Back to Journals » Drug Design, Development and Therapy » Volume 15
Methylcobalamin Protects Melanocytes from H2O2-Induced Oxidative Stress by Activating the Nrf2/HO-1 Pathway
Authors An R ![]() , Li D, Dong Y, She Q, Zhou T
, Li D, Dong Y, She Q, Zhou T ![]() , Nie X, Pan R, Deng Y
, Nie X, Pan R, Deng Y
Received 25 August 2021
Accepted for publication 11 November 2021
Published 30 November 2021 Volume 2021:15 Pages 4837—4848
DOI https://doi.org/10.2147/DDDT.S336066
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Ran An,1,2 Dong Li,1 Yingying Dong,1 Qiuyun She,1 Ting Zhou,1 Xiaoqi Nie,1 Ronghua Pan,1 Yunhua Deng1
1Department of Dermatology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, People’s Republic of China; 2Department of Dermatology, Children’s Hospital of Soochow University, Suzhou, Jiangsu, People’s Republic of China
Correspondence: Yunhua Deng
Department of Dermatology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Ave, Qiaokou, Wuhan, Hubei, People’s Republic of China
Tel +86 027 83663370
Fax +86 027 83663625
Email [email protected]
Purpose: Oxidative stress is considered a major determinant in the pathogenesis of vitiligo. Methylcobalamin (MeCbl) is an activated form of vitamin B12 that regulates inflammatory factors, counters oxidative stress, and reduces apoptosis in many disease models. However, the specific mechanism of MeCbl repigmentation against vitiligo is unknown. In this study, we explored the effect of MeCbl on melanocytes following hydrogen peroxide (H2O2)-induced oxidative stress.
Methods: We established an oxidative stress model using the immortalized human normal melanocyte cell line PIG1. We used a Cell Counting Kit-8 (CCK-8) to detect drug cytotoxicity, and we measured the melanin content of cells using the NaOH method. Intracellular oxidative damage was assessed by flow cytometry and antioxidant enzyme detection kits. In addition, we assessed the presence of apoptosis by flow cytometry and Western blots. We explored the underlying mechanisms of MeCbl during oxidative stress in melanocytes by analyzing the results of experiments based on real-time quantitative polymerase chain reaction (RT-qPCR), Western blotting, and laser scanning confocal immunofluorescence microscopy. Finally, we repeated the experiments after applying an inhibitor to block the Nrf2 pathway.
Results: We found that MeCbl treatment enhanced cell viability, increased melanin content, reduced intracellular reactive oxygen species (ROS) accumulation, increased the activities of antioxidant enzyme superoxide dismutase (SOD) and catalase (CAT), reduced melanocyte apoptosis, and up-regulated the expression of the Nrf2/HO-1 pathway. Moreover, the protective effects of MeCbl were significantly weakened after inhibiting the Nrf2/HO-1 pathway.
Conclusion: Our results indicate that MeCbl attenuated the H2O2-induced oxidative stress in melanocytes by activating the Nrf2/HO-1 pathway, this suggests that MeCbl may be an effective treatment against vitiligo.
Keywords: methylcobalamin, vitiligo, oxidative stress, melanocytes, NF-E2-related factor 2
Introduction
Vitiligo is an acquired chronic depigmenting disorder of the skin, with an estimated prevalence of 0.5–1% worldwide. Vitiligo is clinically characterized by the formation of white macules that arise from the lack of functioning melanocytes.1 The occurrence rates between different genders, phototypes, and ethnicities are similar.2 The exact pathogenesis of vitiligo remains unclear, but multiple mechanisms have been proposed, including genetic, autoimmune, oxidative stress, inflammatory mediator, and melanocyte-detachment mechanisms.3 Among these possible mechanisms, oxidative stress is considered a major determinant of vitiligo. Studies have found increased levels of reactive oxygen species (ROS) in the epidermis (lesions and healthy skin) of patients with vitiligo.4 Moreover, increased H2O2 accumulation has also been observed in the skin of patients with advanced vitiligo.5 Various endogenous or exogenous stimuli break the balance of the oxidation-antioxidant system and may raise the levels of ROS in melanocytes. Ultimately, redundant ROS may trigger multiple mechanisms contributing to the destruction of melanocytes.6
The activation of the nuclear factor erythroid-2-related factor (Nrf2)- Kelch-like ECH-associated protein 1 (Keap1)-antioxidant response element (ARE) pathway is one of the main melanocyte-related protective mechanisms against oxidative injury. This pathway can regulate the expression of a series of antioxidant genes, eliminating oxidative injury through conjugation reactions and enhancing the antioxidant capacity of melanocytes.7 When oxidative stress occurs, keap1 starts the release of Nrf2, which gets translocated into the nucleus and there binds to the ARE to activate the expression of various Phase II antioxidant enzymes such as heme oxygenase-1 (HO-1) and γ-glutamyl cysteine ligase catalytic subunit (GCLC).8 In addition, the intracellular antioxidant enzymes, including catalase (CAT) and superoxide dismutase (SOD), can also resist oxidative injury by catalyzing the conversion of ROS into low-reactive substances.9
Methylcobalamin (MeCbl), the activated form of vitamin B12, is a cofactor of methionine synthase (MS), which can catalyze the methyl group transfer to homocysteine to form methionine and tetrahydrofolate.10 The methyl transfer reaction catalyzed by MS plays three important roles in cells: it synthesizes methionine as a precursor of S-adenosyl methionine (SAM), it produces tetrahydrofolate for DNA synthesis, and it maintains the homeostasis of cellular homocysteine. SAM is a universal donor for the methylation reactions of DNA, RNA, histones, other proteins, and metabolites.11 In an Alzheimer’s disease study, Vitamin B12 (acting on the methionine/SAM cycle) exerted a protective effect by reducing mitochondrial fragmentation and oxidative stress.12 In addition, a study of the Nrf2 gene in the frontal cortex of individuals with autism spectrum disorder found that Nrf2 gene expression was positively correlated with the abundances of MeCbl, methionine, and SAM.13 In vitro experiments have shown that vitamin B12 can act as an effective antioxidant to inhibit the production of intracellular peroxides and prevent the apoptosis caused by H2O2.14 Another Alzheimer’s disease study showed that MeCbl can reduce cell apoptosis by reducing the levels of intracellular ROS.15 In addition, a rat model of diabetic peripheral neuropathy, showed that MeCbl can protect peripheral nerves by reducing oxidative stress damage.16 These studies show that there is a close relationship between vitamin B12, oxidative stress and apoptosis. However, most of the evidence on MeCbl has focused on its neurological effects, and its effects on melanocytes remain unclear.
