Back to Journals » Cancer Management and Research » Volume 11
Metformin induces apoptotic cytotoxicity depending on AMPK/PKA/GSK-3β-mediated c-FLIPL degradation in non-small cell lung cancer
Authors Luo Z ![]() , Zhu T, Luo W, Lv Y, Zhang L, Wang C, Li M
, Zhu T, Luo W, Lv Y, Zhang L, Wang C, Li M ![]() , Wu W
, Wu W ![]() , Shi S
, Shi S ![]()
Received 5 July 2018
Accepted for publication 3 December 2018
Published 11 January 2019 Volume 2019:11 Pages 681—689
DOI https://doi.org/10.2147/CMAR.S178688
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Zhuang Luo,1,* Tingting Zhu,2,* Wei Luo,3,* Yuanyuan Lv,1 Liyan Zhang,1 Chu Wang,1 Min Li,1 Wenjuan Wu,4 Shaoqing Shi1
1Department of Pulmonary and Critical Care Medicine, The First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan 650032, People’s Republic of China; 2Department of Dermatology, The First Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215006, People’s Republic of China; 3Department of Respiratory Medicine, The People’s Hospital of Leshan, Leshan, Sichuan 640000, People’s Republic of China; 4Department of Dermatology, The First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan 650032, People’s Republic of China
*These authors contributed equally to this work
Background: Metformin, a first-line antidiabetic drug, has recently been reported with anticancer activities in various cancers; however, the underlying mechanisms remain elusive. The aim of the present study was to investigate the role of cellular FADD-like IL-1β-converting enzyme (FLICE)-inhibitory protein large (c-FLIPL) in metformin-induced anticancer activity in non-small cell lung cancer (NSCLC) in vitro.
Materials and methods: Cell viability was measured by MTT assay. Quantitative real-time PCR was carried out to detect the level of mRNA of related genes. The expression of related proteins was detected by Western blot. siRNA was used to silence the expression of targeted proteins.
Results: Metformin significantly suppressed proliferation of both A549 and H460 cells in a dose-dependent manner. Mechanistic studies suggested that metformin killed NSCLC cells by inducing apoptotic cell death. Moreover, metformin greatly inhibited c-FLIPL expression and then promoted its degradation. Furthermore, metformin significantly activated Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and its downstream glycogen synthase kinase 3beta (GSK-3β), block the expression of AMPK, and GSK-3β with siRNA partially reversed metformin-induced cytotoxicity and restored the expression of c-FLIPL in lung cancer cells. Metformin also suppressed the activity of AMPK downstream protein kinase A (PKA), PKA activators, both 8-Br-cAMP and forskolin, greatly increased c-FLIPL expression in NSCLC cells.
Conclusion: This study provided evidence that metformin killed NSCLC cells through AMPK/PKA/GSK-3β axis-mediated c-FLIPL degradation.
Keywords: non-small cell lung cancer, c-FLIPL, AMPK, GSK-3β, PKA
Introduction
As one of the most malignant cancers, lung cancer severely endangers the life and health of human beings. The incidence and mortality rates of this disease rank top of different types of tumors in People’s Republic of China.1 Lung cancer mainly includes non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC accounts for about 85% lung cancer, the 5-year survival rate of which is <15%. Surgery, chemotherapy, and radiotherapy are the main options for late-stage NSCLC patients, but the effects are dissatisfying. Immune-oncology has recently changed the paradigms of NSCLC, which greatly improves 5-year survival rate and prognosis of NSCLC patients. Therefore, it is of great significance to explore novel anticancer agents for late-stage NSCLC treatment.
Metformin is a first-line antidiabetic drug and has been reported with antitumor activities in various cancers, including breast cancer,2 hepatocellular cancer,3 gastric cancer,4 lymphoma,5 melanoma,6 esophageal squamous cell carcinoma,7 and lung cancer.8 Increasing studies have demonstrated that the combination of metformin with anticancer agents induced synergic anticancer effects in NSCLC, including radiation,9 gefitinib,10,11 erlotinib,12 sorafenib,13 carboplatin,14 cisplatin,15 and TRAIL.16 Epidemiological studies have suggested that metformin reduces the incidence of lung cancer with diabetes and improves the survival of patients with type II diabetes of lung cancer.17–19 Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) is one of the main mechanisms by which metformin exerts anticancer activities.20 It has been well established that AMPK and its downstream glycogen synthase kinase 3 beta (GSK-3β) and mTOR play a crucial role in metformin-induced anticancer effects.2–4 MicroRNA, c-Myc, and tumor microenvironment contribute to the antitumoral activities of metformin as well.21–23 However, the molecular mechanisms of metformin-induced anticancer effects warrants further investigation.
Cellular FADD-like IL-1β-converting enzyme (FLICE)-inhibitory protein (c-FLIP) is a key negative regulator of apoptosis pathways.24 There are three isoforms of this protein, c-FLIPL, c-FLIPS, and c-FLIPR. c-FLIPL is a 55 kDa protein, including two N-terminal DED domains and a C-terminal caspase-like domain, which is structurally similar to pro-caspase 8.25 c-FLIP is always overexpressed in various tumors and cell lines, including lung cancer.26 Overexpression of c-FLIP confers increased resistance to apoptosis mediated by TRAIL and Fas. Previous studies have demonstrated that high levels of c-FLIP are correlated with poor prognosis in different cancers. Therefore, targeting c-FLIP might be a promising strategy for cancer treatment. Our previous studies have demonstrated that the combination of Chal-24 with anticancer agents results in synergistic cytotoxicity in lung cancer by downregulation of c-FLIPL, including cisplatin27 and TRAIL.28 Previous studies have reported that metformin decreases the expression of c-FLIPL in bladder cancer,29 renal cell carcinoma30 and lung cancer cells;16 however, the molecular mechanisms underlying metformin-mediated c-FLIPL downregulation remain largely unknown.
In the present study, we investigated the role of c-FLIPL in metformin-mediated anticancer effects in NSCLC in vitro. We found that metformin induced apoptotic cell death in NSCLC cells, and it remarkably downregulated the expression of c-FLIPL. Mechanistic studies demonstrated that metformin mainly suppressed c-FLIPL expression through AMPK/PKA/GSK-3β pathway. These results provide novel insight into the role of c-FLIPL in metformin-induced anticancer effects in NSCLC in vitro.
