")
Back to Journals » Vascular Health and Risk Management » Volume 18
Metabolic Pathway of Cardiac Troponins and Its Diagnostic Value
Authors Chaulin A
Received 30 August 2021
Accepted for publication 23 December 2021
Published 25 March 2022 Volume 2022:18 Pages 153—180
DOI https://doi.org/10.2147/VHRM.S335851
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Pietro Scicchitano
Aleksey Chaulin1,2
1Department of Cardiology and Cardiovascular Surgery, Samara State Medical University, Samara, 443099, Russia; 2Department of Histology and Embryology, Samara State Medical University, Samara, 443099, Russia
Correspondence: Aleksey Chaulin Department of Cardiology and Cardiovascular Surgery, Samara State Medical University, 89 Chapaevskaya Street, Samara Region, Samara, 443099, Russia Tel + 7 927 770-25-87 Email [email protected]
Abstract: The study of the metabolic pathway of endo- and exogenous molecules is not only of great fundamental significance, but also of high practical importance, since many molecules serve as drug targets and/or biomarkers for laboratory diagnostics of diseases. Thus, cardiac troponin molecules have long been used as main biomarkers for confirmation of diagnosis of acute myocardial infarction (AMI), and with the introduction of modern (high-sensitivity) test methods many of our ideas about biology and metabolism of these cardiac markers have changed significantly. In clinical practice, there are opening new promising diagnostic capabilities of cardiac troponins, the understanding and justification of which is closely connected with the fundamental principles of the metabolism of these molecules. However, today the metabolic pathway of cardiac troponins has not been properly investigated, in particular, we do not know precise mechanisms of release of these molecules from the myocardium of healthy people and the mechanisms of circulation and elimination of cardiac troponins from the blood stream. Meanwhile, these stages of the metabolic pathway of cardiac troponins may exercise a dominant influence on serum levels of cardiac troponins, and this may be particularly significant when using rapid diagnostic algorithms for AMI. Refinement of some aspects of the metabolism of cardiac troponins, specifically, the understanding of the mechanisms of fragmentation (proteolytic cleavage) in blood serum, as well as of the filtration (transportation) of fragments of troponin molecules through the blood-tissue filters will increase the diagnostic value of troponins in other biological fluids. The purpose of the present article is to examine the metabolic pathway of cardiac troponins and its significance for laboratory diagnostics. The article successively presents the main stages of the metabolic pathway of cardiac troponins (release from cells, circulation, elimination) and potential factors that may influence these mechanisms and, therefore, the diagnostic value of cardiac troponins.
Keywords: metabolic pathway, cardiac troponins, myocardial infarction, release mechanisms, proteolytic cleavage of cardiac troponins, elimination mechanisms, diagnostic value
Introduction
Notwithstanding the solid achievements in the study of etiopathogenesis, diagnostics, and treatment of acute coronary syndrome (ACS), it still remains one of the leading reasons for disability and mortality of the population in all the developed countries of the world. All patients with ACS are under higher risk of the development of acute myocardial infarction (AMI) and death.1–4 According to the results of the reviews of AMI diagnostic criteria conducted in 2012 and 2018 by the European Society of Cardiology (ESC), the American College of Cardiology (ACC), the American Heart Association (AHA) and the World Heart Federation (WHF), the diagnosis verification is based on the presence of myocardial ischemia symptoms (clinical, electrocardiographic, echocardiographic, angiographic) and the positive dynamics of cardiac troponin levels in blood of patients.5–7
Troponins (troponin I, troponin T, troponin C) are proteins being a part of the troponin complex, which is bound to the protein tropomyosin. Tropomyosin, in its turn, together with actin, forms thin filaments of myocytes — the most important component of the contractile apparatus of striated muscle cells (of skeletal and cardiac muscles). All the three troponins participate in calcium-dependent regulation of the striated muscle contraction-relaxation. Each troponin type fulfils specific regulatory functions in contraction-relaxation of striated muscles. Troponin I is the inhibitory subunit of the tropomyosin complex that binds actin during relaxation and inhibits the ATPase activity of actomyosin thus preventing muscle contraction in the absence of calcium ions in the cell cytoplasm. Troponin T is the regulatory subunit, anchoring the troponin complex to thin filaments and, therefore, participating in the calcium-regulated contraction. Troponin C is the calcium-binding subunit. When the action potential is transferred to the muscle cell, calcium channels in the sarcoplasmic reticulum (“the repository of calcium ions”) open and the sarcoplasmic reticulum releases calcium ions into the sarcoplasm. Then, calcium ions bind to troponin C, which leads to the conformational (structural) changes of proteins of the troponin-tropomyosin complex, as a result of which the tropomyosin molecule shifts and releases binding sites for the myosin head on the actin filament. It enables the interaction of the myosin head with actin, which underlies the mechanism of contraction of striated muscles.8–11
Molecules of troponins have a different amino acid structure depending on their localization in muscles, on the basis of which troponin isoforms are distinguished. Thus, troponin I has three isoforms: cardiac troponin I, troponin I of fast-twitch skeletal muscle fibers, and troponin I of slow-twitch skeletal muscle fibers. Troponin T also has three main isoforms: cardiac troponin T, troponin T of fast-twitch skeletal muscle fibers, and troponin T of slow-twitch skeletal muscle fibers. According to molecular genetic studies, the amino acid sequence of cardiac troponin I and cardiac troponin T differs from the amino acid sequences of the corresponding isoforms of skeletal troponins localized in skeletal muscle fibers by approximately 40–60%.12,13 This important structural peculiarity allows for the use of cardiac troponins T and I as specific biomarkers for laboratory diagnostics of myocardial injury in AMI and other non-cardiac pathological conditions. Cardiac troponin C, as opposed to troponins I and T, has completely identical amino acid structure with the muscular (skeletal) troponin C, and increased blood levels of this protein will not let us reliably distinguish the cardiac muscle tissue injury from the damage of skeletal muscles, and, therefore, cardiac troponin С cannot be used as a cardiac marker for AMI diagnostics.14,15
Although the regulatory documents concerning the diagnostics and treatment of different forms of ACS and AMI contain clear recommendations on the time of troponin testing and decision-making levels, and the sensitivity and specificity of most immunoassays approximates 100%, there still remains a number of unsolved problems and issues relating to the application of these markers in clinical practice. Some of these problems are connected with the variety of troponin diagnostic agents, their unequal sensitivity and diagnostic accuracy, different susceptibility to cross-reactive substances (molecules), ie, with different analytical characteristics of test systems.14,16,17 Another range of issues results from the fact that the increase in cardiac troponin levels takes place in case of myocardial necrosis of any etiology and sometimes in the absence of irreversible myocardial injury (for instance, in case of reversible injury induced by physical exercises, renal failure, or the influence of false-positive factors).18–24 Besides, along with the necrosis of cardiomyocytes, there are other mechanisms of cardiac troponin release from myocardial cells and/or the increase of cardiac troponin concentration in blood serum. Thus, several clinical studies give evidence of very frequent increase in cardiac troponin concentration in various pathologies.24–28 At the same time, the mechanism of troponin increase in these diseases is not associated with the ischemic necrosis of myocardial cells — the main mechanism of troponin levels increase in AMI. The study by G. Lindner et al is quite representative in this respect. The researchers have conducted the detailed analysis of the reasons (the diseases) causing increase of cardiac troponin T levels in patients admitted to the emergency department. In total, the study included 1573 patients, and only 10% of which had the increased level of cardiac troponin T associated with AMI, while all the rest (about 90%) showed no signs of AMI, and their increased levels of troponin T were induced by other diseases causing the increase of troponin T serum levels by non-ischemic mechanisms. The most common reasons of troponin T increase were the following ones: pulmonary embolism, renal failure, acute aortic dissection, heart failure, acute myocarditis, rhabdomyolysis, application of cardiotoxic chemotherapeutic agents, acute exacerbation of chronic obstructive pulmonary disease, sepsis and infiltrative cardiac pathologies (for example, amyloidosis). The interesting fact revealed by this study was that in 30% of cases the increased levels of troponin T were not connected with any previously described causes of cardiac troponin increase.25 There is a high probability that these reasons might be connected with the false-positive mechanisms or they have been induced by the factors the researchers and medical practitioners have not paid attention to and have not described yet. Thus, the interpretation of the results showing the increased levels of cardiac troponins is an extremely complicated and sometimes even impossible task of modern clinical practice. Therefore, it is important to remember that the troponin test itself is not “the gold standard test” for AMI diagnostics, but it can become one only for those patients who show typical clinical symptoms of myocardial ischemia, have corresponding ischemic changes on the electrocardiogram, echocardiogram, etc. Generally, when interpreting possible reasons for the increase of cardiac troponins in blood serum, one should be guided by the following schematics (Figure 1).
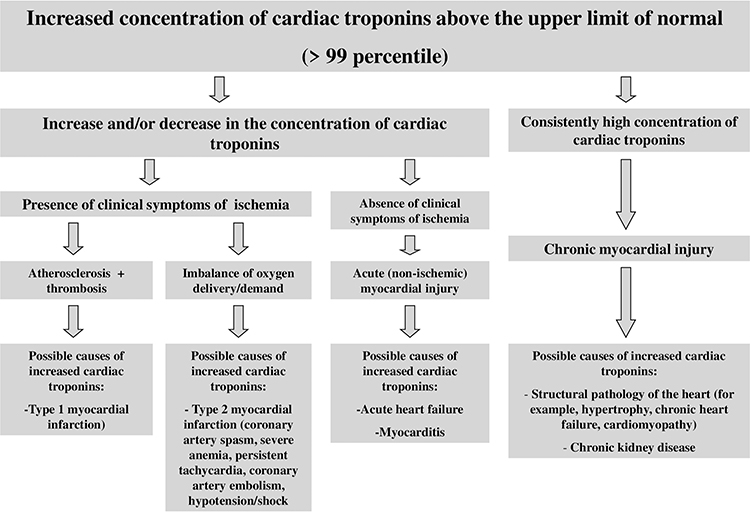 |
Figure 1 Interpretation of possible reasons for myocardial injury and increase of cardiac troponin serum levels. Notes: Adapted from Thygesen K, Alpert JS, Jaffe AS et al6. |
Due to modern ultra-sensitive tests, medical practitioners got the opportunity to early diagnose AMI (within the first two hours from admission of the patient) through the evaluation of dynamic changes of cardiac troponins. The changes (increase) of the concentration of cardiac troponin molecules within the first two hours are very small (may amount to as little as several ng/l) and cannot be detected by moderately sensitive test systems. It should be noted that due to a number of multicenter studies there have been validated algorithms of early diagnostics (0 → 1 hour, and 0 → 2 hours) of non-ST-segment elevation AMI (NSTEMI) for ultra-sensitive test systems of various manufacturers (Table 1).29
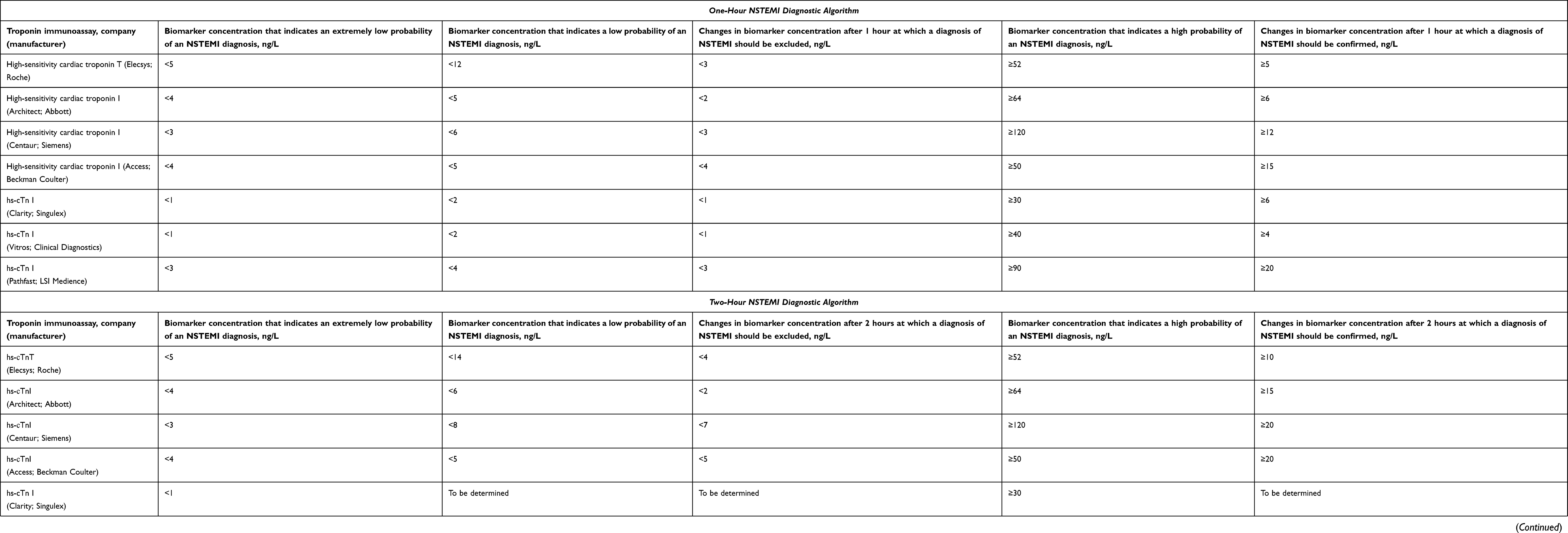 |
Table 1 Current Diagnostic Algorithms for Confirmation/Exclusion of NSTEMI (0 → 1 Hour and 0 → 2 Hours), Approved by the ESC |
According to the data of modern (ultra-sensitive) troponin test methods, the molecules of cardiac troponins are detected in blood and a number of other biological fluids in all healthy people,30–38 which poses new issues and challenges for the scientists in respect of search and explanation of possible mechanisms underlying the release of troponin molecules from intact myocardial cells. Hence, the molecules of cardiac troponins can be considered normal products of cardiac muscle tissue metabolism. However, the precise mechanisms of the release are not clear yet and are hypothetical. Moreover, the factors that may influence and facilitate or, on the contrary, reduce the release of troponins will be of great importance for researchers and medical practitioners. Currently, the most discussable biological factors influencing the degree of troponin release from healthy myocardium are the gender, age, and circadian characteristics.39–48 Gender-related characteristics of cardiac troponins consist in the fact that the myocardium of healthy men releases more molecules of cardiac troponins than that of healthy women. These characteristics are validated by many clinical studies, and practically in all modern test systems, it is recommended to use the threshold values (99th percentile) in accordance with gender.41,49,50 The age-related characteristics of cardiac troponins are that a greater amount of cardiac troponin molecules are released from the myocardium of elderly patients compared to the myocardium of young people.45,51,52 The circadian features of cardiac troponins are that more cardiac troponin molecules are released from the myocardium of healthy people in the morning than from the myocardium of healthy people in the evening-night period.53,54 It should be noted that the age and circadian characteristics of cardiac troponins are not typical for all test systems, and according to some studies, they are contradictory.55–57 Before using these characteristics in rapid diagnostic algorithms, it is necessary to conduct additional large studies to validate the age and circadian characteristics of cardiac troponins.
One of the significant problems of both moderately sensitive and modern highly sensitive immunoassays is the lack of their standardization.58–60 This leads to the fact that different troponin immunoassays detect different values (concentrations) of cardiac troponin molecules in blood and other biological fluids of the same patient. So, in accordance with the data in Table 1, the threshold levels for excluding/confirming NSTEMI differ by several times when using immunoassays from different manufacturers.29 Based on this, we can say that each method detects, in fact, different molecules of cardiac troponins and their fragments in a biological fluid. This creates certain difficulties and problems: 1) the need to validate the threshold concentrations of cardiac troponins for each test system, including newly developed ones, which is associated with additional high costs; 2) the need for a thorough study of interfering factors for each of the known detection methods that also involves additional costs; 3) upon admission of a patient to the hospital, dynamic changes in cardiac troponins to confirm/exclude AMI during the first and subsequent hours can be traced and evaluated only when using the same test system, and immunoassays from different manufacturers cannot be used for this goal. At the same time, different institutions can use different test systems, which will not allow for proper assessment of the dynamic changes in case of urgently required transportation of a patient to another institution, which will be associated with a loss of time for additional examinations and additional economic costs.