From an embryonic development perspective, both melanocytes and nerve cells originate from the neural crest. When exposed to a specific environment, nerve cells can switch to the melanocyte lineage.17 Therefore, we hypothesized that MeCbl could alleviate oxidative damage in melanocytes under oxidative stress in a manner similar to that reported on nerve cells. We explored the effects of MeCbl on H2O2-induced oxidative stress in normal human melanocytes to uncover the mechanisms involved in the hope of providing a new therapeutic avenue for vitiligo.
Materials and Methods
Cell Culture and Treatment
The immortalized human normal melanocyte cell line PIG1 (purchased from Bena Culture Collection, Beijing, China) was cultured in Medium 254 (Gibco, Grand Island, NY) supplemented with Human Melanocyte Growth Supplement (Gibco) and 5% fetal bovine serum at 37 °C with 5% CO2. We established an oxidative stress model in the PIG1 cells by treating them with 1.0 mM H2O2 (Sigma-Aldrich, USA) for 24 h. We added 10 μM MeCbl (Sigma-Aldrich) 48 h before the H2O2 treatment as needed. In addition, we treated PIG1 cells with 1.5 µM ML385 (MedChemExpress, USA), a compound that inhibits the activation of Nrf2, to further explore the MeCbl mechanisms.
Cell Viability Assay
We used a Cell Counting Kit-8 (CCK-8) purchased from Yeasen (Shanghai, China) to measure cell viability. Briefly, to investigate the cytotoxicity of MeCbl, we cultured PIG1 cells in 96-well plates at a density of 1×104 cells/well and then treated them with different concentrations (0, 0.1, 1, 50, 100, and 200 μM) of MeCbl for 0–4 days. To investigate the effect of MeCbl in our oxidative stress model, we inoculated cells into 96-well plates and grew them overnight at a density of 1×104 cells/well before treating them with MeCbl at concentrations ranging from 1 µM to 50 µM for 48 h. After the 1.0 mM H2O2 treatment for another 24 h, we removed the supernatants and added 10 µL of CCK-8 and 100 µL of Medium 254 to each well to culture the cells in the dark at 37 °C for 90 min. We measured OD values at 450 nm in a microplate reader.
Melanin Content Analysis
We detected the melanin contents of PIG1 cells using the NaOH method. Cells were plated into 6-well plates at a density of 2.8×105 cells/well. After treatment with the drugs mentioned above, we collected the cells and washed them twice with PBS. We dissolved the cell pellets in 1 M NaOH at 80 °C for 2 h and then transferred them onto a 96 well plate. We measured the melanin content at 405 nm using a microplate reader.
Intracellular ROS Assay
We determined Intracellular ROS levels using the total ROS assay kit (Invitrogen, Portland, OR, USA). PIG1 cells were plated into 6-well plates overnight and exposed to the relevant drugs. Then, we collected the cells and washed them with PBS. We diluted a CM-H2DCFDA probe in a serum-free medium to prepare a working solution with a final concentration of 1 μM. We added the final solution to each group of cells and incubated them at 37 °C in the dark for 30 min. After that, the cells were washed twice with a serum-free medium, and we measured the fluorescence intensity of DCF in each group by flow cytometry.
Measurement of SOD and CAT Activity Level
We used a SOD assay kit (WST-1 method) and a CAT assay kit (Visible light) (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) to detect SOD and CAT activities. Following the manufacturer’s instructions, we extracted the total proteins of the PIG1 cells, and detected the activities of SOD and CAT in the samples at a wavelength of 450 nm and 405 nm, in a microplate reader.
Apoptosis Assay
We measured cell apoptosis rates using an Annexin V-FITC apoptosis detection kit (BD Biosciences, USA). After treating cells with MeCbl and H2O2, we harvested all cells in each well into test tubes. We washed the cells twice with PBS and then incubated them with Annexin V-FITC and PI at room temperature for 15 min. Finally, we analyzed the samples using flow cytometry.
Western Blot
We lysed PIG1 cells and performed a total protein extraction. We quantified the protein content of the samples using a BCA Protein Assay Kit. We chose 10% or 12% SDS-PAGE gels according to the specific target protein sizes. We loaded 30 μg protein samples onto each well and separated the proteins by electrophoresis. The separated proteins were transferred onto PVDF membranes (Millipore, Billerica, MA). After blocking in 5% nonfat milk diluted in TBST for 1 h, we incubated the membranes with the primary antibodies overnight at 4 °C. The primary antibodies used in the experiment were the following: Bax, Bcl-2, Cleaved Caspase-3, GAPDH, Lamin B (ABclonal Biotechnology, USA), Nrf2 and HO-1 (Proteintech, Wuhan, China). On the following day, we washed the membranes with TBST and then incubated them with fluorescence-conjugated secondary antibodies. Finally, we visualized the target proteins using the Odyssey Infrared Imaging (LI-COR Biosciences, United States) and analyzed the band intensities using the ImageJ software.
Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
According to the manufacturer’s protocol, we extracted total RNA samples from the PIG1 cells using the Trizol reagent. We used a spectrophotometer to determine the concentrations and purity of the purified RNA samples. Next, we obtained cDNA by reverse transcription of eligible samples. RT-qPCR was performed on a LightCycler®96 system (Roche Diagnostics, Mannheim, Germany). The relative mRNA levels of target genes were analyzed by the 2−ΔΔCT method. Table 1 lists the sequences of the primers involved in this study.
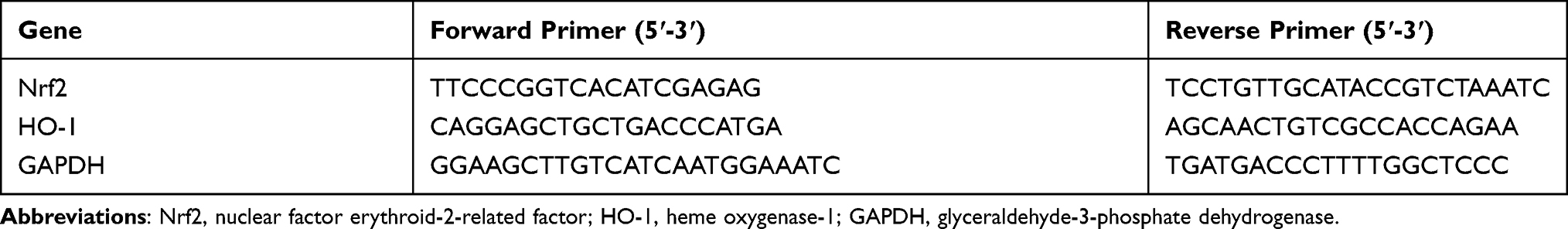 |
Table 1 Primers Used for RT-qPCR |
Laser Scanning Confocal Immunofluorescence Microscopy
We fixed cultured cells in 4% paraformaldehyde for 20 min, permeabilized them with 0.5% Triton X-100 for 20 min, and blocked them using 10% normal goat serum for 1 h. After that, cells were incubated with the diluted primary antibody overnight at 4 °C, followed by incubation with Alexa Fluor 488-conjugated secondary antibody for 1 h at room temperature. Next, we dyed the nuclei with DAPI for 10 min and used laser confocal scanning microscopy (Nikon, Tokyo, Japan) to take fluorescent images.