Materials and methods
Reagents
Metformin (D150959) was purchased from Sigma-Aldrich (St Louis, MO, USA). Anti-phosphor-AMPKα (Thr172) (40H9) (2535) and AMPKα (5832), phosphor-cAMP-response element binding protein (CREB) (Ser133) (87G3) (9198) and CREB (48H2) (9197), phosphor-GSK-3β (Ser9) (9336) and GSK-3β (27C10) (9315), PARP (3493), cleaved Caspase-3 (9664) and GAPDH (2118) were from Cell Signaling Technology (Danvers, MA, USA). Antibody for β-tubulin (200608) was from Zen Bioscience (Chengdu, Sichuan, People’s Republic of China). Anti-Caspase 3 (ab32351) and c-FLIPL were from Abcam (Cambridge, UK). z-VAD (Ome)-FMK (600332) and chlorhexidine were from Cell Signaling Technology.
Cell culture
Lung cancer cell lines A549 and H460 were obtained from America Type Culture Collection (Manassas, VA, USA) and grown in RPMI-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, 100 U penicillin, and 100 µg/mL streptomycin. All cells were cultured in standard incubator conditions at 37°C with 5% CO2. RPMI-1640 medium without glucose and FBS were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA).
Cell viability assay
Cells were seeded in triplicate data density of 5×104/well and after staining with trypan-blue were counted every day for 3 days. In some experiments, cells were seeded in triplicate at a density of 2.5×104/well. The next day and thereafter, cells were incubated with 20 mg/mL MTT for 3 hours. MTT was then dissolved in dimethyl sulfoxide and read at OD 570 nm. The results were expressed as relative cell number against the corresponding readings from the cells on day set to 100.
RNA extraction
Total RNA was extracted from cells using Transzol (Takara Bio Inc., Shiga, Japan), Chloroform (Sangon Biotech, Shanghai, People’s Republic of China), Isopropyl alcohol (Sangon Biotech), DEPC water (Sangon Biotech), and 75% ethanol (Sangon Biotech). The yield of RNA was determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and the integrity was evaluated using agarose gel electrophoresis stained with ethidium bromide.
Real-time quantitative RT-PCR
Quantification was performed with a two-step reaction process: reverse transcription (RT) and PCR. The PrimeScript™ RT reagent Kit with gDNA Eraser (Takara Bio Inc.) was used for each RT reaction in a total volume of 10 µL. Reactions were performed in a GeneAmp® PCR System 9700 (Thermo Fisher Scientific) for 15 minutes at 37°C, followed by heat inactivation of RT for 5 seconds at 85°C. The 10 µL RT reaction mix was then diluted 10× in nuclease-free water and held at –20°C.
Real-time PCR was performed using ABI Prism 7900 Real-time PCR Instrument (Thermo Fisher Scientific) with the TransStart® Top Green qPCR SuperMix (+ Dye I/+ Dye II)-qPCR kit (Transgen Biotech Co. Ltd., Beijing, People’s Republic of China) in a 10 µL PCR reaction mixture. Reactions were incubated in a 96-well optical plate (Transgen Biotech Co. Ltd.) at 94°C for 30 minutes, followed by 40 cycles of 94°C for 50 seconds and 60°C for 30 seconds. Each sample was run in triplicate for analysis. At the end of the PCR cycles, melting curve analysis was performed to validate the specific generation of the expected PCR product. The primer sequences were designed in the laboratory and synthesized by Beijing TSINGKE, based on the mRNA sequences obtained from the NCBI database as follow. Primer sequences for real-time PCR are shown in Table 1.
 | Table 1 Primer sequences for real-time PCR Abbreviations: c-FLIPL, cellular FLICE-inhibitory protein large; FLICE, FADD-like IL-1β-converting enzyme. |
Western blotting
Cell lysates were prepared by suspending cells in M2 buffer (20 mM Tris–HCl pH 7.6, 0.5% NP40, 250 mM NaCl, 3 mM EDTA, 2 mM DTT, 0.5 mM phenylmethylsulfonylfluoride, 20 mM β-glycerophosphate, 1 mM sodium vanadate, and 1 µg/mL leupeptin). The samples were boiled for 5 minutes, equal amounts of proteins from each cell lysate were resolved by 8% or 12% SDS-PAGE and analyzed by Western blot. The proteins were visualized with enhanced chemiluminescence (Millipore) following the instructions of the manufacturer. Each experiment was repeated at least three times and representative results are shown.
Knockdown protein expression by RNAi
siRNA was purchased from RiboBio Co., Ltd (Guangzhou, Guangdong, People’s Republic of China). A549 and H460 cells were seeded in a 12-well plate and 48-well plate the day before transfection at 40%–50% confluence. siRNA was transfected with siRNA INTERFERin (polyplus transfection).31 Twenty-four hours after transfection, metformin (80 mM) was added to the culture for 48 hours and MTT assay was carried out to measure cell viability, or metformin was added to the culture for 16 hours and expression of related proteins were detected by Western blot. Knockdown was confirmed by Western blot.
Statistical analysis
All data were expressed as mean ± SD. All data were analyzed using SPSS 17.0 for Windows (SPSS Inc., Chicago, IL, USA). P<0.05 was considered statistically significant.
Results
Metformin promoted NSCLC cells apoptosis
To investigate the anticancer effects of metformin, A549 and H460 cells were treated with increasing concentrations of metformin for 48 hours; cell viability was detected and quantified by MTT assay. As shown in Figure 1A, metformin suppressed NSCLC cell proliferation in a dose-dependent manner. Consistent with previous studies that metformin induced apoptosis in many types of cancer cells, metformin greatly induced caspase-3 activation and PARP cleavage in both A549 and H460 cell lines (hallmarks of apoptosis), indicating metformin induced apoptosis in NSCLC cells (Figure 1B), which was further validated by pan-caspase inhibitor z-VAD-fmk reversed metformin-induced cytotoxicity (Figure 1C). These results indicate that metformin mainly induces apoptotic cytotoxicity in lung cancer cells.