It should be noted separately that the molecules of cardiac troponins can be affected by very numerous proteolytic enzymes present in blood, which thereby can indirectly affect the levels of troponins in blood of patients. For example, due to an increase in the activity of proteases that cause fragmentation of troponin molecules, the duration of circulation (half-life) of troponins in the bloodstream will decrease, which can potentially lead to false-negative results when using those test systems that have diagnostic antibodies directed against fragmented epitopes of troponin molecules.61,62 A decrease in the activity of such proteolytic enzymes, on the contrary, may hypothetically lead to false-positive results. However, the current specific knowledge about this metabolic stage, in particular, the exact information about all the influencing enzymes and mechanisms of troponin fragmentation, is extremely scarce and is not taken into account in clinical practice. This stage of troponin metabolism and its effect on diagnostics will be considered in more detail in this manuscript below in the paragraph on the circulation of cardiac troponins in blood plasma.
A very interesting direction in studying the diagnostic value of cardiac troponins is the assessment of the possibility of using other biological fluids as biomaterials for detection of troponin molecules. This direction is developing due to an increase in the sensitivity of immunoassays (the creation of highly sensitive and ultra-sensitive test systems), which can detect very low concentrations (at levels of several ng/l) of troponins that circulate in many biological fluids, including non-invasively obtained fluids (urine and oral fluid).34–47,63–69 Moderately sensitive methods, as a rule, cannot detect such low concentrations of cardiac troponin molecules present in these biological fluids.70,71 The mechanisms of penetration/transport of cardiac troponins into these biological fluids should also be considered as one of the stages of the metabolic pathway of cardiac troponins. And the study and understanding of precise mechanisms of penetration/transport of cardiac troponins will increase their diagnostic value and validate new methods for diagnosing cardiovascular diseases through the use of other biological fluids, in particular non-invasively obtained fluids, since their collection has a number of advantages (for example, painlessness and atraumatic nature, lower risk of introduction of blood-borne infections and the possibility of obtaining biomaterial without the involvement of medical personnel) over the use of blood as a biomaterial. In addition, there are prospects for the creation of specialized diagnostic test strips (“dry chemistry” methods) for the detection of troponins in urine and/or oral fluid, which will make it possible to carry out express diagnostics and/or monitoring of cardiovascular diseases at home by patients themselves or by their relatives.
The main biological fluids in which the molecules of cardiac troponins are detected and their diagnostic value are summarized in Table 2.
 |
Table 2 Biological Fluids in Which the Molecules of Cardiac Troponins are Detected and the Diagnostic Value |
Metabolic Pathway of Cardiac Troponins
Conventionally, there can be distinguished three main stages of the metabolic pathway of cardiac troponins (Figure 2): 1) release of cardiac troponins from myocardial cells, 2) circulation of cardiac troponins in blood plasma, 3) removal of cardiac troponins from the bloodstream. Similar key stages of the metabolism of molecules are distinguished for other molecules in order to conveniently and consistently consider the main metabolic characteristics of molecules.
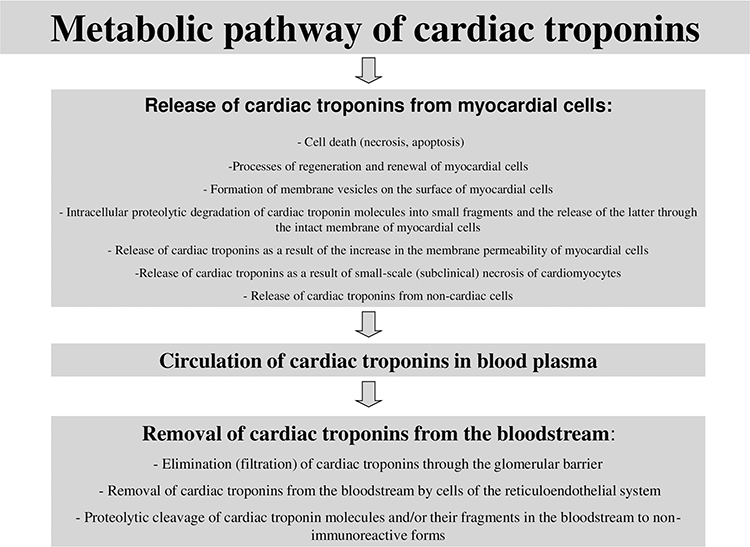 |
Figure 2 Metabolic pathway of cardiac troponins. |
Each of these stages of metabolism can play a decisive role in the regulation of the concentrations of cardiac troponins in blood, ie, their diagnostic value. In addition, there are a number of factors that can have a potential and hypothetical influence on these stages of the metabolic pathway of cardiac troponins. These factors can be physiological conditions, for example, gender, age and circadian characteristics, which have a certain effect on the degree of release of troponin molecules from the myocardium of healthy people. Or, changes in the activity of proteolytic enzymes that target cardiac troponin molecules. The activity of proteolytic enzymes can also change under pathological conditions and/or in case of taking certain medications. Renal failure can be noted as an example of a significant factor affecting the removal of cardiac troponins from the bloodstream. It is important to emphasize that there may be an extremely large number of such factors, and some of them are probably still unknown.
It should be noted that the exact molecular releases, degradation and elimination of cardiac troponins are not yet clear. Further in the course of this manuscript, I will sequentially consider each of the stages of the metabolic pathway of cardiac troponins and note the main known and assumed factors that may affect these mechanisms and the diagnostic value of cardiac troponins.
Release of Cardiac Troponins from Myocardial Cells: Mechanisms and Diagnostic Value
The introduction of highly sensitive test systems into practice made it possible with high accuracy (variability of the analysis within 10%) to detect very low concentrations of cardiac troponins, ranging from 0.001 to 0.01 ng/mL and below the values corresponding to the 99th percentile (upper limit of the norm). As a result, cardiac troponin molecules were found in almost 100% of healthy people, and instead of a clear borderline level typical of AMI, a smooth scale appeared, capable of reflecting subclinical myocardial pathology associated with structural (non-ischemic) damage, stable coronary artery diseases and other pathological conditions that negatively affect myocardial cells.14,38,81–83 Considering the fact that the molecules of cardiac troponins began to be detected in all healthy people, it became necessary to study and explain the mechanisms of the release of cardiac troponins from the intact myocardium. In this regard, the researchers are discussing the following possible mechanisms for the release of troponin molecules and an increase in their serum levels: a) the release of cardiac troponins as a result of the processes of regeneration and renewal of myocardial cells, b) the release of cardiac troponins as a result of apoptosis of myocardial cells, c) the release of cardiac troponins as a result of formation of membrane vesicles on the surface of myocardial cells, d) intracellular proteolytic degradation of cardiac troponin molecules into small fragments and the release of the latter through the intact membrane of myocardial cells, e) release of cardiac troponins as a result of the increase in the membrane permeability of myocardial cells, f) release of cardiac troponins as a result of small-scale (subclinical) necrosis of cardiomyocytes, g) the release of cardiac troponins from non-cardiac cells. Some of the above mechanisms of release can not only explain the detectable concentrations of cardiac troponins in healthy individuals, but also significantly activate/increase under certain physiological conditions and pathological processes.84–87 For example, apoptosis of myocardial cells can increase with an increase in blood pressure,11,88 stretching of the myocardial walls,89,90 increased stimulation of beta-adrenergic receptors91–93 and a number of other mechanisms,94,95 which thereby may facilitate the release of cardiac troponins from cardiomyocytes. And in conditions of chronic renal failure, the expression of cardiac troponins in skeletal muscles is noted,96 which, according to some authors, can lead to an increase in serum concentrations of cardiac troponins in patients with chronic renal failure.96,97
Below, I will consider each of the above mechanisms of cardiac troponin release sequentially and in more detail.
Release of Cardiac Troponins as a Result of the Processes of Regeneration and Renewal of Myocardial Cells
Evidence of the fact that myocardial cells can regenerate/renew has been obtained by studying the metabolism of C14-labeled DNA molecules in myocardial cells. There was carried out a long-term observation of people with the inclusion of a radioactive isotope of carbon (C14) in the DNA of cardiomyocytes, which occurred as a result of nuclear weapons tests. The authors calculated the rate of renewal of cardiomyocytes by studying the rate of DNA synthesis, which was calculated by investigating the rate of accumulation of C14 in myocardial cells. They found the renewal of cardiomyocytes, the intensity of which decreased annually — from 1% per year at the age of 25 years to 0.45% per year at the age of 75 years. In general, about 50% of cardiomyocytes underwent renewal throughout life.98,99 These results indicate the existence of a small regenerative potential in myocardial cells. Some researchers suggest that the process of renewal of myocardial cells may be associated with the release of cardiac troponins from cells,100 however, the specific mechanism underlying this phenomenon remains unknown. As a possible hypothesis, it can be assumed that intracellular molecules of cardiac markers, including cardiac troponins, will be released from gradually aging and naturally dying cardiomyocytes as a result of the gradual destruction of the cell membrane. Since the rate of renewal of cardiomyocytes is low, the degree of increase in serum levels of cardiac troponins is also insignificant (no higher than the 99th percentile). Thus, this mechanism can hypothetically explain the presence of a small amount of cardiac troponin molecules in the bloodstream of all healthy individuals.
In accordance with the data of other researchers, the average rate of cardiomyocyte renewal in mammals is 0.5–2.0% per year and can vary depending on the influence of certain factors, such as physiological conditions (physical activity), trauma and concomitant diseases.101–103 Thus, the rate of renewal of cardiomyocytes increases significantly after myocardial damage. An experimental study conducted by P. Docshin et al has shown that ischemic myocardial injury causes the activation of endogenous stem cells and increases the rate of myocardial cell renewal.104 Two other research groups led by C. Waring et al105 and M. Rovira et al106,107 revealed an increase in the processes of proliferation and differentiation of stem cells in the myocardium of rats and zebrafish (Danio rerio).
However, the assessment of the degree of regeneration and the rate of renewal of myocardial cells can be significantly influenced by inflammatory processes, proliferation of non-myocyte cells and the formation of a connective tissue scar in the myocardium, which often complicate and/or distort investigation results.108,109 Further research is needed to investigate the specific role of myocardial cell regeneration and renewal in the release of cardiac troponins from cells.
Release of Cardiac Troponins as a Result of Apoptosis of Myocardial Cells
To date, a large number of factors have been discovered that can trigger the processes of apoptosis of cardiomyocytes.110,111 Induction of apoptosis leads to an increase in the activity of caspases (proteolytic enzymes of the cysteine protease family), which can fragment (damage) DNA and protein molecules, leading to cell death. In contrast to necrosis, during apoptosis, the cell dies more slowly, the integrity of the cell membrane remains much longer, and the inflammatory reaction around the dead cell is not observed. To study the processes of apoptosis, many methods are used: various types of microscopy (light, electron, fluorescence), flow cytometry, immunohistochemical analysis, the TUNEL method (Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling), etc. The TUNEL method is the most reliable and early method for detecting apoptosis. This method allows visualization of cell nuclei in which the DNA molecule has been fragmented due to increased activity of endonucleases and caspases. This method is most often used in all modern studies aimed at the exploration of the etiopathogenetic mechanisms of apoptosis of various cells, including cardiomyocytes.112–114
An experimental study led by B. Weil et al has shown that short-term ischemia activates apoptosis of myocardial cells in experimental animals, and apoptosis of cardiomyocytes is accompanied by an increase in serum levels of cardiac troponins. Short-term myocardial ischemia was simulated by means of balloon occlusion of a branch of the left coronary artery and the fact of occlusion was confirmed by coronary angiography. The duration of ischemia was 10 minutes, after which reperfusion was carried out by deflation of the balloon. To confirm the apoptosis of myocardial cells, the TUNEL method was used, according to the results of which the number of cardiomyocytes in the state of apoptosis was significantly increased (6 times compared with the control group of animals). At the same time, no histological signs of myocardial necrosis were observed. This suggests that short-term (in this case, 10-minute) ischemia does not cause ischemic necrosis of cardiomyocytes, but enhances apoptotic processes in the myocardium. However, the levels of cardiac troponins began to rise rapidly: 30 minutes after reperfusion, the troponin I concentration approached the upper limit of the norm (38 ng/l) and after 1 hour exceeded it (51 ± 17 ng/l). Two and three hours after reperfusion, the serum levels of cardiac troponin I were 148 ± 88 ng/l and 180 ± 117 ng/l, respectively, which indicated the continuing release of troponin molecules from the myocardium. And, finally, 24 hours after reperfusion, the troponin I concentration reached its peak and amounted to 1021 ± 574 ng/l.115 Thus, this experimental study elegantly demonstrates the role of apoptosis (induced by short-term ischemia) in the release of cardiac troponin molecules from myocardial cells. The limitation of this study is the relatively short interval of investigating the cardiac muscle tissue for the presence of histopathological changes; according to these results, it is impossible to determine the degree and reversibility of damage to cardiac myocytes during apoptosis induced by short-term ischemia. Besides, for detection of cardiac troponin I, there was used a moderately sensitive test system, which is inferior in diagnostic capabilities to modern highly sensitive immunoassays. In this regard, the dynamic changes in the levels of cardiac troponins detected by highly sensitive immunoassays during apoptosis could be significantly different, especially in the first minutes and hours after reperfusion.
The literature also describes other situations when apoptosis of cardiac myocytes is induced by other mechanisms that are not associated with reversible (short-term) ischemia of cardiac muscle tissue. The authors identify the following mechanisms of apoptosis that can promote the release of cardiac troponins from myocardial cells: stretching of the myocardial walls, increased preload on the heart, and increased activity of the sympathoadrenal system.89–91,116 Thus, W. Cheng et al reported that apoptosis of myocardial cells increases with stretching of the myocardial walls.116 This allows us to consider many physiological and pathological conditions that cause stretching of the myocardial walls as possible inducers of apoptosis and thus can, to some extent, explain the increased serum levels in patients after prolonged and intense physical exertion or having arterial hypertension, pulmonary embolism, chronic obstructive pulmonary disease and a number of other pathologies.116–119
In another experimental study, B. Weil et al concluded that an increase in preload on the heart triggers apoptosis and causes an increase in the concentration of troponin I in blood of experimental animals. To increase the preload on the heart, the experimental group of animals received intravenous drug — phenylephrine (300 μg of the drug per minute) for one hour. After the simulation, echocardiography was used to confirm myocardial overload, and to verify apoptosis and determine cardiac troponin levels, there were used histological methods, including the TUNEL method and moderately sensitive immunoassay, respectively. As a result of histological examination of the myocardium of the experimental group of animals, there was noted a significant increase in the number of cardiomyocytes in the state of apoptosis, as compared to the control group. At the same time, no histological signs of myocardial necrosis were recorded. Twenty-four hours after the simulation, the number of cardiomyocytes in the state of apoptosis decreased to the level of the control group, which indicates the reversibility of apoptotic changes. The troponin I concentration exceeded the upper limit of the norm 30 minutes after the simulation, and then the troponin I levels continued to rise sharply and reached a value of 856 ± 956 ng/l one hour after the simulation. Serum levels of cardiac troponin I remained elevated throughout the study period (24 hours) and peaked at 1462 ± 1691 ng/l.120 Since signs of necrosis, unlike apoptosis, were not observed, it should be considered that apoptosis induced by myocardial overload plays an important role in the release of cardiac troponin molecules from cells.121
The degree of release of cardiac troponin molecules from myocardial cells as a result of apoptosis induced by myocardial overload depends on the strength and duration of exposure. For example, relatively small myocardial overload is observed on mild to moderate exertion, in hypertension and non-massive pulmonary embolism, so the increase in serum levels of cardiac troponins in these conditions is also relatively small. But, for example, on high-intensity exertion or in massive pulmonary embolism, myocardial overload becomes much more significant, therefore, these conditions are accompanied by relatively higher serum levels of cardiac troponins.122–124
Another very interesting mechanism for the initiation of apoptosis is an increase in the activity of the sympathoadrenal system. A research group led by K. Singh et al found that stimulation of beta-adrenergic receptors (β-AR) regulates intracellular apoptotic signaling pathways in cardiomyocytes. Moreover, stimulation of β1-AR enhances apoptosis of myocardial cells, while stimulation of β2-AR has the opposite effect.125,126 It was also noted that the density of β-AR subtypes changes significantly with age.127 Thus, in elderly patients, a more pronounced decrease in the number of β2-AR is noted, which may contribute to a weakening of the anti-apoptotic effect and, accordingly, an increase in apoptosis of myocardial cells.127–129 A higher degree of apoptosis in elderly patients can probably be associated with age-related characteristics of cardiac troponin levels: in older people, troponin levels are significantly higher than in young people. The age-related characteristics of cardiac troponins have been demonstrated in a number of clinical studies through blood examination with highly sensitive test systems.43–45,51,52
Considering the above, apoptosis of cardiomyocytes should be considered as a significant mechanism for the release of cardiac troponin molecules from myocardial cells. This mechanism is not associated with necrosis of cardiomyocytes and contributes to a very significant increase in serum levels of cardiac troponins. Thus, apoptosis of myocardial cells can be of great diagnostic value in conditions such as prolonged and intense physical activity, arterial hypertension, pulmonary embolism, heart failure, and, probably, in old age. Further research is needed to clarify the exact role of apoptosis in the release of cardiac troponins in physiological and pathological conditions.
