Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Methionine adenosyltransferase I/III deficiency: beyond the central nervous system manifestations
Authors Nashabat M, Al-Khenaizan S, Alfadhel M ![]()
Received 15 September 2017
Accepted for publication 20 November 2017
Published 2 February 2018 Volume 2018:14 Pages 225—229
DOI https://doi.org/10.2147/TCRM.S151732
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Marwan Nashabat,1 Sultan Al-Khenaizan,2 Majid Alfadhel1
1King Abdullah International Medical Research Centre, King Saud bin Abdulaziz University for Health Sciences, Division of Genetics, Department of Pediatrics, King Abdulaziz Medical City, Ministry of National Guard-Health Affairs (NGHA), Riyadh, Saudi Arabia; 2Department of Dermatology, King Abdullah International Medical Research Center, King Saud bin Abdulaziz University for Health Sciences, King Abdulaziz Medical City, Ministry of National Guard-Health Affairs (NGHA), Riyadh, Saudi Arabia
Abstract: Methionine adenosyltransferase (MAT) I/III deficiency (OMIM # 250850) is caused by a mutation in MAT1A, which encodes the two hepatic MAT isozymes I and III. With the implementation of newborn screening program to discover hypermethioninemia due to cystathionine beta-synthase deficiency, more cases are being discovered. While the majority of patients are asymptomatic, some might have central nervous system (CNS) and extra-CNS manifestations. Although neurologic manifestations and demyelination have been correlated to MAT deficiency in many reported cases, none of the previous reports focused on extra-CNS manifestations associated with the disease. This is a retrospective chart review for a 40-month-old patient with confirmed diagnosis of MAT deficiency. He was found to have a novel homozygous disease-causing variant in MAT1A (NM_000429.2) c.1081G>T (p.Val361Phe). Interestingly, our patient had an unexplained zinc and iron deficiency in addition to mild speech delay. We reviewed the literature and summarized all the reported extra-CNS manifestations. In conclusion, MAT deficiency patients should be thoroughly investigated to check for CNS and extra-CNS manifestations associated with the disease. Keeping in consideration the challenge of assuming correlation, a scrutinized look at extra-CNS manifestations and their course with time might pave the way to understanding the pathophysiology of the disease and MAT1A function.
Keywords: MAT1A, methionine adenosyltransferase, S-adenosyl methionine, central nervous system
Introduction
Methionine adenosyltransferase (MAT) deficiency (OMIM # 250850) was first described by Gaull and Tallan in 1974 in an infant with hypermethioninemia,1 who had normal development on follow-up.2 Many subsequent reports described that patients with hypermethioninemia correlated to MAT deficiency based on decreased activity of the enzyme in liver biopsy. The key factor to identify most of the reported cases in the literature was the implementation of the newborn screening program to discover hypermethioninemia due to cystathionine beta-synthase deficiency.3 The molecular basis of the disease was established for the first time by Ubagai et al.4 Further delineation of the MAT1A mutations and the mode of inheritance were elaborated by Chamberlin et al.5
Clinically, although the majority of patients are asymptomatic, some might have central nervous system (CNS) manifestations.5 Additionally, patients may have extra-CNS manifestations like malodorous breathing.6,7
Biochemical findings include plasma hypermethioninemia ranging from mild elevation to >2,000 μM. Patients usually have decreased or complete loss of MAT I/III activity resulting in low S-adenosyl methionine (AdoMet) level. Furthermore, some patients were reported to have mild elevation in homocysteine level.3
MAT deficiency is inherited either as autosomal recessive or autosomal dominant. MAT1A mutations described previously range from truncating mutations with no residual enzyme activity to mild missense mutations.8
There is no consensus, with a high level of evidence, regarding recommended management plan in the literature.8 Many inputs should be considered like confirmation of the diagnosis, plasma methionine level, and CNS involvement, before starting treatment. The most commonly used treatment modalities are methionine restriction diet and AdoMet supplementation.3,8–10 The most recent recommendations for the diagnosis and management of all inherited methylation disorders were reviewed by Baric et al.11
In this report, we describe the first case in the literature of MAT deficiency in Saudi Arabia with novel mutation not described previously, in addition to a unique presentation with skin features of acrodermatitis enteropathica with severe zinc deficiency necessitating oral supplementation.
