")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 11
Metabolism and Excretion of Intravenous, Radio-Labeled Amisulpride in Healthy, Adult Volunteers
Authors Fox GM , Roffel AF , Hartstra J, Bussian LA, van Marle SP
Received 11 October 2019
Accepted for publication 16 November 2019
Published 2 December 2019 Volume 2019:11 Pages 161—169
DOI https://doi.org/10.2147/CPAA.S234256
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Gabriel M Fox,1 Ad F Roffel,2 Jan Hartstra,2 Linda A Bussian,3 Sjoerd P van Marle2
1Department of Clinical Development, Acacia Pharma Ltd, Cambridge, UK; 2PRA Health Sciences, Groningen, the Netherlands; 3Comedica Ltd, Cambridge, UK
Correspondence: Gabriel M Fox
Department of Clinical Development, Acacia Pharma Ltd, The Officers’ Mess, Duxford, Cambridge CB22 4QH, UK
Tel +44 1223 919760
Fax +44 1223 919769
Email [email protected]
Purpose: Intravenous amisulpride, a dopamine D2/D3 antagonist, has recently been shown in trials to be an effective antiemetic at low doses. This study was conducted to investigate the metabolism and elimination of a single dose of intravenous 14C-labeled amisulpride in healthy, adult volunteers.
Patients and methods: Six healthy male volunteers aged 18–65 years were given a single 10 mg dose of 14C-labeled amisulpride containing not more than 1.8 MBq of radioactivity, infused over 4 mins. Concentrations of amisulpride and total radioactivity were measured in plasma, whole blood, urine and feces at various time points up to 168 hrs after dosing. Metabolites detected in plasma, urine and feces were characterized using liquid chromatography tandem mass spectrometry (LC-MS/MS) with in-line radiometric detection.
Results: The mean recovery of radioactivity in excreta was 96.4% (range 92.0–98.5%), of which 73.6% (range 70.6–79.2%) was recovered from urine and 22.8% (range 18.9–25.7%) from feces. Four metabolites of amisulpride were detected in urine, representing 15.0% of the excreted dose; three of these were also present in feces, representing 6.1% of the excreted dose. No metabolites were detected in plasma. Excretion was initially rapid, with about two-thirds of the drug-related material eliminated within 12 hrs, primarily in the urine. A second, slower phase of excretion was predominantly fecal and was essentially complete by 96 hrs after dosing. The terminal plasma elimination half-life of parent amisulpride was 3.7 hrs and that of total 14C-labeled drug material was 4.2 hrs.
Conclusion: Intravenous amisulpride undergoes limited metabolism and is excreted primarily via the renal route.
Clinical trial registry number: ClinicalTrials.gov NCT02881840.
Keywords: amisulpride, antiemetics, metabolism, elimination, radio-labeled
Introduction
Amisulpride is a substituted benzamide that potently and selectively blocks dopamine D2 and D3 receptors and has been in clinical use since the 1980s as an oral antipsychotic agent. More recently, an intravenous (IV) formulation of amisulpride has been shown in multiple randomized, controlled trials to be an effective antiemetic for the prevention and treatment of nausea and vomiting in the postoperative and emetogenic chemotherapy settings.1–7
Amisulpride is of particular interest therapeutically because, during more than three decades of use in psychiatric practice, it has been reported to have a favorable safety profile, even at high doses and when taken chronically over months and years, with a very low incidence of extrapyramidal, cardiac, central nervous system and gastrointestinal side effects.8,9 It has the major additional benefit of showing minimal QT prolongation at antiemetic doses,10 obviating a significant problem of most other dopaminergic antiemetics.
While the pharmacokinetics of oral amisulpride are well characterized, there is little in the published literature relating to the intravenous route. We, therefore, conducted this single-center, single-cohort, open-label study to assess the mass balance recovery after a single IV dose of carbon-14 (14C)-labeled amisulpride; to identify the chemical structure of major metabolites and to determine the routes and rates of excretion.
Materials and Methods
Study Design and Participants
This study was conducted at a specialist clinical pharmacology unit operated by PRA Health Sciences in the Netherlands in August 2016 (Principal Investigator: Dr Sjoerd van Marle). The study was registered on ClinicalTrials.gov prior to initiation, with the reference identifier NCT02881840. A nationally accredited independent ethics committee, Stichting Beoordeling Ethiek Biomedisch Onderzoek, accredited by the Netherlands Association of Independent Ethics Committees (NVMETC), approved the study, and written informed consent was obtained from all subjects prior to enrolment. The study was conducted in accordance with the principles of the Declaration of Helsinki.
Subjects were eligible to be included in the study if they were male, in good health and aged 18–65 years; had a body mass index in the range 18–30 kg/m2 or, if outside the range, considered by the investigator to be not clinically significant; had regular bowel movements (between one and three stools on average per day); agreed to use an adequate method of contraception; were willing and able to communicate and participate in the whole study; and freely gave written informed consent.
Standard exclusion criteria for a Phase 1 study were applied, including a history of drug or alcohol abuse; regular alcohol consumption above 21 units per week; cigarette smoking; a positive drugs of abuse test; clinically significant abnormal biochemistry, hematology or urinalysis; evidence of renal impairment or positive hepatitis B surface antigen, hepatitis C virus antibody or human immunodeficiency virus results at screening; history of cardiovascular, renal, hepatic, chronic respiratory or gastrointestinal disease; and presence or history of clinically significant allergy requiring treatment, other than not currently active hay fever.