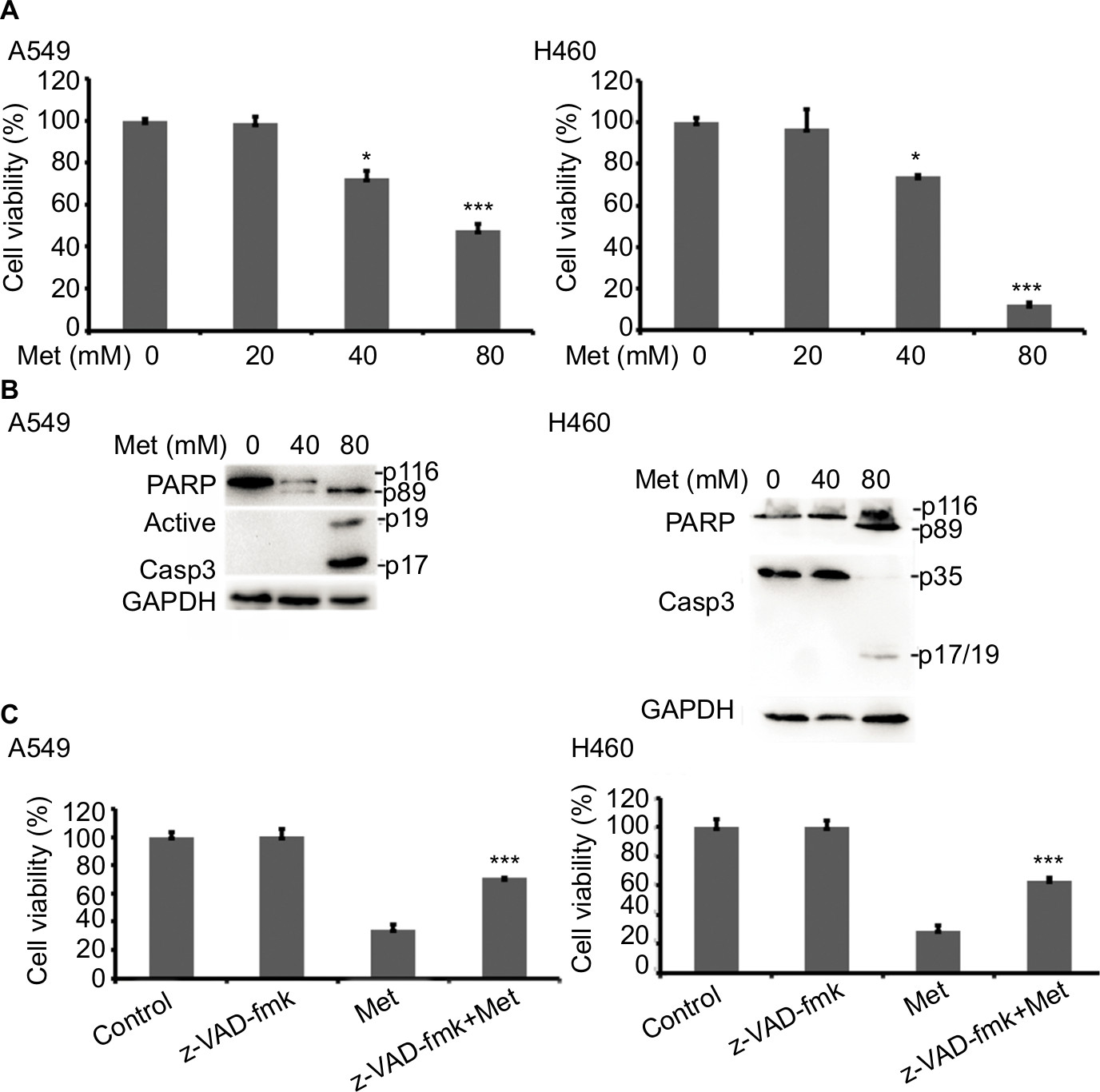 | Figure 1 Metformin induces apoptotic cell death in lung cancer cells. Notes: (A) A549 and H460 cells were treated with metformin (20, 40, and 80 mM) for 48 hours. Cell viability was measured and quantified by MTT assay. (B) A549 and H460 cells were treated with indicated concentrations of metformin for 24 hours. Then, indicated proteins were detected by Western blot, GAPDH or tubulin was detected as an input control. (C) The cells were pretreated with z-VAD-FMK (10 µM) for 1 hour, and then exposed to metformin for 48 hours. Cell viability was measured and quantified by MTT assay. N=3. *P<0.05, ***P<0.001. |
Metformin suppressed the expression of c-FLIPL in NSCLC
It has been well documented that c-FLIPL is one of the main inhibitors of cell death, and previous studies have reported that metformin promoted c-FLIPL degradation in bladder cancer and lung cancer, respectively, thus resulting in synergism of TRAIL-induced cytotoxicity, but whether metformin also influences the expression of c-FLIPL in NSCLC still remains elusive. As shown in Figure 2A, metformin also greatly suppressed c-FLIPL expression in both A549 and H460 cells. To further investigate the mechanism by which metformin suppressed the expression of c-FLIPL in lung cancer cells, we first investigated the c-FLIPL mRNA levels by qRT-PCR. Metformin did not influence the mRNA levels of this protein in both A549 and H460 cell lines (Figure 2B). Then, a cycloheximide chase assay was carried out to investigate the stability of c-FLIPL. Lung cancer cells were pretreated with metformin (80 mM) for 4 hours, then exposed to cycloheximide (200 µg/mL) for indicated times. As shown in Figure 2C, metformin significantly accelerated the degradation of c-FLIPL in both A549 and H460 cells compared with the control group. Taken together, these results strongly suggested that metformin kills lung cancer cells through degradation of c-FLIPL in lung cancer cells.