Release of Cardiac Troponins as a Result of the Formation of Membrane Vesicles on the Surface of Myocardial Cells
This mechanism of release of cardiac troponins from myocardial cells was first described in an experimental study relatively long ago. A research group led by P. Schwartz reported that on the plasma membrane of myocardial cells membrane vesicles (blebbing vesicles) are formed.130,131 During ischemia, the number of blebbing vesicles increases in comparison with intact myocardial cells. A similar trend is also typical for hepatocytes.132 Since these vesicles are formed from fragments of the cell membrane and the cytoplasm of cardiomyocytes, these vesicles may contain some cytoplasmic proteins of myocardial cells, in particular cardiac markers (creatine kinase MB isoform, myoglobin and the cytoplasmic fraction of cardiac troponins, and others). However, since the volume of the cytoplasmic fraction of cardiac troponins is small (approximately 3–4% for troponin I and 7–8% for troponin T of the total amount of troponins in the cardiomyocyte),53,133 the contribution of this mechanism to the degree of increase in serum levels of cardiac troponins will also be limited. Based on the peculiarities of the formation of blebbing vesicles (a significant increase in ischemia), it can be assumed that this mechanism is involved in the release of cardiac troponins in those pathological conditions that are accompanied by ischemia of myocardial cells at an early stage. For example, the initial (prenecrotic) stage of myocardial ischemia can provoke the formation of blebbing vesicles and the release of troponins into the bloodstream, which will lead to the formation of the first peak in serum concentrations of cardiac troponins. Subsequently, two main scenarios are possible: 1) with a decrease of ischemia of myocardial cells, the formation of blebbing vesicles stops and troponin concentrations quickly return to normal, 2) with the continuation/intensification of ischemia (as, for example, during AMI), the formation of blebbing vesicles increases, and, in addition to this, there occur the destruction of the plasma membrane of myocardial cells and proteolysis (fragmentation) of troponin proteins, which are part of the main (structural or contractile) troponin fraction, which will lead to the formation of the second peak in serum concentrations of cardiac troponins. From a pathogenetic point of view, any physiological or pathological condition that will lead to ischemia of myocardial cells (even reversible myocardial ischemia) can activate this mechanism of cardiac troponin release. For instance, some physiological conditions (physical activity)134 or pathological conditions (sepsis)135 can cause an increase in the oxygen demand of myocardial cells, which, accordingly, will be accompanied by ischemia of the cardiac muscle tissue.
Intracellular Proteolytic Degradation of Cardiac Troponin Molecules into Small Fragments and the Release of the Latter Through the Intact Membrane of Myocardial Cells
The size and location of intracellular molecules are two key factors that affect the transport (release) of molecules across the cell membrane. Low molecular weight biomarkers are much more intensively released across the plasma membrane, which plays a role in the diagnostics of many diseases, including cardiovascular pathologies. So, for example, with the development of AMI, the concentration of low molecular weight cardiac markers (myoglobin) in blood serum rises much earlier than the concentration of high molecular weight cardiac markers (lactate dehydrogenase-1).136–138 This is due to the fact that myoglobin molecules are small and can be released at the initial stages of ischemia during the development of AMI (when the plasma membrane of myocardial cells is still relatively insignificantly damaged). A larger molecule (lactate dehydrogenase-1) can leave the cardiomyocyte only when its cell membrane is significantly damaged. Biomarkers that are freely localized in the cytoplasm of cells (for example, myoglobin, cytoplasmic (non-contractile) fraction of troponins) also have advantages when released from the cell, in contrast to those biomarkers that are localized in organelles (nucleus or mitochondria of cells) or are tightly bound to structural components of sarcoplasm (for example, the structural fraction of troponins involved in the regulation of the contractile fraction of the myocardium). So, during the development of AMI, the primarily released molecules are troponin molecules that are part of the cytoplasmic fraction of cardiac troponins, and only then there takes place the destruction of sarcomeres, in particular of the troponin-tropomyosin complex, and the release of structural cardiac troponins.
The most important factor that can affect the size of a molecule (biomarker) and, accordingly, the possibility of its release, is the degree of activity of enzymes that cause proteolysis (fragmentation) of this molecule.139,140 The activity of proteolytic enzymes can change both under physiological and pathological conditions. In an experimental study conducted by J. Feng et al, it was demonstrated that an increase in preload on cardiac muscle tissue activates the enzyme calpain, which fragments the cardiac troponin I molecule, which could potentially play a role in the release of this biomarker from myocardial cells and an increase in its level in blood serum.140 Thus, physiological and pathological conditions causing an increase in the preload on the myocardial wall can promote the release of cardiac troponins from myocardial cells by this mechanism.
In addition to the enzyme calpain, the cleavage of cardiac troponin molecules can be catalyzed by some types of matrix metalloproteinases (MMP 2 and MMP 14)140–143 and the enzyme thrombin.144–146 The activity of these enzymes can also be influenced by pathological processes and some drugs, which thereby hypothetically can affect the serum levels of cardiac troponins. For example, an increase of thrombin activity in patients with dilated cardiomyopathy147,148 can contribute to the fragmentation of cardiac troponin T, which can have both pathogenetic significance (damage to troponin T, which is one of the main components of the contractile apparatus of cardiomyocytes), and diagnostic value: a decrease in the size of the troponin T molecule as a result of fragmentation and a possible increase in the release of these fragments into the bloodstream.
Changes in acidity (pH) can also modulate the activity of intracellular proteolytic enzymes.149,150 So, pathological conditions that disrupt myocardial metabolism, in particular myocardial ischemia, lead to a switch from aerobic myocardial metabolism to anaerobic metabolism and an increase in the formation of lactic acid, which will shift the pH towards acidosis. Under conditions of acidosis, then, proteolytic and proapoptotic enzymes will be activated,69,140,150,151 which, through fragmentation, will promote the formation of many small fragments (molecules) of cardiac troponins, which will increase the likelihood of their release from myocardial cells into the bloodstream.
Release of Cardiac Troponins as a Result of Increased Membrane Permeability of Myocardial Cells
The membrane permeability of myocardial cells is an important factor that plays a role in the release of cardiac marker molecules from myocardial cells into the bloodstream. Based on the analysis of the results of existing experimental data, two main mechanisms can be distinguished that underlie the change (increase) in the membrane permeability of myocardial cells: 1) an increase in the membrane permeability of myocardial cells as a result of an increase in the load on the myocardium and stretching of its walls; 2) an increase in the membrane permeability of myocardial cells as a result of myocardial ischemia and activation of proteolytic enzymes that can damage the cell membrane.
The first mechanism for the release of cardiac troponins was studied by M. Hessel et al.152 In their experimental study, the authors stimulated special glycoprotein receptors of myocardial cells (integrins) that are sensitive to myocardial stretching. To model myocardial stretching and activation of integrins, the researchers used the RGD (Arg–Gly–Asp) tripeptide, which is a potent integrin agonist and is part of fibronectin and other regulatory proteins of the extracellular matrix.153 The authors particularly note that myocardial stretching is not associated with ischemic and necrotic processes in the cardiac muscle tissue, which indicates that it was the specific mechanism of myocardial wall stretching and the activation of integrins that ensured the release of cardiac troponins from viable myocardial cells.152
The second mechanism for increasing membrane permeability is associated with membrane damage during ischemia of myocardial cells. As already described above, myocardial ischemia initiates changes in the metabolism of cardiomyocytes and acidification (acidosis) of the intracellular space of myocardial cells, which, in turn, will lead to the activation of proteolytic and proapoptotic enzymes. These enzymes have many targets and in addition to the specific action (fragmentation of cardiac troponins), they can obviously catalyze the proteolysis of proteins that make up cell organelles and membranes.154–156 Thus, this mechanism of troponin increase is closely interrelated with the above-described mechanism (troponin increase due to increased proteolytic degradation into small fragments). In general, increased membrane permeability and intracellular fragmentation of cardiac troponins can be considered as two interrelated and synergistic mechanisms underlying the release of cardiac troponin molecules from myocardial cells. The degree of activity of these mechanisms is probably related to the severity of pathological processes. For example, short term and/or reversible ischemia of myocardial cells during exercise or in uncomplicated sepsis may be associated with a relatively small increase in the activity of intracellular proteolytic enzymes. In this regard, the degree of increase in serum levels of cardiac troponins will also be relatively small and dependent only on the cytoplasmic fraction of cardiac troponins (their fragmentation into small molecular fragments) and reversible membrane damage/increased membrane permeability. In pathological conditions that cause irreversible ischemia of myocardial cells (for example, AMI or severe/complicated sepsis), serum troponin levels increase much more significantly and the main contribution to total serum levels of cardiac troponins will be made by the structural fraction of cardiac troponins. Both the proteins of the troponin-tropomyosin complex and the proteins of the membranes of cardiomyocytes will be more actively fragmented (cleaved) and therefore the degree of release of cardiac troponins in these pathologies will be higher. The further prognosis of patients suffering from both cardiac and non-cardiac pathologies is also associated with the degree of increase in serum levels of cardiac troponins, which indicates the depth and nature of damage to cardiac muscle tissue.
Release of Cardiac Troponins as a Result of Small-Scale (Subclinical) Necrosis of Cardiomyocytes
A possible mechanism underlying the release of cardiac troponins is small-scale necrotic processes, which can be caused by both ischemia and inflammatory-toxic processes, imbalances in the neurohumoral system.
So, according to some researchers, regular heavy physical exertion, myocardites and stressful situations can cause subclinical damage to myocardial tissue (death of single cardiomyocytes), which can subsequently be associated with the formation of relatively small areas of fibrosis and an increased risk of sudden cardiac death.157 So, for example, the adverse effect of serious and/or intense physical activity is confirmed by a number of studies and described clinical cases in which sudden cardiac death was recorded in athletes.158–160
Some studies registered extremely high levels of cardiac markers, including cardiac troponins in the blood serum of athletes after serious and prolonged physical activity,161–163 which is also a reason for discussing possible small-scale necrotic processes. A contradictory argument is a clinical study using magnetic resonance imaging with gadolinium (contrast) that revealed no signs of necrosis and sclerosis in the cardiac muscle tissue of athletes.164 However, the limitation of this method is its relatively lower sensitivity compared to laboratory biomarkers of myocardial necrosis and fibrosis.
Although during psycho-emotional stress the level of troponin increase is relatively small (rarely exceeds the levels of the 99th percentile in the isolated effect of stress), it cannot be considered a safe process.165,166 The constant influence of stress is considered as a risk of developing cardiovascular diseases and may be one of the triggers of AMI.167,168 A number of molecules released during stress (for example, cortisol, catecholamines) increase myocardial oxygen demand, thereby contributing to the development of relative ischemia of myocardial cells.
Release of Cardiac Troponins from Non-Cardiac Cells
One of the controversial but hypothetically possible mechanisms underlying the increase in serum levels of cardiac troponins is the release of these molecules from non-cardiac cells. Several experimental and clinical studies indicate the expression of cardiac troponin molecules in skeletal muscle cells96,169,170 and the walls of large vessels,171,172 which allows us to consider these organs as possible sources of serum levels of cardiac troponins. Thus, American biochemists (V. Ricchiuti and F. Apple), using polymerase chain reaction (PCR), revealed the expression of messenger RNA of cardiac troponin T in the skeletal muscle tissue of adults suffering from end-stage chronic renal failure (CRF) and hereditary skeletal myopathy (Duchenne muscular dystrophy). Cardiac troponin I messenger RNA was not detected in skeletal muscles of patients suffering from these pathologies and in skeletal muscles of healthy people. In addition, no signs of cardiac troponin T expression were detected in the skeletal muscles of healthy people,96 which indicates the possible expression of one type of cardiac troponin (T) only in the presence of the indicated pathologies. In another study, B. Messner et al confirmed the possibility of extracardiac expression of cardiac troponin T in patients with skeletal myopathies. The researchers, using PCR, found messenger RNA of cardiac troponin T in patients with primary sarcoglycanopathy and Duchenne muscular dystrophy.170 In some patients with skeletal myopathies, in addition to cardiac troponin T messenger RNA, the expression of cardiac troponin I messenger RNA was observed.170 However, in these studies, the authors did not measure serum levels of cardiac troponins in patients with myopathies and renal failure. This is an important limitation of these studies because it does not answer the question: can the expression of cardiac troponins in skeletal muscles lead to an increase in serum levels of cardiac troponins in patients with CRF or hereditary skeletal myopathies? In addition, there should be mentioned several other studies, the results of which contradict the above data on non-cardiac expression.173–175 For example, G. Bodor et al conducted a study and concluded that cardiac troponins are not expressed in skeletal muscle tissue in patients with Duchenne muscular dystrophy and polymyosites.173 Other research groups led by A. Hammerer-Lercher and J. Schmid also did not find signs of expression of cardiac troponins in skeletal muscles.174,175
A second potential non-cardiac source of cardiac troponin release is the walls of large veins (venae cavae and pulmonary veins). Some studies report only the presence of expression of cardiac troponins in the walls of these veins, but do not describe the possible role of these troponins in diagnostics.171,172 Hypothetically, it can be believed that damage or stretching of the walls of these large veins can lead to the release of cardiac troponin molecules into the bloodstream.
Thus, due to the fact that data on extracardiac expression are either insufficient or contradictory, further research is needed to validate this mechanism.
The mechanisms of cardiac troponin release described above and their diagnostic value are summarized in Table 3.