Subjects were screened for eligibility to participate in the study within 28 days before dosing. Eligible subjects were admitted to the study unit in the afternoon prior to dosing (Day −1). Following a standardized breakfast, subjects received study medication on the morning of Day 1. Subjects remained resident in the clinic until at least 96 hrs after dosing. At or after that time, it was planned that each subject would be released when <1% of the dose administered had been collected in urine and feces within two separate, consecutive 24-hr periods (assessed by quick counts). In that case, the collection of all samples (blood, urine and feces) was stopped. If this criterion had not been met by day 5 (96-hr postdose), the subject could remain in the clinic for up to a further 72 hrs, until the criterion had been met. If the criterion had not been met after 168 hrs of collections, home collection of urine and feces could be requested at the discretion of the investigator.
Study medication was prepared by dissolving appropriate quantities of unlabeled amisulpride (Icrom SpA, Monza, Italy) and 14C-labeled amisulpride, with a specific activity of 3.6 MBq/mg (Moravek Inc, Brea, CA, USA), in water and standard excipients, in order to provide a sterile 100 mL solution containing 250 mg amisulpride and 44.6 MBq of radioactivity, which was kept refrigerated at 2–8°C. Each subject was given a 4 mL infusion of the study drug, amounting to a 10 mg dose with an average 1.784 MBq of radioactivity.
Vital signs and ECGs were recorded, and blood, urine and fecal samples were taken prior to dosing and then at multiple postdosing time points. Blood samples were drawn via an in-dwelling venous cannula or venepuncture at the following times after the start of the study drug infusion: 4 mins (end of infusion), 8 mins, 30 mins and 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72 and 96 hrs and 24-hourly thereafter until completion criteria were met. Urine was collected for the intervals 0–12 hrs, 12–24 hrs and then 24-hourly; and feces were collected 24-hourly. Any other accidental sources of elimination, eg, emesis, were collected as voided.
Analysis of amisulpride levels in whole blood, plasma, urine and feces was done by liquid chromatography tandem mass spectrometry (LC-MS/MS) (LCG Ltd, Fordham, UK), and radioactivity was quantified using liquid scintillation counting (PRA Health Sciences Ltd, Assen, The Netherlands). Characterization of the metabolites of 14C-amisulpride in plasma, urine and feces was done by LC-MS/MS with in-line radiometric detection (Sekisui Xenotech Inc, Kansas City, USA).
Primary endpoints for the study were: mass balance recovery of total radioactivity in urine, feces and all excreta; chemical structure of major metabolites of 14C-amisulpride, defined as those representing at least 10% of the area under the concentration versus time curve of the parent compound; and routes and rates of excretion of 14C-amisulpride. Primary variables evaluated were amount excreted (Ae) and Ae as a percentage of the administered dose (%Ae), cumulative recovery (Cum Ae) and cumulative recovery expressed as a percentage of the dose (Cum %Ae), in total and in urine and feces separately.
Key plasma pharmacokinetic parameters evaluated for amisulpride and for total radioactivity in plasma were: maximum observed plasma concentration (Cmax); time from dosing to Cmax (Tmax); area under the concentration–time curve (AUC) from dosing (time zero) to the last observed concentration, calculated using the linear-up log-down trapezoidal method, and extrapolated to infinity by adding the last observed concentration divided by the terminal elimination rate constant, representing total drug exposure (AUC(0–∞)); terminal phase half-life (t½); clearance (CL) and volume of distribution at steady state (Vss). In addition, the ratio of whole blood-to-plasma concentration for total radioactivity was calculated at 0.5, 4 and 12 hrs after dosing.
Statistics
No formal sample size calculation was made. Based on experience from previous studies of a similar design, a total of six subjects enrolled to achieve a minimum of four evaluable subjects (subjects who had provided biological samples for at least 48 hrs after study drug administration) was considered sufficient. Descriptive statistics (eg, mean, standard deviation, range) were considered adequate for a study of this type.
Results
Six adult, male subjects were enrolled in the study, having signed the informed consent form. All six subjects met all inclusion and exclusion criteria, received treatment and completed the study. Baseline characteristics are summarized in Table 1.
