Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Metabolic and Energy Imbalance in Dysglycemia-Based Chronic Disease
Authors Kalra S, Unnikrishnan AG, Baruah MP, Sahay R ![]() , Bantwal G
, Bantwal G
Received 15 October 2020
Accepted for publication 2 December 2020
Published 15 January 2021 Volume 2021:14 Pages 165—184
DOI https://doi.org/10.2147/DMSO.S286888
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Sanjay Kalra,1,2 Ambika Gopalakrishnan Unnikrishnan,3 Manash P Baruah,4 Rakesh Sahay,5 Ganapathi Bantwal6
1Department of Endocrinology, Bharti Hospital, Karnal, India; 2Department of Endocrinology, All India Institute of Medical Sciences, Rishikesh, Uttarakhand, India; 3Department of Endocrinology and Diabetes, Chellaram Diabetes Institute, Pune, Maharashtra, India; 4Department of Endocrinology, Excel Hospitals, Guwahati, India; 5Department of Endocrinology, Osmania Medical College, Hyderabad, Telangana, India; 6Department of Endocrinology, St. John’s Medical College and Hospital, Bangalore, Karnataka, India
Correspondence: Sanjay Kalra
Bharti Hospital, Kunjpura Road, Karnal, 132001 Haryana, India
Tel +919896048555
Email [email protected]
Abstract: Metabolic flexibility is the ability to efficiently adapt metabolism based on nutrient availability and requirement that is essential to maintain homeostasis in times of either caloric excess or restriction and during the energy-demanding state. This regulation is orchestrated in multiple organ systems by the alliance of numerous metabolic pathways under the master control of the insulin-glucagon-sympathetic neuro-endocrine axis. This, in turn, regulates key metabolic enzymes and transcription factors, many of which interact closely with and culminate in the mitochondrial energy generation machinery. Metabolic flexibility is compromised due to the continuous mismatch between availability and intake of calorie-dense foods and reduced metabolic demand due to sedentary lifestyle and age-related metabolic slowdown. The resultant nutrient overload leads to mitochondrial trafficking of substrates manifesting as mitochondrial dysfunction characterized by ineffective substrate switching and incomplete substrate utilization. At the systemic level, the manifestation of metabolic inflexibility comprises reduced skeletal muscle glucose disposal rate, impaired suppression of hepatic gluconeogenesis and adipose tissue lipolysis manifesting as insulin resistance. This is compounded by impaired β-cell function and progressively reduced β-cell mass. A consequence of insulin resistance is the upregulation of the mitogen-activated protein kinase pathway leading to a pro-hypertensive, atherogenic, and thrombogenic environment. This is further aggravated by oxidative stress, advanced glycation end products, and inflammation, which potentiates the risk of micro- and macro-vascular complications. This review aims to elucidate underlying mechanisms mediating the onset of metabolic inflexibility operating at the main target organs and to understand the progression of metabolic diseases. This could potentially translate into a pharmacological tool that can manage multiple interlinked conditions of dysglycemia, hypertension, and dyslipidemia by restoring metabolic flexibility. We discuss the breadth and depth of metabolic flexibility and its impact on health and disease.
Keywords: metabolic flexibility, DBCD, insulin resistance, prediabetes, diabetes, microvascular and macrovascular complication
Introduction
There is an alarming surge in the prevalence of diabetes globally, and it has become a significant public and economic health burden. According to the 2019 International Diabetes Federation (IDF) estimates, the global prevalence of Type 2 diabetes mellitus (T2DM) is 463 million (9.3%) and is projected to reach 578 million (10.2%) by 2030.1 Among these, the second-highest number of people with diabetes after China is in India (69.2 million), and it is estimated to increase to 123.5 million by 2040. In the latest report of the Indian Council of Medical Research (ICMR), the estimated prevalence of diabetes and prediabetes was 7.3% and 10.3%, respectively, indicating a large pool of individuals at the risk of T2DM.2
To add to the complexity, the young Asian Indian population has a unique phenotype distinguishable from the western population characterized by the higher incidence of insulin resistance (IR), increased levels of triglyceride (TG) and small low-density lipoprotein (LDL) and, decreased levels of high-density lipoprotein (HDL), adiponectin, and β-cell mass.3,4 ICMR-INDIAB study estimates the prevalence rate of low HDL, hypertriglyceridemia, and high LDL to be 72.3%, 29.5%, and 11.8%, respectively.5 Similarly, Yadav et al noted an increased occurrence of dyslipidemia (64%) and hypertension (49%) in patients with T2DM.6 Collectively, the evidence suggests that young Asian Indians are at a higher risk for IR despite a low body max index (BMI) due to increased waist circumference, visceral fat, and increased prevalence of dyslipidemia.7,8 This observation is of grave concern considering that IR triggers not only an increased risk of prediabetes and, subsequently, T2DM but also carries an inherent risk of micro- and macro-vascular diseases. Consequently, there is a recognition of the need for a renewed focus on prevention by intervening in the early phase of the disease.9
For decades, studies related to metabolic disorders have focused on the metabolic pathways or target organs that involved regulation of metabolism at different levels independent of each other. Lifestyle and drug interventions, such as those focusing on simultaneously reversing multiple pathways, had limited success. Besides, these interventions failed to stop the progression of β-cell dysfunction and reduction of micro- and macro-vascular complications. Interestingly, these conditions frequently co-exist in individuals at the stage when they are not frankly diabetic, ie, prediabetes.10,11 Prediabetes predisposes individuals to a higher risk of T2DM than the general population.12 Observational studies have also shown the association between prediabetes and the development of microvascular changes manifesting as neuropathy, nephropathy, retinopathy, and macro-vascular complications.13–16 In general, the onset of IR plays a fundamental role in the early development of the disease spectrum of prediabetes, T2DM, micro- and macro-vascular complications.17–19 Recently, the disease spectrum has been redefined as Dysglycemia-Based Chronic Disease (DBCD).20 The DBCD model positions prediabetes and T2DM along a continuous spectrum of IR-prediabetes-T2DM-vascular complications to bring focus onto diagnosing an actionable condition and developing means to address it more efficiently and in a cost-effective manner (Figure 1).