Case report
A 40-month-old male patient, who was a product of spontaneous vaginal delivery after full-term uneventful pregnancy to a healthy 32-year-old woman and her 34-year-old first cousin. Birth weight was 3.28 kg, length at 50 cm, and head circumference at 34.5 cm, all were around the median, with APGAR scores of 9 and 9 at 5 and 10 minutes, respectively. The patient underwent newborn screening tests as part of the routine care for all newborns, which showed elevated methionine at 105 (11–27) μmol/L. A repeated sample confirmed the elevation at 85 μmol/L. Clinically the patient was stable with unremarkable complete systemic examination.
Other metabolic investigations were requested including homocysteine, and urine for organic acids and methylmalonic acid but all were unremarkable. Renal and liver functions were unremarkable.
At the age of 2 months, the patient developed irritability and skin eruption consisting of multiple scaly and erosive hyperkeratotic plaques all over the body but most pronounced on lower extremities. In addition to leukotrichia and positive hair pull test (Figure 1), his zinc level was very low at 3.4 (9.18–18.36) μmol/L. All skin eruptions started to resolve after oral zinc supplementation over 2 weeks (Figure 1).
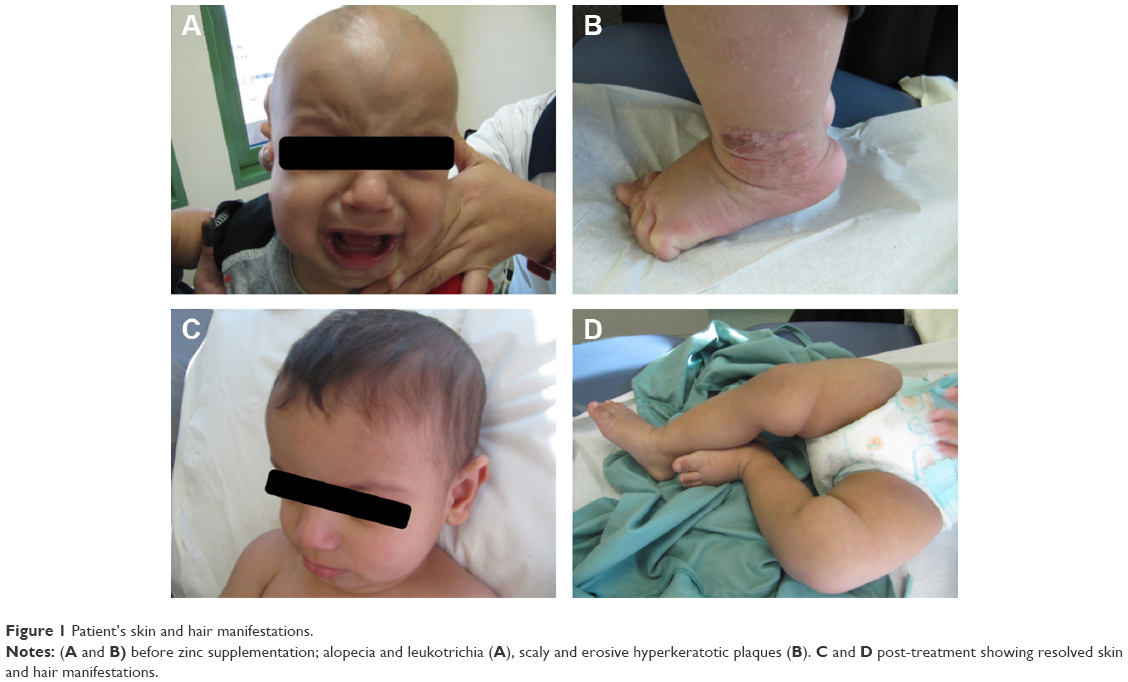 | Figure 1 Patient’s skin and hair manifestations. |
At the age of 6 months, the final diagnosis was reached by the genetic study of MAT1A (NM_000429.2), which revealed a homozygous variant in c.1081G>T (p.Val361Phe). In silico study predictions were as follows: Mutation taster12 predicted a disease-causing variant, PolyPhen2:13 possibly damaging (score: 0.889) and SIFT:14 damaging variant. Further genetic testing to rule out acrodermatitis enteropathica (OMIM # 201100) caused by SLC39A4 (NM_130849) mutation was negative. All the genetic tests requested for the patient were single gene sequencing done at commercial accredited labs.