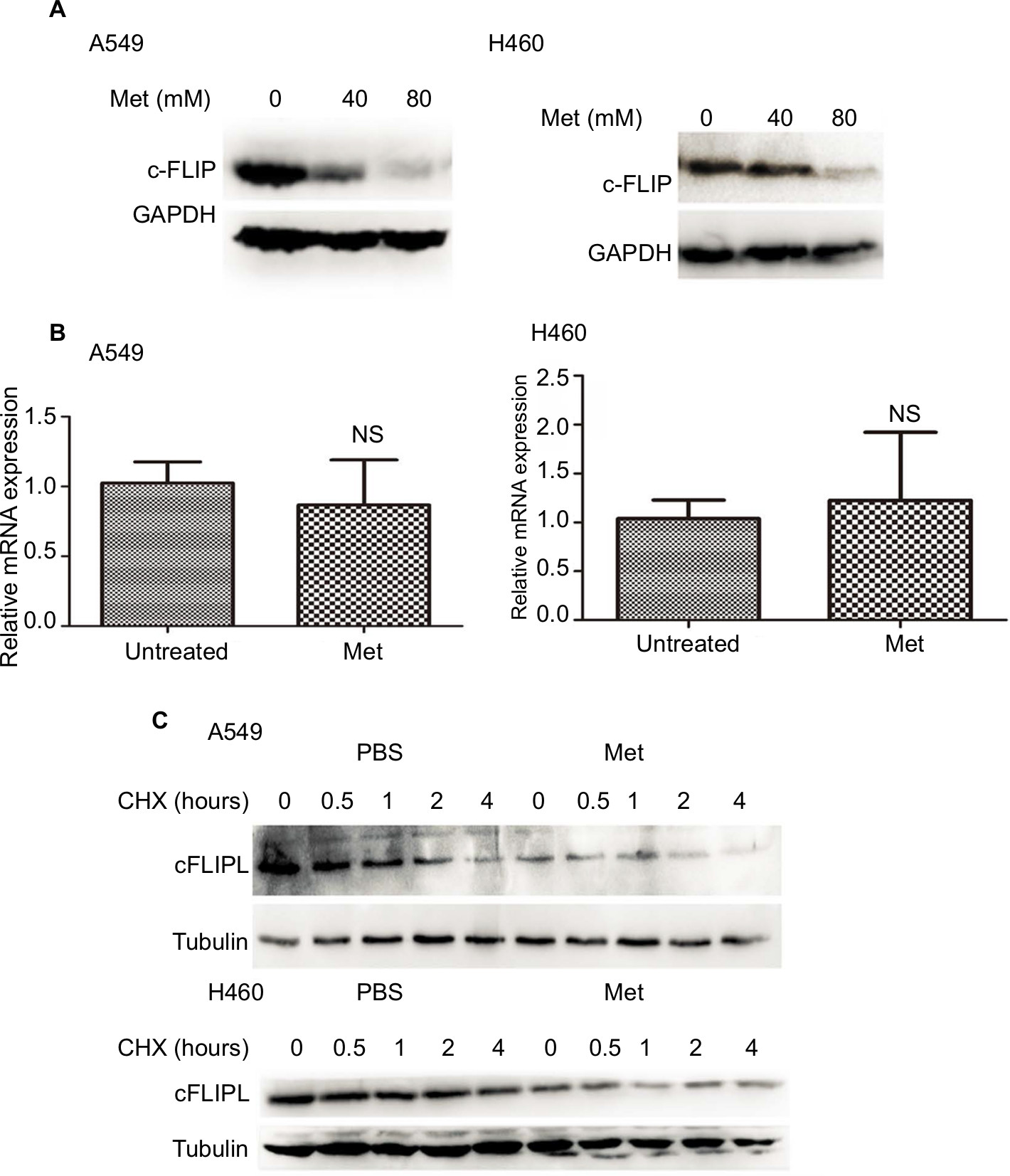 | Figure 2 Metformin downregulated the expression of c-FLIPL in NSCLC. Notes: (A) A549 and H460 cells were treated with metformin (80 mM) for 16 hours. Then, indicated proteins were detected by Western blot, and GAPDH or tubulin was detected as an input control. (B) Lung cancer cells were treated with metformin for 8 hours. Then, total RNA were extracted, and the mRNA level of c-FLIPL was examined by qRT-PCR, β-actin was detected as an input control. (C) A549 and H460 cells were pretreated with metformin (80 mM) or PBS for 4 hours, and then treated with CHX (200 µg/mL) for indicated times. Then, indicated proteins were detected by Western blot and tubulin was detected as an input control. N=3. Abbreviations: c-FLIPL, cellular FLICE-inhibitory protein large; FLICE, FADD-like IL-1β-converting enzyme; NSCLC, non-small cell lung cancer. |
Metformin downregulated c-FLIPL expression depending on AMPK activation
It has been well documented that AMPK activation is the main action of metformin in various cancer cells. However, the part of this pathway in apoptosis via c-FLIPL remains elusive in NSCLC. To elucidate whether metformin downregulates c-FLIPL expression through AMPK, we examined the AMPK activation by Western blotting. Metformin indeed induced AMPK activation in both A549 and H460 cell lines in a dose-dependent manner (Figure 3A). To further confirm the role of AMPK in metformin-induced c-FLIPL degradation in lung cancer cells, siRNA was used to silence the expression of AMPK. As shown in Figure 3B, siRNA partially reversed metformin-induced cell death in both A549 and H460 cell lines. Consistently, siRNA restored the expression of c-FLIPL in NSCLC cells (Figure 3C). These results support that metformin promotes c-FLIPL degradation in an AMPK-dependent manner.