 |
Figure 1 Key features of dysglycemia-based chronic disease and the insulin resistance-prediabetes-type 2 diabetes spectrum. Insulin resistance is the driving factor leading to prediabetes, diabetes, micro- and macro-vascular complications. |
Evidence is now gathering, which suggests metabolic inflexibility as the underlying mechanism that interlinks the defect across the disease spectrum, and which lies in the human evolutionary history. Adaptive obesity was deemed advantageous in the harsh environment aiding “survival of the fittest,” and provided the human organism tools to cope with metabolic challenges induced by food deprivation due to the regular occurrence of famine. With limited food availability, adaptive obesity enabled humans to adjust their energy needs to develop a flexible metabolism, ie, efficiently adapt metabolism depending on demand and supply.21 The term “Metabolic flexibility” is the ability to efficiently adapt substrate utilization, ie, glucose or fatty acids (FA), based on nutrient availability and requirements. However, in the modern era of nutrition excess, adaptive obesity has become an adaptation that has led to negative consequences.22 Overnutrition, at the supply end and sedentary lifestyle coupled with an age-related slowing of basal metabolism on the demand side, has resulted in a state of “metabolic inflexibility.”23 In general, the metabolically “inflexible” individual cannot switch between FA and glucose for fuel oxidation in response to metabolic demands thereby leading to nutrient overload and dysregulation of energy homeostasis.24 Extensive research suggests that IR is the key component of metabolic inflexibility that encompasses defects in skeletal muscle, white adipose tissue (WAT), and liver causing dysglycemia, hypertension, and hyperlipidemia.10,25,26
Therefore, the pathophysiological basis of metabolic flexibility linking multiple components of the DBCD spectrum provides a more holistic approach in managing the disease. Surprisingly, very few studies have investigated the vicious synergy between multiple independent and interdependent mechanisms involving glycemic and hemodynamic dysfunction, lipotoxicity, glucotoxicity, and oxidative and non-oxidative inflammatory state in DBCD. This review discusses the cellular, biochemical, physiological, and morphological consequences of metabolic inflexibility leading to the disease spectrum of IR-prediabetes-T2DM-vascular complications and causative linkage to hypertension and dyslipidemia seen in this population.
Metabolic Flexibility and Its Physiological Relevance in Maintaining Energy Homeostasis
Mitochondria, the powerhouse of the cell, plays a critical role in the generation of energy from the three sources, namely glucose, FA, and amino acids. These macromolecules are metabolized through the pathways that are compartmentalized within the cell. Metabolic processes such as glycolysis, glycogenesis, glycogenolysis, pentose phosphate pathway, and lipogenesis occur in the cytosol. On the other hand, enzymes of the citric acid cycle, β-oxidation of FA, respiratory chain, and adenosine triphosphate (ATP) synthase are present in mitochondria. Besides, the endoplasmic reticulum contain the enzymes for several other processes, including triacylglycerol synthesis. Among these metabolic reactions, acetyl coenzyme A (Acetyl CoA) serves as an intermediate substrate that feeds into the tricarboxylic cycle (TCA), generating high energy molecules nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). These reducing equivalents generate ATP after transversing through a series of reactions in the mitochondrial complexes through the electron transport chain (ETC) located in the inner mitochondrial membrane through the process of oxidative phosphorylation (OXPHOS) (Figure 2).
 |
Figure 2 Overvie Abbreviations: ADP, adenosine diphosphate; ATP, adenosine triphosphate; TCA, tricarboxylic acid cycle. |
Under normal circumstances, a diurnal oscillation between the usage of substrates, namely FAs and glucose, occur based on the nutritional supply and physiological demands. The intake of meals determines the supply side, and so the mitochondria flexibly utilize these substrates under different physiological conditions, namely, fed and fasting states.
