2019
Accepted for publication 1 September 2019
Published 23 October 2019 Volume 2019:12 Pages 8621—8635
DOI https://doi.org/10.2147/OTT.S211904
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Nicola Silvestris
Myoung Seok Jeong,* Ji Hoon Jung,* Hyemin Lee, Chang Geun Kim, Sung-Hoon Kim
College of Korean Medicine, Kyung Hee University, Dongdaemun-Gu, Seoul 02447, Republic of Korea
*These authors contributed equally to this work
Correspondence: Sung-Hoon Kim
College of Korean Medicine, Kyung Hee University, 26, Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Republic of Korea
Tel +82-2-961-9233
Fax +82-2-961-9598
Email [email protected]
Background: To develop a potent anticancer agent similar to oleanolate, the underlying mechanisms of its derivative, methyloleanolate, in the apoptosis and autophagy of A549 and H1299 cells were elucidated.
Purpose: The aim of the present study was to investigate the effect of methyloleanolate in inducing apoptotic and autophagic cell death in cancer cells.
Materials and methods: Flow cytometric analysis with Annexin V/PI staining, Western blot analysis, and immunofluorescence analysis were conducted in A549 and H1299 cells.
Results: Methyloleanolate increased the fraction of Annexin V/PI apoptotic cells and activated caspase-8, caspase-3, and death receptor 5 (DR5) more than oleanolate in A549 and H1299 cells pretreated with pancaspase inhibitor z-VAD-fmk and DR5 depletion. Also, methyloleanolate induced autophagic features of microtubule-associated protein light chain 3 3BII (LC3BII) conversion and puncta in A549 and H1299 cells, along with autophagosomes and vacuoles. Methyloleanolate blocked autophagy flux for impaired autophagy and chloroquine (CQ)-enhanced microtubule-associated protein LC3BII accumulation and cytotoxicity in A549 and H1299 cells, although 3-methyladenine (3-MA) did not. Interestingly, LC3BII accumulation was detected only in methyloleanolate-treated autophagy-related gene 5 (ATG5)+/+ mouse embryonic fibroblast (MEF) cells but not in ATG5−/- MEF cells. Methyloleanolate reduced p-mTOR but activated p-c-Jun N-terminal kinases and reactive oxygen species production in A549 and H1299 cells. Conversely, n-acetyl-l-cysteine and SP600125 blocked apoptotic and autophagic cascades caused by methyloleanolate in A549 and H1299 cells.
Conclusion: Overall, the findings suggest that methyloleanolate induces apoptotic and autophagic cell death in non–small cell lung cancers via reactive oxygen species generation and c-Jun N-terminal kinase phosphorylation.
Keywords: methyloleanolate, apoptosis, autophagy, JNK, ROS
Introduction
Lung cancer causes 1.6 million deaths annually worldwide.1 Eighty percent of lung cancers are non–small cell lung cancers (NSCLCs), which have a poor prognosis.2 In particular, NSCLC is known to develop drug resistance by means of epidermal growth factor receptor mutation and anaplastic lymphoma kinase translocation.3 Epidermal growth factor receptor inhibitor gefitinib4,5 and anaplastic lymphoma kinase inhibitor crizotinib6 have been applied to combat drug resistance. Recently, natural compounds such as cucurbitacin7 1, fisetin,8 solamargine,9 and protocatechuic acid10 have been reported to increase antitumor efficacy and reduce chemoresistance in NSCLCs.
Programmed cell death is important in determining the fate of a cell under physiological and pathological conditions. Programmed cell death is classified into three categories based on morphological changes:11,12 apoptosis (type I), autophagic cell death (type II), and necrosis (type III). Apoptosis is characterized by cell shrinkage, loss of adhesion to the extracellular matrix or neighbors, nuclear condensation, and fragmentation.13 Autophagy is a catabolic process in which cells succumb to nutrient starvation, damage, and various stresses.14 Misfolded proteins and organelles are usually sequestered into autophagosomes and then degraded by lysozyme after the autophagosome fuses with a lysosome.15 In contrast, excessive accumulation of autophagic vacuoles in the cytoplasm can induce autophagic cell death16–18 in a caspase-independent pathway19 different from apoptosis or necrosis.20,21
Oleanolic acid (3β-hydroxyolean-12-en-28-oic acid; OA), a pentacyclic triterpenoid contained in Salvia glutinosa, Phytolacca americana, Rosa woodsii, Oldenladia diffusa, Kochia scoparia, and other plants, is known to have multiple biological activities, such as anti-inflammatory, antiviral, antifungal, hepatoprotective, and antitumor effects.22–24 The antitumor effect of oleanolic acid has been considered to be limited in several cancers. The present study aimed to explore the underlying antitumor mechanism of methyloleanolate (MO), a derivative of OA, in A549 and H1299 NSCLC cells and to determine MO’s association with apoptotic and autophagic cell death signaling pathways.
Materials And Methods
Reagents And Antibodies
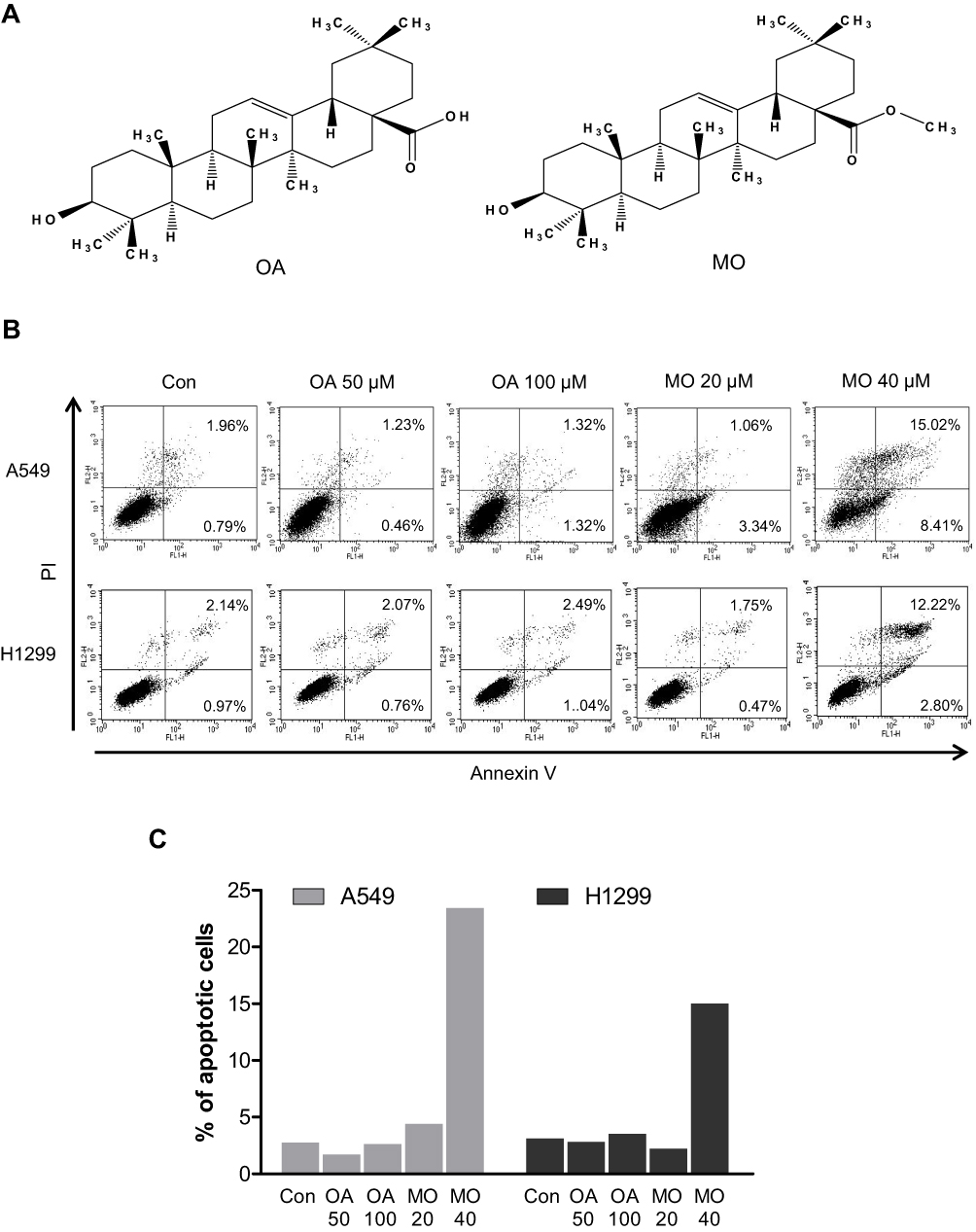
OA and MO (Figure 1) were isolated from Salvia glutinosa25 by Dr Namin Baek, a pharmacognosist and professor at Kyung Hee University, and purified up to 98% by means of high-performance liquid chromatography. Dulbecco’s Modified Eagle’s Medium, Roswell Park Memorial Institute (RPMI) 1640 medium, and fetal bovine serum (FBS) were purchased from Welgene (Gyeongsan, Korea). 3-Methyladenine (3-MA), N-Acetyl-L-cysteine (NAC), 2′,7′-dichlorofluorescin diacetate (DCF-DA) and β-actin were purchased from Sigma-Aldrich (St Louis, MO, USA). Lipofectamine 2000 reagent was purchased from Invitrogen (Carlsbad, CA, USA). Antibodies for poly (ADP-ribose) polymerase (PARP), caspase-3, caspase-8, FasL, DR4, Bid, mammalian or mechanistic target of rapamycin (mTOR), p-mTOR (Ser2448), SAPK/JNK, p-SAPK/JNK (Thr183/Tyr185), ATG5, ATG7 p62 sequestosome (p62/SQSTM1), and beclin 1 were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies for LCB3B and DR5 were purchased from Novous (San Diego, CA, USA) and Santa Cruz Biotechnology (Santa Cruz, CA, USA), respectively. SP600125 and z-VAD-fmk were obtained from Calbiochem (San Diego, CA, USA).