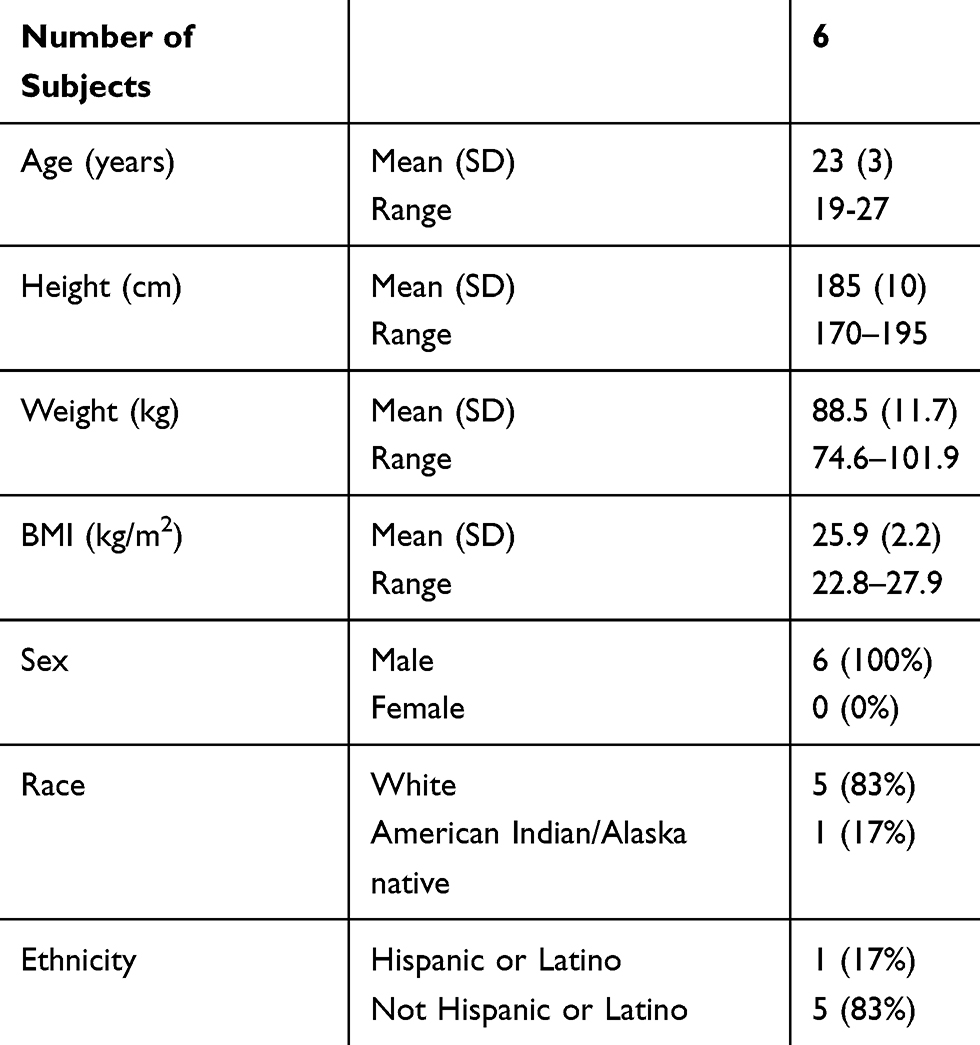 |
Table 1 Baseline Characteristics of Study Subjects |
Mass Balance
The mean recovery of radioactivity in excreta was 96.4% (range 92.0–98.5%), of which 73.6% (range 70.6–79.2%) was recovered from urine and 22.8% (range 18.9–25.7%) from feces (see Figure 1).
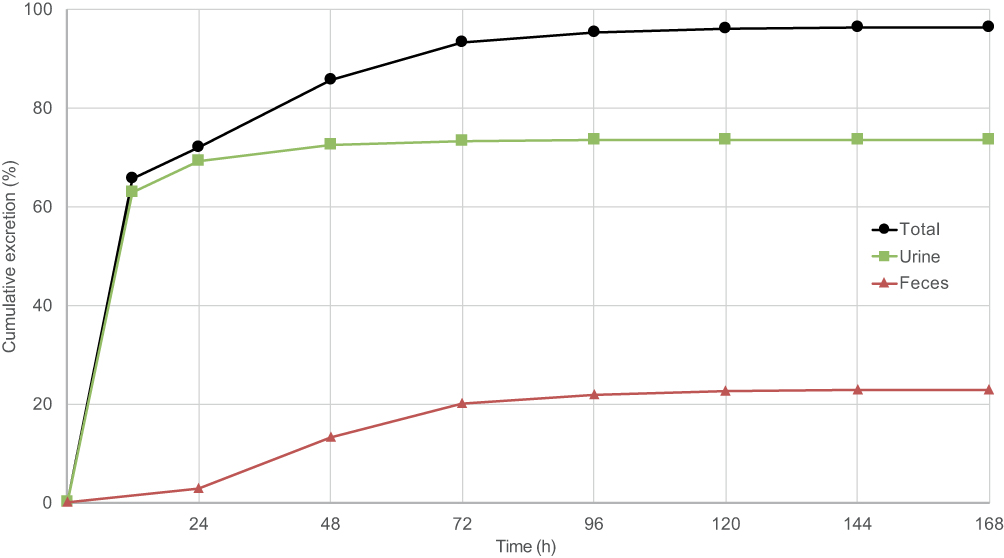 |
Figure 1 Mean cumulative total radioactivity excretion over time in urine, feces and total. |
Metabolite Profiling and Identification
The only radioactive peak detected in plasma was 14C-amisulpride. This was also detected in urine and feces. In addition, four related components (C1, C2, C3 and C4) were associated with detectable radioactivity in pooled human urine and three (C1, C2 and C4) in pooled human feces.
These components were formed by oxygenation (C1), N dealkylation (C2), oxygenation plus di-dehydrogenation or methylation plus dehydrogenation (C3) and oxygenation plus dehydrogenation (C4) (see Table 2 and Figure 2).