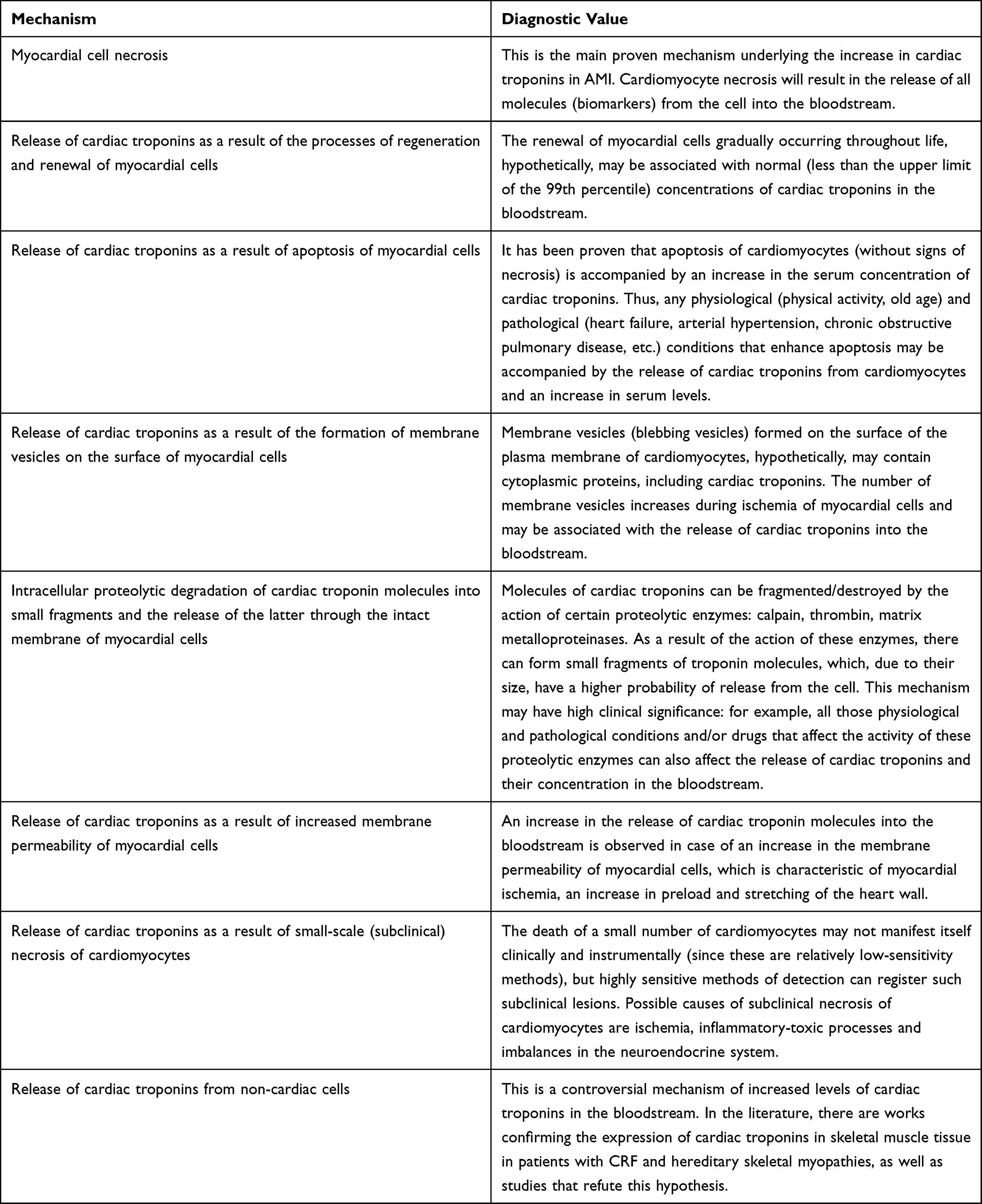 |
Table 3 Release of Cardiac Troponins from Myocardial Cells: Mechanisms and Diagnostic Value |
Circulation of Cardiac Troponins in Blood Plasma: Influencing Factors and Diagnostic Value
The second major stage of the metabolic pathway of cardiac troponins is circulation in the bloodstream. At this stage, the molecules of cardiac troponins are influenced by a number of factors (activity of proteolytic enzymes, kinases, phosphatases, the state of renal function and the reticuloendothelial system, etc.), which can affect serum levels of cardiac troponins and, therefore, their diagnostic value. The molecules of cardiac troponins released into the bloodstream are represented by a heterogeneous fraction (a significant variety of different forms of troponin molecules): free troponins; combined complexes consisting of several free forms of cardiac troponins (for example, cardiac troponin I + troponin C, cardiac troponin T + cardiac troponin I, etc.) and small fragments of cardiac troponins.61,144,145,176–180 All of the above forms of troponin molecules can undergo oxidation, glycosylation, phosphorylation and dephosphorylation processes, which leads to the formation of very diverse forms (varieties) of troponin proteins. Modifications of troponin proteins can affect such an important parameter as the half-life (half-decay) of cardiac troponins. This parameter has not only fundamental, but also high practical importance, since with an intensification of the breakdown of troponin proteins, their concentration and the “diagnostic window” can decrease, and with a weakening of the breakdown of cardiac troponins, their serum levels and the duration of the diagnostic window can increase. The researchers estimate that the half-life of cardiac troponin T in the bloodstream is approximately 2 hours, however, for many other forms, the half-life is controversial and unknown.181–183 The cardiac troponin I molecule is much less stable in the bloodstream, since it actively undergoes the processes of oxidation, phosphorylation and fragmentation.184,185 The latter, in turn, as noted above for cardiac troponin T, depend on the activity of these enzymes, the presence of concomitant pathologies that can affect the activity of these enzymes, the intake of drugs that affect the catalytic activity of proteolytic enzymes and the functional state of the organs responsible for the elimination of molecules of cardiac troponins. For example, increasing the activity of the enzyme thrombin (which has been shown to cause specific fragmentation of cardiac troponin T)145 can reduce the half-life of cardiac troponin T and its concentration in the bloodstream. It is logical that taking drugs that reduce thrombin activity (for example, direct thrombin inhibitors, direct and indirect anticoagulants) can increase the half-life of cardiac troponin T and the duration of the diagnostic window. As examples of the influence of other factors on the duration of the circulation of cardiac troponins in the bloodstream, there can be named the functional state of the kidneys and the reticuloendothelial system. Thus, protein molecules of cardiac markers, including cardiac troponins T and I, can be captured by the reticuloendothelial system (macrophages) and are destroyed there.67,186,187 Based on this, the following point of view comes out: an increase in the activity of the reticuloendothelial system (for example, with hypersplenism and splenomegaly) may be accompanied by an increase in the cleavage of cardiac troponins and a decrease in the half-life; and a decrease in the activity of the reticuloendothelial system, on the contrary, will lead to a weakening of the cleavage of cardiac troponins and an increase in the half-life. Renal function is also significantly associated with cardiac troponin levels and increased blood protease activity. Thus, an increase in the activity of proteolytic enzymes can lead to the formation of a large number of small fragments of troponin molecules, which, like many low molecular weight proteins, can be filtered through the three-layer glomerular (filtration) barrier of nephrons. However, the filtration rate can change both under physiological and pathological conditions, which can have a significant effect on the rate of removal of troponin fragments. With pronounced drops in filtration rate (for example, with CRF or a decrease in blood pressure), the molecules of cardiac troponins will not be filtered (removed) from the bloodstream into the urine, but will accumulate in the blood, which will lead to an increase in the half-life of cardiac troponins and prolongation of the diagnostic window.188–190
Clear evidence that cardiac troponin molecules can pass (filter) through the glomerular filter has been presented in several recent clinical studies due to the use of highly sensitive troponin immunoassays.34,67,69 A similar transport mechanism is probably characteristic of the filtration of cardiac troponins into the oral fluid through the blood-salivary barrier, which is also supported by several pilot studies that have established a correlation between serum and salivary troponin levels.35–37,68
The search for specific mechanisms of proteolytic cleavage of cardiac troponins in the bloodstream is of great practical importance, since it will optimize laboratory diagnostics: in particular, there is a possibility of developing antibodies directed against individual fragments of cardiac troponins or introducing inhibitors of the main proteolytic enzymes that catalyze troponin proteolysis into diagnostic test systems to reduce interference and more thoroughly interpret the test results, taking into account comorbidities that affect the activity of enzymes breaking down cardiac troponins, etc. Unfortunately, the number of fundamental studies devoted to the investigation of the processes of proteolytic cleavage of cardiac troponins in the bloodstream is extremely small. And to date, only one specific mechanism is known, described in the study by I. Katrukha et al61,145 According to the results of this study, the enzyme thrombin catalyzes the cleavage of the full-length molecule of cardiac troponin T (the molecular weight is 35 kDa) in the region of the peptide bond between amino acids 68 and 69 into two fragments, one of which is larger (the molecular weight is 29 kDa), and the second—smaller (the molecular weight is 6 kDa).145 As noted above, any significant effect on thrombin activity (for instance, the use of anticoagulants) can influence the fragmentation of troponin T and, accordingly, its diagnostic value.
In general, a number of research groups studying the processes of proteolytic cleavage in the bloodstream report the presence of a very large number of fragments (approximately several tens) of cardiac troponin molecules, which have different sizes (molecular weights from several kDa to 30 or more kDa), stability and half-life in the bloodstream (from several hours to a day) and conditions of formation (physiological conditions, the degree of severity and progression of ischemia, reperfusion time, etc.).144,145,176–180 A. Vylegzhanina et al studied the composition of troponin complexes in patients with AMI.180 The researchers have identified the following main forms of cardiac troponins in AMI: a ternary complex consisting of full-size cardiac troponins T and I and troponin C; a ternary complex consisting of truncated cardiac troponin I and integral troponins T and C; a binary complex consisting of truncated cardiac troponin I and troponin C, as well as a number of short fragments of cardiac troponin T and troponin I, formed mainly from the central part of the molecules. As AMI progressed, there was a decrease in the number of ternary complexes consisting of full-size cardiac troponins and an increase in the number of ternary and binary complexes consisting of truncated troponins, as well as an increase in the level of fragments of cardiac troponins.180 Such changes in the heterogeneous fraction of cardiac troponins are most likely due to an increase in the activity of proteolytic enzymes, which increase with the progression of ischemia and AMI, and, accordingly, cause fragmentation (truncation) of troponin proteins.
Very interesting data are presented by the researchers S. Zahran et al, who studied the degree of proteolytic degradation of cardiac troponin I in patients with varying degrees of ischemia and damage to cardiac muscle tissue.179 The researchers noted that the degree of proteolytic cleavage of cardiac troponin I increases with an increase in the severity of ischemia and myocardial injury: the highest degree of proteolytic cleavage of cardiac troponin I was characteristic of patients with ST-segment elevation AMI, while in patients with non-ST-segment elevation AMI the degree of troponin I degradation was significantly lower. The authors also found a decrease in the degree of proteolytic degradation of cardiac troponin I after reperfusion, which can probably be used to assess the quality of reperfusion. It is quite remarkable that the degree of proteolytic degradation of cardiac troponin I had a higher diagnostic value in AMI than the total serum concentration of cardiac troponin I.179
Summing up the role of the stage of cardiac troponin circulation, we should once again emphasize its potentially high diagnostic value for practical medicine. At the moment, this stage is a relatively poorly studied area of the biology of cardiac troponins. The main directions of further work in this area, in my opinion, should be as follows:
1) Study of the fundamental specific mechanisms of proteolytic degradation of cardiac troponins in the bloodstream both under normal conditions and under the conditions of simulated concomitant pathologies. This requires a targeted and thorough study of the potential effect of individual serum proteolytic enzymes (for example, specific thrombin-mediated degradation of cardiac troponin T).
2) Search for specific fragments of cardiac troponins, which are released at the earliest possible time after the onset of myocardial ischemia and the creation of antibodies to them, which will increase the sensitivity and specificity of troponin immunoassays.
3) Search for specific fragments of cardiac troponins, which have a small molecular weight and are able to pass through the glomerular and blood-salivary barriers. The creation of antibodies to these fragments will make it possible to develop specific highly sensitive test systems for the analysis of non-invasive biological fluids (urine and oral fluid) and for the introduction of new methods of non-invasive diagnostics and monitoring of cardiovascular pathologies, including AMI, into routine clinical practice.
4) Study and identification of potentially possible specific mechanisms of proteolytic cleavage of cardiac troponins under the action of other (non-ischemic) factors. This will allow the development of specific troponin immunoassays to identify those fragments that, for example, will increase exclusively with stretching of the myocardium or exclusively with an increase in the activity of the adrenergic nervous system and an increase in β-AR stimulation, etc. Thus, it will be possible to carry out a more specific diagnosis of non-ischemic myocardial damage in some physiological and pathological conditions not associated with ischemia of the cardiac muscle tissue.
Removal of Cardiac Troponins from the Bloodstream: Mechanisms and Diagnostic Value
The final stage of the metabolic pathway of cardiac troponins in the bloodstream is as important as the other two stages (release and circulation). Both of these stages are closely related to the terminal stage of the metabolic pathway of cardiac troponins. So, for example, when small fragments are released from cardiomyocytes (as a result of intracellular proteolytic cleavage of cardiac troponins), they will obviously be almost immediately removed from the bloodstream by filtration through the glomerular and blood-salivary barriers. When larger fragments of cardiac troponins and/or binary and ternary complexes are released, filtration of these molecules is unlikely due to their large size (molecular weight).
The circulation of cardiac troponins is equally closely related to the removal of these molecules. So, for example, with a higher activity of serum proteolytic enzymes, the process of degradation of cardiac troponin molecules will be more active, which will lead to more rapid formation of small troponin molecules and their filtration (removal) from the bloodstream.
In general, today, the mechanism of filtering cardiac troponins through the glomerular barrier is one of the main and definitively proven ways to remove cardiac troponins from the bloodstream. The inferential (indirect) evidence is that when the filtration rate decreases (for example, in CRF), cardiac troponin molecules accumulate in the bloodstream and their serum levels begin to rise sharply in those patients who do not have any signs of cardiovascular pathology and damage of myocardial cells. The more the renal function is suppressed (ie, the lower the filtration rate is), the higher the concentration of cardiac troponins in the bloodstream rises.189–191 In other pathological conditions that are accompanied by inhibition of the filtration rate, for example, in sepsis, serum levels of cardiac troponins are positively correlated with serum creatinine levels,192,193 which also accumulate due to a decrease in the filtration capacity of nephrons. From a pathogenetic point of view, any conditions accompanied by a drop in the filtration rate can contribute to the accumulation of troponins. This fact, of course, should be taken into account by medical practitioners when interpreting the results.
Recent clinical studies by several research groups can be considered as valuable evidence of the existence of a mechanism for the elimination of cardiac troponins across the filtration barrier.34,67 A key feature of these studies is the use of highly sensitive troponin immunoassays, which can detect small concentrations (from several ng/l to several tens of ng/l) of cardiac troponins in urine. According to these studies, it is also noteworthy that there is a possibility of non-invasive assessment of myocardial damage in arterial hypertension and diabetes mellitus,34,67 which is very convenient for non-hospital and outpatient settings. This will allow monitoring the patient’s condition, assessing the prognosis, and, on its basis, choosing/correcting the tactics of further management of patients, including their treatment. However, it should be noted that these methods have not yet been completely validated and research work in this direction should be continued before introducing new non-invasive methods for diagnosing and monitoring cardiovascular pathologies in routine clinical practice.
One of the key and very labile factors affecting the glomerular filtration rate (including the rate of removal of cardiac troponins from the bloodstream) is blood pressure. So, with a decrease in blood pressure, the filtration rate will slow down and the degree of removal of cardiac troponins from the bloodstream will decrease. This mechanism, in particular, can contribute to the fact that the molecules of cardiac troponins will increase much higher and circulate in the bloodstream for longer in pathological conditions accompanied by a sharp drop in blood pressure. This can be typical for large-focal myocardial infarctions, which are often accompanied by a sharp decrease in blood pressure (cardiogenic shock), and the degree of increase/duration of circulation of cardiac troponins in the bloodstream can be considered as a prognostically unfavorable sign.194 With an increase in blood pressure, the filtration rate may increase, and more cardiac troponin molecules will be filtered from the bloodstream into the urine. The evidence for a possible role of this mechanism comes from a clinical study showing that urinary troponin levels are higher in hypertensive patients than in those with normal blood pressure or those taking antihypertensive drugs.67
Another way of cardiac troponins removal is associated with the activity of the reticuloendothelial system, the cells of which capture protein molecules of cardiac markers from the bloodstream and cause their intracellular proteolytic cleavage.186,187,195,196 The clinical significance of this mechanism for removing cardiac troponins (as opposed to the mechanism for removing cardiac troponins through the glomerular filter) is difficult to judge, since there are no similar well-controlled clinical studies confirming the possibility of a significant increase in serum levels of cardiac troponins in case of the reticuloendothelial system dysfunction. In addition, in contrast to renal failure, dysfunctions of the components of the reticuloendothelial system are much less common.
As noted earlier, the proteolytic cleavage of troponin molecules in the bloodstream is an extremely understudied mechanism for the elimination of cardiac troponins. As a result of this mechanism, a large number of small fragments of cardiac troponins are formed, which can be immunoreactive (can interact with antibodies and be detected by immunoassays) and non-immunoreactive fragments (which will not interact with antibodies). From the point of view of laboratory diagnostics, non-immunoreactive fragments of troponins can be considered already removed from the bloodstream, since they will not bind to antibodies and thus will not have any effect on the result of laboratory diagnostics of AMI or any other pathology. To elucidate the mechanism of cardiac troponin removal by proteases, well-controlled basic research is needed to thoroughly investigate the role of individual serum proteolytic enzymes in the degradation of cardiac troponin molecules in the bloodstream.
Thus, there can be distinguished 3 main mechanisms of elimination of cardiac troponins from the bloodstream: 1) elimination (filtration) of cardiac troponins through the glomerular barrier, 2) removal of cardiac troponins from the bloodstream by cells of the reticuloendothelial system, 3) proteolytic cleavage of cardiac troponin molecules and/or their fragments in the bloodstream to non-immunoreactive forms. Taking into account the analysis of the literature, the main mechanism for the removal of cardiac troponins, in my opinion, is the elimination of troponins through a three-layer filtration (glomerular) barrier. This mechanism can have a significant impact on the diagnostics of cardiovascular diseases, including AMI, since impaired removal of cardiac troponins from the bloodstream is often accompanied by a significant increase in serum levels of cardiac troponins. In addition, many patients have comorbid pathologies, among which kidney damage (chronic renal failure) is relatively common. Many other common diseases, for example, diabetes mellitus, sepsis, are also often complicated by renal failure. Thus, they may increase serum levels of cardiac troponins in patients having no signs of cardiovascular diseases.
Removal of cardiac troponins from the bloodstream by glomerular filtration may have an important impact on rapid algorithms for diagnostics/exclusion of AMI. Thus, the research group led by P. Kavsak reported that the currently established upper threshold levels of troponins (99th percentile) for the diagnostics/exclusion of AMI can be used only for patients with an optimal glomerular filtration rate (≥90 mL/min).197 In patients who have lower glomerular filtration rate values, cardiac troponin levels will increase due to impaired elimination, which can lead to overdiagnosis of AMI if medical practitioners do not take renal function (filtration rate value) into account. Thus, it is necessary to stratify the threshold values of cardiac troponins taking into account different values of the filtration rate and, in particular, to develop special algorithms for diagnostics/exclusion of AMI for patients who suffer from concomitant CRF.