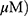
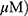
Cell Cultures
Non–small cell lung cancer (NSCLC) cell lines A549 (ATCC® ccl-185™) and H1299 (ATCC® CRL-5803™) and normal lung fibroblast HEL299 cells (ATCC® CCL-137™) were purchased from American Type Culture Collection (Manassas, VA, USA). ATG5+/+ and ATG5−/- MEF cells were obtained from Professor Guillermina Lozano (The University of Texas MD Anderson Cancer Center, USA) and were approved (19–084) by the Living Modified Organism (LMO) committee of Kyung Hee University. The cells were maintained in RPMI 1640 supplemented with 10% FBS (Gibco, Carlsbad, CA, USA), 1% penicillin/streptomycin, and 2 µM L-glutamine (WelGene, Deagu, South Korea) at 37 °C in 5% CO2. The cells were incubated in the growth medium under a humidified, 5% CO2 atmosphere at 37 °C.
Cell Viability Assay
The cytotoxic effects of OA and MO were evaluated by means of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In brief, A549 and H1299 cells (1 × 104 cells per well) were seeded onto 96-well culture plates and exposed to various concentrations of OA or MO for 24 or 48 hrs. The cells were incubated with MTT (1 mg/mL) for 2 hrs and then treated with MTT lysis solution for 1 hr. Optical density was measured with a microplate reader (Molecular Devices Co, San Jose, USA) at 570 nm. Cell viability was calculated as the number of viable cells in the groups treated with OA or MO as a percentage of the number of viable cells in the untreated control.
Apoptosis Detection By Annexin V/PI Double Staining
A549 and H1299 cells were exposed to OA (50 or 100 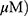 or MO (20 or 40
or MO (20 or 40 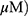 for 12 hrs, washed with phosphate-buffered saline (PBS), and harvested by trypsinization. The cells were gently resuspended in a binding buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) and incubated with Annexin V-FITC and propidium iodide for 15 mins at room temperature in the dark with the Annexin V-Apoptosis Detection kit (Biovision, Mipitas, USA). Apoptotic cells were analyzed by FACSCalibur (Becton Dickinson, San Jose, USA) and were defined as the number of cells positively stained by Annexin V/PI as apoptotic portion.
for 12 hrs, washed with phosphate-buffered saline (PBS), and harvested by trypsinization. The cells were gently resuspended in a binding buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) and incubated with Annexin V-FITC and propidium iodide for 15 mins at room temperature in the dark with the Annexin V-Apoptosis Detection kit (Biovision, Mipitas, USA). Apoptotic cells were analyzed by FACSCalibur (Becton Dickinson, San Jose, USA) and were defined as the number of cells positively stained by Annexin V/PI as apoptotic portion.
Western Blot Analysis
A549 and H1299 cells were seeded onto 60-mm dishes at 5 × 105 cells per dish in RPMI 1640 with 10% FBS and exposed to OA (50 or 100 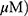 or MO (20 or 40
or MO (20 or 40 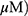 for 12 hrs. The cells were then washed with cold PBS and harvested by centrifugation. The pellets were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer containing RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, and 1% TritonX-100) containing protease inhibitors (Roche, Basel, Switzerland). Cell lysates were centrifuged at 13,000 rpm for 15 mins at 4 °C, and the supernatant was collected. Protein content was quantified with the DC Protein Assay Kit (Bio-Rad) according to the manufacturer’s instructions. An equal amount of protein (30 μg) was subjected to 10% to 15% sodium dodecyl sulfate and polyacrylamide gel SDS-PAGE) and transferred onto 0.45 μm nitrocellulose membranes. The membranes were blocked with 5% skim milk or 3% Bovine Serum Albumin (BSA) in PBS with 0.1% Tween-20 and incubated with primary antibody at 4 °C. The membranes were washed and incubated with secondary antibodies. Proteins were detected using the enhanced chemiluminescence (ECL) system (Amersham, Buckinghamshire, England) and analyzed by densitometry with Image J (Bethesda, USA) software.
for 12 hrs. The cells were then washed with cold PBS and harvested by centrifugation. The pellets were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer containing RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, and 1% TritonX-100) containing protease inhibitors (Roche, Basel, Switzerland). Cell lysates were centrifuged at 13,000 rpm for 15 mins at 4 °C, and the supernatant was collected. Protein content was quantified with the DC Protein Assay Kit (Bio-Rad) according to the manufacturer’s instructions. An equal amount of protein (30 μg) was subjected to 10% to 15% sodium dodecyl sulfate and polyacrylamide gel SDS-PAGE) and transferred onto 0.45 μm nitrocellulose membranes. The membranes were blocked with 5% skim milk or 3% Bovine Serum Albumin (BSA) in PBS with 0.1% Tween-20 and incubated with primary antibody at 4 °C. The membranes were washed and incubated with secondary antibodies. Proteins were detected using the enhanced chemiluminescence (ECL) system (Amersham, Buckinghamshire, England) and analyzed by densitometry with Image J (Bethesda, USA) software.