Metabolism in Post-Prandial Condition
After the consumption of a meal, there is an ample supply of carbohydrates, amino acids, and fats. Increased blood glucose concentration in the portal blood stimulates pancreatic β cells and the parasympathetic nervous system leading to insulin secretion into the bloodstream.27 Under insulin-stimulated conditions, glucose is a major fuel for oxidation in most tissues, and the excess supply is converted into and stored, mainly as triacylglycerol in WAT. Under the influence of insulin, the absorption of glucose is initiated in the peripheral tissues, including skeletal muscles, WAT, liver, cardiomyocytes, and the brain, and conversely, the mobilization of endogenous carbohydrate and lipid stores is suppressed under these conditions28 (Figure 3).
 |
Figure 3 Regulation of blood glucose via insulin and glucagon feedback. |
After absorption of nutrients, the liver receives a wide range of nutrients, including carbohydrates, amino acids, and short-chain FAs. Among these, glucose enters the hepatocytes directly, independent of insulin via glucose transporter, GLUT2.29 In hepatocytes, regulation happens at the level of glucokinase (GK) enzyme under the influence of insulin, which catalyzes the phosphorylation of glucose to glucose-6-phosphate (G-6-P), which is essential for committing glucose to glycolytic, glycogenesis, or lipogenic pathways.30 Subsequently, G-6-P may follow a number of metabolic pathways, including glycogen synthesis, hexosamine pathway, pentose phosphate pathway, and oxidative routes.31 Amongst these, insulin stimulates glycogen synthesis and suppresses hepatic gluconeogenesis and glycogenolysis.32 When the liver glycogen stores are replenished, glucose undergoes glycolysis, and generates pyruvate, which gets oxidized in the mitochondria through pyruvate dehydrogenase (PDH) to generate acetyl-CoA. In the presence of excess energy as in the fed state, acetyl-CoA is transferred to the cytosol and then carboxylated to form malonyl CoA by acetyl CoA carboxylase (ACC).33 The malonyl CoA, an essential regulator of de novo lipogenesis, is utilized through fatty acid synthase (FAS) reactions to generate FAs, which is then utilized to synthesize TGs. Besides, malonyl-CoA inhibits the carnitine palmitoyltransferase-I (CPT1), thereby suppressing fatty acid oxidation (FAO), which is not required at this stage due to the abundance of carbohydrate. In these pathways, insulin is involved in inducing these rate-limiting enzymes; namely, FAS and ACC, to drive carbohydrate utilization, de novo lipogenesis, inhibit fat utilization, and promote storage (glycogen and triglycerides) of excess nutrients (Figure 4). Consequently, newly formed endogenous TGs are incorporated into very low density lipoprotein (VLDL) for transport to WAT for storage34 (Figure 5).
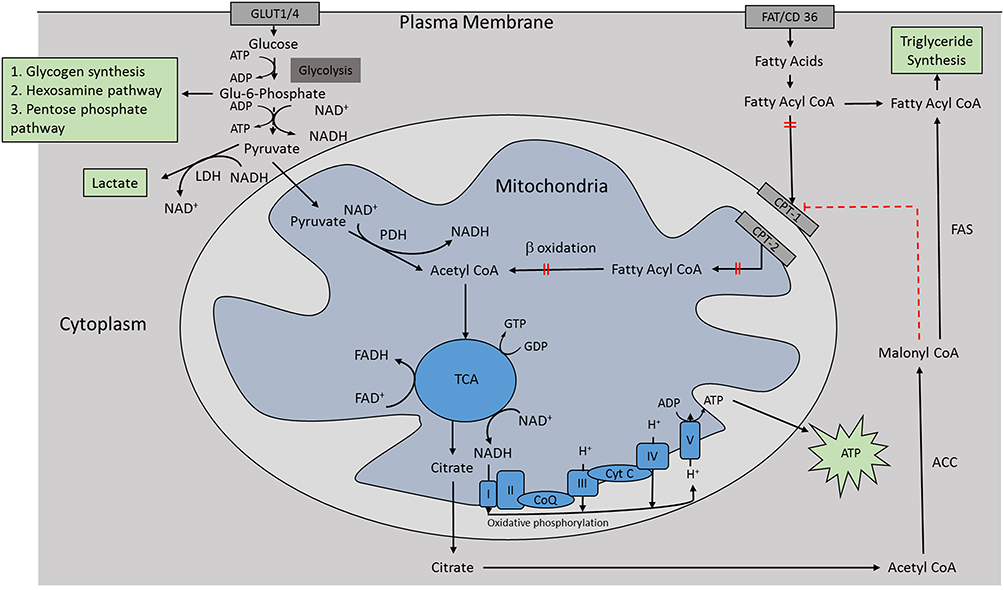 |