Finally, the filtration of troponin fragments through the blood-brain barrier into the cerebrospinal fluid and through the blood-salivary barrier into saliva can be considered as additional potential mechanisms for the removal of cardiac troponins. As evidence of the existence of these mechanisms, one can consider studies,35–37,68,74–76 which reported on the detection of cardiac troponins in the cerebrospinal fluid and saliva. The investigation of cardiac troponins in the cerebrospinal fluid can be used in forensic medicine, and the investigation of cardiac troponins in saliva—in clinical practice for diagnostics and monitoring of cardiovascular pathologies, including AMI. Overall, more research is needed to validate these diagnostic capabilities.
Circadian Rhythms of Cardiac Troponins: Possible Mechanisms of Formation and Diagnostic Role
The activity of many systems (organs, tissues, and cells) of our body changes cyclically during the day (with the change of day and night), which is commonly called circadian or diurnal rhythms. Circadian rhythms are an evolutionarily developed mechanism necessary to maintain optimal functioning of the body and adapt to changing environmental conditions.198
Due to the fact that the tissues and cells of our body change, there is a change in the concentration of a number of molecules (for example, hormones, metabolic products), which are produced or metabolized by these tissues and cells. Many of these molecules are laboratory biomarkers, the concentration of which is used to diagnose diseases.199,200 This must be taken into account in routine clinical practice, since changes in the concentration of biomarkers caused by natural circadian rhythms can be mistakenly interpreted as diagnostic signs and, accordingly, lead to diagnostic errors. Certain hormones, the release of which varies from day to night, can affect a number of other laboratory parameters that must also be considered when interpreting laboratory diagnostic results.
Recent clinical studies have reported that bloodstream levels of cardiac troponins are dependent on circadian rhythms. These studies used highly sensitive troponin immunoassays able to detect small fluctuations in the concentration of cardiac troponins in the bloodstream (at the level of several ng/l).46,47,201,202 For example, L. Klinkenberg et al revealed changes in troponin T concentration (detected by a highly sensitive method) in patients without signs of cardiovascular diseases. At the same time, the maximum levels of troponins were recorded in the morning (16.2 ng/l at 8:30), and the minimum—in the evening (12.1 ng/l at 19:30). In addition, when analyzing the hourly curve of serum levels of cardiac troponins, the researchers found very regular and gradual changes: for example, from the maximum morning concentrations of troponins there was a gradual decrease to the evening (minimum) concentrations of cardiac troponins, and then there was a gradual increase in concentrations to the maximum morning values.201 However, the researchers noted that such relatively minor circadian fluctuations in troponins would not have a significant impact on diagnostic algorithms for AMI, but should be considered for screening purposes. The levels of cardiac troponin I (also detected by a highly sensitive immunoassay) changed over a 24-hour period by no more than 1 ng/l, ie, had no significant circadian rhythms.201 However, this study investigated the circadian rhythms of troponins only in healthy people, but in conditions of concomitant pathologies (especially with damage to those organs that affect the metabolism of cardiac troponins), fluctuations in the circadian rhythms of troponins can be much more pronounced. Thus, in patients with concomitant CRF, troponin T and troponin I concentrations changed more significantly during the day. N. van der Linden et al202 reported that the maximum fluctuations in troponin T concentration in a patient with CRF during the 24-hour investigation period were about 50 ng/l, while the fluctuations in troponin T levels during one hour were about 20 ng/l, which, by the way, is a very significant contribution to the laboratory diagnosis of AMI. So, for example, if we take into account modern algorithms for diagnostics of non-ST-segment elevation AMI (Table 1)29 (where the change in the levels of cardiac troponins within 1–2 hours by only 5–10 ng/l is diagnostically significant), we can say that that troponin T circadian rhythms may affect the diagnostics of AMI and contribute to overdiagnosis.202 Troponin I levels showed slightly higher fluctuations in concentration during the day compared to the study by L. Klinkenberg et al,201 however, they would not reach the thresholds of 5–10 ng/l and thus would not have a significant effect on one- and two-hour algorithms of AMI diagnostics.
The precise mechanisms of the formation of circadian rhythms of cardiac troponins are unknown, however, it can be assumed that they will be associated with changes in the functional activity of those organs, tissues and cells that can somehow affect the metabolic pathway of cardiac troponins, in particular the stages of their release into the bloodstream, the stage of circulation (for example, the effect on the activity of proteolytic enzymes that cause the cleavage of troponins in the bloodstream) or the elimination stage (for example, the effect on the functional state of the kidneys). Among the most probable mechanisms for the formation of circadian rhythms of cardiac troponins, in my opinion, there are circadian fluctuations in the activity of the cortex and medulla of the adrenal glands, of the thyroid gland, and the activity of enzymes of the hemostatic system.54,203–206 A possible rationale for the formation of circadian rhythms is that peak troponin concentrations occur in the morning period, being the period of the maximum activity of the adrenal glands (producing elevated levels of catecholamines, cortisol), the thyroid gland (producing thyroid hormones, which can enhance the effects of catecholamines on myocardial cells). The increased activity of these organs also coincides with their main effects on the cardiovascular system, namely, in the morning period, patients have the highest heart rate, and the blood pressure is higher than in the evening-night period. In general, the increased activity of these organs is a kind of adaptive and evolutionary developed mechanism, which is necessary to maintain the period of wakefulness. However, we should take into account the negative impact of these organs and their metabolic products (for example, catecholamines, cortisol) on myocardial cells. The evidence of the adverse effect of cortisol on myocardial cells is a clinical study that demonstrates that increased levels of the stress hormone (cortisol) are associated with increased levels of cardiac troponin T.165 In addition, a number of researchers associate the increased high activity of the sympathoadrenal system with a larger size of the focus of myocardial necrosis in AMI, the incidence of acute cardiovascular diseases and an unfavorable prognosis.206–209
A possible explanation of the reason for the fact that the circadian rhythms of troponin T are more significant than the circadian rhythms of troponin I consists in their biochemical features, in particular, the volume of the cytoplasmic fraction of troponin T is almost twice the volume of the cytoplasmic fraction of troponin I (approximately 7–8% versus 3–4%).53,54 Thus, the cytoplasmic fraction of troponin T is more “mobile” and can be released into the bloodstream with an increase in the effect of a number of factors on the myocardium.210,211 There is a need for further clinical studies validating the circadian rhythms of cardiac troponins and their effect on the diagnostics of cardiovascular diseases, including AMI, and for fundamental research clarifying the molecular mechanisms of the formation of circadian rhythms of cardiac troponins.
Conclusion
The metabolic pathway of cardiac troponins includes three main stages (release of troponins from myocardial cells, circulation of cardiac troponins in blood plasma, removal of cardiac troponins from the bloodstream), each of which can have a very significant effect on serum levels of cardiac troponins, ie, on their diagnostic value.
It should be noted that many new views on the metabolism and diagnostic value of cardiac troponins (in particular, the role of circadian rhythms, gender- and age-related characteristics of concentrations, the possibility of detecting troponins in urine and saliva, etc.) were formed as a result of an increase in the sensitivity of troponin immunoassays.
Unfortunately, today many stages of the metabolic pathway of cardiac troponins and factors influencing the metabolic pathway of cardiac troponins are extremely poorly understood and are hypothetical and/or contradictory. In particular, the specific mechanisms of the release of cardiac troponins from the myocardium into the bloodstream as affected by physiological conditions and characteristics (physical exertion, stress, circadian fluctuations in the activity of organs and tissues that influence the release of cardiac troponin molecules) and non-ischemic pathologies, which are often accompanied by an increase in the concentration of cardiac troponins in the bloodstream, are little known. And factors affecting the circulation of cardiac troponin molecules in the bloodstream, in particular enzymes involved in the metabolism (fragmentation) of troponin molecules, remain unknown. The mechanisms of filtration (transport) of cardiac troponin molecules from the bloodstream to other biological fluids are not investigated, and, accordingly, these possibilities of non-invasive diagnostics have not been validated. Thus, the study of the metabolic pathway of cardiac troponins and potential factors influencing it is a relatively large and poorly studied area for further research that is needed to optimize diagnostics and validate new diagnostic capabilities.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Smith JN, Negrelli JM, Manek MB, Hawes EM, Viera AJ. Diagnosis and management of acute coronary syndrome: an evidence-based update. J Am Board Fam Med. 2015;28(2):283–293. PMID: 25748771. doi:10.3122/jabfm.2015.02.140189
2. Henderson RA. Acute coronary syndrome: optimising management through risk assessment. Clin Med. 2013;13(6):602–606. PMID: 24298110; PMCID: PMC5873665. doi:10.7861/clinmedicine.13-6-602
3. Makki N, Brennan TM, Girotra S. Acute coronary syndrome. J Intensive Care Med. 2015;30(4):186–200. Epub 2013 Sep 18. PMID: 24047692. doi:10.1177/0885066613503294
4. Chaulin AM, Grigorieva V, Pavlova TV, Duplyakov DV. Diagnostic significance of complete blood count in cardiovascular patients; Samara State Medical University. Russian J Cardiol. 2020;25(12):3923. doi:10.15829/1560-4071-2020-3923
5. Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60(16):1581–1598. Epub 2012 Sep 5. PMID: 22958960. doi:10.1016/j.jacc.2012.08.001
6. Thygesen K, Alpert JS, Jaffe AS, et al.; Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth Universal Definition of Myocardial Infarction (2018). Circulation. 2018;138(20):e618–e651. PMID: 30571511. doi:10.1161/CIR.0000000000000617
7. Eckner D, Pauschinger M, Ademaj F, Martinovic K. Universellen Definition des Myokardinfarkts [Clinical implications of the fourth universal definition of myocardial infarction]. Herz. 2020. 45(6):520–527. German. PMID: 32514585. doi:10.1007/s00059-020-04948-6
8. Takeda S. Crystal structure of troponin and the molecular mechanism of muscle regulation. J Electron Microsc. 2005;54 Suppl 1:i35–41. PMID: 16157639. doi:10.1093/jmicro/54.suppl_1.i35
9. Katrukha IA. Human cardiac troponin complex. Structure and functions. Biochemistry (Mosc). 2013;78(13):1447–1465. PMID: 24490734. doi:10.1134/S0006297913130063
10. Henderson CA, Gomez CG, Novak SM, Mi-Mi L, Gregorio CC. Overview of the Muscle Cytoskeleton. Compr Physiol. 2017;7(3):891–944. PMID: 28640448; PMCID: PMC5890934. doi:10.1002/cphy.c160033
11. Chaulin A. Clinical and diagnostic value of highly sensitive cardiac troponins in arterial hypertension. Vasc Health Risk Manag. 2021;17:431–443. PMID: 34366667; PMCID: PMC8336985. doi:10.2147/VHRM.S315376
12. Wei B, Jin JP. Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch Biochem Biophys. 2011;505(2):144–154. Epub 2010 Oct 18. PMID: 20965144; PMCID: PMC3018564. doi:10.1016/j.abb.2010.10.013
13. Jin JP. Evolution, Regulation, and Function of N-terminal Variable Region of Troponin T: modulation of Muscle Contractility and Beyond. Int Rev Cell Mol Biol. 2016;321:1–28. Epub 2015 Nov 4. PMID: 26811285. doi:10.1016/bs.ircmb.2015.09.002
14. Chaulin A. Cardiac troponins: contemporary biological data and new methods of determination. Vasc Health Risk Manag. 2021;17:299–316. PMID: 34113117; PMCID: PMC8184290. doi:10.2147/VHRM.S300002
15. Wang XY, Zhang F, Zhang C, Zheng LR, Yang J. The biomarkers for acute myocardial infarction and heart failure. Biomed Res Int. 2020;2020:2018035. PMID: 32016113; PMCID: PMC6988690. doi:10.1155/2020/2018035
16. Apple FS, Sandoval Y, Jaffe AS, Ordonez-Llanos J, Task IFCC. Force on Clinical applications of cardiac bio-markers. cardiac troponin assays: guide to understanding analytical characteristics and their impact on clinical care. Clin Chem. 2017;63(1):73–81. Epub 2016 Oct 10. PMID: 28062612. doi:10.1373/clinchem.2016.255109
17. Aakre KM, Saeed N, Wu AHB, Kavsak PA. Analytical performance of cardiac troponin assays - Current status and future needs. Clin Chim Acta. 2020;509:149–155. Epub 2020 Jun 12. PMID: 32540128. doi:10.1016/j.cca.2020.06.021
18. Chaulin AM, Duplyakov DV. MicroRNAs in atrial fibrillation: pathophysiological aspects and potential biomarkers. Int J Biomed. 2020;10:198–205. doi:10.21103/Article10(3)_RA3
19. Chaulin AM, Abashina OE, Duplyakov DV. Pathophysiological mechanisms of cardiotoxicity in chemotherapeutic agents. Russ Open Med J. 2020;9:e0305. doi:10.15275/rusomj.2020.0305
20. Chuang AM, Nguyen MT, Kung WM, Lehman S, Chew DP. High-sensitivity troponin in chronic kidney disease: considerations in myocardial infarction and beyond. Rev Cardiovasc Med. 2020;21(2):191–203. PMID: 32706208. doi:10.31083/j.rcm.2020.02.17
21. Chaulin AM, Duplyakov DV. Arrhythmogenic effects of doxorubicin. Complex Issues Cardiovasc Dis. 2020;9:69–80. doi:10.17802/2306-1278-2020-9-3-69-80
22. Chaulin AM, Duplyakov DV. Increased natriuretic peptides not associated with heart failure. Russ J Cardiol. 2020;25:4140. doi:10.15829/1560-4071-2020-4140
23. Stavroulakis GA, George KP. Exercise-induced release of troponin. Clin Cardiol. 2020;43(8):872–881. Epub 2020. PMID: 31975465; PMCID: PMC7403670. doi:10.1002/clc.23337
24. Chauin A. The Main Causes and Mechanisms of Increase in Cardiac Troponin Concentrations Other Than Acute Myocardial Infarction (Part 1): physical Exertion, Inflammatory Heart Disease, Pulmonary Embolism, Renal Failure, Sepsis. Vasc Health Risk Manag. 2021;17:601–617. PMID: 34584417; PMCID: PMC8464585. doi:10.2147/VHRM.S327661
25. Lindner G, Pfortmueller CA, Braun CT, Exadaktylos AK. Non-acute myocardial infarction-related causes of elevated high-sensitive troponin T in the emergency room: a cross-sectional analysis. Intern Emerg Med. 2014;9(3):335–339. Epub 2013 Dec 11. PMID: 24326466. doi:10.1007/s11739-013-1030-y
26. Wu W, Li DX, Wang Q, Xu Y, Cui YJ. Relationship between high-sensitivity cardiac troponin T and the prognosis of elderly inpatients with non-acute coronary syndromes. Clin Interv Aging. 2018;13:1091–1098. PMID: 29922047; PMCID: PMC5995414. doi:10.2147/CIA.S157048
27. Askin L, Tanriverdi O, Turkmen S. Clinical importance of high- sensitivity troponin T in patients without coronary artery disease. North Clin Istanb. 2020;7(3):305–310. PMID: 32478307; PMCID: PMC7251271. doi:10.14744/nci.2019.71135
28. Qin ZJ, Wu QY, Deng Y, et al. Association Between High-Sensitivity Troponin T on Admission and Organ Dysfunction During Hospitalization in Patients Aged 80 Years and Older with Hip Fracture: a Single-Centered Prospective Cohort Study. Clin Interv Aging. 2021;16:583–591. PMID: 33854308; PMCID: PMC8039433. doi:10.2147/CIA.S303246
29. Collet JP, Thiele H, Barbato E; ESC Scientific Document Group. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J. 2021;42(14):1289–1367. PMID: 32860058. doi:10.1093/eurheartj/ehaa575.