Immunofluorescence Analysis For LC3 Puncta
A549 and H1299 cells were seeded at 5 × 104 cells onto glass discs, and one disc was placed in each well of 24-well plates and exposed to OA or MO for 12 hrs. The cells were fixed with 4% paraformaldehyde in PBS for 15 mins at room temperature and the discs were treated with 0.01% Triton X-100 for 10 mins and blocked with 2% BSA for 30 mins at room temperature. The cells were incubated with anti-LC3 antibody at 4 °C overnight and cultured with Alexa Fluor 488-conjugated secondary antibody for 1 hr. Nuclear staining was then performed with DAPI (Sigma) and fluorescence images of the cells were observed under a Zeiss LSM 710 microscope (Boston, USA). Also, the number of green fluorescent protein (GFP)-LC3 puncta per cell was counted from randomly selected photographs.
Autophagic Flux Assay With Tandem Sensor Red And Green Fluorescent Protein LC3 Constructs
To investigate whether autophagy flux was impaired by MO, an autophagy flux assay was conducted. A549 and H1299 cells were transfected with tandem sensor LC3 conjugated to red fluorescent protein and green fluorescent protein (RFP-GFP-LC3) constructs (a gift from Professor Hongbo Hu from the Department of Nutrition and Health, College of Food Science and Nutritional Engineering, China Agricultural University) by a transient transfection method using Lipofectamine 2000 (Thermo Fisher Scientific, New York, USA). The cells were then exposed to 40 µM MO for 12 hrs. Then, the merged red or green channel signals were observed in MO-treated A549 and H1299 cells under the Zeiss LSM 710 confocal microscope.
RNA Interference
A549 and H1299 cells were transfected with scrambled siRNA or DR5 siRNA plasmids with Lipofectamine 2000 and InterferinTM transfection reagent (Polyplus-transfection Inc, New York, NY, USA). The mixtures of DR5 siRNA (40 nM) and transfection reagent were incubated for 10 mins and then the cells were incubated at 37 °C for 48 hrs before exposure to MO (40 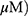 for 12 hrs.
for 12 hrs.
Measurement Of Reactive Oxygen Species Production
A549 and H1299 cells were seeded at 3 × 105 per well in 6-well plates in a phenol red–free RPMI 1640 with 10% FBS. The cells were pretreated with or without NAC for 1 hr and then exposed to MO (10, 20, 30, or 40  for 12 hrs. After incubation, fluorescent 2ʹ,7ʹ –dichlorofluorescin diacetate (DCF-DA) was added at 37 °C for 30 mins before cells were harvested. Reactive oxygen species (ROS) production was analyzed by means of flow cytometry and confocal microscopy.
for 12 hrs. After incubation, fluorescent 2ʹ,7ʹ –dichlorofluorescin diacetate (DCF-DA) was added at 37 °C for 30 mins before cells were harvested. Reactive oxygen species (ROS) production was analyzed by means of flow cytometry and confocal microscopy.
Statistical Analysis
Values were presented as the mean plus or minus standard deviation. Statistical significance was evaluated by Student’s t-test using SigmaPlot software (Systat Software Inc, San Jose, USA) or ANOVA followed by Tukey post-hoc test when the F statistic was significant. P < 0.05 was considered statistically significant. At least three independent experiments were performed in duplicate for each assay.
Results
Effect Of MO On The Fraction Of Apoptotic Cells In A549 And H1299 Non–Small Cell Lung Cancer Cells
To compare the apoptotic effect of MO and OA (Figure 1A), a flow cytometry assay with Annexin V/PI staining was conducted on A549 and H1299 NSCLC cells. MO increased the fraction of apoptotic A549 and H1299 cells at concentrations of 20 and 40 μM, while OA only weakly increased the fraction of apoptotic cells even at 50 and 100 μM (Figure 1B and C). But MO and OA showed weak cytotoxicity in normal lung fibroblast HEL 299 cells (Supplementary Figure 1).
Effect Of MO On Extrinsic Apoptosis Through DR5 Activation In A549 And H1299 Cells
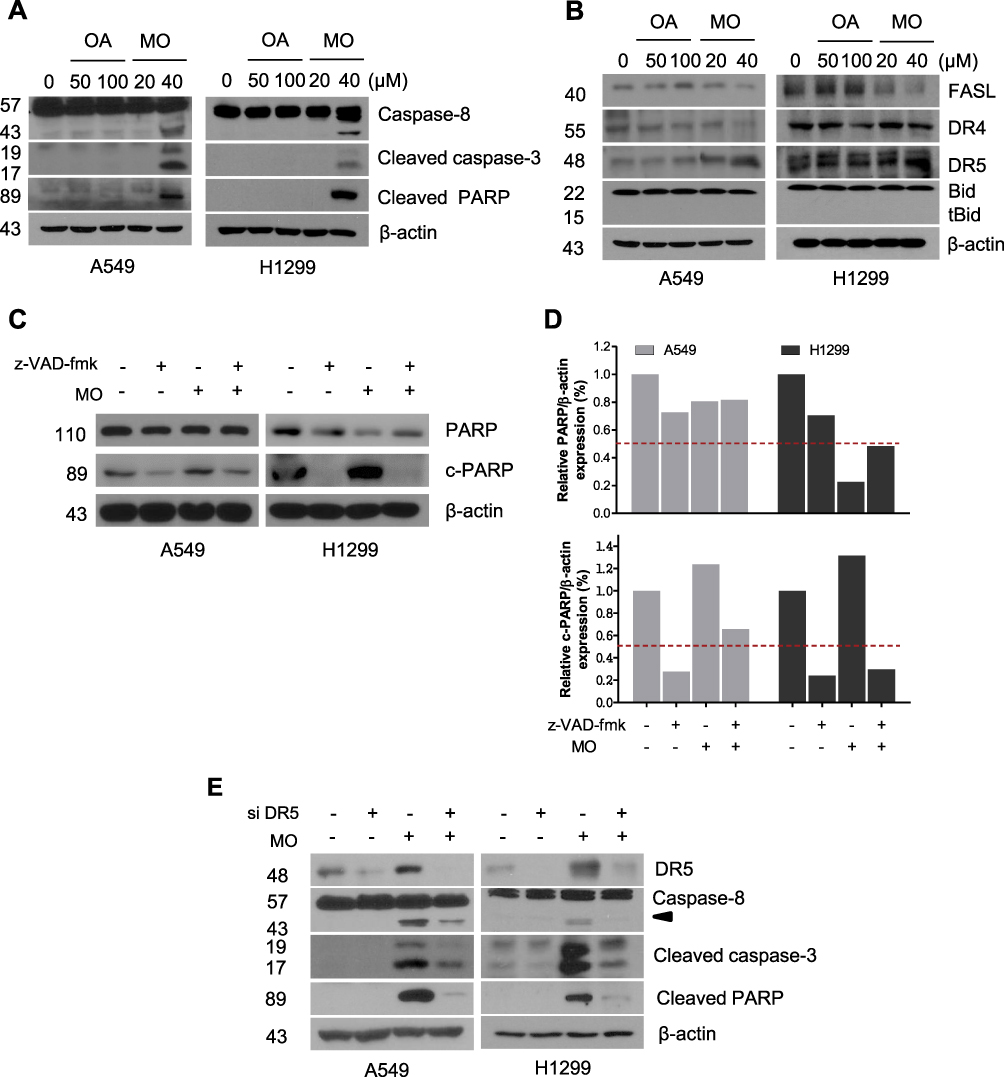
To determine whether the cytotoxic effect of MO is due to apoptosis induction, Western blotting was performed on A549 and H1299 cells. MO markedly activated caspase-8 and caspase-3 and cleaved PARP in MO-treated A549 and H1299 cells (Figure 2A), while DR5 was upregulated and no effect was seen on FasL, DR4, and tBid (Figure 2B). However, pretreatment with pancaspase inhibitor z-VAD-fmk or knockdown of DR5 reduced cytotoxicity and cleavage of PARP and caspase-3 in MO-treated A549 and H1299 cells (Figure 2C and D).