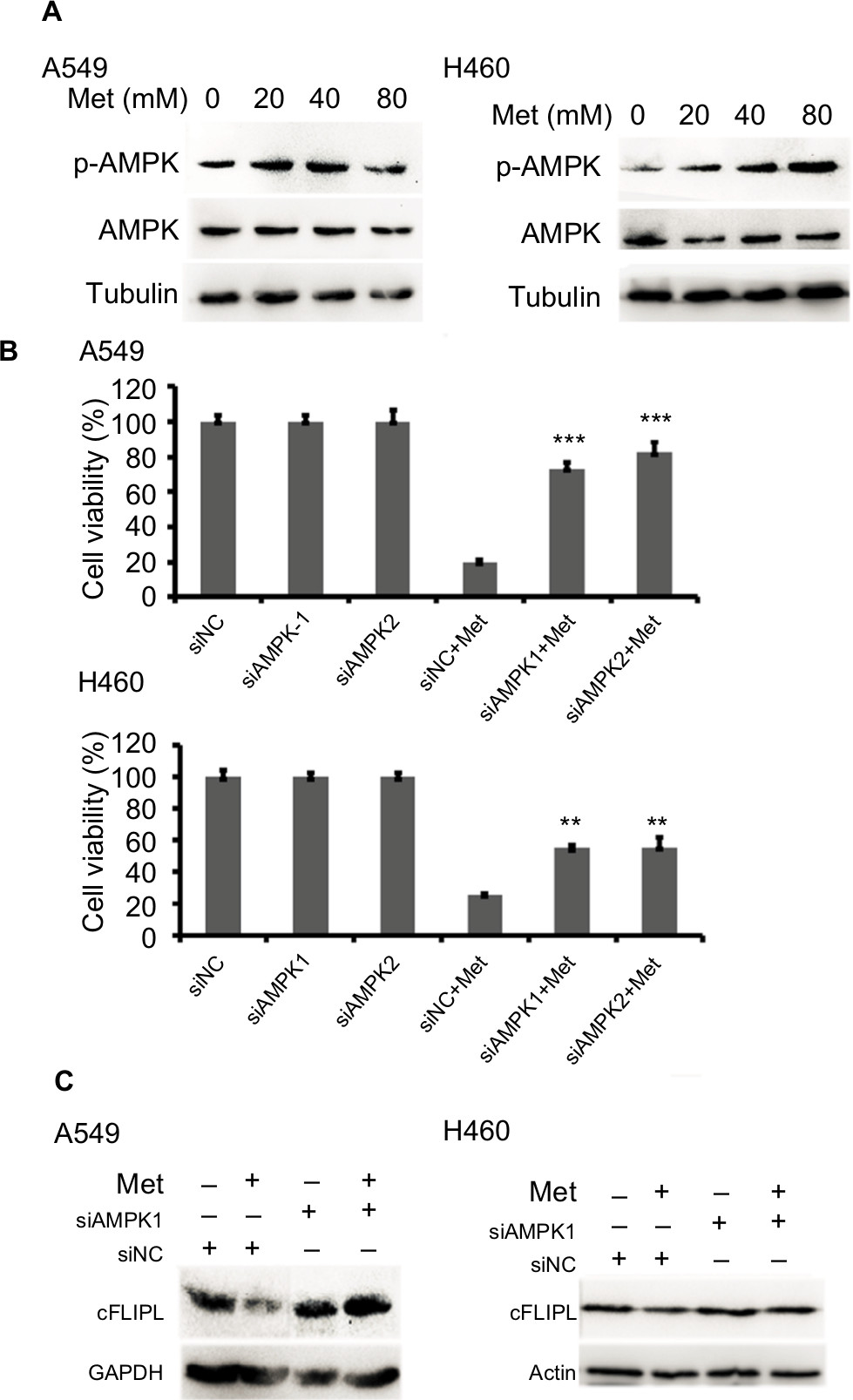 | Figure 3 Metformin inhibits c-FLIPL expression through AMPK. Notes: (A) A549 and H460 cells were treated with metformin (20, 40, 80 mM) for 2 hours. Then, indicated proteins were detected by Western blot, and tubulin was detected as an input control. (B) The cells were transfected with siRNA (10 nM) for 24 hours, and then treated with metformin (80 mM) for 24 hours. Then, cell viability was detected by MTT assay. (C) A549 and H460 cells were transfected with siRNA (10 nM) for 24 hours, and then treated with metformin (80 mM) for 16 hours. Then, indicated proteins were detected by Western blot, and tubulin was detected as an input control. N=3. **P<0.01, ***P<0.001. Abbreviations: AMPK, adenosine 5′-monophosphate-activated protein kinase; c-FLIPL, cellular FLICE-inhibitory protein large; FLICE, FADD-like IL-1β-converting enzyme. |
Metformin downregulated the expression of c-FLIPL through the inhibition of PKA
We further explored the mechanism by which metformin promoted c-FLIPL degradation in lung cancer cells. It has been reported that metformin increases the level of AMPK, then leads to inhibition of cAMP production and protein kinase A (PKA) activity.20,32,33 We then examined the PKA activity by investigating the phosphorylation level of CREB, which is a downstream substrate of PKA. Metformin indeed suppressed the activity of PKA in lung cancer cells, and it greatly decreased the level of p-CREB (Figure 4A). To further confirm the role of PKA in metformin-mediated c-FLIPL suppression, PKA activators, 8-Br-cAMP and forskolin, remarkably increased the activity of PKA, meanwhile significantly increased the protein levels of c-FLIPL (Figure 4B,C). Meanwhile, both 8-Br-cAMP and forskolin greatly promoted the proliferation rate of lung cancer cells under microscope (data not shown). These results suggest that metformin downregulates c-FLIPL expression by inhibition of PKA activity.
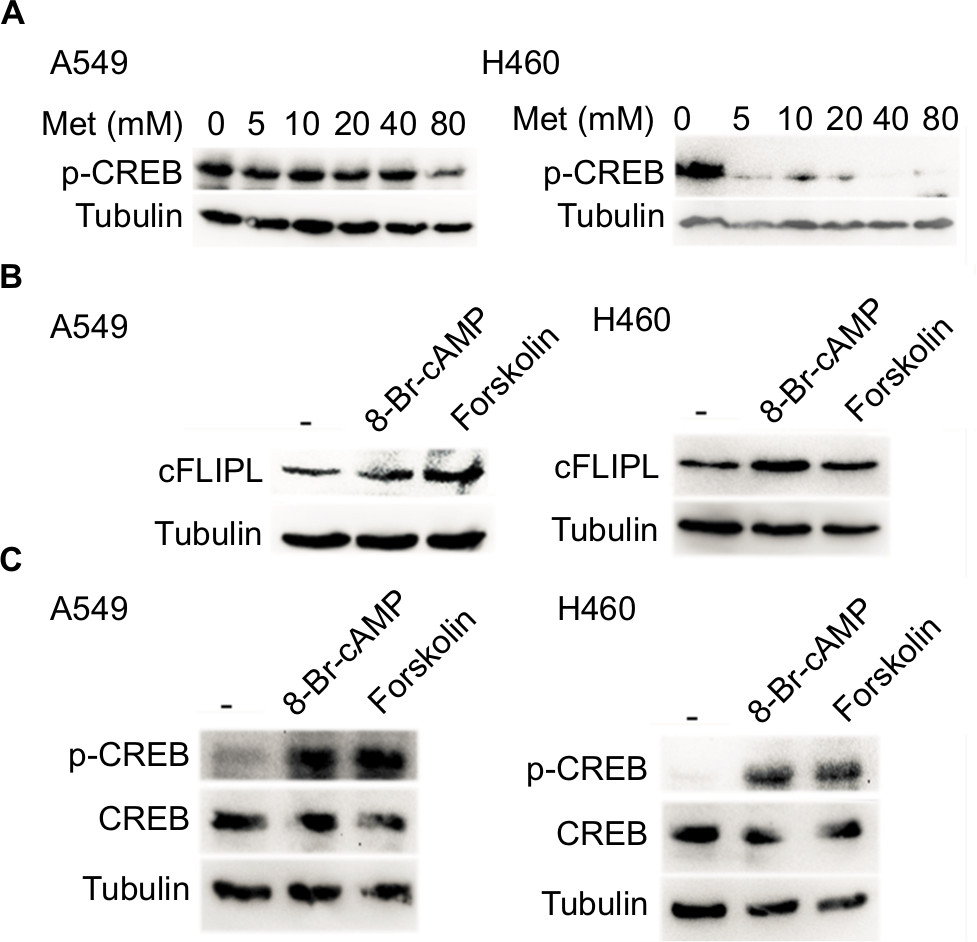 | Figure 4 Metformin suppressed the expression of c-FLIPL by inhibition of PKA activity. Notes: (A) A549 and H460 cells were treated with metformin (5, 10, 20, 40, 80 mM) for 8 hours. Then, indicated proteins were detected by Western blot, and GAPDH was detected as an input control. (B, C) Lung cancer cells were treated with 8-Br-cAMP (1 mM) and forskolin (10 µM) for 8 hours. Then, indicated proteins were detected by Western blot, and tubulin was detected as an input control. Abbreviations: c-FLIPL, cellular FLICE-inhibitory protein large; FLICE, FADD-like IL-1β-converting enzyme; PKA, protein kinase A. |