In addition, the patient was found to have iron deficiency, which was discovered at 9 months of age; hemoglobin level was 90 (110–145) g/L, mean corpuscular volume (MCV) 62.4 (75–89) fL, mean corpuscular hemoglobin (MCH) 17 (25–30) pg, serum iron 4.8 (9–21.5) μmol/L, and ferritin 7 (21.8–274.6) μg/L. Consequently, ferrous sulfate was prescribed.
Of note, the patient’s nutritional history was unremarkable during the time he developed the symptoms. He had combined breast and formula feeding initially then he was receiving regular diet. His feeding was well and he had acceptable feeding habits. The family history was negative for MAT, zinc, or iron deficiency.
Currently, the patient is 40 months old with normal developmental milestones except for mild speech delay in the form of dysarthria. All his growth parameters are around the median. He is still on zinc and iron supplementation. All the skin and hair symptoms improved. The patient’s zinc level is still low at 6.79 (9.18–18.36) μmol/L. The serum ferritin is still low at 13.7 (21.8–274.6) μg/L, hemoglobin was normalized at 128 g/L, while MCV and MCH are slightly low at 71.1 (75–89) fL and 24.2 (25–30) pg, respectively. Latest lab investigations showed methionine at 34 μmol/L and homocysteine at 6.1 (≤15) μmol/L. The highest methionine reading reported for our patient was 110 μmol/L and the average was at 59.2 μmol/L.
Discussion
MAT1A encodes the two MAT isozymes I and III.4 MAT acts predominantly in liver but also present in other tissues.15,16 MAT catalyzes the first step in the methionine cycle to synthesize AdoMet.17 MAT deficiency results in variable ranges of hypermethioninemia and decreased AdoMet level, based on the type of gene mutation. Although truncating mutations are thought to cause a severe phenotype with high methionine level and neurologic derangement, this was not true in all cases. Some of the patients harbor truncating mutations and had a markedly elevated methionine level, but still they developed normally.4,8 In our patient, although the methionine level was mildly elevated, he had signs of developmental delay.
MAT deficiency is inherited either as autosomal-dominant- or autosomal-recessive trait like our patient. Initially, it was thought that the autosomal recessive is the most common way of inheritance, but after the implementation of newborn screening, more heterozygous mild cases were discovered. This set the fact that the autosomal-dominant inherited genotype is the most common.8 Our patient had a novel homozygous mutation c.1081G>T (p.Val361Phe) in MAT1A gene that was not reported before.
Additionally, our patient had unexplained zinc deficiency with temporary features of acrodermatitis enteropathica. This may raise the suspicion of impaired zinc absorption. Bauchart-Thevret et al studied the effect of dietary sulfur amino acids deficiency on neonatal pigs and concluded that it results in suppression of the epithelial growth, which was manifested as villous atrophy and caused upregulation of the methionine cycle in the intestine.18 Villous atrophy is a well-known cause of zinc and iron deficiency in many gastrointestinal diseases.19–21 However, our patient did not have clinical symptoms of villus atrophy.
Zinc deficiency could be due to a wide spectrum of causes, usually categorized in either decreased intake, malabsorption, increased loss, or increased demand.22 None of these categories fit our patient, and accordingly, we tried to investigate for the other genetic causes including SLC39A, which was negative.
A long list of causes can result in iron deficiency, among which are the four categories mentioned above. Our patient was completely symptomatic, with balanced diet, no signs of malabsorption, and no signs of increased loss and even no increase in the demand.23 Although the cause of iron and zinc deficiency in our patient is still obscure, it is not clear whether they could be correlated with MAT1A mutation or not.