Figure 4 Mechanism of inhibition of fatty acid oxidation in mitochondria in post-prandial state. Malonyl CoA generated from acetyl CoA derived from utilization of carbohydrates through glycolysis and TCA cycle by acetyl CoA carboxylase, inhibits the entry of long-chain fatty acyl CoA into mitochondria. Abbreviations: ATP, adenosine triphosphate; ACC, acetyl CoA carboxylase; CPT, carnitine palmitoyltransferase; FAS, fatty acyl synthase; LDH, lactate dehydrogenase; TCA, tricarboxylic acid cycle; PDH, pyruvate dehydrogenase. |
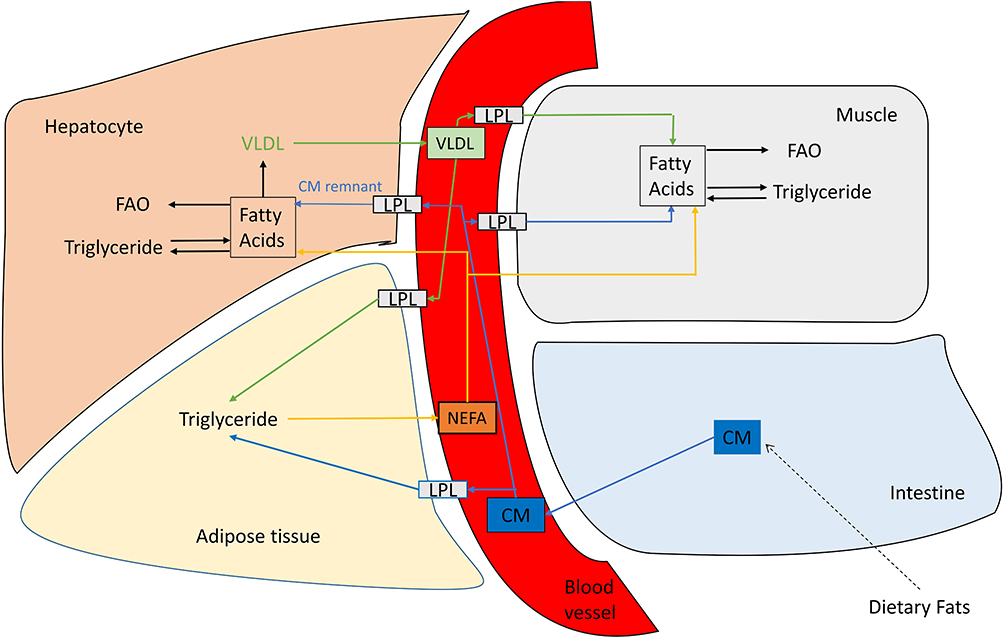 |
Figure 5 Lipid transport and storage. |
In the WAT and skeletal muscle, insulin binds to insulin receptor substrate (IRS), leading to active recruitment of glucose transporters (GLUT), mainly GLUT1/4, to the plasma membrane, allowing rapid removal of glucose from circulation35 (Figure 4). Approximately 80% of the glucose utilization is accounted for by skeletal muscle due to its large mass compared to other tissues, and this glucose is then incorporated into glycogen.36 These storage reserves serve as a local energy source in the muscle for exercise and not for fasting as muscles lack glucose 6-phosphatase essential for conversion to glucose, wherein it depends on hepatic gluconeogenesis to provide the fuel source. In fact, during exercise, these muscle glycogen stores are broken down to lactate, which is then transported to the liver to be recycled to glucose and maintain euglycemia.37
Besides glucose, blood also has a high concentration of FAs available from chylomicrons for systemic utilization or storage. These FAs are delivered to the tissue expressing lipoprotein lipase (LPL) bound to endothelial cells of capillaries, including skeletal muscle and WAT. The LPL is activated in response to insulin and increases the re-esterification of FA into TG for storage in WAT.38 The storage of TGs differs between males and females.39 Females preferentially store TG derived FAs in the gluteo-femoral subcutaneous WAT. In contrast, males store meal derived FAs in the visceral WAT.40 Some of the non-esterified fatty acids (NEFA) are released in the plasma and are bound to albumin. The liver takes up FAs in various forms and packages and liberates TGs and FA as VLDL particles.41 Similarly, skeletal muscles can take up FAs from the plasma circulating TGs in chylomicron or VLDL and from the albumin-bound NEFA pool through the action of LPL. The FAs enter muscle through facilitated diffusion or the fatty acid transport protein (FATP) and are stored as lipid droplets, often in close contact with mitochondria34 (Figure 5).
In cardiomyocytes, glucose uptake occurs through insulin-independent (GLUT1) and insulin-dependent (GLUT4) transporters.42 After glucose uptake, it is phosphorylated by hexokinase to G-6-P, which can then undergo a metabolic pathway similar to the hepatocyte. Pyruvate derived from the glycolysis can either be converted to lactate by lactate dehydrogenase (LDH) or transferred to the mitochondria matrix, where PDH catalyzes the conversion of pyruvate to acetyl CoA. The fate of acetyl CoA appears to be similar, as described above, for the liver, while many of the features of FA metabolism in the fed state are similar to skeletal muscle.