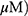
Effect Of MO On Autophagy In A549 And H1299 Cells
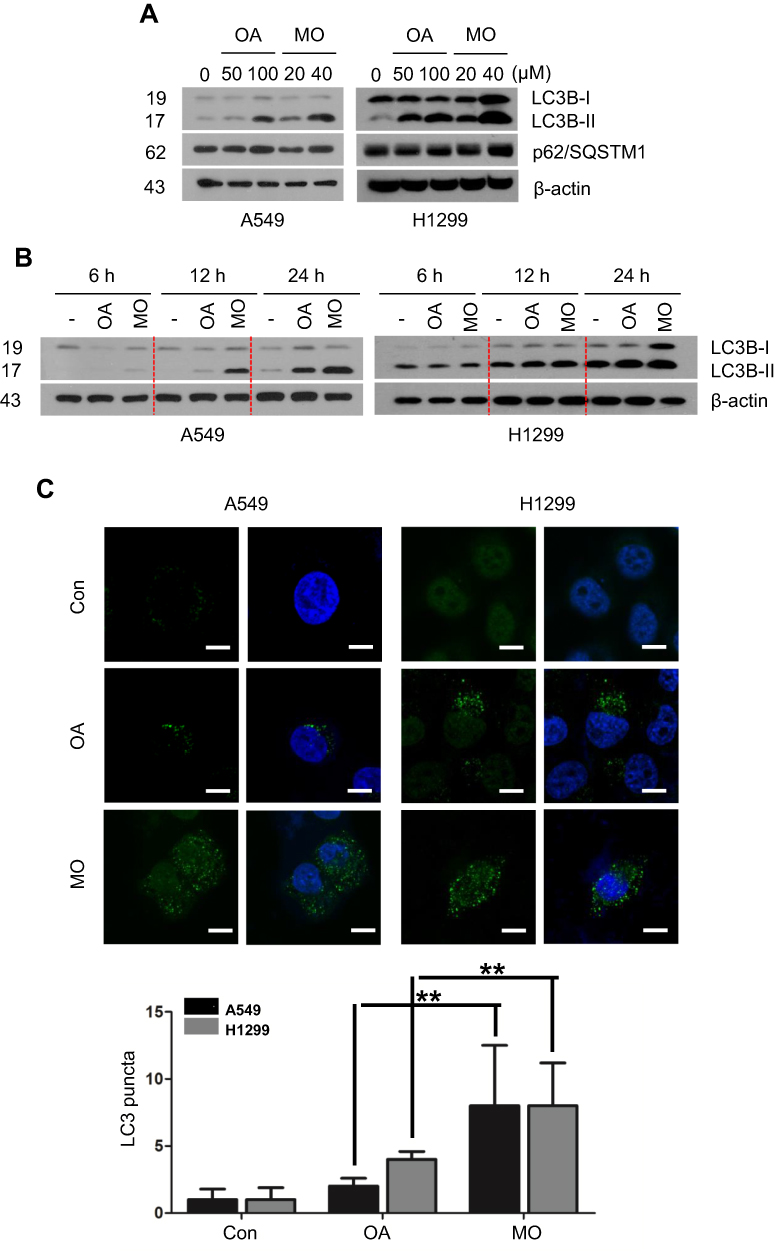
Based on findings that OA can induce protective autophagy in A549 cells26 at 100 µg/mL, the effect of MO on autophagy was evaluated in A549 and H1299 cells. MO increased LC3B-II accumulation in a concentration- and time-dependent manner without significant effect on p62 in A549 and H1299 cells more than OA did (Figure 3A and B). MO consistently enhanced the formation of GFP-LC3 puncta and autophagic vacuoles more than OA did in A549 and H1299 cells (Figure 3C, Supplementary Figure 2).
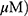
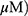
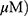
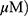
Effect Of MO On Incomplete Autophagy Flux In A549 And H1299 Cells
To evaluate whether the elevation of LC3 lipidation induced by MO was due to fusion with autolysosomes or increased degradation, an autophagy flux assay was conducted in A549 and H1299 cells transfected with RFP-GFP-LC3 constructs. As shown in Figure 4A, immunofluorescence revealed the merged yellow color in MO-treated A549 and H1299 cells, whereas more red puncta were observed in OA-treated A549 and H1299 cells. Of note, an early stage autophagy inhibitor, 3-MA, did not significantly affect the cytotoxicity and weakly attenuated the expression of LC3BII in MO-treated A549 and H1299 cells (Figure 4B and C), while a late-stage autophagy inhibitor, chloroquine (CQ), enhanced the cytotoxicity of MO in A549 and H1299 cells (Figure 4D).