Neurologic manifestations and demyelination have been correlated to MAT deficiency in many reported cases.24,25 But none of the previous reports shed light on extra-CNS manifestations associated with the disease. Table 1 summarizes all the previously reported cases with extra-CNS manifestations.
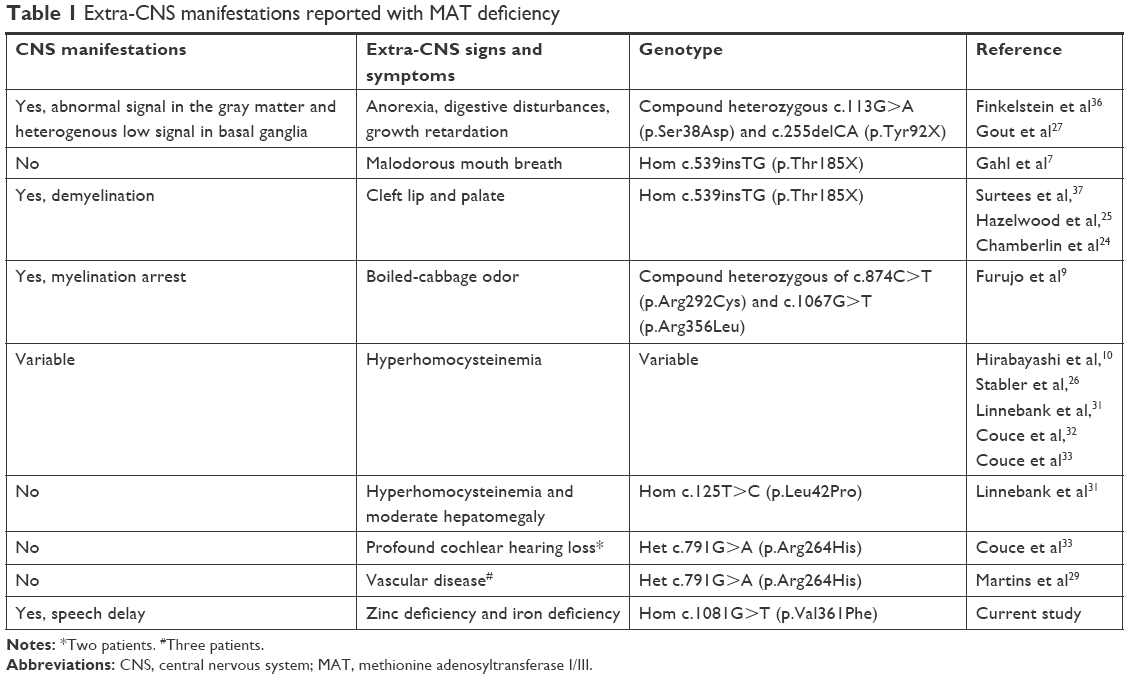 | Table 1 Extra-CNS manifestations reported with MAT deficiency |
By reviewing all the reported cases in the literature, we found that many extra-CNS manifestations were reported sporadically along with MAT deficiency. Some of these signs and symptoms could be explained by the pathophysiology of the disease like the malodorous mouth breath and the boiled-cabbage odor.6 On the other hand, the remaining manifestations were not completely explained. The pathophysiology of the MAT deficiency is centered around two themes: elevated methionine level and deprived AdoMet level.26 AdoMet is involved in tremendous cellular functions,16 which, in theory, might have variable clinical consequences.
Gout et al reported anorexia, digestive disturbances, and growth retardation in their case, in addition to the CNS manifestations.27 Subsequent reports mentioning the same patient did not comment on any of these symptoms. Instead, they reported a normal cognitive function,5,28 which could be a clue of improvement. Similarly, our patient’s skin and hair symptoms improved with time.