Metformin downregulated c-FLIPL expression depending on GSK-3β activation
It has been reported that metformin induces AMPK activation, leading to PKA suppression; PKA phosphorylated GSK-3β at S9, then activates GSK-3β.34,35 Then, we continued to investigate whether metformin decreased the expression of c-FLIPL through GSK-3β in lung cancer cells. Metformin indeed decreased the levels of inhibitory p-GSK-3β (S9), indicating metformin induced activation of GSK-3β signaling pathway (Figure 5A). To further clarify whether metformin downregulated the expression of c-FLIPL through GSK-3β, siRNA was used to silence the expression of GSK-3β. As shown in Figure 5B, siRNA partially reversed metformin-induced anticancer activities in NSCLC cells (Figure 5C). Meanwhile, siRNA restored the expression of c-FLIPL in both A549 and H460 cell lines (Figure 5D). Collectively, these results strongly indicate that metformin promotes c-FLIPL degradation through AMPK/PKA/GSK-3β pathway in NSCLC.
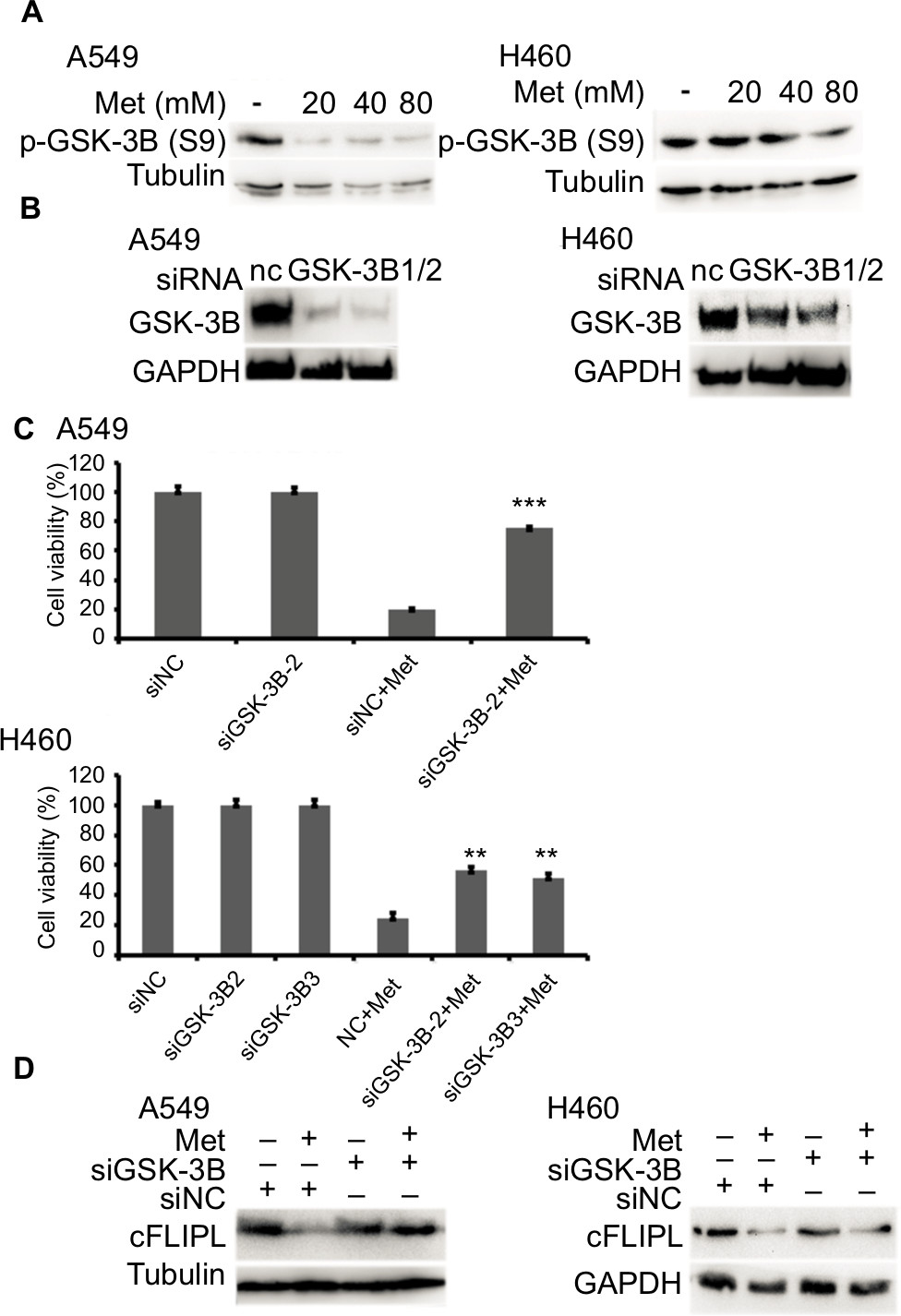 | Figure 5 Metformin promotes c-FLIPL degradation via GSK-3β. Notes: Metformin inhibits c-FLIPL expression through AMPK. (A) A549 and H460 cells were treated with metformin (20, 40, 80 mM) for 8 hours. Then, indicated proteins were detected by Western blot, and tubulin was detected as an input control. (B, C) The cells were transfected with siRNA (10 nM) for 24 hours, and then treated with metformin (80 mM) for 24 hours. Then, cell viability was detected by MTT assay. (D) A549 and H460 cells were transfected with siRNA (10 nM) for 24 hours, and then treated with metformin (80 mM) for 16 hours. Then, indicated proteins were detected by Western blot, and tubulin was detected as an input control. N=3. **P<0.01, ***P<0.001. Abbreviations: AMPK, adenosine 5′-monophosphate-activated protein kinase; c-FLIPL, cellular FLICE-inhibitory protein large; FLICE, FADD-like IL-1β-converting enzyme; GSK-3β, glycogen synthase kinase 3 beta. |
Discussion
In the present study, we explored the role of c-FLIPL in metformin-induced anticancer activities in NSCLC in vitro. The results demonstrated that metformin induced apoptotic cytotoxicity in several human lung cancer cell lines through downregulation of c-FLIPL. These results established a novel mechanism for metformin-induced antitumor activity which involves a series of processes: metformin promoted c-FLIPL degradation to induce apoptotic cytotoxicity mainly through AMPK/PKA/GSK-3β axis.