Statistical Analysis
We performed the statistical analysis using the GraphPad Prism 8 software. We used a Shapiro–Wilk test to check whether the data conformed to the normal distribution. The normally distributed data were tested for homogeneity of variance via one-way ANOVA. We applied Tukey’s multiple comparison tests for multiple group comparisons for normally distributed data without significant variance inhomogeneity between groups. Otherwise, we used the Mann–Whitney U-test for non-normally distributed or variance inhomogeneous data. All data were presented as means ± SEM, and experiments were carried out independently and in triplicate. We considered adjusted p < 0.05 as statistically significant.
Results
MeCbl Protected Melanocytes from H2O2-Induced Cytotoxicity
To investigate the protective effects of MeCbl against H2O2 treatment, we established an oxidative stress model using PIG1 cells. We proceeded by stimulating P1G1 cells using different concentrations of exogenous H2O2 for 24 h and assessing cell viability with CCK-8 assays. As shown in Figure 1A, the melanocytes treated with 0.4 mM–1.2 mM H2O2 displayed reduced cell viabilities in a concentration-dependent manner. We chose a standard 1.0 mM H2O2 concentration for our subsequent experiments, consistent with the results of a published report.18 At this concentration, most cells survived the oxidative damage. We wondered if MeCbl could affect cell proliferation and treated melanocytes with different MeCbl concentrations (Figure 1B) to test this possibility. We found that the proliferation of melanocytes treated with 200 μM MeCbl was reduced in contrast to the proliferation of the untreated cells. Based on these results, we set a concentration gradient between 1 μM and 50 μM to further investigate the effects of MeCbl. Melanocytes were pretreated with different concentrations of MeCbl and then treated with H2O2 for 24 h (Figure 1C). Our results showed that addition of MeCbl at concentrations between 1 μM and 30 μM could ameliorate H2O2-induced cytotoxicity, with 10 μM MeCbl performing the best. Moreover, this concentration of MeCbl also increased the melanin content of the H2O2-treated cells compared with the content in the control cells (Figure 1D). Interestingly, MeCbl lost its protective effect on melanocytes at concentrations ≥ 50 μM. Consequently, our results suggest that an appropriate concentration of MeCbl may protect melanocytes from H2O2-induced cytotoxicity. Thus, we chose a concentration of 10 μM MeCbl for subsequent experiments.
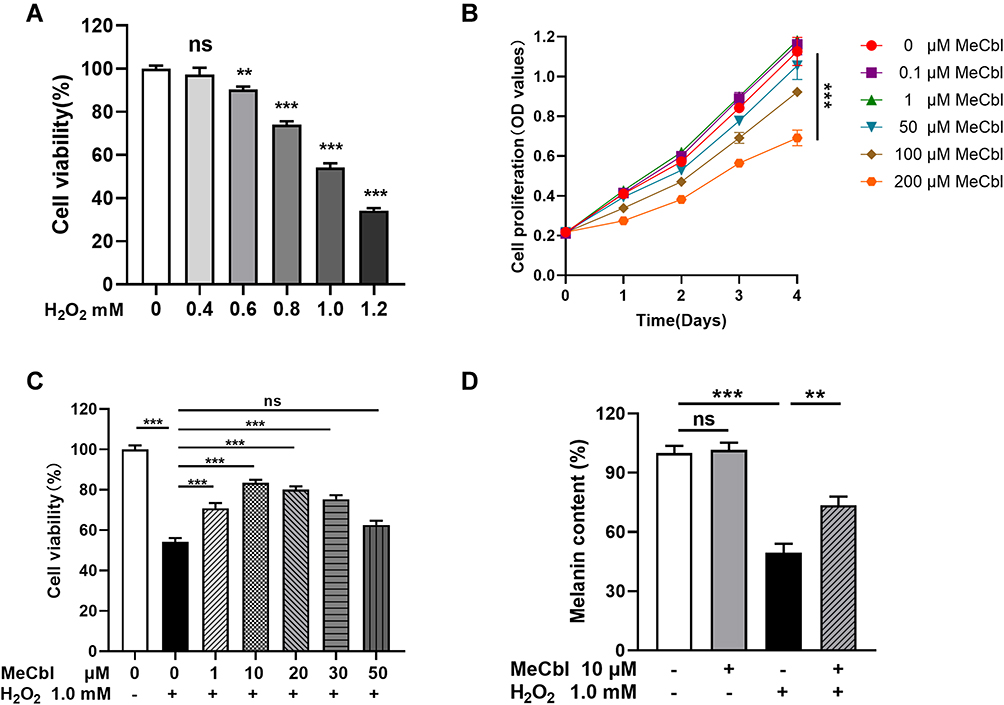 |
Figure 1 Methylcobalamin (MeCbl) protected melanocytes from H2O2-induced cytotoxicity. (A) We tested the cell viability of melanocytes treated with different concentrations of H2O2 for 24 h using a CCK-8 assay. (B) Melanocytes were treated with different concentrations of MeCbl for 0–4 days. We detected cell proliferation using a CCK-8 assay. (C) Melanocytes were pretreated with different concentrations of MeCbl for 48 h and then treated with 1.0 mM H2O2 for 24 h. We determined cell viability by CCK-8 assay. (D) Melanocytes were pretreated with 10 μM MeCbl for 48 h and then treated with 1.0 mM H2O2 for 24 h. We quantified the melanin content using the NaOH method. **P < 0.01; ***P < 0.001. Abbreviation: ns, non-significant. |
MeCbl Reduced H2O2-Induced Oxidative Injury of Melanocytes
To evaluate the effects of MeCbl reducing oxidative damage, we pretreated melanocytes with 10 μM MeCbl for 48 h before adding 1.0 mM H2O2. The level of intracellular ROS, an important marker of oxidative stress, increased significantly after exposure to H2O2, but treatment with MeCbl reduced ROS accumulation (Figure 2A and B). The critical antioxidant enzymes SOD and CAT can reduce intracellular ROS production. Therefore, we measured their enzyme activity levels and found that MeCbl could partially restore their H2O2-induced inhibition (Figure 2C and D). However, melanocytes treated only with MeCbl displayed similar intracellular ROS and antioxidant enzyme activity levels to those of control melanocytes. Our results suggest that MeCbl was able to reduced H2O2-induced oxidative injury in melanocytes.