30. Odsæter IH, Grenne B, Hov GG, Laugsand LE, Wiseth R, Mikkelsen G. Establishing the 99th percentile of a novel assay for high-sensitivity troponin I in a healthy blood donor population. Clin Chem Lab Med. 2020;58(9):1557–1563. PMID: 32286238. doi:10.1515/cclm-2019-1023
31. Bahadur K, Ijaz A, Salahuddin M, Alam A. Determination of high sensitive cardiac troponin I 99th percentile upper reference limits in a healthy Pakistani population. Pak J Med Sci. 2020;36(6):1303–1307. PMID: 32968398; PMCID: PMC7501037. doi:10.12669/pjms.36.6.2328
32. Koerbin G, Tate J, Potter JM, Cavanaugh J, Glasgow N, Hickman PE. Characterisation of a highly sensitive troponin I assay and its application to a cardio-healthy population. Clin Chem Lab Med. 2012;50(5):871–878. PMID: 22628331. doi:10.1515/cclm-2011-0540
33. Abe N, Tomita K, Teshima M, et al. Distribution of cardiac troponin I in the Japanese general population and factors influencing its concentrations. J Clin Lab Anal. 2018;32(3):e22294. Epub 2017 Aug 1. PMID: 28763113; PMCID: PMC5888119. doi:10.1002/jcla.22294
34. Chen JY, Lee SY, Li YH, Lin CY, Shieh MD, Ciou DS. Urine high-sensitivity troponin I predict incident cardiovascular events in patients with diabetes mellitus. J Clin Med. 2020;9(12):3917. PMID: 33276667; PMCID: PMC7761585. doi:10.3390/jcm9123917
35. Chaulin AM, Karslyan LS, Bazyuk EV, Nurbaltaeva DA, Duplyakov DV. [Clinical and diagnostic value of cardiac markers in human biological fluids]. Kardiologiia. 2019. 59(11):66–75. Russian. PMID: 31849301. doi:10.18087/cardio.2019.11.n414
36. Chaulin AM, Duplyakova PD, Bikbaeva GR, Tukhbatova AA, Grigorieva EV, Duplyakov DV. Concentration of high-sensitivity cardiac troponin I in the oral fluid in patients with acute myocardial infarction: a pilot study. Russian J Cardiol. 2020;25(12):3814. doi:10.15829/1560-4071-2020-3814
37. Mirzaii-Dizgah I, Riahi E. Salivary high-sensitivity cardiac troponin T levels in patients with acute myocardial infarction. Oral Dis. 2013;19(2):180–184. Epub 2012 Jul 27. PMID: 22834943. doi:10.1111/j.1601-0825.2012.01968.x
38. Garcia-Osuna A, Gaze D, Grau-Agramunt M, et al. Ultrasensitive quantification of cardiac troponin I by a Single Molecule Counting method: analytical validation and biological features. Clin Chim Acta. 2018;486:224–231. Epub 2018 Aug 12. PMID: 30110608. doi:10.1016/j.cca.2018.08.015
39. Giannitsis E, Mueller-Hennessen M, Zeller T, et al. Gender-specific reference values for high-sensitivity cardiac troponin T and I in well-phenotyped healthy individuals and validity of high-sensitivity assay designation. Clin Biochem. 2020;78:18–24. Epub 2019 Nov 28. PMID: 31786204. doi:10.1016/j.clinbiochem.2019.11.013
40. Rocco E, La Rosa G, Liuzzo G, Biasucci LM. High-sensitivity cardiac troponin assays and acute coronary syndrome: a matter of sex? J Cardiovasc Med. 2019;20(8):504–509. PMID: 31259857. doi:10.2459/JCM.0000000000000811
41. Romiti GF, Cangemi R, Toriello F, et al. Sex-specific cut-offs for high-sensitivity cardiac troponin: is less more? Cardiovasc Ther. 2019;2019:9546931. PMID: 31772621; PMCID: PMC6739766. doi:10.1155/2019/9546931
42. Monneret D, Gellerstedt M, Bonnefont-Rousselot D. Determination of age- and sex-specific 99th percentiles for high-sensitive troponin T from patients: an analytical imprecision- and partitioning-based approach. Clin Chem Lab Med. 2018;56(5):818–829. PMID: 29176015. doi:10.1515/cclm-2017-0256
43. Bohn MK, Higgins V, Kavsak P, Hoffman B, Adeli K. High-sensitivity generation 5 cardiac troponin t sex- and age-specific 99th percentiles in the caliper cohort of healthy children and adolescents. Clin Chem. 2019;65(4):589–591. Epub 2019. PMID: 30737206. doi:10.1373/clinchem.2018.299156
44. Boeddinghaus J, Nestelberger T, Twerenbold R, et al.; TRAPID-AMI Investigators. Impact of age on the performance of the ESC 0/1h-algorithms for early diagnosis of myocardial infarction. Eur Heart J. 2018;39(42):3780–3794. PMID: 30169752. doi:10.1093/eurheartj/ehy514
45. Gore MO, Seliger SL, Defilippi CR, et al. Age- and sex-dependent upper reference limits for the high-sensitivity cardiac troponin T assay. J Am Coll Cardiol. 2014;63(14):1441–1448. Epub 2014 Feb 12. PMID: 24530665; PMCID: PMC3984900. doi:10.1016/j.jacc.2013.12.032
46. Fournier S, Iten L, Marques-Vidal P, et al. Circadian rhythm of blood cardiac troponin T concentration. Clin Res Cardiol. 2017;106(12):1026–1032. Epub 2017 Aug 30. PMID: 28856443. doi:10.1007/s00392-017-1152-8
47. Klinkenberg LJ, van Dijk JW, Tan FE, van Loon LJ, van Dieijen-visser MP, Meex SJ. Circulating cardiac troponin T exhibits a diurnal rhythm. J Am Coll Cardiol. 2014;63(17):1788–1795. Epub 2014 Feb 26. PMID: 24583293. doi:10.1016/j.jacc.2014.01.040
48. Chaulin AM, Duplyakov DV. High-sensitivity cardiac troponins: circadian rhythms. Cardiovasc Therapy Prevent. 2021;20(1):2639. doi:10.15829/1728-8800-2021-2639
49. Chaulin AM, Abashina OE, Duplyakov DV. High-sensitivity cardiac troponins: detection and central analytical characteristics. Cardiovascular Therapy and Prevention. J Chem Med. 2021;20(2):2590. doi:10.15829/1728-8800-2021-2590
50. Eggers KM, Lindahl B. Impact of Sex on Cardiac Troponin Concentrations-A Critical Appraisal. Clin Chem. 2017;63(9):1457–1464. Epub 2017 Jun 19. PMID: 28630238. doi:10.1373/clinchem.2017.271684
51. Sedighi SM, Prud’Homme P, Ghachem A, et al. Increased level of high-sensitivity cardiac Troponin T in a geriatric population is determined by comorbidities compared to age. Int J Cardiol Heart Vasc. 2019;22:187–191. PMID: 30963093; PMCID: PMC6437284. doi:10.1016/j.ijcha.2019.02.015
52. Hickman PE, Abhayaratna WP, Potter JM, Koerbin G. Age-related differences in hs-cTnI concentration in healthy adults. Clin Biochem. 2019;69:26–29. Epub 2019 Apr 25. PMID: 31028731. doi:10.1016/j.clinbiochem.2019.04.014
53. Chaulin AM, Duplyakova PD, Duplyakov DV. Circadian rhythms of cardiac troponins: mechanisms and clinical significance. Russian J Cardiol. 2020;25(3S):4061. doi:10.15829/1560-4071-2020-4061
54. Chaulin AM, Duplyakov DV. On the potential effect of circadian rhythms of cardiac troponins on the diagnosis of acute myocardial infarction. Signa Vitae. 2021;17:79–84. doi:10.22514/sv.2021.050
55. Vogiatzis I. Circadian rhythm of cardiac troponins. Does it really exist? Int J Cardiol. 2018;270:72–73. Epub 2018 Jul 7. PMID: 30017523. doi:10.1016/j.ijcard.2018.07.035
56. Zaninotto M, Padoan A, Mion MM, Marinova M, Plebani M. Short-term biological variation and diurnal rhythm of cardiac troponin I (Access hs-TnI) in healthy subjects. Clin Chim Acta. 2020;504:163–167. Epub 2020 Feb 5. PMID: 32035091. doi:10.1016/j.cca.2020.02.004
57. Wildi K, Singeisen H, Twerenbold R, et al.; APACE Investigators. Circadian rhythm of cardiac troponin I and its clinical impact on the diagnostic accuracy for acute myocardial infarction. Int J Cardiol. 2018;270:14–20. Epub 2018 Jun 4. PMID: 29891238. doi:10.1016/j.ijcard.2018.05.136
58. Tate JR, Bunk DM, Christenson RH, et al.; Group on Standardization of Troponin I. Standardisation of cardiac troponin I measurement: past and present. Pathology. 2010;42(5):402–408. PMID: 20632814. doi:10.3109/00313025.2010.495246
59. Panteghini M, Bunk DM, Christenson RH, et al.; Group on Standardization of Troponin I. Standardization of troponin I measurements: an update. Clin Chem Lab Med. 2008;46(11):1501–1506. PMID: 18778218. doi:10.1515/CCLM.2008.291
60. Jarolim P. High sensitivity cardiac troponin assays in the clinical laboratories. Clin Chem Lab Med. 2015;53(5):635–652. PMID: 25252753. doi:10.1515/cclm-2014-0565
61. Katrukha IA, Kogan AE, Vylegzhanina AV, et al. Full-size cardiac troponin I and its proteolytic fragments in blood of patients with acute myocardial infarction: antibody selection for assay development. Clin Chem. 2018;64(7):1104–1112. Epub 2018 Apr 9. PMID: 29632125. doi:10.1373/clinchem.2017.286211
62. Streng AS, de Boer D, van der Velden J, van Dieijen-visser MP, Wodzig WK. Posttranslational modifications of cardiac troponin T: an overview. J Mol Cell Cardiol. 2013;63:47–56. Epub 2013 Jul 18. PMID: 23871791. doi:10.1016/j.yjmcc.2013.07.004
63. Mirzaii-Dizgah I, Riahi E. Salivary troponin I as an indicator of myocardial infarction. Indian J Med Res. 2013;138(6):861–865. PMID: 24521627; PMCID: PMC3978973.
64. Bahbah EI, Noehammer C, Pulverer W, Jung M, Weinhaeusel A. Salivary biomarkers in cardiovascular disease: an insight into the current evidence. FEBS J. 2021;288(22):6392–6405. Epub 2021. PMID: 33370493. doi:10.1111/febs.15689
65. Abdul Rehman S, Khurshid Z, Hussain Niazi F, et al. Role of salivary biomarkers in detection of cardiovascular diseases (CVD). Proteomes. 2017;5(3):21. PMID: 28783097; PMCID: PMC5620538. doi:10.3390/proteomes5030021
66. Klichowska-Palonka M, Załęska-Chromińska K, Bachanek T. [Possibility of using saliva as a diagnostic test material in cardiovascular diseases]. Wiad Lek. 2015;68(3 pt 2):354–357. Polish. PMID: 28501835.
67. Pervan P, Svaguša T, Prkačin I, Savuk A, Bakos M, Perkov S. Urine high sensitive Troponin I measuring in patients with hypertension. Signa Vitae. 2017;13:62–64. doi:10.22514/SV133.062017.13
68. Mishra V, Patil R, Khanna V, et al. Evaluation of salivary cardiac troponin-I as potential marker for detection of acute myocardial infarction. J Clin Diagn Res. 2018;12:44–47. doi:10.7860/JCDR/2018/32109.11791
69. Chaulin AM. Phosphorylation and fragmentation of the cardiac troponin T: mechanisms, role in pathophysiology and laboratory diagnosis. Int J Biomed. 2021;11:250–259. doi:10.21103/Article11(3)_RA2
70. Ziebig R, Lun A, Hocher B, et al. Renal elimination of troponin T and troponin I. Clin Chem. 2003;49(7):1191–1193. PMID: 12816921. doi:10.1373/49.7.1191
71. Ellis K, Dreisbach AW, Lertora JL. Plasma elimination of cardiac troponin I in end-stage renal disease. South Med J. 2001;94(10):993–996. PMID: 11702827. doi:10.1097/00007611-200194100-00011
72. Zhu BL, Ishikawa T, Michiue T, et al. Postmortem cardiac troponin I and creatine kinase MB levels in the blood and pericardial fluid as markers of myocardial damage in medicolegal autopsy. Leg Med. 2007;9(5):241–250. doi:10.1016/j.legalmed.2007.01.010
73. González-Herrera L, Valenzuela A, Ramos V, Blázquez A, Villanueva E. Cardiac troponin T determination by a highly sensitive assay in postmortem serum and pericardial fluid. Forensic Sci Med Pathol. 2016;12(2):181–188. doi:10.1007/s12024-016-9749-1
74. Maeda H, Michiue T, Zhu BL, Ishikawa T, Quan L. Analysis of cardiac troponins and creatine kinase MB in cerebrospinal fluid in medicolegal autopsy cases. Leg Med. 2009;11(Suppl 1):S266–8. doi:10.1016/j.legalmed.2009.01.005
75. Wang Q, Michiue T, Ishikawa T, Zhu BL, Maeda H. Combined analyses of creatine kinase MB, cardiac troponin I and myoglobin in pericardial and cerebrospinal fluids to investigate myocardial and skeletal muscle injury in medicolegal autopsy cases. Leg Med. 2011;13(5):226–232. doi:10.1016/j.legalmed.2011.05.002
76. Chen JH, Inamori-Kawamoto O, Michiue T, Ikeda S, Ishikawa T, Maeda H. Cardiac biomarkers in blood, and pericardial and cerebrospinal fluids of forensic autopsy cases: a reassessment with special regard to postmortem interval. Leg Med. 2015;17(5):343–350. doi:10.1016/j.legalmed.2015.03.007
77. Stefanovic V, Loukovaara M. Amniotic fluid cardiac troponin T in pathological pregnancies with evidence of chronic fetal hypoxia. Croat Med J. 2005;46(5):801–807. PMID: 16158475.
78. Yoshida M, Matsuda H, Yoshinaga Y, et al. Analysis about the influence on the fetus infected with parvovirus B19 using amniotic erythropoietin and troponin-T. Arch Gynecol Obstet. 2013;288(3):521–525. doi:10.1007/s00404-013-2815-y
79. Blohm ME, Arndt F, Fröschle GM, et al. Cardiovascular biomarkers in amniotic fluid, umbilical arterial blood, umbilical venous blood, and maternal blood at delivery, and their reference values for full-term, singleton, cesarean deliveries. Front Pediatr. 2019;7:271. doi:10.3389/fped.2019.00271
80. Van Mieghem T, Doné E, Gucciardo L, et al. Amniotic fluid markers of fetal cardiac dysfunction in twin-to-twin transfusion syndrome. Am J Obstet Gynecol. 2010;202(1):
81. Chaulin AM, Duplyakov DV. Cardiac troponins: current data on the diagnostic value and analytical characteristics of new determination methods. Cor Vasa. 2021;63:486–493. doi:10.33678/cor.2021.041
82. Kavsak PA. Should detectable cardiac troponin concentrations in a healthy population be the only criterion for classifying high-sensitivity cardiac troponin assays? Clin Biochem. 2018;56:1–3. Epub 2018 May 24. PMID: 29803619. doi:10.1016/j.clinbiochem.2018.05.015
83. Ji M, Moon HW, Hur M, Yun YM. Determination of high-sensitivity cardiac troponin I 99th percentile upper reference limits in a healthy Korean population. Clin Biochem. 2016;49(10–11):756–761. Epub 2016 Apr 9. PMID: 27067595. doi:10.1016/j.clinbiochem.2016.01.027
84. Tjora S, Hall TS, Larstorp AC, Hallen J, Atar D. Increases in circulating cardiac troponin are not always associated with myocardial cell death. Clin Lab. 2018;64(11). PMID: 30549980. doi:10.7754/Clin.Lab.2018.180615
85. Jaffe AS, Wu AH. Troponin release–reversible or irreversible injury? Should we care? Clin Chem. 2012;58(1):148–150. Epub 2011 Oct 28. doi:10.1373/clinchem.2011.173070
86. Mair J, Lindahl B, Hammarsten O, et al. How is cardiac troponin released from injured myocardium? Eur Heart J Acute Cardiovasc Care. 2018;7(6):553–560. Epub 2017 Dec 27. PMID: 29278915. doi:10.1177/2048872617748553
87. Hammarsten O, Mair J, Möckel M, Lindahl B, Jaffe AS. Possible mechanisms behind cardiac troponin elevations. Biomarkers. 2018; (8):725–734. Epub 2018 Aug 23. PMID: 29976112. doi:10.1080/1354750X.2018.1490969
88. Gumprecht J, Domek M, Lip GYH, Shantsila A. Invited review: hypertension and atrial fibrillation: epidemiology, pathophysiology, and implications for management. J Hum Hypertens. 2019;33(12):824–836. Epub 2019 Nov 5. PMID: 31690818. doi:10.1038/s41371-019-0279-7
89. Liao XD, Wang XH, Jin HJ, Chen LY, Chen Q. Mechanical stretch induces mitochondria-dependent apoptosis in neonatal rat cardiomyocytes and G2/M accumulation in cardiac fibroblasts. Cell Res. 2004;14(1):16–26. PMID: 15040886. doi:10.1038/sj.cr.7290198
90. Cheng WP, Wang BW, Lo HM, Shyu KG. Mechanical Stretch Induces Apoptosis Regulator TRB3 in. Cardiomyocytes C, Heart V-O. PLoS One. 2015;10(4):e0123235. PMID: 25898323; PMCID: PMC4405267. doi:10.1371/journal.pone.0123235
91. Jiang S, Huo D, Wang X, et al. β-adrenergic Receptor-stimulated Cardiac Myocyte Apoptosis: role of Cytochrome P450 ω-hydroxylase. J Cardiovasc Pharmacol. 2017;70(2):94–101. PMID: 28768289. doi:10.1097/FJC.0000000000000499
92. Communal C, Colucci WS. The control of cardiomyocyte apoptosis via the beta-adrenergic signaling pathways. Arch Mal Coeur Vaiss. 2005;98(3):236–241. PMID: 15816327.