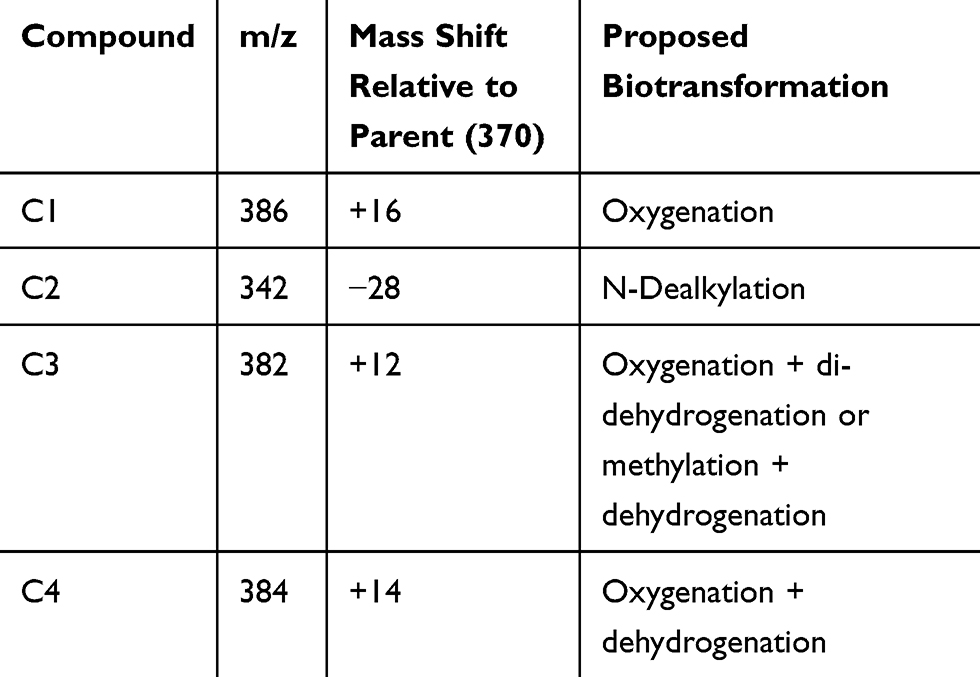 |
Table 2 Characterization of Metabolites of Amisulpride |
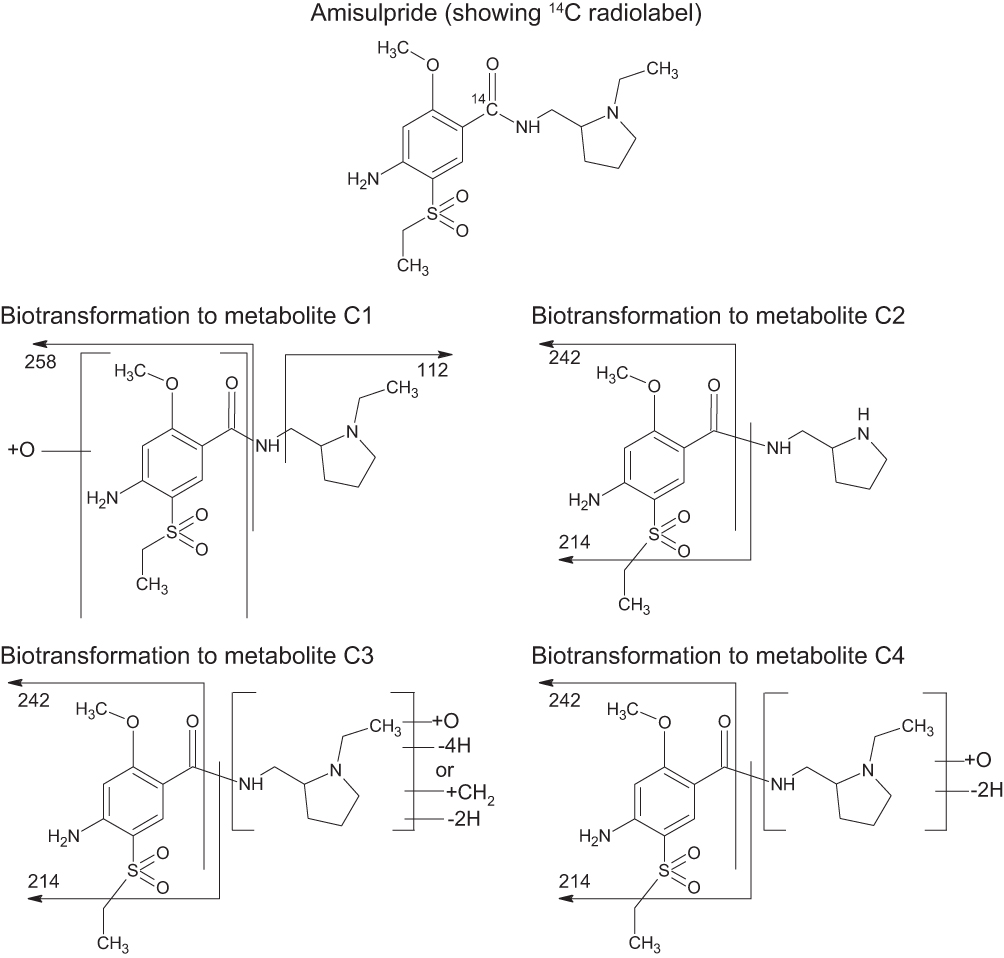 |
Figure 2 Proposed biotransformations of amisulpride. |
Elimination
Excretion of amisulpride was initially rapid, with approximately two-thirds of the radioactive dose eliminated within 12 hrs, primarily in urine (see Table 3). Around 85% of urinary excretion of total radioactivity occurred in the first 12 hrs and 94% in the first 24 hrs after dosing. After the first 24 hrs, excretion was slower and predominantly in the feces. Only 12% of the total radioactivity detected in feces was found in the first 24 hrs after dosing, while 46% was found in the period 24–48 hrs and a further 30% in the period 48–72 hrs. Excretion of the single dose of radioactivity was essentially complete by 96 hrs after dosing.