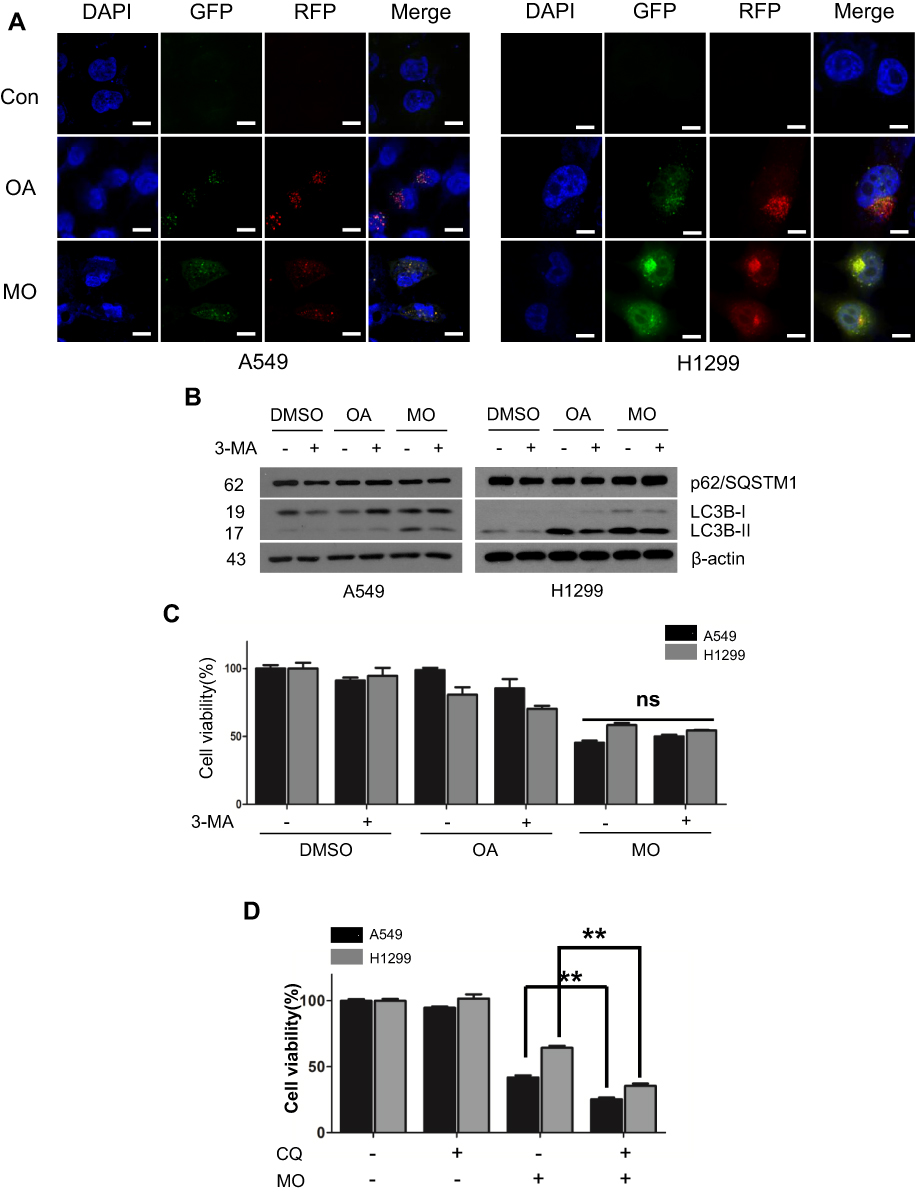 |
Figure 4 Methyloleanolate (MO)-impaired autophagy flux in A549 and H1299 cells. (A) Effect of oleanolate (OA) and MO on autophagy flux in A549 and H1299 cells. A549 and H1299 cells were transfected with LC3 conjugated to red fluorescent protein and green fluorescent protein constructs and exposed to OA (100 μM) or MO (40 μM) for 12 hrs. Autophagy flux was evaluated with confocal microscopy. (B) Effect of 3-methyladenine (3-MA) on 1A/1B-light chain 3BII (LC3BII) and p62/SQSTM1 in OA- or MO-treated A549 and H1299 cells. The cells were pretreated with 3-MA for 1 hr and incubated with OA (100 μM) or MO (40 μM) for 12 hrs. Then western botting was conducted and antibodies of LC3BII and p62/SQSTM1 were used for detection. (C) Effect of 3-MA on the viability of OA- or MO-treated A549 and H1299 cells. The cells were pretreated with 3-MA for 1 hr, exposed to OA (100 μM) or MO (40 μM) for 12 hrs, and an MTT assay was performed. (D) Effect of chloroquine (CQ) on the viability of MO-treated A549 and H1299 cells. The cells were pretreated with CQ for 1 hr and exposed to MO (40 μM) for 12 hrs and a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay was performed. Data are shown as means ±SD from three independent experiments. **P < 0.01 between CQ- and MO-treated groups. N = 6 per group. Abbreviation: NS, not significant. |
Effect Of MO On ATG5-Dependent Autophagy In A549 And H1299 Cells
To elucidate the mechanism of MO-induced autophagy, the effect of MO on beclin 1, ATG5, and ATG7 was examined in A549 and H1299 cells. Interestingly, MO attenuated the expression of beclin 1 and ATG7 but increased the expression of ATG5 in A549 and H1299 cells more than OA did (Figure 5A). Furthermore, MO reduced the expression of beclin 1 in a time-dependent fashion in A549 and H1299 cells (Figure 5B). To confirm the role of ATG5 in MO-induced autophagy, Western blotting was performed in ATG5+/+ and ATG5−/- MEF cells. As shown in Figure 5C, MO induced LC3II conversion and upregulation of ATG5 in ATG5+/+ MEF cells but it did not in ATG5−/- MEF cells.
 |
Figure 5 Autophagy-related gene 5 (ATG5) plays a critical role in methyloleanolate (MO)-induced autophagy in A549 and H1299 cells. (A) Concentration-dependent effect of oleanolate (OA) or MO on beclin 1, ATG5 and ATG7 in A549 and H1299 cells. Cells were incubated with OA (50, 100 µM) or MO (20, 40 µM) for 12 hrs and subjected to Western blotting and antibodies of beclin 1, ATG5, and ATG7 were used for detection. (B) Time-dependent effect of MO (40 μM) on beclin 1 and 1A/1B-light chain 3BII (LC3BII) in A549 and H1299 cells. (C) Effect of endogenous ATG5 on beclin 1, LC3BII, and ATG5 in ATG5+/+ or ATG5−/- MEF cells. ATG5+/+ or ATG5−/- MEF cells were treated with MO (40 μM) for 12 hrs and Western blotting was performed with LC3BII and ATG5 antibodies. |
Effect Of ROS Generation On MO-Induced Autophagy In A549 And H1299 Cells
Emerging evidence shows that ROS play a critical role in apoptotic and autophagic cell death.12,27–29 To confirm whether MO is associated with ROS generation, ROS production was measured by flow cytometric analysis with DCFDA staining in A549 and H1299 cells. MO significantly induced ROS production in a concentration- and time-dependent manner (Figure 6A and B). Conversely, ROS inhibitor NAC suppressed the number of green fluorescent ROS particles observed by immunofluorescence after DCFDA staining (Figure 6C). ROS inhibitor NAC blocked phosphorylation of c-Jun N-terminal kinases (JNK), activation of DR5, LC3BII conversion, and PARP cleavage induced by MO in A549 and H1299 cells (Figure 6D). NAC consistently suppressed cytotoxicity and LC3 puncta by MO in A549 and H1299 cells (Figure 6E and F).