As described above, the system functions efficiently in the feeding state in the presence of an adequate supply of glucose to meet the immediate energy demand through the glycolytic pathway, thereby generating reducing equivalents. These reducing equivalents are used for ATP generation in the mitochondria and for converting excess glucose into FA by the FA synthetic pathway in different tissue.
Metabolism in Fasting Conditions
The metabolic rate is not substantially affected in that there is a continued need for oxidative metabolism to meet basal energy demands. In a fasted state, the metabolic fuel switches from glucose to FA in most tissues except the brain and red blood cells.43 During the initial stages of fasting (8–12 hours), the glucose level in the portal blood coming from the intestine declines, leading to a drop in insulin levels. In response to dropping blood glucose level, α-cells of the pancreas release hormone glucagon,44 and adrenal glands stimulate epinephrine release.45 There is less glucose available for target tissues, ie, skeletal muscle and WAT, and simultaneously, the conversion of glucose to glycogen and TG storage slows down. As a result, there is the mobilization of TGs contained within WAT. Hormone-sensitive lipase is activated by glucagon resulting in hydrolysis of TGs to give NEFAs and glycerol.46 The circulating NEFAs are bound to albumin and/or released from triacylglycerols (TAG) contained in chylomicrons or very-low-density lipoproteins (VLDL). There is also an increased release of glycerol from the WAT, which serves as a precursor for gluconeogenesis in the liver.
In the liver, glucagon inhibits glycogenesis and stimulates glycogenolysis to increase blood glucose levels.47 The glycerol released from hydrolysis is converted into glucose; however, this remains a minor source of glucose. The peripheral tissue and the brain use the glucose released from the liver. As blood glucose levels continue to fall, insulin secretion remains suppressed while the glucagon and epinephrine stimulate glycogenolysis through activation of glycogen phosphorylase, catalyzing the breakdown of glycogen. Concurrently, gluconeogenesis also begins in the liver to replace the glucose that has been used by the peripheral tissues.48 There is concomitant suppression of lipogenesis and activation of FAO. Inhibition of glucose utilization by FAO is mediated by inhibition of pyruvate dehydrogenase and phosphofructokinase. PDH inhibition is caused by acetyl CoA, NADH accumulation, and inactivation by Pyruvate dehydrogenase kinase (PDK) resulting from FAO, while citrate causes inhibition of PFK. Glucagon mobilizes glucose from the available sources including, FAs, glycogen, glycerol, amino acids, and lactate through gluconeogenesis.
In other peripheral organs such as skeletal muscle and cardiomyocytes, the extracellular FAs are transported into the cell through passive diffusion or via FATP or fatty acid translocase. Once FAs are in the cytosol, they are esterified to acyl-CoA and are shuttled directly into the mitochondria for FAO. Consequently, FAO increases the proportion of acetyl CoA, which allosterically inhibits PDH and activates PDK leading to suppression of glucose oxidation as this is not the fuel of choice under fasting condition due to its limited availability.49 Besides, activation of AMP-activated kinase (AMPK) due to increased AMP/ATP ratio leads to simultaneous activation of CPT-1 and inhibition ACC, resulting in increased FAO (Figure 6). Simultaneously, as gluconeogenesis depletes oxaloacetic acid levels in the TCA cycle, acetyl CoA can no longer be used by the mitochondria and therefore is diverted to ketone body (KB) production. From then on, FAO and KBs meet the whole-body energy requirements, especially during prolonged fasting.50
 |
Figure 6 Metabolism in fasting condition. Inhibition of glucose utilization by fatty acid oxidation mediated by inhibition of pyruvate dehydrogenase and phosphofructokinase. Abbreviations: ATP, adenosine triphosphate; ACC, acetyl CoA carboxylase; Cyt C, cytochrome C; CoQ, coenzyme Q; CPT, carnitine palmitoyltransferase; FAS, fatty acyl synthase; DHAP, dihydroxyacetone phosphate; LDH, lactate dehydrogenase; NAD/NADH, nicotinamide adenine dinucleotide; PFK, phosphofructokinase; PEPCK, phosphoenolpyruvate carboxykinase; PDH, pyruvate dehydrogenase; TCA, tricarboxylic acid cycle. |
Thus, the switch between the fuel substrate to meet energy demands is mainly mediated through counter-regulatory hormones, with mitochondria being a critical driving force from the nutritional and physiological perspective. In the fed state, mitochondria sense the fuel source availability and activate appropriate enzymes to utilize glucose preferentially in most tissue to generate adequate reducing equivalents to meet ATP generation requirement and store the excess. On the other hand, in the fasting state, the mitochondrial requirement for reducing equivalents for ATP generation by the ETC drives FAs mobilization from the WAT stores. With glycolysis no longer generating NADH and NADPH, there is the release of inhibition of CPT-1 to drive FA towards generating reducing equivalents through the TCA cycle to provide for the ETC cycle to continue generating the ATP to meet energy demand.