93. Dalal S, Foster CR, Das BC, Singh M, Singh K. Β-adrenergic receptor stimulation induces endoplasmic reticulum stress in adult cardiac myocytes: role in apoptosis. Mol Cell Biochem. 2012;364(1–2):59–70. Epub 2012. PMID: 22270541; PMCID: PMC3320150. doi:10.1007/s11010-011-1205-7
94. Chen QM, Tu VC. Apoptosis and heart failure: mechanisms and therapeutic implications. Am J Cardiovasc Drugs. 2002;2(1):43–57. PMID: 14727998. doi:10.2165/00129784-200202010-00006
95. Kunapuli S, Rosanio S, Schwarz ER. “How do cardiomyocytes die?” apoptosis and autophagic cell death in cardiac myocytes. J Card Fail. 2006;12(5):381–391. PMID: 16762802. doi:10.1016/j.cardfail.2006.02.002
96. Ricchiuti V, Apple FS. RNA expression of cardiac troponin T isoforms in diseased human skeletal muscle. Clin Chem. 1999;45(12):2129–2135. PMID: 10585344. doi:10.1093/clinchem/45.12.2129
97. Ricchiuti V, Voss EM, Ney A, Odland M, Anderson PA, Apple FS. Cardiac troponin T isoforms expressed in renal diseased skeletal muscle will not cause false-positive results by the second generation cardiac troponin T assay by Boehringer Mannheim. Clin Chem. 1998;44(9):1919–1924. PMID: 9732977. doi:10.1093/clinchem/44.9.1919
98. Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324(5923):98–102. PMID: 19342590; PMCID: PMC2991140. doi:10.1126/science.1164680
99. Bergmann O, Zdunek S, Frisén J, Bernard S, Druid H, Jovinge S. Cardiomyocyte renewal in humans. Circ Res. 2012;110(1):e17. PMID: 22223215. doi:10.1161/CIRCRESAHA.111.259598
100. White HD. Pathobiology of troponin elevations: do elevations occur with myocardial ischemia as well as necrosis? J Am Coll Cardiol. 2011;57(24):2406–2408. PMID: 21658560. doi:10.1016/j.jacc.2011.01.029
101. Nakada Y, Canseco DC, Thet S, et al. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541(7636):222–227. Epub 2016 Oct 31. PMID: 27798600. doi:10.1038/nature20173
102. Lázár E, Sadek HA, Bergmann O. Cardiomyocyte renewal in the human heart: insights from the fall-out. Eur Heart J. 2017;38(30):2333–2342. PMID: 28810672; PMCID: PMC5837331. doi:10.1093/eurheartj/ehx343
103. Foglia MJ, Poss KD. Building and re-building the heart by cardiomyocyte proliferation. Development. 2016;143(5):729–740. PMID: 26932668; PMCID: PMC4813344. doi:10.1242/dev.132910
104. Docshin PM, Karpov AA, Eyvazova SD, et al. Activation of Cardiac Stem Cells in Myocardial Infarction. Cell Tissue Biol. 2018;12:175–182. doi:10.1134/S1990519X18030045
105. Waring CD, Vicinanza C, Papalamprou A, et al. The adult heart responds to increased workload with physiologic hypertrophy, cardiac stem cell activation, and new myocyte formation. Eur Heart J. 2014;35(39):2722–2731. Epub 2012 Oct 25. PMID: 23100284; PMCID: PMC4196078. doi:10.1093/eurheartj/ehs338
106. Rovira M, Borràs DM, Marques IJ, Puig C, Planas JV. Physiological responses to swimming-induced exercise in the adult zebrafish regenerating heart. Front Physiol. 2018;9:1362. PMID: 30327615; PMCID: PMC6174316. doi:10.3389/fphys.2018.01362
107. Schüttler D, Clauss S, Weckbach LT, Brunner S. Molecular mechanisms of cardiac remodeling and regeneration in physical exercise. Cells. 2019;8(10):1128. PMID: 31547508; PMCID: PMC6829258. doi:10.3390/cells8101128
108. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016;365(3):563–581. Epub 2016 Jun 21. PMID: 27324127; PMCID: PMC5010608. doi:10.1007/s00441-016-2431-9
109. Isomi M, Sadahiro T, Ieda M. Progress and Challenge of Cardiac Regeneration to Treat Heart Failure. J Cardiol. 2019;73(2):97–101. Epub 2018 Nov 9. PMID: 30420106. doi:10.1016/j.jjcc.2018.10.002
110. Zhang J, Liu D, Zhang M, Zhang Y. Programmed necrosis in cardiomyocytes: mitochondria, death receptors and beyond. Br J Pharmacol. 2019;176(22):4319–4339. Epub 2018 Jun 25. PMID: 29774530; PMCID: PMC6887687. doi:10.1111/bph.14363
111. Lee Y, Gustafsson AB. Role of apoptosis in cardiovascular disease. Apoptosis. 2009;14(4):536–548. PMID: 19142731. doi:10.1007/s10495-008-0302-x
112. Kyrylkova K, Kyryachenko S, Leid M, Kioussi C. Detection of apoptosis by TUNEL assay. Methods Mol Biol. 2012;887:41. PMID: 22566045. doi:10.1007/978-1-61779-860-3_5
113. Zorc-Pleskovic R, Alibegović A, Zorc M, Milutinović A, Radovanović N, Petrović D. Apoptosis of cardiomyocytes in myocarditis. Folia Biol. 2006;52(1–2):6–9. PMID: 17007104.
114. Zhang Q, Yu N, Yu BT. MicroRNA-298 regulates apoptosis of cardiomyocytes after myocardial infarction. Eur Rev Med Pharmacol Sci. 2018;22(2):532–539. PMID: 29424914. doi:10.26355/eurrev_201801_14206
115. Weil BR, Young RF, Shen X, et al. Brief myocardial ischemia produces cardiac troponin I release and focal myocyte apoptosis in the absence of pathological infarction in swine. JACC Basic Transl Sci. 2017;2(2):105–114. Epub 2017 Mar 29. PMID: 28979949; PMCID: PMC5624553. doi:10.1016/j.jacbts.2017.01.006
116. Cheng W, Li B, Kajstura J, et al. Stretch-induced programmed myocyte cell death. J Clin Invest. 1995;96(5):2247–2259. PMID: 7593611; PMCID: PMC185875. doi:10.1172/JCI118280
117. Gherasim L. Troponins in heart failure - a perpetual challenge. Maedica. 2019;14(4):371–377. PMID: 32153668; PMCID: PMC7035435. doi:10.26574/maedica.2019.14.4.371
118. Aengevaeren VL, Baggish AL, Chung EH, et al. Exercise-Induced Cardiac Troponin Elevations: from Underlying Mechanisms to Clinical Relevance. Circulation. 2021;144(24):1955–1972. Epub 2021 Dec 13. PMID: 34898243; PMCID: PMC8663527. doi:10.1161/CIRCULATIONAHA.121.056208
119. Park KC, Gaze DC, Collinson PO, Marber MS. Cardiac troponins: from myocardial infarction to chronic disease. Cardiovasc Res. 2017;113(14):1708–1718. PMID: 29016754; PMCID: PMC5852618. doi:10.1093/cvr/cvx183
120. Weil BR, Suzuki G, Young RF, Iyer V, Canty JM. Troponin release and reversible left ventricular dysfunction after transient pressure overload. J Am Coll Cardiol. 2018;71(25):2906–2916. PMID: 29929614; PMCID: PMC6020832. doi:10.1016/j.jacc.2018.04.029
121. Felker GM, Fudim M. Unraveling the mystery of troponin elevation in heart failure. J Am Coll Cardiol. 2018;71(25):2917–2918. PMID: 29929615. doi:10.1016/j.jacc.2018.03.537
122. Sanchez O, Planquette B, Wermert D, Marié E, Meyer G. Embolies pulmonaires graves [Massive pulmonary embolism]. Presse Med. 2008. 37(10):1439–1446. French. Epub 2008 Sep 4. PMID: 18775637. doi:10.1016/j.lpm.2008.07.003
123. El-Menyar A, Sathian B, Al-Thani H. Elevated serum cardiac troponin and mortality in acute pulmonary embolism: systematic review and meta-analysis. Respir Med. 2019;157:26–35. Epub 2019 Aug 23. PMID: 31476570. doi:10.1016/j.rmed.2019.08.011
124. Daquarti G, March Vecchio N, Mitrione CS, et al. High-sensitivity troponin and right ventricular function in acute pulmonary embolism. Am J Emerg Med. 2016;34(8):1579–1582. Epub 2016 May 29. PMID: 27306263. doi:10.1016/j.ajem.2016.05.071
125. Singh K, Communal C, Sawyer DB, Colucci WS. Adrenergic regulation of myocardial apoptosis. Cardiovasc Res. 2000;45(3):713–719. PMID: 10728393. doi:10.1016/s0008-6363(99)00370-3
126. Colucci WS, Sawyer DB, Singh K, Communal C. Adrenergic overload and apoptosis in heart failure: implications for therapy. J Card Fail. 2000;6(2 Suppl 1):1–7. PMID: 10908092.
127. Xiao RP, Tomhave ED, Wang DJ, et al. Age-associated reductions in cardiac beta1- and beta2-adrenergic responses without changes in inhibitory G proteins or receptor kinases. J Clin Invest. 1998;101(6):1273–1282. PMID: 9502768; PMCID: PMC508681. doi:10.1172/JCI1335
128. Mougenot N, Mika D, Czibik G, et al. Cardiac adenylyl cyclase overexpression precipitates and aggravates age-related myocardial dysfunction. Cardiovasc Res. 2019;115(12):1778–1790. PMID: 30605506; PMCID: PMC6755357. doi:10.1093/cvr/cvy306
129. de Lucia C, Eguchi A, Koch WJ. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front Pharmacol. 2018;9:904. PMID: 30147654; PMCID: PMC6095970. doi:10.3389/fphar.2018.00904
130. Schwartz P, Piper HM, Spahr R, Spieckermann PG. Ultrastructure of cultured adult myocardial cells during anoxia and reoxygenation. Am J Pathol. 1984;115(3):349–361. PMID: 6731585; PMCID: PMC1900509.
131. Siegmund B, Koop A, Klietz T, Schwartz P, Piper HM. Sarcolemmal integrity and metabolic competence of cardiomyocytes under anoxia-reoxygenation. Am J Physiol. 1990;258(2 Pt 2):H285–91. PMID: 2309898. doi:10.1152/ajpheart.1990.258.2.H285
132. Piper HM, Schwartz P, Spahr R, Hütter JF, Spieckermann PG. Absence of reoxygenation damage in isolated heart cells after anoxic injury. Pflugers Arch. 1984;401(1):71–76. PMID: 6473067. doi:10.1007/BF00581535
133. Chaulin AM. Updated information about methods of identification and diagnostic opportunities of cardiac troponins. Medicina di Lab. 2016;17(3):154–164. doi:10.23736/S1825-859X.21.00116-X
134. Aakre KM, Omland T. Physical activity, exercise and cardiac troponins: clinical implications. Prog Cardiovasc Dis. 2019;62(2):108–115. Epub 2019 Feb 22. PMID: 30797799. doi:10.1016/j.pcad.2019.02.005
135. Sheyin O, Davies O, Duan W, Perez X. The prognostic significance of troponin elevation in patients with sepsis: a meta-analysis. Heart Lung. 2015;44(1):75–81. Epub 2014 Nov 18. PMID: 25453390. doi:10.1016/j.hrtlng.2014.10.002
136. Gibler WB, Gibler CD, Weinshenker E, et al. Myoglobin as an early indicator of acute myocardial infarction. Ann Emerg Med. 1987;16(8):851–856. PMID: 3619163. doi:10.1016/s0196-0644(87)80521-8
137. Bhayana V, Henderson AR. Biochemical markers of myocardial damage. Clin Biochem. 1995;28(1):1–29. PMID: 7720223. doi:10.1016/0009-9120(94)00065-4
138. Chen Y, Tao Y, Zhang L, Xu W, Zhou X. Diagnostic and prognostic value of biomarkers in acute myocardial infarction. Postgrad Med J. 2019;95(1122):210–216. Epub 2019 Apr 4. PMID: 30948439. doi:10.1136/postgradmedj-2019-136409
139. McDonough JL, Arrell DK, Van Eyk JE. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ Res. 1999;84(1):9–20. PMID: 9915770. doi:10.1161/01.res.84.1.9
140. Feng J, Schaus BJ, Fallavollita JA, Lee TC, Canty JM
141. Gao CQ, Sawicki G, Suarez-Pinzon WL, et al. Matrix metalloproteinase-2 mediates cytokine-induced myocardial contractile dysfunction. Cardiovasc Res. 2003;57(2):426–433. PMID: 12566115. doi:10.1016/s0008-6363(02)00719-8
142. Lin NN, Cheng CC, Lee YF, et al. Early activation of myocardial matrix metalloproteinases and degradation of cardiac troponin I after experimental subarachnoid hemorrhage. J Surg Res. 2013;179(1):e41–8. Epub 2012 Mar 28. PMID: 22475348. doi:10.1016/j.jss.2012.02.008
143. Parente JM, Blascke de Mello MM, Silva PHLD. MMP inhibition attenuates hypertensive eccentric cardiac hypertrophy and dysfunction by preserving troponin I and dystrophin. Biochem Pharmacol. 2021;193:114744. Epub 2021 Aug 25. PMID: 34453903. doi:10.1016/j.bcp.2021.114744
144. Streng AS, de Boer D, van Doorn WP, Kocken JM, Bekers O, Wodzig WK. Cardiac troponin T degradation in serum is catalysed by human thrombin. Biochem Biophys Res Commun. 2016;481(1–2):165–168. Epub 2016 Nov 2. PMID: 27816455. doi:10.1016/j.bbrc.2016.10.149
145. Katrukha IA, Kogan AE, Vylegzhanina AV, et al. Thrombin-Mediated Degradation of Human Cardiac Troponin T. Clin Chem. 2017;63(6):1094–1100. Epub 2017 Apr 20. PMID: 28428352. doi:10.1373/clinchem.2016.266635
146. Bodor GS. Cardiac Troponins: molecules of Many Surprises. Clin Chem. 2017;63(6):1059–1060. Epub 2017 Apr 20. PMID: 28428360. doi:10.1373/clinchem.2017.273094
147. Ito K, Date T, Ikegami M, et al. An immunohistochemical analysis of tissue thrombin expression in the human atria. PLoS One. 2013;8(6):e65817. doi:10.1371/journal.pone.0065817
148. Ito K, Hongo K, Date T, et al. Tissue thrombin is associated with the pathogenesis of dilated cardiomyopathy. Int J Cardiol. 2017;228:821–827. doi:10.1016/j.ijcard.2016.11.176
149. Matsukura U, Okitani A, Nishimuro T, Kato H. Mode of degradation of myofibrillar proteins by an endogenous protease, cathepsin L. Biochim Biophys Acta. 1981;662(1):41–47. PMID: 7306557. doi:10.1016/0005-2744(81)90221-7
150. Peng K, Liu H, Yan B, et al. Inhibition of cathepsin S attenuates myocardial ischemia/reperfusion injury by suppressing inflammation and apoptosis. J Cell Physiol. 2021;236(2):1309–1320. Epub 2020 Jul 13. PMID: 32657442. doi:10.1002/jcp.29938
151. Hickman PE, Potter JM, Aroney C, et al. Cardiac troponin may be released by ischemia alone, without necrosis. Clin Chim Acta. 2010;411(5–6):318–323. Epub 2009 Dec 28. PMID: 20036224. doi:10.1016/j.cca.2009.12.009
152. Hessel MH, Atsma DE, van der Valk EJ, Bax WH, Schalij MJ, van der Laarse A. Release of cardiac troponin I from viable cardiomyocytes is mediated by integrin stimulation. Pflugers Arch. 2008;455(6):979–986. Epub 2007 Oct 2. PMID: 17909848; PMCID: PMC2226063. doi:10.1007/s00424-007-0354-8
153. Ross RS, Borg TK. Integrins and the myocardium. Circ Res. 2001;88(11):1112–1119. PMID: 11397776. doi:10.1161/hh1101.091862
154. Khabbaz KR, Feng J, Boodhwani M, Clements RT, Bianchi C, Sellke FW. Nonischemic myocardial acidosis adversely affects microvascular and myocardial function and triggers apoptosis during cardioplegia. J Thorac Cardiovasc Surg. 2008;135(1):139–146. PMID: 18179930. doi:10.1016/j.jtcvs.2007.07.035
155. Thatte HS, Rhee JH, Zagarins SE, et al. Acidosis-induced apoptosis in human and porcine heart. Ann Thorac Surg. 2004;77(4):1376–1383. PMID: 15063270. doi:10.1016/j.athoracsur.2003.07.047
156. Graham RM, Frazier DP, Thompson JW, et al. A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J Exp Biol. 2004;207(Pt18):3189. PMID: 15299040. doi:10.1242/jeb.01109
157. Wasfy MM, Hutter AM, Weiner RB. Sudden Cardiac Death in Athletes. Methodist Debakey Cardiovasc J. 2016;12(2):76–80. PMID: 27486488; PMCID: PMC4969030. doi:10.14797/mdcj-12-2-76
158. Aune D, Schlesinger S, Hamer M, Norat T, Riboli E. Physical activity and the risk of sudden cardiac death: a systematic review and meta-analysis of prospective studies. BMC Cardiovasc Disord. 2020;20(1):318. PMID: 32631241; PMCID: PMC7336483. doi:10.1186/s12872-020-01531-z
159. DeFroda SF, McDonald C, Myers C, Cruz AI, Owens BD, Daniels AH. Sudden Cardiac Death in the Adolescent Athlete: history, Diagnosis, and Prevention. Am J Med. 2019;132:1374–1380. Epub 2019 Jun 12. PMID: 31199891. doi:10.1016/j.amjmed.2019.05.025
160. Sollazzo F, Palmieri V, Gervasi SF, et al. Sudden Cardiac Death in Athletes in Italy during 2019: internet-Based Epidemiological Research. Medicina. 2021;57(1):61. PMID: 33445447; PMCID: PMC7827560. doi:10.3390/medicina57010061
161. Klinkenberg LJ, Luyten P, Linden N, et al. Cardiac Troponin T and I Release After a 30-km Run. Am J Cardiol. 2016;118(2):281–287. Epub 2016 May 4. PMID: 27282835. doi:10.1016/j.amjcard.2016.04.030.