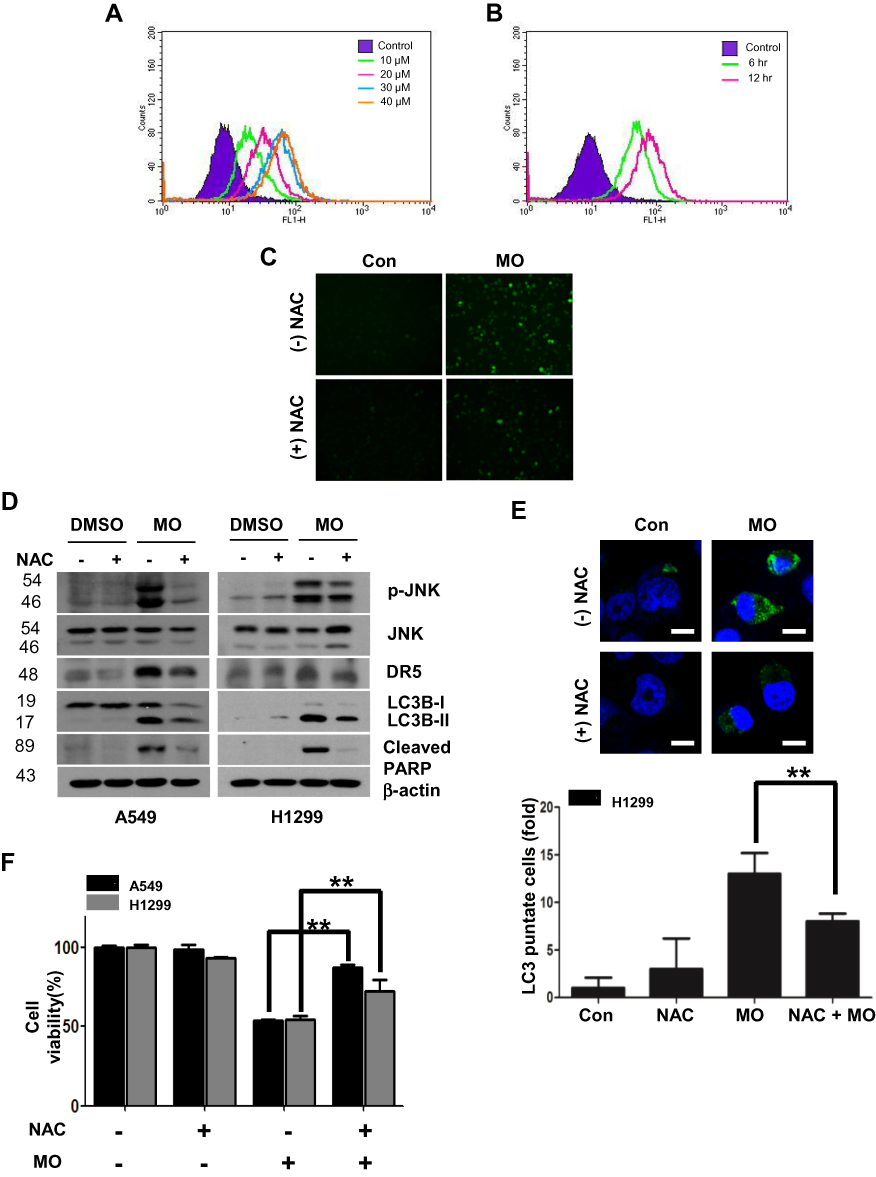 |
Figure 6 Reactive oxygen species (ROS) mediate cytotoxicity and cell death induced by methyloleanolate (MO) in H1299 cells. (A) Effect of MO on ROS production in H1299 cells. ROS level was measured as DCFDA by flow cytometry in H1299 cells treated with various concentrations of MO (10, 20, 30, 40 μM) for 12 hrs. (B) Time-dependent effect of MO on ROS production in H1299 cells. The cells were exposed to 40 μM MO for 6 and 12 hrs, and ROS production was analyzed by flow cytometry after DCFDA staining for 30 mins. (C) ROS were generated by MO (40 μM) with or without N-acetyl-L-cysteine (NAC) by using DCFDA, and the green fluorescence images were analyzed by fluorescence microscopy. (D) Effect of NAC on pJNK DR5, LC3BII, and c-PARP in A549 and H1299 cells. Cells were treated with MO (40 μM) for 12 hrs, after pretreatment with 4 mM NAC, and subjected to Western blotting with p-JNK, JNK, DR5, LC3BII, and cleaved PARP antibodies. (E) Effect of NAC on LC3 puncta in MO-treated H1299 cells. H1299 cells were exposed to MO (40 μM) for 12 hrs and LC3B puncta were detected by confocal microscopy. **P <0.01 versus control. Bar: 10 μm. (F) Effect of NAC on the viability of MO-treated A549 and H1299 cells. Cells were exposed to MO (40 μM) for 12 hrs and a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay was performed. Results represent means ±SD from three independent experiments. **P < 0.01 versus control. N = 6 per group. |
Effect Of JNK On MO-Induced Autophagy In A549 And H1299 Cells
It is well documented that JNK and the mammalian or mechanistic target of rapamycin (mTOR) play crucial roles in autophagy induction.30–32 Here, Western blots revealed that MO weakly attenuated the expression of mTOR and remarkably activated the phosphorylation of JNK in a time- and concentration-dependent fashion more than OA did in A549 and H1299 cells (Figure 7A). Conversely, JNK inhibitor SP600125 blocked phosphorylation of JNK, LC3BII accumulation, DR5 activation, and PARP cleavage induced by MO in A549 and H1299 cells (Figure 7B). SP600125 consistently reduced the number of LC3 puncta and cytotoxicity by MO in H1299 cells (Figure 7C and D).

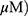
Discussion
Lung cancer has a high death rate because it is difficult to detect at an early stage and thus has often metastasized to other organ tissues before it is diagnosed and treated.33 In the current study, the underlying antitumor mechanism of MO was comparatively assessed in A549 and H1299 NSCLCs with OA. There have been no previous reports on the antitumor effect of MO, although there have been reports on its antidiabetic activity.34 The results of this study showed that MO increased the fraction of Annexin V/PI–positive apoptotic cells and activated caspases (8 and 3) and DR5 in A549 and H1299 cells, and did not affect Bid. This implies that MO has an extrinsic apoptotic effect in NSCLCs. Pancaspase inhibitor z-VAD-fmk consistently reduced the cytotoxicity and cleavage of PARP and caspase-3. DR5 knockdown also attenuated the cleavage of PARP and caspase-8 and caspase-3 in MO-treated A549 and H1299 cells, which indicates that the cytotoxicity of MO mainly occurs via an extrinsic apoptosis signaling pathway.