Metabolism in Caloric Restriction or Prolonged Fasting
During caloric restriction (CR) or prolonged fasting, besides FAs, ketone bodies (KB) and branched-chain amino acids (BCAAs) also provide an excellent source of energy. KBs synthesized in the liver through ketogenesis play a critical role in survival during the energy crisis by serving as a substrate for the brain and heart as the body glucose levels are low, and free fatty acid (FFA) cannot cross the blood-brain barrier. The synthesis of KBs, acetoacetate, and 3-beta-hydroxybutyrate is markedly increased due to excess supply of acetyl CoA from the breakdown of FA. Also, BCAAs are consumed in response to a protein-rich diet and prolonged fasting in the liver.51 The branched-chain α-ketoacid dehydrogenase (BCKD) complex, the rate-limiting enzyme in mitochondria, is inhibited by acyl-CoA and NADH, ensuring conservation of cellular proteins during short interval fasting or light exercise.52,53
Within 2 days of CR, whole body FA metabolism changes occur, and there is a shift to a diurnal cyclic pattern. In the initial phase lasting for 4–6 hours after food intake, there is elevated endogenous FA synthesis in the WAT, followed by a prolonged period of FA oxidation.54,55 In the second phase, FAs are effectively mobilized from WAT and oxidized in the mitochondria in the peripheral organs. Since this occurs where energy demand exceeds supply, the efficiency of FAO is explicitly increased in the muscle and the liver. Ratios of NAD+/NADH and the resultant redox state of the cell trigger activation of nuclear-encoded genes, including FoxO1, sirtuin-1 (SIRT 1), and Peroxisome proliferator-activated receptor-gamma co-activator (PGC-1α) activity, which in turn regulate enzymes in the metabolic pathway to facilitate utilization of the available fuel substrate and mitochondrial oxidative function regulating energy homeostasis.56
Furthermore, the shift to FAO with efficient mitochondrial function reduces the production of reactive oxygen species (ROS), attenuating oxidative damage and maintaining the cellular redox balance.57,58 Energy depletion is marked by the accumulation of adenosine monophosphate (AMP), an impending energy crisis activates the serine/threonine AMP-activated protein kinase (AMPK). AMPK interrupts ATP-consuming reactions and activates ATP-generating pathways, thereby promoting mitochondrial biogenesis. Energy homeostasis by the mitochondria in the face of changes in nutrient availability is regulated in part by the NAD-dependent deacetylase, SIRT1. During nutrient depletion, SIRT1, in response to increased NAD+/NADH ratio, deacetylates PGC-1α, allowing the co-activator to facilitate target gene transcription, which regulates lipid homeostasis in the liver during fasting and starvation through nuclear peroxisome proliferator-activated receptors (PPAR).59 Simultaneously, it also directly regulates the activity of acetyl-CoA synthetases through deacetylation, thereby regulating upstream pathways providing reducing equivalents for the generation of ATP.60 Therefore, CR may be another beneficial way of improving mitochondrial function inducing changes in macronutrient metabolism, specifically FA synthesis and FAO, thereby restoring metabolic flexibility.
During exercise, the oxidative metabolism of glucose and FAs provides almost all of the ATP required for the exercise, the two substrates being utilized differentially depending on the intensity and duration of the increased energy demand. Among all tissues, skeletal muscles use more than 95% of energy requirement, and the major substrates for oxidation are glycogen, TGs, FAs, and plasma glucose.61 The glucose is derived from liver glycogenolysis and gluconeogenesis and also directly through diet. FAs derived from both WAT and intramuscular TG breakdown is another source of the substrate.
In the skeletal muscle, the substrate uptake and oxidation are highly dependent on the intensity and duration of the exercise. FAO dominates in the low-intensity exercise up to ~60– 65% VO2 max.62 With increasing intensity of exercise, the fuel switches to muscle glycogen and glucose to meet energy demand efficiently, reduced FA delivery and lower mitochondrial FAO secondary to glycolytic flux.63 The increase in skeletal muscle glucose uptake is due to enhanced liver glucose output, initially from glycogenolysis, but later from gluconeogenesis with increasing intensity of exercise.
However, there is increased pyruvate production with accelerated glycolysis, more than mitochondrial intake capacity. So the pyruvate is converted into lactate in the cytosol, resulting in the replenishment of NAD+. Increased NAD+-NADH ratio generation promotes substrate flux for sustained glycolysis for ATP generation.31 Sustained endurance training diminishes lactate formation due to the increased efficiency of OXPHOS.64 Skeletal muscle senses limitation in glucose availability and adapt metabolically by SIRT1-dependent deacetylation of transcriptional regulators PGC-1α and FOXO1, culminating in transcriptional modulation of mitochondrial and lipid utilization genes.65 Interestingly, with longer duration of exercise, FAs from WAT (subcutaneous > visceral) make more significant contribution to overall energy supply due to exercise mediated adrenergic activation.66 Therefore, sustained regular exercise can upregulate glucose and FA metabolism and improve the state of metabolic inflexibility.