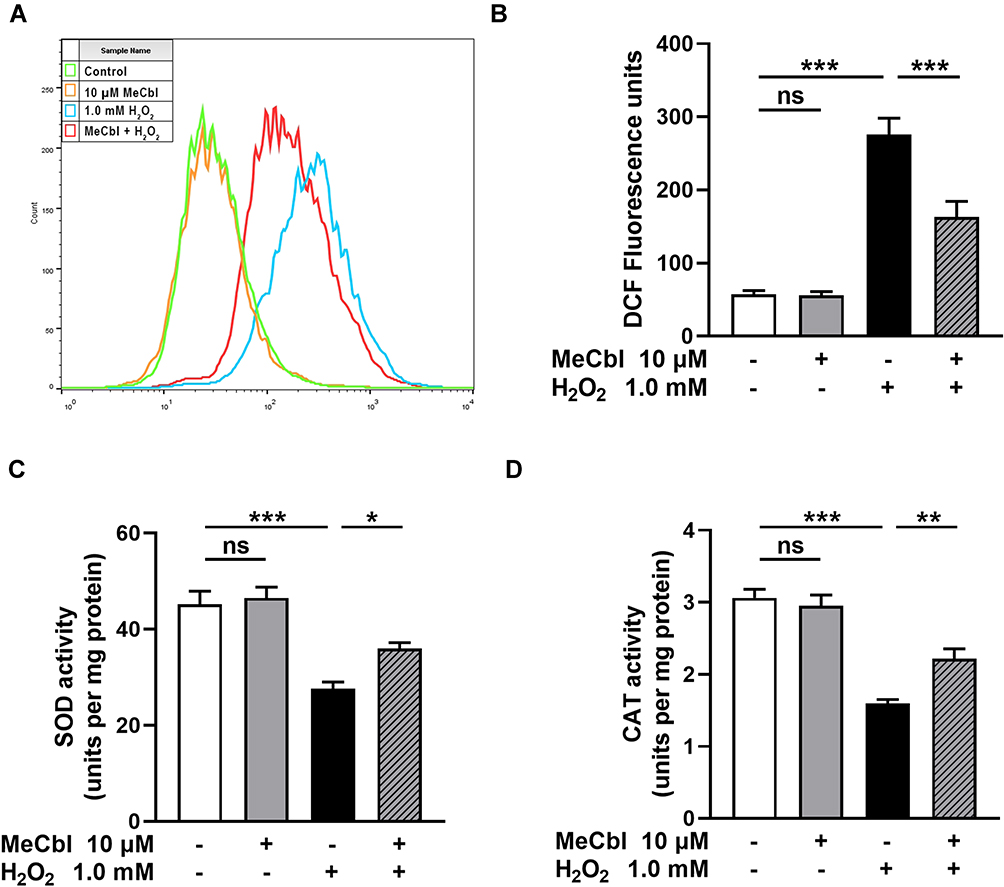 |
Figure 2 Methylcobalamin (MeCbl) reduced the H2O2-induced oxidative injury of melanocytes. (A) Melanocytes were pretreated with 10 μM MeCbl for 48 h and then treated with 1.0 mM H2O2 for 24 h. We detected the intracellular reactive oxygen species (ROS) production by flow cytometry. (B) Statistical analysis of fluorescence intensity of the ROS level. (C) Activity of the antioxidant enzyme superoxide dismutase (SOD). (D) Activity of the antioxidant enzyme catalase (CAT). *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviation: ns, non-significant. |
MeCbl Reduced H2O2-Induced Apoptosis in Melanocytes
Excessive ROS leads to the apoptosis of melanocytes, therefore, we investigated the effect of MeCbl on apoptosis. To that end, we used flow cytometry to quantify the specific percentage of apoptosis in each group of cells. The apoptosis rate in the MeCbl group was similar to that in the control group, but the rate was increased significantly in the H2O2 group. Moreover, pretreatment with MeCbl markedly reduced the proportion of apoptosis (Figure 3A and B). In addition, we used Western blots to detect the expression of apoptosis-associated indicators. Studies have suggested that H2O2 treatment decreases the production of anti-apoptotic protein Bcl-2 and increases the production of pro-apoptotic proteins Bax and Caspase-3.19 Based on these indicators, we showed that the melanocytes of the MeCbl group expressed similar apoptosis biomarkers to those in the melanocytes of the control group. After being exposed to H2O2, melanocytes showed a decreased level of Bcl-2 and increased levels of Bax and Cleaved Caspase-3. However, pretreatment with MeCbl reversed this situation and led to a decrease in the Bax/Bcl-2 ratio (Figure 3C and D), which was consistent with our flow cytometry results. Together, these results suggest that MeCbl may decrease H2O2-induced apoptosis in melanocytes.