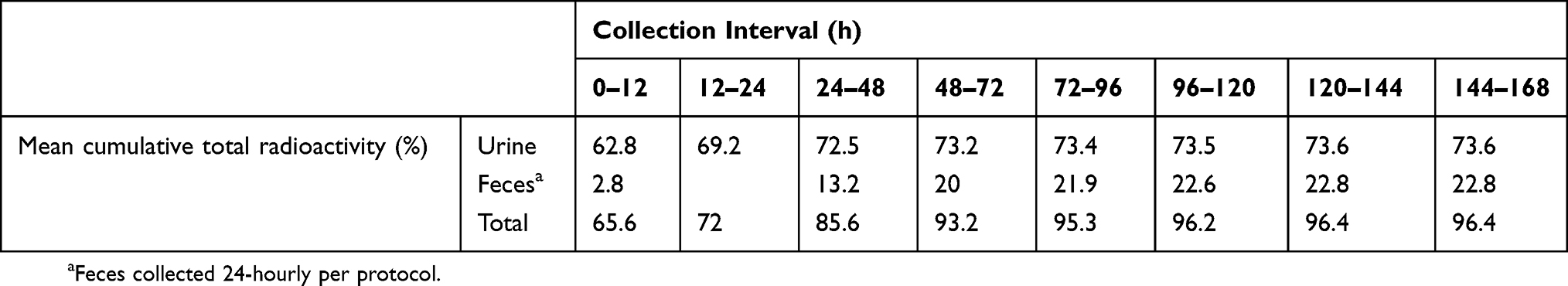 |
Table 3 Cumulative Total Radioactivity Excretion Over Time |
Amisulpride was predominantly excreted unchanged in urine and feces. Of the total amount of radioactivity excreted, 78.1% was associated with 14C-amisulpride (57.5% in urine and 20.6% in feces). The metabolites together represented 21.1% of the excreted radioactivity, 15.0% in urine and 6.1% in feces (see Table 4).
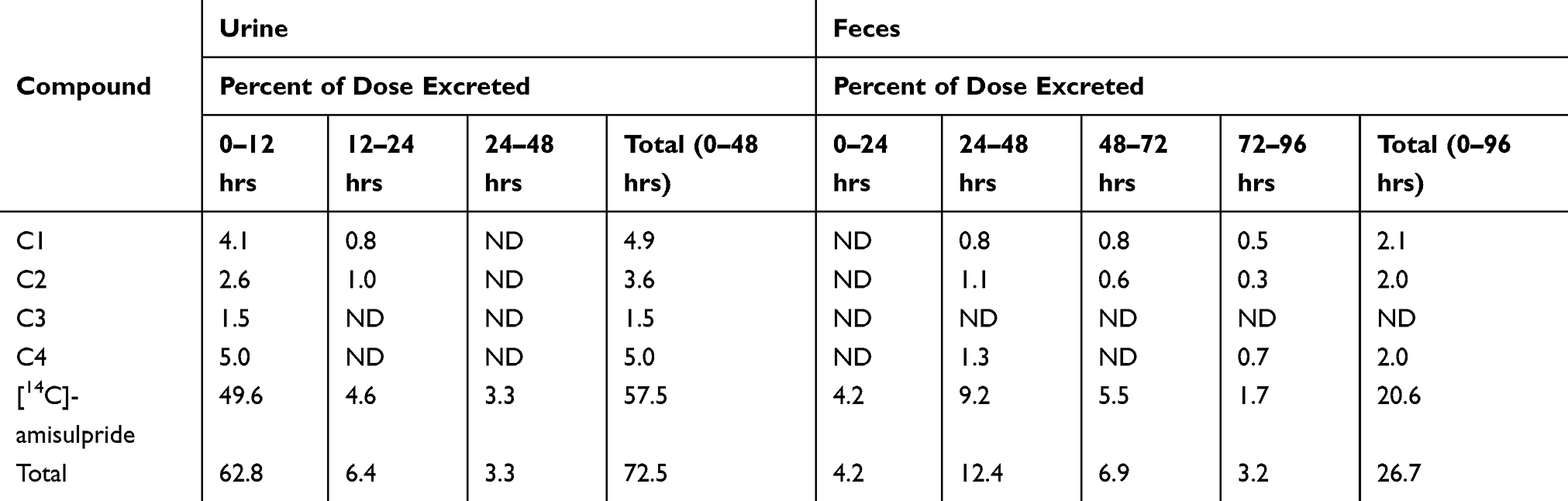 |
Table 4 Amisulpride and Metabolites in Urine and Feces |
Plasma Pharmacokinetics
A single 10 mg dose of amisulpride delivered a mean peak plasma concentration of 357 ng/mL (SD 149) and a mean total exposure of 228 ng.h/mL (SD 18.9), see Table 5. Most of the total radioactivity in plasma at each time point was associated with parent amisulpride (Figure 3). The mean ratio of plasma to whole blood radioactivity was very close to 1 at all three time points assessed: 0.5 hrs, 4 hrs and 12 hrs.