Hormones Involved in Maintaining Energy Homeostasis
Insulin
Insulin is an anabolic hormone in energy homeostasis. In healthy, non-obese individuals, post-meal, glucose enters β-cells of the pancreas through the GLUT2 transporter, and go through the glycolytic pathway and subsequently through OXPHOS to generate ATP. The rise in the ATP-ADP ratio leads to the closure of ATP-sensitive potassium channels, which in turn, depolarizes cell membranes. Consequently, it opens voltage-gated Ca2+ channels, resulting in Ca2+ influx into the cells triggering the initial phase of insulin secretion from prestored insulin in intracellular secretory vesicles.67 In the second phase, 1–2 hours after a meal, there is new recruitment of insulin secretory vesicles to the β-cell membrane resulting in slow and sustained insulin release from the pancreas in a pulsatile manner until blood glucose remains elevated.68 Insulin binds to IRS and triggers intracellular signaling cascade by activation of two pathways; the phosphatidylinositol-3-kinase (PI3K) and Mitogen-Activated Protein Kinase (MAPK).69 The PI3K pathway is responsible for metabolic action, while the MAPK pathway regulates gene expression and mitogenic effect.70 The metabolic actions primarily result in post-meal utilization of carbohydrates as preferred fuel and storage of excess as glycogen and after conversion to fat.
Besides metabolic fuel disposal and storage in tissues, insulin plays a crucial role in cardiac contractility and vascular tone.71 It causes vasodilation through the release of endothelium-derived nitrous oxide (NO) and increases capillary recruitment through its action on vascular endothelium leading to increased delivery of the hormone as well as glucose.72,73 Besides, insulin also exerts vasoconstriction through the Endothelin-1 (ET-1)74 and increased expression of vascular cell adhesion molecule (VCAM-1), intracellular adhesion molecule (ICAM), and E-selectin on endothelium mediated by MAPK pathway.75,76
Glucagon
Glucagon, a key catabolic hormone secreted by the α-cells of the pancreas, maintains the glucose concentration in the narrow range. The hormone mediates its glucoregulatory effects, mostly in the liver.77 When the glucose concentration falls below the threshold, the glucagon hormone stimulates hepatic glucose production, initially through glycogenolysis followed by gluconeogenesis. It also induces amino acid uptake except for BCAA to generate glucose through gluconeogenesis.78 Alternatively, the alanine released from the skeletal muscle during prolonged fasting is also used for gluconeogenesis. Glucagon also inhibits glycogenesis. Besides inducing FAO, it also stimulates ketogenesis, increasing β-hydroxybutyrate and acetoacetate production in the prolonged fasting state.79 Similarly, in WAT, glucagon increases lipolysis in elevating the plasma level of free FAs and induces FAO in the liver.80 As a result, glucagon is one of the major driving forces for metabolic adaptation by facilitating fuel availability in conditions such as starvation, exercise, and metabolic stress.
Renin-Angiotensin-Aldosterone System (RAAS)
There is evidence demonstrating the role of RAAS in the regulation of metabolic and hemodynamic function. RAAS interacts with insulin at extra- and intra-cellular level.81 Extracellularly, the RAAS controls bradykinin release that enhances insulin sensitivity in adipocytes via a NO-dependent pathway.82 Intracellularly, angiotensin II (ANG) opposes the action of insulin by inhibition of the PI3K pathway and its downstream kinases mediating both metabolic and hemodynamic effects, including vasoconstriction, adrenal aldosterone release, and glucose uptake in the target organs. Besides, ANG binding to angiotensin-1 (AT1) receptor inhibits insulin-induced NO production by rapid phosphorylation of tyrosine residues in Janus kinase-signal transducer and activator of transcription (JAK/STAT) pathway, activation of ERK1/2 pathway, and JNK pathway.83,84 As a result, ANG impairs the vasodilatory effects of insulin-mediated via the PI3K pathway.