162. Martínez-Navarro I, Sánchez-Gómez J, Sanmiguel D, et al. and 24-h post-marathon cardiac troponin T is associated with relative exercise intensity. Eur J Appl Physiol. 2020;120(8):1723–1731. Epub 2020 May 28. PMID: 32468283. doi:10.1007/s00421-020-04403-8
163. Marshall L, Lee KK, Stewart SD, et al. Effect of Exercise Intensity and Duration on Cardiac Troponin Release. Circulation. 2020;141(1):83–85. Epub 2019 Dec 30. PMID: 31887079; PMCID: PMC6940024. doi:10.1161/CIRCULATIONAHA.119.041874
164. O’Hanlon R, Wilson M, Wage R, et al. Troponin release following endurance exercise: is inflammation the cause? a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson. 2010;12(1):38. PMID: 20598139; PMCID: PMC2908607. doi:10.1186/1532-429X-12-38
165. Lazzarino AI, Hamer M, Gaze D, Collinson P, Steptoe A. The association between cortisol response to mental stress and high-sensitivity cardiac troponin T plasma concentration in healthy adults. J Am Coll Cardiol. 2013;62(18):1694–1701. Epub 2013 Jun 27. PMID: 23810896; PMCID: PMC3807660. doi:10.1016/j.jacc.2013.05.070
166. Eggers KM. Mental stress and cardiac troponin: keep calm and carry on? J Am Coll Cardiol. 2013;62(18):1702–1703. Epub 2013 Jun 27. PMID: 23810870. doi:10.1016/j.jacc.2013.06.010
167. Yamaji M, Tsutamoto T, Kawahara C, et al. Serum cortisol as a useful predictor of cardiac events in patients with chronic heart failure: the impact of oxidative stress. Circ Heart Fail. 2009;2(6):608–615. Epub 2009 Sep 28. PMID: 19919986. doi:10.1161/CIRCHEARTFAILURE.109.868513
168. Iwaszczuk P, Łosiak W, Szczeklik W, Musiałek P. Patient periprocedural stress in cardiovascular medicine: friend or foe? Postepy Kardiol Int. 2021;17(3):259–271. Epub 2021 Sep 14. PMID: 34819962; PMCID: PMC8596718. doi:10.5114/aic.2021.109176
169. Bakay M, Zhao P, Chen J, Hoffman EP. A web-accessible complete transcriptome of normal human and DMD muscle. Neuromuscul Disord. 2002;12(Suppl 1):S125–41. doi:10.1016/s0960-8966(02)00093-7
170. Messner B, Baum H, Fischer P, Quasthoff S, Neumeier D. Expression of messenger RNA of the cardiac isoforms of troponin T and I in myopathic skeletal muscle. Am J Clin Pathol. 2000;114(4):544–549. PMID: 11026100. doi:10.1309/8KCL-UQRF-6EEL-36XK
171. Rusakov DY, Yamshcikov NV, Tulayeva ON, Suvorova LA, Metlenko OI. Histogenesis and peculiarities of structural organization of the cardiac muscle tissue in the walls of human caval and pulmonary veins. Morphology. 2015;148(6):38–42.
172. Rusakov DY, Vologdina NN, Tulayeva ON. The development of striated cardiac muscle tissue in the walls of the caval and pulmonary veins. J Anatomy Histopathol. 2015;4(3):105. doi:10.18499/2225-7357-2015-4-3-105-105
173. Bodor GS, Porterfield D, Voss EM, Smith S, Apple FS. Cardiac troponin-I is not expressed in fetal and healthy or diseased adult human skeletal muscle tissue. Clin Chem. 1995;41(12 Pt 1):1710–1715. PMID: 7497610. doi:10.1093/clinchem/41.12.1710
174. Hammerer-Lercher A, Erlacher P, Bittner R, et al. Clinical and experimental results on cardiac troponin expression in Duchenne muscular dystrophy. Clin Chem. 2001;47(3):451–458. PMID: 11238296. doi:10.1093/clinchem/47.3.451
175. Schmid J, Liesinger L, Birner-Gruenberger R, et al. Elevated Cardiac Troponin T in Patients With Skeletal Myopathies. J Am Coll Cardiol. 2018;71(14):1540–1549. PMID: 29622161. doi:10.1016/j.jacc.2018.01.070
176. Anderson PA, Greig A, Mark TM, et al. Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circ Res. 1995;76(4):681–686. doi:10.1161/01.res.76.4.681
177. Bates KJ, Hall EM, Fahie-Wilson MN, et al. Circulating immunoreactive cardiac troponin forms determined by gel filtration chromatography after acute myocardial infarction. Clin Chem. 2010;56(6):952–958. doi:10.1373/clinchem.2009.133546
178. Maekawa A, Lee JK, Nagaya T, et al. Overexpression of calpastatin by gene transfer prevents troponin I degradation and ameliorates contractile dysfunction in rat hearts subjected to ischemia/reperfusion. J Mol Cell Cardiol. 2003;35(10):1277–1284. doi:10.1016/s0022-2828(03)00238-4
179. Zahran S, Figueiredo VP, Graham MM, Schulz R, Hwang PM. Proteolytic Digestion of Serum Cardiac Troponin I as Marker of Ischemic Severity. J Appl Lab Med. 2018;3(3):450–455. doi:10.1373/jalm.2017.025254
180. Vylegzhanina AV, Kogan AE, Katrukha IA, et al. Full-Size and Partially Truncated Cardiac Troponin Complexes in the Blood of Patients with Acute Myocardial Infarction. Clin Chem. 2019;65(7):882–892. doi:10.1373/clinchem.2018.301127
181. Katus HA, Remppis A, Looser S, Hallermeier K, Scheffold T, Kubler W. Enzyme linked immune assay of cardiac troponin T for the detection of acute myocardial infarction in patients. J Mol Cell Cardiol. 1989;21(12):1349–1353. doi:10.1016/0022-2828(89)90680-9
182. Labugger R, Organ L, Collier C, Atar D, Van Eyk JE. Extensive troponin I and T modification detected in serum from patients with acute myocardial infarction. Circulation. 2000;102(11):1221–1226. PMID: 10982534. doi:10.1161/01.cir.102.11.1221
183. Gaze DC, Collinson PO. Multiple molecular forms of circulating cardiac troponin: analytical and clinical significance. Ann Clin Biochem. 2008;45(Pt 4):349–355. doi:10.1258/acb.2007.007229
184. Katrukha AG, Bereznikova AV, Esakova TV, et al. Troponin I is released in bloodstream of patients with acute myocardial infarction not in free form but as complex. Clin Chem. 1997;43(8 Pt 1):1379–1385. PMID: 9267317. doi:10.1093/clinchem/43.8.1379
185. Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PA. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997;96(5):1495–1500. doi:10.1161/01.cir.96.5.1495
186. Hayashi T, Notkins AL. Clearance of LDH-5 from the circulation of inbred mice correlates with binding to macrophages. Int J Exp Pathol. 1994;75(3):165–168. PMID: 8086313.
187. Prabhudas M, Bowdish D, Drickamer K, et al. Standardizing scavenger receptor nomenclature. J Immunol. 2014;192(5):1997–2006. doi:10.4049/jimmunol.1490003
188. De Zoysa JR. Cardiac troponins and renal disease. Nephrology. 2004;9(2):83–88. PMID: 15056267. doi:10.1111/j.1440-1797.2003.00235.x
189. Dubin RF, Li Y, He J, et al.; CRIC Study Investigators. Predictors of high sensitivity cardiac troponin T in chronic kidney disease patients: a cross-sectional study in the chronic renal insufficiency cohort (CRIC). BMC Nephrol. 2013;14:229. PMID: 24148285; PMCID: PMC4016297. doi:10.1186/1471-2369-14-229
190. Di Lullo L, Barbera V, Santoboni A, et al. Malattia renale cronica e sindrome coronarica acuta: il ruolo della troponina [Troponins and chronic kidney disease]. G Ital Nefrol. 2015;32(4):515. Italian. PMID: 26252257.
191. Han X, Zhang S, Chen Z, et al. Cardiac biomarkers of heart failure in chronic kidney disease. Clin Chim Acta. 2020;510:298–310. Epub 2020 Jul 23. PMID: 32710942. doi:10.1016/j.cca.2020.07.040
192. Wilhelm J, Hettwer S, Schuermann M, et al. Elevated troponin in septic patients in the emergency department: frequency, causes, and prognostic implications. Clin Res Cardiol. 2014;103(7):561–567. Epub 2014 Feb 18. PMID: 24535379. doi:10.1007/s00392-014-0684-4
193. Røsjø H, Varpula M, Hagve TA, et al.; FINNSEPSIS Study Group. Circulating high sensitivity troponin T in severe sepsis and septic shock: distribution, associated factors, and relation to outcome. Intensive Care Med. 2011;37(1):77–85. Epub 2010 Oct 12. PMID: 20938765; PMCID: PMC3020309. doi:10.1007/s00134-010-2051-x.
194. Daly M, Long B, Koyfman A, Lentz S. Identifying cardiogenic shock in the emergency department. Am J Emerg Med. 2020;38(11):2425–2433. Epub 2020 Sep 23. PMID: 33039227. doi:10.1016/j.ajem.2020.09.045
195. Muslimovic A, Fridén V, Tenstad O, et al. The Liver and Kidneys mediate clearance of cardiac troponin in the rat. Sci Rep. 2020;10(1):6791. doi:10.1038/s41598-020-63744-8
196. Fridén V, Starnberg K, Muslimovic A, et al. Clearance of cardiac troponin T with and without kidney function. Clin Biochem. 2017;50(9):468–474. doi:10.1016/j.clinbiochem.2017.02.007
197. Kavsak PA, Worster A, Shortt C, et al. Performance of high-sensitivity cardiac troponin in the emergency department for myocardial infarction and a composite cardiac outcome across different estimated glomerular filtration rates. Clin Chim Acta. 2018;479:166–170. Epub 2018 Feb 3. PMID: 29366835. doi:10.1016/j.cca.2018.01.034
198. Patke A, Young MW, Axelrod S. Molecular mechanisms and physiological importance of circadian rhythms. Nat Rev Mol Cell Biol. 2020;21(2):67–84. doi:10.1038/s41580-019-0179-2
199. Cribbet MR, Logan RW, Edwards MD, et al. Circadian rhythms and metabolism: from the brain to the gut and back again. Ann N Y Acad Sci. 2016;1385(1):21–40. doi:10.1111/nyas.13188
200. Thosar SS, Butler MP, Shea SA. Role of the circadian system in cardiovascular disease. J Clin Invest. 2018;128(6):2157–2167. doi:10.1172/JCI80590
201. Klinkenberg LJJ, Wildi K, van der Linden N, et al. Diurnal rhythm of cardiac troponin: consequences for the diagnosis of acute myocardial infarction. Clin Chem. 2016;62(12):1602–1611. doi:10.1373/clinchem.2016.257485
202. van der Linden N, Cornelis T, Klinkenberg LJJ, Kimenai DM, Hilderink JM, Litjens EJR. Strong diurnal rhythm of troponin T, but not troponin I, in a patient with renal dysfunction. Int J Cardiol. 2016;221:287–288. doi:10.1016/j.ijcard.2016.06.268
203. Tofler GH, Brezinski D, Schafer AI, et al. Concurrent morning increase in platelet aggregability and the risk of myocardial infarction and sudden cardiac death. N Engl J Med. 1987;316(24):1514–1518. PMID: 3587281. doi:10.1056/NEJM198706113162405
204. Panza JA, Epstein SE, Quyyumi AA. Circadian variation in vascular tone and its relation to alpha-sympathetic vasoconstrictor activity. N Engl J Med. 1991;325(14):986–990. PMID: 1886635. doi:10.1056/NEJM199110033251402
205. Tsareva YO, Mayskova EA, Fedotov EA, Shvarts YG. [Circadian rhythms of thyroid hormones in patients with ischemic heart disease, arterial hypertension, and atrial fibrillation]. Kardiologiia. 2019. 59(3S):23–29. Russian. PMID: 30990149. doi:10.18087/cardio.2506
206. Chaulin AM, Grigorieva JV, Suvorova GN, Duplyakov DV. Experimental Modeling of Hypothyroidism: Principles, Methods, Several Advanced Research Directions in Cardiology. Russian Open Medical Journal. 2021;10:e0311. doi:10.15275/rusomj.2021.0311
207. Suárez-Barrientos A, López-Romero P, Vivas D, et al. Circadian variations of infarct size in acute myocardial infarction. Heart. 2011;97(12):970–976. Epub 2011 Apr 27. PMID: 21525526. doi:10.1136/hrt.2010.212621
208. Arroyo Úcar E, Dominguez-Rodriguez A, Abreu-Gonzalez P. Influencia de la variabilidad diurna en el tamaño del infarto agudo de miocardio [Influence of diurnal variation in the size of acute myocardial infarction]. Med Intensiva. 2012. 36(1):11–14. Spanish. Epub 2011 Sep 6. PMID: 21899925. doi:10.1016/j.medin.2011.07.002
209. Seneviratna A, Lim GH, Devi A, et al. Circadian Dependence of Infarct Size and Acute Heart Failure in ST Elevation Myocardial Infarction. PLoS One. 2015;10(6):e0128526. PMID: 26039059; PMCID: PMC4454698. doi:10.1371/journal.pone.0128526
210. Manfredini R, Boari B, Bressan S, et al. Influence of circadian rhythm on mortality after myocardial infarction: data from a prospective cohort of emergency calls. Am J Emerg Med. 2004;22(7):555–559. PMID: 15666260. doi:10.1016/j.ajem.2004.08.014
211. Chaulin AM. [Main analytical characteristics of laboratory methods for the determination of cardiac troponins: a review from the historical and modern points of view]. Orv Hetil. 2022 Jan 2;16:3(1)12–20. doi:10.1556/650.2021.32296. PMID: 34974429.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.