 |
Table 5 Principal Amisulpride Plasma PK Parameters |
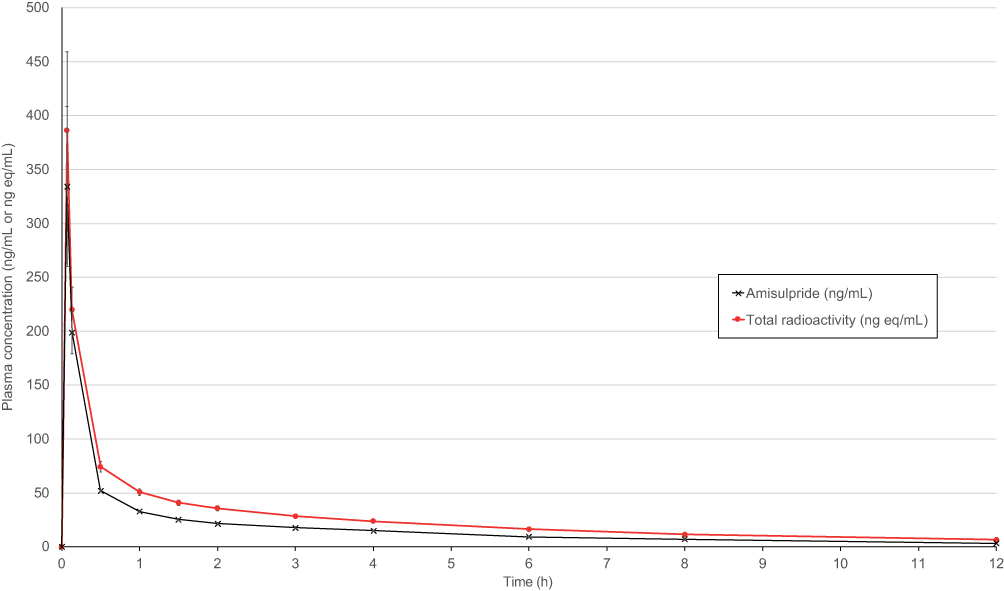 |
Figure 3 Plasma concentration of amisulpride and total radioactivity over time. Note: Concentrations at time points after 12 hrs all below the limit of quantification (<2 ng/mL or ng eq/mL). |
Safety
Seventeen treatment-emergent adverse events were reported in the study, 15 mild and two moderate. All six subjects reported mild (N=4) or moderate (N=2) pain on infusion which resolved rapidly and did not require interruption of the infusion. The other adverse events considered possibly or probably related to study drug were one instance each of dizziness, headache and somnolence.
Plasma prolactin levels increased from a mean (SD) of 9.2 (3.4) ng/mL prior to dosing to 104.6 (37.1) ng/mL at 30 mins after dosing, falling back to 19.8 (5.7) ng/mL, which is around the upper limit of normal for adult males, by 12 hrs after dosing. No clinical sequelae were noted. There were no material changes in any other hematology or clinical chemistry parameters tested.
No clinically significant ECG abnormalities were recorded in the study. The QT interval showed little change after dosing: the mean change in QTcF from baseline was –5 msec at the end of infusion (4 mins), +7 msec at 30 mins and 0 msec at 1 hr after the start of infusion. The highest absolute QTcF value recorded was 410 msec.
Discussion
In this study, IV amisulpride was excreted largely unchanged, primarily by the renal route. This corroborates data on the metabolism and elimination of IV amisulpride previously published in abstract form.11
The minimal degree of metabolism, indicated by around 80% of the eliminated drug being in the parent form and by metabolites not being detectable in plasma at any time, is of clinical relevance for a number of reasons.
First, the generation of metabolites can have important safety consequences. For example, haloperidol, another dopamine antagonist used as an antiemetic, undergoes substantial metabolism, with only 1% of the drug excreted unchanged. Inactive (50–60%), active (23%) and toxic (20–30%) metabolites are generated, the latter adding materially to its toxicity profile.12
Second, the limited metabolism of amisulpride implies that it is not a substrate for the cytochrome P450 isoenzyme system. Previously published data indicate that it is also not an inhibitor of those enzymes.13 Taken together, these properties suggest a greatly reduced risk of drug–drug interactions. Furthermore, the close concordance of plasma and whole blood radioactivity at all time points suggests that amisulpride passes freely in and out of red blood cells, which is consistent with a low degree of plasma protein binding, previously reported to be 17%.14 Thus, it is unlikely that amisulpride would displace or be displaced by any other plasma protein-bound drug. In a published review, amisulpride has been described as being “almost devoid of clinically relevant metabolic interactions.”15 Given the number of medications used perioperatively, these characteristics may well represent a benefit in clinical practice.
Third, given the low degree of metabolism, the pharmacokinetics of amisulpride are likely to show little inter-patient variability, as the potential impact of cytochrome P450 isoenzyme polymorphisms is avoided. This assertion is corroborated by the consistency of key pharmacokinetic parameters between this study and others in the literature.10,11,14 This contrasts with one of the most commonly used antiemetic agents, ondansetron, which is substantially metabolized by isoenzyme CYP2D6, resulting in reduced drug levels, and possibly impaired efficacy, in patients overexpressing CYP2D6.16
Compared with oral amisulpride, for which extensive data are available in the literature,14,17–20 IV amisulpride reaches a much higher and more rapid Cmax – for example, a 200 mg oral capsule delivered a mean Cmax of 510 ng/mL at a mean Tmax of 3 hrs – but total exposure is only about double for the same dose, representing an absolute oral bioavailability of 50%. Because of the much slower absorption and distribution of oral amisulpride, leading to lower drug concentrations circulating for a longer time, its excretion is shifted more to the fecal route (about 65%) than the urinary (35%).