Interestingly, MO induced autophagic features in A549 and H1299 cells, including LC3BII accumulation and puncta, along with autophagosomes and vacuoles (observed by transmission electron microscopy). These results may indicate that MO causes autophagic cell death, because excessive accumulation of autophagic vacuoles in the cytoplasm may indicate hyperactivation of autophagy which can lead to autophagic cell death.16–18 However, the effects of different concentrations of MO may be complex. Research has shown that low concentrations of flavonoids are protective in rat H4IIE cells whereas high concentrations cause DNA damage and apoptosis.35 In addition, low concentrations of nonsteroidal anti-inflammatory drugs can induce autophagy at early stages, whereas higher concentrations of nonsteroidal anti-inflammatory drugs can inhibit autophagy and induce apoptosis at later stages.36 In light of these results, further study on the effect of MO on autophagy and apoptosis at low and high concentrations is warranted.
Autophagy is a catabolic process consisting of four distinct steps, initiation, nucleation, elongation, and degradation, which are regulated by autophagy-related genes (ATGs).37 Among the ATG proteins, autophagy-related gene 5 (ATG5) is one of the key regulators of autophagic cell death,38 while ATG6 and beclin 1 have multiple functions are not autophagy-specific39 and ATG7 is an important protein in the elongation of the autophagosome membranes.40 The results reported here show that MO attenuated the expression of mTOR, but activated ATG5 in A549 and H1299 cells. However, depletion or overexpression of ATG6 or beclin 1 did not affect LC3BII accumulation in A549 and H1299 cells (data not shown). In contrast, MO-induced LC3BII accumulation in ATG5+/+ MEF cells but not in ATG5−/- MEF cells, which indicates the pivotal role of ATG5 in the MO-induced autophagic effect in A549 and H1299 cells.
Emerging evidence reveals that autophagy has dual effects: it can have a cytoprotective effect on healthy cells and can cause autophagic cell death in cancer cells.41–44 To confirm the exact autophagic mechanism of MO, an autophagy flux assay and an autophagy inhibitor study were carried out in the current study. Immunofluorescence revealed a yellowish color in MO-treated A549 and H1299 cells transfected with RFP-GFP-LC3 constructs, which indicates incomplete autophagy because the green fluorescent protein fluorescence was not reduced under the acidic lysosomal environment. Furthermore, early-stage autophagy inhibitor 3-MA did not significantly affect the cytotoxicity and protein expression of LC3BII and p62/SQSTM-1, whereas late-stage autophagy inhibitor CQ enhanced the cytotoxicity of MO in A549 and H1299 cells. This implies that the cytotoxicity of MO is mainly caused by autophagic cell death via autophagosome accumulation, which occurs despite the impaired autophagy process in A549 and H1299 cells.
There is accumulating evidence that excessive ROS generation can be toxic and induce cell death.45 Herein, MO significantly induced ROS production in a concentration- and time-dependent fashion and ROS inhibitor NAC blocked cytotoxicity, PARP cleavage, and LC3BII accumulation with puncta and activation of p-JNK and DR5 induced by MO in A549 and H1299 cells. This demonstrates the crucial role of ROS in activating p-JNK in MO-induced apoptotic and autophagic cell death in NSCLCs. Similarly, Song et al46 revealed that OA derivative SZC017 induced ROS-dependent apoptosis in A549 cells through inactivation of Akt and JAK2/STAT3 signaling.
It is well documented that transient activated JNK is associated with cell survival, sustained JNK activation induces apoptosis,47 and chemoresistance is acquired through autophagy mediated by JNK and mTOR pathways.45,48,49 Our study showed that MO-induced phosphorylation of JNK in A549 and H1299 cells. Conversely, JNK inhibitor SP600125 blocked cytotoxicity, PARP cleavage, LC3BII accumulation with puncta and activation of p-JNK and DR5 induced by MO in A549 and H1299 cells, which indicates a critical role of p-JNK in MO-induced apoptotic and autophagic cell death in NSCLCs. Similarly, Liu et al50 demonstrated OA-induced apoptosis in A549 cells via activation of ROS/ASK1/p38 MAPK pathways, but not JNK signaling. Further in vitro, in vivo, and clinical studies are required to elucidate the detailed mechanisms, pharmacokinetics, and toxicology of MO, especially given that MO showed better anticancer potential in NSCLC cells than OA did.
Conclusion
MO increased the fraction of Annexin V/PI apoptotic cells, activated caspase-3, caspase-8, DR5, ATG5, p-JNK, and ROS production, and induced autophagic features of LC3BII conversion. NAC and JNK inhibitor SP600125 blocked the apoptotic and autophagic cell death by MO in A549 and H1299 cells. Overall, the findings reported here suggest that MO induces apoptotic and autophagic cell death in NSCLC cells more effectively than OA does through ROS generation and JNK phosphorylation.
Acknowledgments
We thank the Kim lab members for active discussion and suggestions. The work reported here was supported by the National Research Foundation of Korea Grant funded by the Korean Government (no. 2017R1A2A1A17069297).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA. 2015;65(2):87–108. doi:10.3322/caac.21262
2. Zhou C, Yao LD. Strategies to improve outcomes of patients with EGRF-mutant non-small cell lung cancer: review of the literature. J Thorac Oncol. 2016;11(2):174–186. doi:10.1016/j.jtho.2015.10.002
3. Shi SB, Wang M, Tian J, Li R, Chang CX, Qi JL. MicroRNA 25, microRNA 145, and microRNA 210 as biomarkers for predicting the efficacy of maintenance treatment with pemetrexed in lung adenocarcinoma patients who are negative for epidermal growth factor receptor mutations or anaplastic lymphoma kinase translocations. Transl Res. 2016;170:1–7. doi:10.1016/j.trsl.2015.11.006
4. Liu Z, Yao L, Tan B, Li L, Chen B. Detection of microRNA-200b may predict the inhibitory effect of gefitinib on non-small cell lung cancer and its potential mechanism. Oncol Lett. 2016;12(6):5349–5355. doi:10.3892/ol.2016.5365
5. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi:10.1126/science.1099314
6. You L, Shou J, Deng D, et al. Crizotinib induces autophagy through inhibition of the STAT3 pathway in multiple lung cancer cell lines. Oncotarget. 2015;6(37):40268–40282. doi:10.18632/oncotarget.5592
7. Hsu HS, Huang PI, Chang YL, et al. Cucurbitacin I inhibits tumorigenic ability and enhances radiochemosensitivity in nonsmall cell lung cancer-derived CD133-positive cells. Cancer. 2011;117(13):2970–2985. doi:10.1002/cncr.25869