As a novel anticancer agent, metformin has attracted great attention since it greatly decreases the incidence of different types of cancers in clinical trials. In vitro and in vivo studies have shown that metformin not only inhibits proliferation, but also suppresses metastasis in various cancers. Moreover, combination therapies of metformin and other anticancer drugs always result in synergic cytotoxicity, including radiation,9 gefitinib,10,11 erlotinib,12 sorafenib,13 carboplatin,14 cisplatin,15 and TRAIL.16 However, until now, the underlying mechanisms of metformin-induced anticancer activities remain unclear.
As one crucial negative regulator of cell death, c-FLIP is able to block caspase 8 activation induced by chemotherapy-induced apoptosis. c-FLIP is often highly expressed in different type of cancers, which confers poor prognosis of patients.26,36 Accumulating evidence has suggested that downregulation of c-FLIP restores sensitivity to anticancer agents, including TRAIL, Fas, and cisplatin.31,36 Previous studies have reported that metformin decreases the expression of c-FLIPL in bladder cancer,29 renal cell carcinoma,30and lung adenocarcinoma cells.16 Consistently, in the present study, we demonstrated that metformin significantly suppressed the expression of c-FLIPL by decreasing the stability of c-FLIPL protein with little influence of mRNA level of c-FLIPL gene (Figure 2).
Shumei Meng and Jia Cao have reported that low concentrations of metformin suppress glucose production by activating AMPK.37,38 AMPK is downregulated in hepatocellular carcinoma, low p-AMPK expression is associated with poor prognosis, and metformin inhibits hepatocellular carcinoma by inducing AMPK activation.3 It has been reported that AMPK activation is the main pathway of metformin in cancer cells. Storozhuk et al have reported that metformin inhibits NSCLC cell growth depending on AMPK; suppression of AMPK with siRNA reversed metformin-induced anticancer effect.9 The role of AMPK and its downstream pathway in metformin-mediated anticancer activities has also been reported in various cancers, including GSK-3β2,39 and mTOR.4,5 Tao Zhang et al firstly reported that metformin downregulated c-FLIPL expression in bladder cancer cells mainly through mTOR/S6K1 pathway.29 Nazim et al demonstrated that metformin promoted c-FLIPL autophagic degradation in lung adenocarcinoma cells.16 Recently, Jang et al reported that metformin promoted c-FLIPL degradation in renal cell carcinoma without affecting the mRNA level of c-FLIPL.30 However, the role of c-FLIPL in metformin-induced anticancer activities remains largely unknown. In the present study, unlike previous report that metformin decreases the expression of c-FLIPL independent of AMPK in bladder cancer,29 we demonstrated that metformin mainly downregulated c-FLIPL expression through AMPK and its downstream GSK-3β pathway since siRNA-targeted AMPK and GSK-3β not only reversed metformin-induced cell death, but also restored c-FLIPL expression (Figures 3 and 5). Pei-guo Shi et al have reported that metformin inhibits the activity of PKA, which is a downstream substrate of AMPK, then leading to the suppression of TNBC stem cell.2 Consistently, we also found that metformin suppressed the activity of PKA in NSCLC cells, PKA activators, both 8-Br-cAMP and forskolin, significantly increased the expression of c-FLIPL without affecting the level of c-FLIPL mRNA (Figure 4). Taken together, the part of c-FLIPL in metformin-mediated anticancer activities may be specific in different types of cancers.
Conclusion
Our results established a novel mechanism for killing cancer cells by metformin that involves AMPK/PKA/GSK-3β signaling pathways-mediated c-FLIPL degradation and extended our knowledge of metformin-induced anticancer activities. Further investigation in vivo should be designed to confirm these observations.
Disclosure
This study was supported in part by National Natural Science Foundation of China (No. 81860412) to S. Shi, Yunnan Natural Science Foundation of China (2017FE468 [-151] to S. Shi, 2017FE468[-200] to Z. Luo, 2018FE001 [-031] to C. Wang), Yunnan Provincial Fund for High Level Reserve Talents in Health Science (H2017063) to Z. Luo and also partly supported by 100 young and middle-aged academic and technical backbone of Kunming Medical University (60117190451 to S. Shi, 60117190423 to Z. Luo). The authors report no other conflicts of interest in this work.