The primarily renal route of elimination of IV amisulpride means that consideration must be given to the possibility of increased toxicity in patients with significant renal impairment. However, given the low effective dose of IV amisulpride as an antiemetic, 5 or 10 mg, it is questionable whether even severe renal impairment would present a significant clinical risk, especially in the context of single-dose use. Peak plasma concentration occurs at the end of the infusion, whereupon drug is rapidly distributed from plasma into total body water, as indicated by the high volume of distribution. The plasma concentration of amisulpride decreases by half within about 5 mins, which is far too quick for renal elimination, however much impaired, to have any material effect. Reduced renal clearance would, though, increase the total exposure, as measured by the area under the concentration–time curve. However, the safety margin is very large: the 50% absolute bioavailability of amisulpride means that a 10 mg IV infusion delivers about one-twentieth the total exposure of a 400 mg oral dose, which has been widely and safely used in psychiatric practice for decades.21 Previously published data on oral amisulpride indicate that severe renal impairment (creatinine clearance 10–20 mL/min) results in an approximately tenfold increase in total exposure.22 Even allowing for the lower ratio of urinary to fecal excretion seen with oral amisulpride, a 10 mg IV dose of amisulpride in a patient with severe renal impairment may be expected to deliver a total exposure well below the 8800 ng.h/mL reported for a 400 mg tablet in normal individuals.23 The risk is further mitigated by the expected single-dose use of IV amisulpride as an antiemetic in the perioperative setting.
The safety profile of a single 10 mg IV dose of amisulpride appeared benign. The observed short-lived increase in plasma prolactin is an expected result of the D2-receptor antagonism of amisulpride, since prolactin secretion by the anterior pituitary is under dopaminergic control. Although a tenfold increase was seen at 1 hr, the level had fallen back to within the normal range by 12 hrs, making it unlikely for any clinically meaningful effects to occur. Only male subjects took part in this study, but previous data in male and female surgical patients confirm that prolactin levels return to within the normal range in both sexes within 24 hrs.2 No prolactin-associated adverse events have been reported to date in any clinical trial of IV amisulpride. With oral amisulpride, the incidence of events such as galactorrhea, nonpuerperal lactation, gynecomastia and menstrual disorder was not significantly higher with amisulpride than placebo in randomized, double-blind, placebo-controlled trials.24,25
Short-lived, mild or moderate pain on infusion of study drug was reported by all six subjects. In previous double-blind studies in conscious patients, this effect has been reported by fewer than 10% of amisulpride-treated participants and a similar proportion of those receiving the placebo, suggesting that the effect may not be related to the amisulpride itself.4,5 Possible contributory factors could include the acidity (pH 5.0), citrate content and temperature of the drug solution. In this study, the radioactive drug solution was kept refrigerated until shortly before dosing, as is typical for a Phase 1 unit handling a radio-labeled study drug, and was likely to be at a considerably lower temperature than was the case in the patient studies conducted in busy hospitals, where study drug was often not refrigerated (as it is not required), or had more time to reach ambient temperature before administration.
It is important to recognize the usual limitation of a Phase 1 study conducted in healthy volunteers in terms of generalizability to a patient population, which may have a range of comorbidities and may be receiving multiple medications. In the case of amisulpride, this limitation is mitigated somewhat by the low risk of drug–drug interactions and the wide safety margin evident from its existing higher dose oral use. Further reassurance is given by the comparability of the adverse event profile in these healthy subjects with that reported in both surgical and oncology patients.1–7
Conclusion
Amisulpride, given intravenously, undergoes little metabolism and is primarily eliminated by the renal route. The pharmacokinetics show low interpatient variability. Its single-dose use as an antiemetic at 5 or 10 mg is unlikely to be associated with clinically significant drug–drug interactions or toxicity issues.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Specifically
GMF served as the study director, led study design and protocol development, oversaw study operational activities, reviewed final data and drafted the manuscript.
AFR was involved in study design and protocol development and critically reviewed the manuscript. JH was involved in study design and protocol development, conducted biostatistical analyses and critically reviewed the manuscript. LAB was involved in protocol development and study conduct and critically reviewed the manuscript. SPvM served as the principal investigator, was involved in study design and protocol development, enrolled subjects and critically reviewed the manuscript. All authors approved the final manuscript and take responsibility for its contents.
Disclosure
GMF is an employee of Acacia Pharma Ltd. AFR, JH and SPvM are employees of PRA Health Sciences which received funding from Acacia Pharma to conduct the study. LAB is an employee of Comedica Ltd which received funding from Acacia Pharma to conduct the study. The authors report no other conflicts of interest in this work.