Martins et al reported vasculopathy in three families with MAT deficiency, which was correlated to the elevated total homocysteine as a possible risk factor for vasculopathy.29,30 Although elevated total homocysteine was reported in many other cases of MAT deficiency, vasculopathy was not mentioned in any of them.10,31–33
Among all the reported MAT deficiency cases, only one patient was reported to have unexplained moderate hepatomegaly.31 None of the other reported cases, including our patient, had a liver injury or developed hepatocellular carcinoma, which is against that observed in MAT1A knocked-out mice models.34,35
To conclude, MAT deficiency patients should be thoroughly investigated to check for CNS and extra-CNS manifestations associated with the disease. It is still not easy to correlate the genotype and phenotype of this disease. A scrutinized look at the extra-CNS manifestations and their course might pave the way to better understand the pathophysiology of the disease and MAT1A function.
Ethics approval
This study was approved by King Abdullah International Medical Research Center Institutional Review Board (RC 16/113/R). A written informed consent was taken from the parents to have the case details and any accompanying images published.
Acknowledgments
The authors would like to thank the patient and his family for their participation. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
Gaull GE, Tallan HH. Methionine adenosyltransferase deficiency: new enzymatic defect associated with hypermethioninemia. Science.1974;186(4158):59–60. | ||
Mudd SH, Tangerman A, Stabler SP, et al. Maternal methionine adenosyltransferase I/III deficiency: reproductive outcomes in a woman with four pregnancies. J Inherit Metab Dis.2003;26(5):443–458. | ||
Mudd SH. Hypermethioninemias of genetic and non-genetic origin: a review. Am J Med Genet C Semin Med Genet.2011;157C(1):3–32. | ||
Ubagai T, Lei KJ, Huang S, Mudd SH, Levy HL, Chou JY. Molecular mechanisms of an inborn error of methionine pathway. Methionine adenosyltransferase deficiency. J Clin Invest.1995;96(4):1943–1947. | ||
Chamberlin ME, Ubagai T, Mudd SH, et al. Methionine adenosyltransferase I/III deficiency: novel mutations and clinical variations. Am J Hum Genet.2000;66(2):347–355. | ||
Gahl WA, Bernardini I, Finkelstein JD, et al. Transsulfuration in an adult with hepatic methionine adenosyltransferase deficiency. J Clin Invest.1988;81(2):390–397. | ||
Gahl WA, Finkelstein JD, Mullen KD, et al. Hepatic methionine adenosyltransferase deficiency in a 31-year-old man. Am J Hum Genet.1987;40(1):39–49. | ||
Chien YH, Abdenur JE, Baronio F, et al. Mudd’s disease (MAT I/III deficiency): a survey of data for MAT1A homozygotes and compound heterozygotes. Orphanet J Rare Dis.2015;10:99. | ||
Furujo M, Kinoshita M, Nagao M, Kubo T. S-adenosylmethionine treatment in methionine adenosyltransferase deficiency, a case report. Mol Genet Metab.2012;105(3):516–518. | ||
Hirabayashi K, Shiohara M, Yamada K, et al. Neurologically normal development of a patient with severe methionine adenosyltransferase I/III deficiency after continuing dietary methionine restriction. Gene.2013;530(1):104–108. | ||
Baric I, Staufner C, Augoustides-Savvopoulou P, et al. Consensus recommendations for the diagnosis, treatment and follow-up of inherited methylation disorders. J Inherit Metab Dis.2017;40(1):5–20. | ||
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. Mutation Taster evaluates disease-causing potential of sequence alterations. Nat Methods.2010;7(8):575–576. | ||
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods.2010;7(4):248–249. | ||
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res.2001;11(5):863–874. | ||
Fernandez-Irigoyen J, Santamaria E, Chien YH, et al. Enzymatic activity of methionine adenosyltransferase variants identified in patients with persistent hypermethioninemia. Mol Genet Metab.2010;101(2–3):172–177. | ||