References
Liu Y, He C, Huang X, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Shi P, Liu W, Tala, et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 2017;3:17010. | ||
Zheng L, Yang W, Wu F, et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res. 2013;19(19):5372–5380. | ||
Han G, Gong H, Wang Y, Guo S, Liu K. AMPK/mTOR-mediated inhibition of survivin partly contributes to metformin-induced apoptosis in human gastric cancer cell. Cancer Biol Ther. 2015;16(1):77–87. | ||
Shi WY, Xiao D, Wang L, et al. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012;3:e275. | ||
Tomic T, Botton T, Cerezo M, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011;2:e199. | ||
Hsieh SC, Tsai JP, Yang SF, Tang MJ, Hsieh YH. Metformin inhibits the invasion of human hepatocellular carcinoma cells and enhances the chemosensitivity to sorafenib through a downregulation of the ERK/JNK-mediated NF-κB-dependent pathway that reduces uPA and MMP-9 expression. Amino Acids. 2014;46(12):2809–2822. | ||
Pennati M, Folini M, Zaffaroni N. Targeting survivin in cancer therapy: fulfilled promises and open questions. Carcinogenesis. 2007;28(6):1133–1139. | ||
Storozhuk Y, Hopmans SN, Sanli T, et al. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. 2013;108(10):2021–2032. | ||
Li KL, Li L, Zhang P, et al. A multicenter double-blind phase II study of metformin with gefitinib as first-line therapy of locally advanced non-small-cell lung cancer. Clin Lung Cancer. 2017;18(3):340–343. | ||
Morgillo F, Sasso FC, Della Corte CM, et al. Synergistic effects of metformin treatment in combination with gefitinib, a selective EGFR tyrosine kinase inhibitor, in LKB1 wild-type NSCLC cell lines. Clin Cancer Res. 2013;19(13):3508–3519. | ||
Wang X, Chen K, Yu Y, et al. Metformin sensitizes lung cancer cells to treatment by the tyrosine kinase inhibitor erlotinib. Oncotarget. 2017;8(65):109068–109078. | ||
Groenendijk FH, Mellema WW, van der Burg E, et al. Sorafenib synergizes with metformin in NSCLC through AMPK pathway activation. Int J Cancer. 2015;136(6):1434–1444. | ||
Liu Y, He C, Huang X. Metformin partially reverses the carboplatin-resistance in NSCLC by inhibiting glucose metabolism. Oncotarget. 2017;8(43):75206–75216. | ||
Lin CC, Yeh HH, Huang WL, et al. Metformin enhances cisplatin cytotoxicity by suppressing signal transducer and activator of transcription-3 activity independently of the liver kinase B1-AMP-activated protein kinase pathway. Am J Respir Cell Mol Biol. 2013;49(2):241–250. | ||
Nazim UM, Moon JH, Lee JH, et al. Activation of autophagy flux by metformin downregulates cellular FLICE-like inhibitory protein and enhances TRAIL-induced apoptosis. Oncotarget. 2016;7(17):23468–23481. | ||
Lin J, Gill A, Zahm SH, et al. Metformin use and survival after non-small cell lung cancer: a cohort study in the US Military health system. Int J Cancer. 2017;141(2):254–263. | ||
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305. | ||
Tsai MJ, Yang CJ, Kung YT, et al. Metformin decreases lung cancer risk in diabetic patients in a dose-dependent manner. Lung Cancer. 2014;86(2):137–143. | ||
Pernicova I, Korbonits M. Metformin – mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143–156. | ||
Pulito C, Mori F, Sacconi A, et al. Metformin-induced ablation of microRNA 21-5p releases Sestrin-1 and CAB39L antitumoral activities. Cell Discov. 2017;3:17022. | ||
Blandino G, Valerio M, Cioce M, et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun. 2012;3:865. | ||
Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci USA. 2015;112(6):1809–1814. | ||
Micheau O. Cellular FLICE-inhibitory protein: an attractive therapeutic target? Expert Opin Ther Targets. 2003;7(4):559–573. | ||
Irmler M, Thome M, Hahne M, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388(6638):190–195. | ||
Shirley S, Micheau O. Targeting c-FLIP in cancer. Cancer Lett. 2013;332(2):141–150. | ||
Shi S, Wang Q, Xu J, et al. Synergistic anticancer effect of cisplatin and Chal-24 combination through IAP and c-FLIPL degradation, Ripoptosome formation and autophagy-mediated apoptosis. Oncotarget. 2015;6(3):1640–1651. | ||
Xu J, Xu X, Shi S, et al. Autophagy-mediated degradation of IAPs and c-FLIP(L) potentiates apoptosis induced by combination of TRAIL and Chal-24. J Cell Biochem. 2016;117(5):1136–1144. | ||
Zhang T, Wang X, He D, Jin X, Guo P. Metformin sensitizes human bladder cancer cells to TRAIL-induced apoptosis through mTOR/S6K1-mediated downregulation of c-FLIP. Anticancer Drugs. 2014;25(8):887–897. | ||
Jang JH, Song IH, Sung EG, Lee TJ, Kim JY. Metformin-induced apoptosis facilitates degradation of the cellular caspase 8 (FLICE)-like inhibitory protein through a caspase-dependent pathway in human renal cell carcinoma A498 cells. Oncol Lett. 2018;16(2):2030–2038. | ||
He W, Wang Q, Xu J, et al. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy. 2012;8(12):1811–1821. | ||
Beristain AG, Molyneux SD, Joshi PA, et al. PKA signaling drives mammary tumorigenesis through Src. Oncogene. 2015;34(9):1160–1173. | ||
Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260. | ||
Fang X, Yu SX, Lu Y, Bast RC, Woodgett JR, Mills GB. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci USA. 2000;97(22):11960–11965. | ||
Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol Cell Biol. 2000;20(24):9356–9363. | ||
Subramaniam K, Hirpara JL, Tucker-Kellogg L, Tucker-Kellogg G, Pervaiz S. FLIP: a flop for execution signals. Cancer Lett. 2013;332(2):151–155. | ||
Cao J, Meng S, Chang E, et al. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J Biol Chem. 2014;289(30):20435–20446. | ||
Meng S, Cao J, He Q, et al. Metformin activates AMP-activated protein kinase by promoting formation of the αβγ heterotrimeric complex. J Biol Chem. 2015;290(6):3793–3802. | ||
Gwak H, Kim Y, An H, Dhanasekaran DN, Song YS. Metformin induces degradation of cyclin D1 via AMPK/GSK3β axis in ovarian cancer. Mol Carcinog. 2017;56(2):349–358. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.