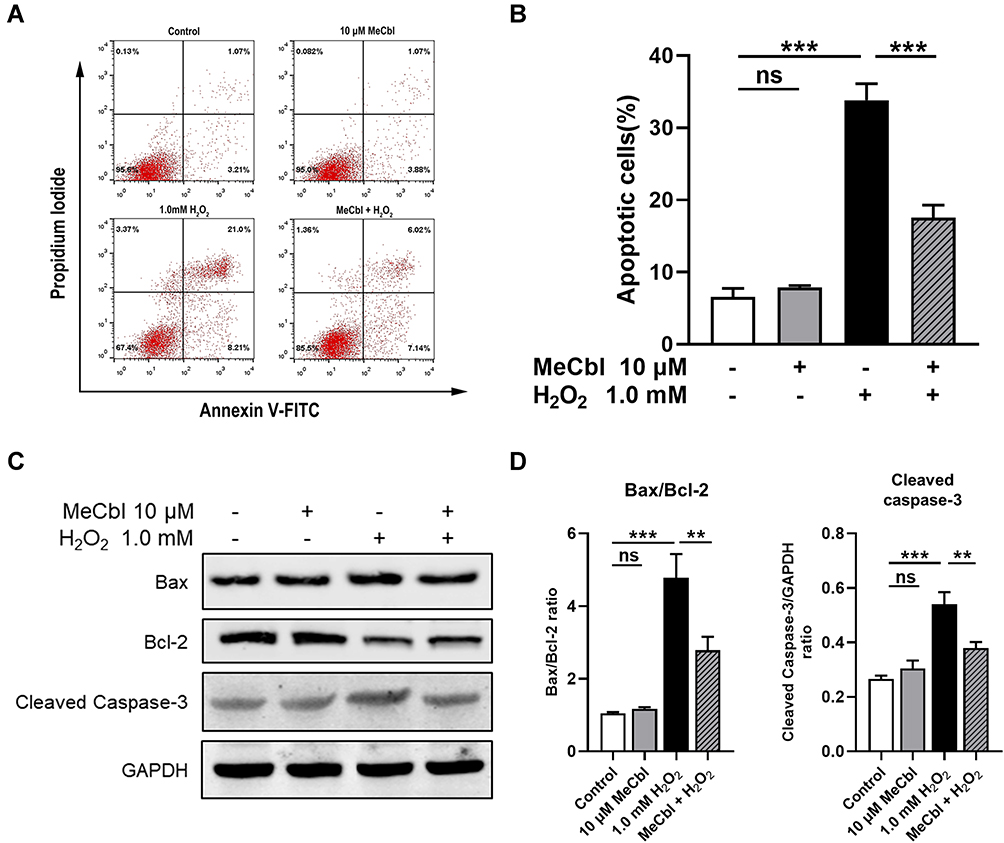 |
Figure 3 Methylcobalamin (MeCbl) reduced H2O2-induced apoptosis of melanocytes. (A) Melanocytes were exposed to 10 μM MeCbl for 48 h and treated with 1.0 mM H2O2 for another 24 h. We quantified their level of apoptosis by flow cytometry. (B) Statistical analysis of the specific apoptosis percentages. (C) Representative Western blots for Bax, Bcl-2, and Cleaved Caspase-3. (D) Quantitative analysis of the apoptosis-associated indicators. **P < 0.01; ***P < 0.001. Abbreviation: ns, non-significant. |
MeCbl Activated the Nrf2/HO-1 Pathway to Protect Melanocytes from H2O2-Induced Injury
To confirm whether MeCbl protects melanocytes through its activation of the Nrf2/HO-1 pathway, we measured the mRNA levels of Nrf2 and HO-1. The melanocytes pretreated with MeCbl showed increased mRNA levels of Nrf2 and HO-1 than the melanocytes treated only with H2O2 (Figure 4A). Also, we found that the protein expressions of Nrf2 and HO-1 increased significantly in the melanocytes treated with MeCbl (Figure 4B and C). The activation of the Nrf2/HO-1 pathway depends on the translocation of Nrf2 from the cytoplasm into the nucleus; therefore, we also visualized the distribution of Nrf2 in melanocytes. Our quantitative analysis of nuclear and cytosolic Nrf2 revealed that MeCbl pretreatment raised the Nrf2 nuclear/cytosolic ratio (Figure 4D and E). We also used laser confocal scanning microscopy to localize Nrf2 in melanocytes. As shown in Figure 4F, the melanocytes pretreated with MeCbl had a significantly increased fluorescence intensity of Nrf2 in the nucleus, indicating that nuclear translocation of Nrf2 had occurred. These results show that MeCbl may activate the Nrf2/HO-1 pathway to protect melanocytes from H2O2-induced injury.
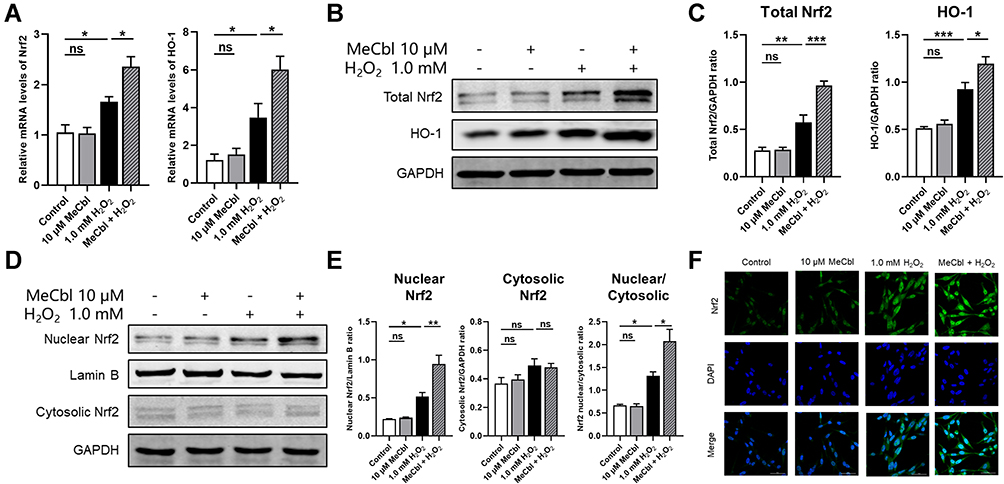 |
Figure 4 Methylcobalamin (MeCbl) activated the Nrf2/HO-1 pathway to protect melanocytes from H2O2-induced injury. Melanocytes were treated with 10 μM MeCbl for 48 h and then exposed to 1.0 mM H2O2 for 24 h. (A) Measured mRNA levels of Nrf2 and HO-1. (B) Protein levels of total Nrf2 and HO-1. (C) Quantitative analysis of total Nrf2 and HO-1. (D) Representative Western blots for nuclear Nrf2 and cytosolic Nrf2. (E) Quantitative analysis of nuclear Nrf2, cytosolic Nrf2, and Nrf2 nuclear/cytosolic. (F) We visualized the cellular distribution of Nrf2 in melanocytes by laser confocal scanning microscopy. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviation: ns, non-significant. |
Inhibition of Nrf2 Reduced the Protective Effect of MeCbl on H2O2-Induced Melanocytes
To further verify that MeCbl treatment protects melanocytes by activating the Nrf2 pathway, we used ML385 (a Nrf2 inhibitor) for follow-up experiments. We first tested the influence of inhibitors on the Nrf2/HO-1 pathway and found that Nrf2 inhibitors down-regulated the Nrf2/HO-1 pathway activated by MeCbl (Figure 5A and B). In addition, after inhibiting Nrf2, the protective effect of MeCbl on H2O2-induced cytotoxicity decreased considerably (Figure 5C) and the melanin content of the same cells was reduced (Figure 5D). In terms of anti-oxidative damage, the decrease in intracellular ROS induced by MeCbl was significantly attenuated by the Nrf2 inhibitor (Figure 5E and F); also, the activities of SOD and CAT were reduced in the Nrf2 inhibitor-treated cells (Figure 5G). In addition, inhibition of Nrf2 weakened the protection of MeCbl against H2O2-induced apoptosis (Figure 5H and I). The Bax/Bcl-2 ratio and protein expression of Cleaved Caspase-3 were both higher in the melanocytes treated with MeCbl, H2O2 and the Nrf2 inhibitor than in those treated with MeCbl and H2O2 (Figure 5J and K). These results suggest that inhibition of Nrf2 reduces the protective effect of MeCbl on H2O2-induced PIG1 cells and further substantiate our findings suggesting that MeCbl protects melanocytes under oxidative stress by activating the Nrf2/HO-1 pathway.