8. Kang KA, Piao MJ, Hyun JW. Fisetin induces apoptosis in human nonsmall lung cancer cells via a mitochondria-mediated pathway. In Vitro Cell Dev Biol Anim. 2015;51(3):300–309. doi:10.1007/s11626-014-9830-6
9. Liang CH, Shiu LY, Chang LC, Sheu HM, Tsai EM, Kuo KW. Solamargine enhances HER2 expression and increases the susceptibility of human lung cancer H661 and H69 cells to trastuzumab and epirubicin. Chem Res Toxicol. 2008;21(2):393–399. doi:10.1021/tx700310x
10. Tsao SM, Hsia TC, Yin MC. Protocatechuic acid inhibits lung cancer cells by modulating FAK, MAPK, and NF-kappaB pathways. Nutr Cancer. 2014;66(8):1331–1341. doi:10.1080/01635581.2014.956259
11. Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl). 1990;181(3):195–213. doi:10.1007/bf00174615
12. Baehrecke EH. How death shapes life during development. Nat Rev Mol Cell Biol. 2002;3(10):779–787. doi:10.1038/nrm931
13. Nishida K, Yamaguchi O, Otsu K. Crosstalk between autophagy and apoptosis in heart disease. Circ Res. 2008;103(4):343–351. doi:10.1161/CIRCRESAHA.108.175448
14. Towers CG, Thorburn A. Therapeutic targeting of autophagy. EBioMedicine. 2016. doi:10.1016/j.ebiom.2016.10.034
15. Song S, Tan J, Miao Y, Li M, Zhang Q. Crosstalk of autophagy and apoptosis: involvement of the dual role of autophagy under ER stress. J Cell Physiol. 2017. doi:10.1002/jcp.25785
16. Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8(6):569–581. doi:10.1038/sj.cdd.4400852
17. Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol. 2001;2(7):545–550. doi:10.1038/35080097
18. Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16(6):663–669. doi:10.1016/j.ceb.2004.09.011
19. de Bruin EC, Medema JP. Apoptosis and non-apoptotic deaths in cancer development and treatment response. Cancer Treat Rev. 2008;34(8):737–749. doi:10.1016/j.ctrv.2008.07.001
20. Bialik S, Zalckvar E, Ber Y, Rubinstein AD, Kimchi A. Systems biology analysis of programmed cell death. Trends Biochem Sci. 2010;35(10):556–564. doi:10.1016/j.tibs.2010.04.008
21. Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16(7):966–975. doi:10.1038/cdd.2009.33
22. Sheng H, Sun H. Synthesis, biology and clinical significance of pentacyclic triterpenes: a multi-target approach to prevention and treatment of metabolic and vascular diseases. Natural Product Reports. 2011;28(3):543–593. doi:10.1039/c0np00059k
23. Liby KT, Sporn MB. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev. 2012;64(4):972–1003. doi:10.1124/pr.111.004846
24. Pollier J, Goossens A. Oleanolic acid. Phytochemistry. 2012;77:10–15. doi:10.1016/j.phytochem.2011.12.022
25. Topcu G, Tan N, Kokdil G, Ulubelen A. Terpenoids from Salvia glutinosa. Phytochemistry. 1997;45(6):1293–1294. doi:10.1016/s0031-9422(97)00042-3
26. Liu J, Zheng L, Zhong J, Wu N, Liu G, Lin X. Oleanolic acid induces protective autophagy in cancer cells through the JNK and mTOR pathways. Oncol Rep. 2014;32(2):567–572. doi:10.3892/or.2014.3239
27. Zhang L, Wang K, Lei Y, Li Q, Nice EC, Huang C. Redox signaling: potential arbitrator of autophagy and apoptosis in therapeutic response. Free Radic Biol Med. 2015;89:452–465. doi:10.1016/j.freeradbiomed.2015.08.030
28. Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015;4:184–192. doi:10.1016/j.redox.2014.12.003
29. Kaminskyy VO, Zhivotovsky B. Free radicals in cross talk between autophagy and apoptosis. Antioxid Redox Signal. 2014;21(1):86–102. doi:10.1089/ars.2013.5746
30. Zhou YY, Li Y, Jiang WQ, Zhou LF. MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep. 2015;35:3. doi:10.1042/BSR20150111
31. He W, Wang Q, Srinivasan B, et al. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene. 2014;33(23):3004–3013. doi:10.1038/onc.2013.256
32. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32. doi:10.1172/JCI73939
33. Simmons CP, Koinis F, Fallon MT, et al. Prognosis in advanced lung cancer–A prospective study examining key clinicopathological factors. Lung Cancer. 2015;88(3):304–309. doi:10.1016/j.lungcan.2015.03.020
34. Yoshikawa M, Matsuda H. Antidiabetogenic activity of oleanolic acid glycosides from medicinal foodstuffs. Biofactors. 2000;13(1–4):231–237.
35. Watjen W, Michels G, Steffan B, et al. Low concentrations of flavonoids are protective in rat H4IIE cells whereas high concentrations cause DNA damage and apoptosis. J Nutr. 2005;135(3):525–531. doi:10.1093/jn/135.3.525
36. Yu C, Li WB, Liu JB, Lu JW, Feng JF. Autophagy: novel applications of nonsteroidal anti-inflammatory drugs for primary cancer. Cancer Med. 2018;7(2):471–484. doi:10.1002/cam4.1287
37. Kohli L, Roth KA. Autophagy: cerebral home cooking. Am J Pathol. 2010;176(3):1065–1071. doi:10.2353/ajpath.2010.090850
38. Yousefi S, Simon HU. Apoptosis regulation by autophagy gene 5. Crit Rev Oncol Hematol. 2007;63(3):241–244. doi:10.1016/j.critrevonc.2007.06.005
39. Wirawan E, Lippens S, Vanden Berghe T, et al. Beclin1: a role in membrane dynamics and beyond. Autophagy. 2012;8(1):6–17. doi:10.4161/auto.8.1.16645
40. Gomez-Puerto MC, Folkerts H, Wierenga AT, et al. Autophagy proteins ATG5 and ATG7 are essential for the maintenance of human CD34(+) hematopoietic stem-progenitor cells. Stem Cells. 2016;34(6):1651–1663. doi:10.1002/stem.2347
41. Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14(3):500–510. doi:10.1038/sj.cdd.4402039
42. Claerhout S, Verschooten L, Van Kelst S, et al. Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int J Cancer. 2010;127(12):2790–2803. doi:10.1002/ijc.25300
43. Ren JH, He WS, Nong L, et al. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother Radiopharm. 2010;25(1):75–80. doi:10.1089/cbr.2009.0701
44. O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7(5):509–524. doi:10.4161/auto.7.6.15066
45. Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007;72(1):29–39. doi:10.1124/mol.106.033167
46. Song Y, Kong L, Sun B, et al. Induction of autophagy by an oleanolic acid derivative, SZC017, promotes ROS-dependent apoptosis through Akt and JAK2/STAT3 signaling pathway in human lung cancer cells. Cell Biol Int. 2017;41(12):1367–1378. doi:10.1002/cbin.10868
47. Kyriakis JM, Banerjee P, Nikolakaki E, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369(6476):156–160. doi:10.1038/369156a0
48. Xavier CP, Lima CF, Pedro DF, Wilson JM, Kristiansen K, Pereira-Wilson C. Ursolic acid induces cell death and modulates autophagy through JNK pathway in apoptosis-resistant colorectal cancer cells. J Nutr Biochem. 2013;24(4):706–712. doi:10.1016/j.jnutbio.2012.04.004
49. Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006;13(5):730–737. doi:10.1038/sj.cdd.4401830
50. Liu J, Wu N, Ma LN, et al. p38 MAPK signaling mediates mitochondrial apoptosis in cancer cells induced by oleanolic acid. Asian Pac J Cancer Prev. 2014;15(11):4519–4525. doi:10.7314/apjcp.2014.15.11.4519
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.