Reytor E, Perez-Miguelsanz J, Alvarez L, Perez-Sala D, Pajares MA. Conformational signals in the C-terminal domain of methionine adenosyltransferase I/III determine its nucleocytoplasmic distribution. FASEB J.2009;23(10):3347–3360. | ||
Kotb M, Geller AM. Methionine adenosyltransferase: structure and function. Pharmacol Ther.1993;59(2):125–143. | ||
Bauchart-Thevret C, Stoll B, Chacko S, Burrin DG. Sulfur amino acid deficiency upregulates intestinal methionine cycle activity and suppresses epithelial growth in neonatal pigs. Am J Physiol Endocrinol Metab.2009;296(6):E1239–E1250. | ||
Skrovanek S, DiGuilio K, Bailey R, et al. Zinc and gastrointestinal disease. World J Gastrointest Pathophysiol.2014;5(4):496–513. | ||
Rawal P, Thapa BR, Prasad R, Prasad KK, Nain CK, Singh K. Zinc supplementation to patients with celiac disease–is it required? J Trop Pediatr.2010;56(6):391–397. | ||
Hershko C, Patz J. Ironing out the mechanism of anemia in celiac disease. Haematologica.2008;93(12):1761–1765. | ||
Corbo MD, Lam J. Zinc deficiency and its management in the pediatric population: a literature review and proposed etiologic classification. J Am Acad Dermatol.2013;69(4):616.e611–624.e611. | ||
Lopez A, Cacoub P, Macdougall IC, Peyrin-Biroulet L. Iron deficiency anaemia. Lancet.2016;387(10021):907–916. | ||
Chamberlin ME, Ubagai T, Mudd SH, Wilson WG, Leonard JV, Chou JY. Demyelination of the brain is associated with methionine adenosyltransferase I/III deficiency. J Clin Invest.1996;98(4):1021–1027. | ||
Hazelwood S, Bernardini I, Shotelersuk V, et al. Normal brain myelination in a patient homozygous for a mutation that encodes a severely truncated methionine adenosyltransferase I/III. Am J Med Genet.1998;75(4):395–400. | ||
Stabler SP, Steegborn C, Wahl MC, et al. Elevated plasma total homocysteine in severe methionine adenosyltransferase I/III deficiency. Metabolism.2002;51(8):981–988. | ||
Gout JP, Serre JC, Dieterlen M, et al. [Still another cause of hypermethioninemia in children: S-adenosylmethionine synthetase deficiency]. Arch Fr Pediatr.1977;34(5):416–423. French. | ||
Mudd SH, Levy HL, Tangerman A, et al. Isolated persistent hypermethioninemia. Am J Hum Genet.1995;57(4):882–892. | ||
Martins E, Marcao A, Bandeira A, Fonseca H, Nogueira C, Vilarinho L. Methionine adenosyltransferase I/III deficiency in Portugal: high frequency of a dominantly inherited form in a small area of Douro High Lands. JIMD Rep.2012;6:107–112. | ||
Smulders YM, Blom HJ. The homocysteine controversy. J Inherit Metab Dis.2011;34(1):93–99. | ||
Linnebank M, Lagler F, Muntau AC, et al. Methionine adenosyltransferase (MAT) I/III deficiency with concurrent hyperhomocysteinaemia: two novel cases. J Inherit Metab Dis.2005;28(6):1167–1168. | ||
Couce ML, Boveda MD, Castineiras DE, et al. Hypermethioninaemia due to methionine adenosyltransferase I/III (MAT I/III) deficiency: diagnosis in an expanded neonatal screening programme. J Inherit Metab Dis.2008;31(Suppl 2):S233–S239. | ||
Couce ML, Boveda MD, Garcia-Jimemez C, et al. Clinical and metabolic findings in patients with methionine adenosyltransferase I/III deficiency detected by newborn screening. Mol Genet Metab.2013;110(3):218–221. | ||
Lu SC, Alvarez L, Huang ZZ, et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci U S A.2001;98(10):5560–5565. | ||
Lu SC, Mato JM. Role of methionine adenosyltransferase and S-adenosylmethionine in alcohol-associated liver cancer. Alcohol.2005;35(3):227–234. | ||
Finkelstein JD, Kyle WE, Martin JJ. Abnormal methionine adenosyltransferase in hypermethioninemia. Biochem Biophys Res Commun.1975;66(4):1491–1497. | ||
Surtees R, Leonard J, Austin S. Association of demyelination with deficiency of cerebrospinal-fluid S-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet.1991;338(8782–8783):1550–1554. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.