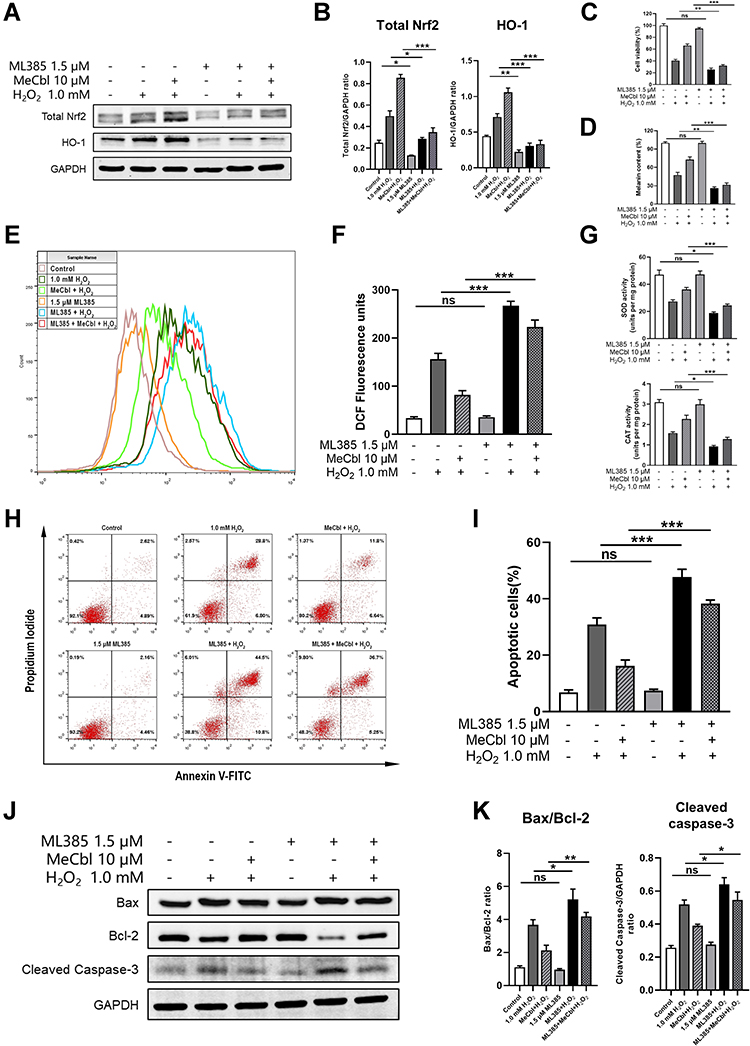 |
Figure 5 Inhibition of Nrf2 inhibited the protective effect of methylcobalamin (MeCbl) on H2O2-induced melanocytes. (A) After using ML385 to inhibit Nrf2, we quantified the protein expressions of Nrf2 and HO-1 in melanocytes by Western blot. (B) Quantitative analysis of Nrf2 and HO-1. (C) We determined the cell viability of each group of treated cells using CCK-8 assays. (D) Melanin contents determined by the NaOH method. (E) We quantified the intracellular reactive oxygen species (ROS) levels by flow cytometry. (F) Statistical analysis of fluorescence intensity of ROS levels. (G) Activities of antioxidant enzymes superoxide dismutase (SOD) and catalase (CAT) in each group of cells as assessed by the relevant kits. (H) We quantified the levels of apoptosis by flow cytometry after the inhibition of Nrf2. (I) Statistical analysis of the apoptosis percentages in each cell group. (J) Representative Western blots for Bax, Bcl-2, and Cleaved Caspase-3. (K) Quantitative analysis of apoptosis-associated biomarkers. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviation: ns, non-significant. |
Discussion
In this study, we explored the effects of MeCbl on melanocytes under oxidative stress. We found that MeCbl concentrations between 1 μM and 30 μM could ameliorate H2O2-induced cytotoxicity, and that a concentration of 10 μM yielded the best protective effect. Treatment with 10 μM MeCbl increased the melanin content in PIG1 cells and protected the melanocytes from oxidative damage by reducing the levels of intracellular ROS and increasing the activities of the antioxidant enzymes SOD and CAT. In addition, MeCbl reduced the Bax/Bcl-2 ratio and the expression of Cleaved Caspase-3; and, it decreased H2O2-induced apoptosis in melanocytes. The nuclear translocation of Nrf2 might mediate these effects by up-regulating the Nrf2/HO-1 pathway (Figure 6). After inhibiting Nrf2, the protective effect of MeCbl was significantly weakened.
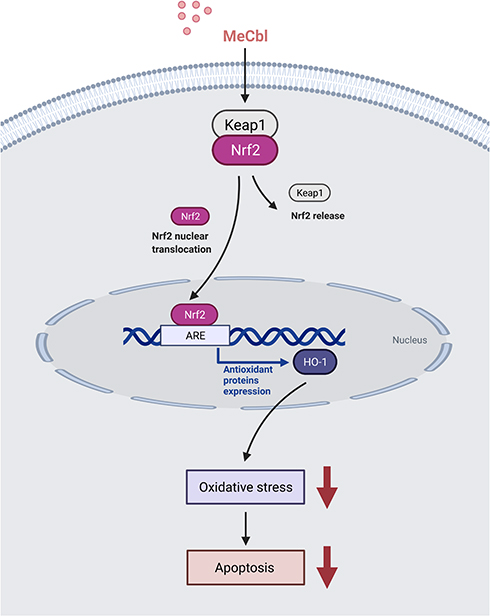 |
Figure 6 Mechanism of methylcobalamin (MeCbl) protecting melanocytes from oxidative damage induced by H2O2. In our experiments MeCbl promoted the nuclear translocation of Nrf2, thereby activating the expression of its signaling pathway. The up-regulation of this pathway increased the activity of the antioxidant enzymes superoxide dismutase (SOD) and catalase (CAT) and reduced the level of intracellular reactive oxygen species (ROS), thereby enhancing the ability of melanocytes to resist oxidative damage. Oxidative stress can promote cell apoptosis by destroying lipids, nucleic acids, proteins and other macromolecular substances leading to an altered integrity of the respiratory chain and activating the unfolded protein response (UPR); inhibiting the intracellular oxidative stress reduces the apoptosis of melanocytes. Figure created with BioRender.com. |
Oxidative stress, which initiate and lead to the progression of vitiligo, is considered one of the pathogenesis mechanisms of the disorder.20 Oxidative stress occurs when the production of ROS exceeds the scavenging ability of the antioxidant system and leads to ROS accumulation and the breakdown of the intracellular oxidation-antioxidant system balance.21 Various endogenous and exogenous factors may cause excessive ROS production, such as ultraviolet radiation, infection, stress, malignant tumors, cell proliferation, differentiation, and melanin metabolism.22–24 To resist oxidative stress, human cells have evolved a corresponding antioxidant defense system that includes small molecular antioxidants (such as vitamin C, vitamin E, and glutathione) and antioxidant enzymes (such as SOD and CAT). These molecules can directly remove ROS or catalyze its conversion into low-reactivity substances.9 SOD can remove superoxide anions in cells by removing or adding electrons, while CAT can catalyze the decomposition of H2O2 into oxygen and water, molecules that can both reduce the level of ROS in cells.25 Our findings indicate that MeCbl could reverse the increase in intracellular ROS and the decrease in the antioxidant enzymes SOD and CAT activity caused by H2O2. In brief, MeCbl may protect melanocytes from H2O2-induced oxidative stress by directly reducing ROS accumulation and increasing the vitality of the antioxidant defense system.