References
1. Kranke P, Eberhart L, Motsch J, et al. I.V. APD421 (amisulpride) prevents postoperative nausea and vomiting: a randomized, double-blind, placebo-controlled, multicentre trial. Br J Anaesth. 2013;111(6):938–945. doi:10.1093/bja/aet251
2. Gan TJ, Kranke P, Minkowitz HS, et al. Intravenous amisulpride for the prevention of postoperative nausea and vomiting: two concurrent, randomized, double-blind, placebo-controlled trials. Anesthesiology. 2017;126(2):268–275. doi:10.1097/ALN.0000000000001458
3. Kranke P, Bergese SD, Minkowitz HS, et al. Amisulpride prevents postoperative nausea and vomiting in patients at high risk: a randomized, double-blind, placebo-controlled trial. Anesthesiology. 2018;128(6):1099–1106. doi:10.1097/ALN.0000000000002133
4. Candiotti KA, Kranke P, Bergese SD, et al. Randomized, double-blind, placebo-controlled study of intravenous amisulpride as treatment of established postoperative nausea and vomiting in patients who have had no prior prophylaxis. Anesth Analg. 2019;128(6):1098–1105. doi:10.1213/ANE.0000000000003733
5. Habib AS, Kranke P, Bergese SD, et al. Amisulpride for the rescue treatment of postoperative nausea or vomiting in patients failing prophylaxis: a randomized, placebo-controlled Phase III trial. Anesthesiology. 2019;130(2):203–212. doi:10.1097/ALN.0000000000002509
6. Herrstedt J, Summers Y, Jordan K, et al. Amisulpride prevents nausea and vomiting associated with highly emetogenic chemotherapy: a randomised, double-blind, placebo-controlled, dose-ranging trial. Support Care Cancer. 2019;27(7):2699–2705. doi:10.1007/s00520-018-4564-8
7. Herrstedt J, Summers Y, Daugaard G, et al. Amisulpride in the prevention of nausea and vomiting induced by cisplatin-based chemotherapy: a dose-escalation study. Support Care Cancer. 2018;26(1):139–145. doi:10.1007/s00520-017-3825-2
8. Coulouvrat C, Dondey-Nouvel L. Safety of amisulpride (Solian): a review of 11 clinical studies. Int Clin Psychopharmacol. 1999;14(4):209–218. doi:10.1097/00004850-199907000-00002
9. Juruena MF, de Sena EP, de Oliveira IR. Safety and tolerability of antipsychotics: focus on amisulpride. Drug Healthc Patient Saf. 2010;2:205–211. doi:10.2147/DHPS.S6226
10. Taubel J, Ferber G, Fox G, Fernandes S, Lorch U, Camm AJ. Thorough QT study of the effect of intravenous amisulpride on QTc interval in Caucasian and Japanese healthy subjects. Br J Clin Pharmacol. 2017;83(2):339–348. doi:10.1111/bcp.13128
11. Canal M, Espie P, Thenot JP. Amisulpride: metabolic and pharmacokinetic profile after 14C intravenous administration. Eur Neuropsychopharmacol. 2002;12(Supplement 3):310. doi:10.1016/S0924-977X(02)80458-8
12. Smith HS, Cox LR, Smith BR. Dopamine receptor antagonists. Ann Palliat Med. 2012;1(2):137–142. doi:10.3978/j.issn.2224-5820.2012.07.09
13. Gillet G, Dormerque L, Canal M, Thenot JP. Amisulpride does not inhibit cytochrome P450 isozymes. Eur Neuropsychopharmacol. 2000;10(Supplement 3):331–332. doi:10.1016/S0924-977X(00)80407-1
14. Dufour A, Desanti C. Pharmacocinétique et métabolisme de l’amisulpride. Ann Psychiatr. 1988;3(3bis):298–305.
15. Spina E, de Leon J. Metabolic drug interactions with newer antipsychotics: a comparative review. Basic Clin Pharmacol Toxicol. 2007;100(1):4–22. doi:10.1111/pto.2007.100.issue-1
16. Candiotti KA, Birnbach DJ, Lubarsky DA, et al. The impact of pharmacogenomics on postoperative nausea and vomiting: do CYP2D6 allele copy number and polymorphisms affect the success or failure of ondansetron prophylaxis? Anesthesiology. 2005;102(3):543–549. doi:10.1097/00000542-200503000-00011
17. Canal M, Fraisse J, Thenot JP. Amisulpride: metabolic and pharmacokinetic profile after 14C oral administration. Eur Neuropsychopharmacol. 2002;12(Supplement 3):319. doi:10.1016/S0924-977X(02)80480-1
18. Rosenzweig P, Canal M, Patat A, Bergougnan L, Zieleniuk I, Bianchetti G. A review of the pharmacokinetics, tolerability and pharmacodynamics of amisulpride in healthy volunteers. Hum Psychopharmacol. 2002;17(1):1–13.
19. Hamon-Vilcot B, Chaufour S, Deschamps C, et al. Safety and pharmacokinetics of a single oral dose of amisulpride in healthy elderly volunteers. Eur J Clin Pharmacol. 1998;54(5):405–409.
20. Coukell AJ, Spencer CM, Benfield P. Amisulpride: a review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of schizophrenia. CNS Drugs. 1996;6(3):237–256. doi:10.2165/00023210-199606030-00006
21. Rein W, Coulouvrat C, Dondey-Nouvel L. Safety profile of amisulpride in short- and long-term use. Acta Psychiatr Scand Suppl. 2000;400:23–27. doi:10.1111/j.0065-1591.2000.007s021[dash]5.x
22. Canal M, MacMahon M, Kwan J, Dubruc C. Amisulpride: kinetics in patients with renal failure. Eur Neuropsychopharmacol. 2000;10(Supplement 3):330.
23. Medicines and Healthcare products Regulatory Agency. UKPAR on Amisulpride 50mg, 100mg and 200mg Tablets PL 19364/0044-6; Amisulpride 400mg Film-Coated Tablets PL 19364/0047. London; 2010.
24. Lecrubier Y, Quintin P, Bouhassira M, Perrin E, Lancrenon S. The treatment of negative symptoms and deficit states of chronic schizophrenia: olanzapine compared to amisulpride and placebo in a 6-month double-blind controlled clinical trial. Acta Psychiatr Scand. 2006;114(5):319–327. doi:10.1111/acp.2006.114.issue-5
25. Danion JM, Rein W, Fleurot O. Improvement of schizophrenic patients with primary negative symptoms treated with amisulpride. Amisulpride Study Group. Am J Psychiatry. 1999;156(4):610–616. doi:10.1176/ajp.156.4.610
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.