Oxidative stress induces apoptosis of melanocytes in a variety of ways. ROS can destroy macromolecular substances such as lipids, nucleic acids, and proteins, leading to lipid peroxidation, DNA fragmentation, oxidative decomposition of proteins, and various enzymes’ activations or inactivations.26 Moreover, ROS may also cause a decrease in the mitochondrial membrane potential, abnormalities in the mitochondrial membrane lipid composition, and impairment of the integrity of the respiratory chain, situations that can make melanocytes more sensitive to apoptosis stimuli and more damageable.27 In addition, oxidative stress destroys the folding mechanism of the endoplasmic reticulum and activates the unfolded protein response (UPR). If the adaptive mechanisms in melanocytes cannot resolve the protein-folding defect, the cells undergo apoptosis.2 Also, the amount of E-cadherin in the cell membrane is down-regulated by oxidative damage, resulting in impaired the adhesion of the melanocytes to the basement membrane and triggering melanocyte apoptosis.4 Compared with healthy people and individuals with stable vitiligo, patients with advanced vitiligo have a higher expression of Caspase-3 in their melanocytes.28 Studies on the oxidative stress model of human primary melanocytes have found that H2O2 can increase the ratio of Bax/Bcl-2 and the activity of Caspase-3.19 Our experiments yielded similar results on the apoptosis of melanocytes: the overall apoptosis rate of PIG1 cells treated with H2O2 was increased significantly, and correspondingly, the Bax/Bcl-2 ratio and the expression of Cleaved Caspase-3 increased. MeCbl pretreatment partially eliminated these effects, suggesting that MeCbl may alleviate H2O2-induced apoptosis in melanocytes.
The Nrf2-ARE pathway is an early sensor of oxidative stress and plays a vital role in regulating skin homeostasis under oxidative stress.29 HO-1 is one of the antioxidant genes regulated downstream of Nrf2. It increases the rate of free heme catabolism, thereby avoiding the oxidative damage caused by free heme groups, and its metabolites also have antioxidant effects.30,31 In our study, the expressions of Nrf2 and HO-1 in PIG1 cells were increased after treatment with 1.0 mM H2O2 for 24 h, in agreement with previous reports.32,33 After pretreating the melanocytes with MeCbl, the mRNA and protein expressions of Nrf2/HO-1 increased, and the Nrf2 nuclear/cytosolic ratio was raised. We also observed nuclear translocation of Nrf2 by laser confocal scanning microscopy. Therefore, the protective mechanism of MeCbl against oxidative stress may be mediated through up-regulation of the Nrf2/HO-1 pathway.
As an inhibitor of Nrf2, ML385 inhibits the binding of Nrf2 and ARE and reduces the transcriptional activity of Nrf2. In addition, ML385 also reduces the activity of the Nrf2 promoter, which leads to a decrease in the expression of Nrf2 itself.34 In our study, we repeated our experiments on the Nrf2/HO-1 pathway, cell viability, melanin content, intracellular ROS levels, antioxidant enzyme SOD and CAT viability, cell apoptosis rate, and apoptosis-related indicators after inhibiting Nrf2 with ML385. Our results suggest that MeCbl does not alleviate the oxidative damage induced by H2O2 if Nrf2 is inhibited, and these findings support the idea that MeCbl protects melanocytes mainly through the Nrf2/HO-1 pathway.
Conclusion
Taken together, our results suggest that MeCbl attenuates H2O2-induced oxidative stress in melanocytes by activating the Nrf2/HO-1 pathway. Other studies have proved that activating the expression of Nrf2 and its downstream antioxidant genes can improve the ability of melanocytes to resist oxidative stress.35–37 Our study corroborates the importance of this pathway in the oxidative stress of melanocytes. More importantly, our results suggest that MeCbl may be a potential treatment for vitiligo. However, whether MeCbl affects melanin synthesis and metabolism or it eliminates oxidative damage through its effects on the methionine/SAM cycle, and/or whether it is effective and safe in vivo are topics that remain unanswered and warrant further in-depth studies.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.81773306).
Disclosure
The authors declare no conflict of interest.
References
1. Ezzedine K, Eleftheriadou V, Whitton M, van Geel N. Vitiligo. Lancet. 2015;386(9988):74–84. doi:10.1016/S0140-6736(14)60763-7
2. Boniface K, Seneschal J, Picardo M, Taieb A. Vitiligo: focus on clinical aspects, immunopathogenesis, and therapy. Clin Rev Allergy Immunol. 2018;54(1):52–67. doi:10.1007/s12016-017-8622-7
3. Bergqvist C, Ezzedine K. Vitiligo: a review. Dermatology. 2020;236(6):571–592. doi:10.1159/000506103
4. Delmas V, Larue L. Molecular and cellular basis of depigmentation in vitiligo patients. Exp Dermatol. 2019;28(6):662–666. doi:10.1111/exd.13858
5. Schallreuter KU, Moore J, Wood JM, et al. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J Investig Dermatol Symp Proc. 1999;4(1):91–96. doi:10.1038/sj.jidsp.5640189
6. Xie H, Zhou F, Liu L, et al. Vitiligo: how do oxidative stress-induced autoantigens trigger autoimmunity? J Dermatol Sci. 2016;81(1):3–9. doi:10.1016/j.jdermsci.2015.09.003
7. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284(20):13291–13295. doi:10.1074/jbc.R900010200
8. Lin X, Meng X, Song Z, Lin J. Nuclear factor erythroid 2-related factor 2 (Nrf2) as a potential therapeutic target for vitiligo. Arch Biochem Biophys. 2020;696:108670. doi:10.1016/j.abb.2020.108670
9. Lei XG, Zhu JH, Cheng WH, et al. Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol Rev. 2016;96(1):307–364. doi:10.1152/physrev.00010.2014
10. Matthews RG. Cobalamin-dependent methyltransferases. Acc Chem Res. 2001;34(8):681–689. doi:10.1021/ar0000051
11. Gueant JL, Gueant-Rodriguez RM, Kosgei VJ, Coelho D. Causes and consequences of impaired methionine synthase activity in acquired and inherited disorders of vitamin B12 metabolism. Crit Rev Biochem Mol Biol. 2021;1–23. doi:10.1080/10409238.2021.1979459
12. Lam AB, Kervin K, Tanis JE. Vitamin B12 impacts amyloid beta-induced proteotoxicity by regulating the methionine/S-adenosylmethionine cycle. Cell Rep. 2021;36(13):109753. doi:10.1016/j.celrep.2021.109753
13. Schrier MS, Zhang Y, Trivedi MS, Deth RC. Decreased cortical Nrf2 gene expression in autism and its relationship to thiol and cobalamin status. Biochimie. 2021. doi:10.1016/j.biochi.2021.09.006
14. Birch CS, Brasch NE, McCaddon A, Williams JH. A novel role for vitamin B(12): cobalamins are intracellular antioxidants in vitro. Free Radic Biol Med. 2009;47(2):184–188. doi:10.1016/j.freeradbiomed.2009.04.023
15. Wang M, Xu T. Methyl B12 protects PC12 cells against cytotoxicity induced by Abeta25-35. J Cell Biochem. 2019. doi:10.1002/jcb.28475
16. Mizukami H, Ogasawara S, Yamagishi S, Takahashi K, Yagihashi S. Methylcobalamin effects on diabetic neuropathy and nerve protein kinase C in rats. Eur J Clin Invest. 2011;41(4):442–450. doi:10.1111/j.1365-2362.2010.02430.x
17. Vandamme N, Berx G. From neural crest cells to melanocytes: cellular plasticity during development and beyond. Cell Mol Life Sci. 2019;76(10):1919–1934. doi:10.1007/s00018-019-03049-w
18. Jian Z, Li K, Liu L, et al. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J Invest Dermatol. 2011;131(7):1420–1427. doi:10.1038/jid.2011.56
19. Jiang L, Guo Z, Kong Y, Liang J, Wang Y, Wang K. Protective effects of glutamine on human melanocyte oxidative stress model. Indian J Dermatol Venereol Leprol. 2018;84(3):269–274. doi:10.4103/ijdvl.IJDVL_106_17
20. Manga P, Elbuluk N, Orlow SJ. Recent advances in understanding vitiligo. F1000Res. 2016;5:2234. doi:10.12688/f1000research.8976.1
21. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1–15. doi:10.1002/jcp.10119
22. Deo SS, Bhagat AR, Shah RN. Study of oxidative stress in peripheral blood of Indian vitiligo patients. Indian Dermatol Online J. 2013;4(4):279–282. doi:10.4103/2229-5178.120637
23. Trouba KJ, Hamadeh HK, Amin RP, Germolec DR. Oxidative stress and its role in skin disease. Antioxid Redox Signal. 2002;4(4):665–673. doi:10.1089/15230860260220175
24. Denat L, Kadekaro AL, Marrot L, Leachman SA, Abdel-Malek ZA. Melanocytes as instigators and victims of oxidative stress. J Invest Dermatol. 2014;134(6):1512–1518. doi:10.1038/jid.2014.65
25. Valko M, Jomova K, Rhodes CJ, Kuca K, Musilek K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol. 2016;90(1):1–37. doi:10.1007/s00204-015-1579-5
26. Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. J Invest Dermatol. 2006;126(12):2565–2575. doi:10.1038/sj.jid.5700340
27. Chen J, Li S, Li C. Mechanisms of melanocyte death in vitiligo. Med Res Rev. 2021;41(2):1138–1166. doi:10.1002/med.21754
28. Kumar R, Parsad D, Kanwar AJ. Role of apoptosis and melanocytorrhagy: a comparative study of melanocyte adhesion in stable and unstable vitiligo. Br J Dermatol. 2011;164(1):187–191. doi:10.1111/j.1365-2133.2010.10039.x
29. Qiu L, Song Z, Setaluri V. Oxidative stress and vitiligo: the Nrf2-ARE signaling connection. J Invest Dermatol. 2014;134(8):2074–2076. doi:10.1038/jid.2014.241
30. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016;73(17):3221–3247. doi:10.1007/s00018-016-2223-0
31. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–354. doi:10.1146/annurev.pharmtox.010909.105600
32. Zhang S, Yi X, Su X, et al. Ginkgo biloba extract protects human melanocytes from H2 O2 -induced oxidative stress by activating Nrf2. J Cell Mol Med. 2019;23(8):5193–5199. doi:10.1111/jcmm.14393
33. Jiang W, Li S, Chen X, et al. Berberine protects immortalized line of human melanocytes from H2O2-induced oxidative stress via activation of Nrf2 and Mitf signaling pathway. J Dermatol Sci. 2019;94(1):236–243. doi:10.1016/j.jdermsci.2019.03.007
34. Singh A, Venkannagari S, Oh KH, et al. {Wu, 2021 #119}. ACS Chem Biol. 2016;11(11):3214–3225. doi:10.1021/acschembio.6b00651
35. Hu Y, Huang J, Li Y, et al. Cistanche deserticola polysaccharide induces melanogenesis in melanocytes and reduces oxidative stress via activating NRF2/HO-1 pathway. J Cell Mol Med. 2020;24(7):4023–4035. doi:10.1111/jcmm.15038
36. Li XS, Tang XY, Su W, Li X. Vitexin protects melanocytes from oxidative stress via activating MAPK-Nrf2/ARE pathway. Immunopharmacol Immunotoxicol. 2020;42(6):594–603. doi:10.1080/08923973.2020.1835952
37. Mou K, Pan W, Han D, et al. Glycyrrhizin protects human melanocytes from H2O2induced oxidative damage via the Nrf2 dependent induction of HO1. Int J Mol Med. 2019;44(1):253–261. doi:10.3892/ijmm.2019.4200
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.