Further, activation of the ANG – AT1 pathway rapidly generates ROS, impairing glucose uptake as well as causing endothelial and B-cell dysfunction.85,86 In contrast, Ang (1-7) induces vasodilation, diuresis, and natriuresis and inhibits cell growth and nor-epinephrine release.87,88 Aldosterone might exert direct effects on insulin receptors, and recent experiments indicate that aldosterone might decrease insulin sensitivity in human adipocytes.89 Conclusively, RAAS acts directly in WAT, skeletal muscle, liver, and the pancreas modulating both central and peripheral insulin sensitivity.90
Other Factors Regulating Metabolic Flexibility
Besides insulin and glucagon, several other glucoregulatory hormones also influence metabolic flexibility, such as growth hormone (GH), thyroid hormones (TH), glucocorticoids, catecholamines, adiponectin, aldosterone, glucagon-like peptide 1 (GLP-1), gastric inhibitory peptide (GIP), inflammatory factors secreted by the WAT, skeletal muscle, and the liver. These other hormones play a substantial role and maintain circulating glucose concentrations in the narrow range by controlling lipolysis through direct or indirect pathways, especially under stress. GH can exert lipolytic and ketogenic effects in the exercise and fasting over 2–3 hours.91 Strangely, glucocorticoids exert a similar effect to GH, exerting catabolic actions acutely by increasing the availability of substrate for mitochondrial oxidation.92 In the liver, it counterbalances the insulin action by promoting hepatic gluconeogenesis and increase glycogen storage. Conversely, in the skeletal muscle and WAT, it inhibits glucose uptake and utilization, which is critical during periods of stress, for instance, during fasting/starvation.93 It affects the function of the pancreas by increasing glucagon secretion.94
Similar to GH, the effect of TH is pervasive across various tissues via several mechanisms. Its action is mediated by cytoplasmic or mitochondrial receptors or through non-specific binding proteins that alter signaling cascade.95–97 TH regulates metabolic rate and adiposity through its direct effect on receptors that are expressed in the hypothalamus, WAT, brown adipose tissue (BAT), skeletal muscles, and liver, modulating glucose and lipid metabolism. In BAT tissue, TH regulates uncoupling protein (UCP1) expression, mitochondrial biogenesis, adipocyte differentiation, increases FAO and lipogenesis.98 Interestingly, browning (differentiation from WAT to BAT) has been reported on exposure to cold.99,100 Like BAT, in WAT, TH mobilizes fat, leading to increased FAs in the blood.101 It also enhances FAO in the liver and the muscle through AMPK dependent and independent mechanism.102 In the pancreas, TH is required for the physiological maturation of pancreatic β-cells to glucose-stimulated insulin-secreting cells.103 Besides, it also enhances the insulin-dependent entry of glucose via GLUT 4 and induces skeletal muscle fiber shift, thereby increasing energy expenditure.104
GLP-1 and GIP are insulinotropic hormones released from the gastrointestinal tract and stimulate pancreatic β-cells to release insulin and decrease glucagon secretion from α-cells.105 Together these gastrointestinal hormones promote β-cell proliferation and inhibit apoptosis, thereby countering hyperglycemia.106 The hormone, amylin, secreted by β-cells, complements the effects of insulin-lowering effect of glucose concentration by delaying stomach emptying and suppressing glucagon secretion. The hormone leptin secreted from adipocytes exerts its physiological effect by suppressing food intake and increasing energy expenditure via the hypothalamic circuit. It reduces lipogenesis and increases the FAO of non-adipocyte tissues.107 Another hormone from WAT, adiponectin, enhances insulin sensitivity through FAO and inhibition of hepatic gluconeogenesis.108
Amongst catecholamine, nor-adrenaline modulates metabolic homeostasis directly by lipolysis through adrenergic receptors expressed on WAT and indirectly by regulating blood flow to WAT.109 Besides catecholamine, aldosterone secreted from the adrenal cortex regulates sodium homeostasis controlling blood pressure and volume. It modulates insulin function and glucose metabolism through the mineralocorticoid receptor, adversely affecting vascular function by reducing NO production and increasing ROS causing endothelial dysfunction and VSM remodeling.110,111
Epigenetic Regulation of Energy Homeostasis
Besides hormonal regulation, epigenetics, ie, the complex interaction between environment and genome, also influences critical periods of embryonic, fetal, and early postnatal life that can affect metabolic flexibility with a permanent consequence.112 Research data on animal models and human studies indicate that epigenetic dysregulation can cause obesity. For example, comparisons of siblings before and after maternal bariatric surgery showed that maternal obesity before and during pregnancy promotes obesity in offspring.113,114 Maternal undernutrition and offspring obesity in the Dutch Winter Famine of 1944 and the thrifty gene hypothesis are likely inherited epigenetic alterations. These observations are also supported by vast experimental animal models.115
From an epigenetics perspective, hypothalamus, WAT, the liver, and skeletal muscles have been widely explored. Overall these studies suggest that the maternal obesity during pregnancy may induce epigenetic changes, enhancing adipogenesis, lowering IRS expression, altering hepatic gene expression and neuroanatomic architecture, consequently affecting intracellular signaling, hypothalamic function, and gene expression.116–119 There is also a strong compelling evidence suggesting that epigenetic changes are also induced via alteration in spontaneous physical activity.120,121 Twin association studies estimate approximately 0.3 to 0.8 heritability risk of genetic component to human physical activity.122,123 However, the extent to which epigenetic mechanisms regulate energy balance has not been identified. Further studies are required that convincingly demonstrate that epigenetic inheritance of environmental induced effects can alter metabolic flexibility. Metabolic regulation in an efficient manner results from a set of complex gene–environment interactions and is an evolutionarily preserved trait. However, its modification with associated disease susceptibility is probably predominantly determined by epigenetic variations with the potential to be inherited across generations.
Sex Difference in Metabolic Flexibility
Males and females distinctly differ in WAT distribution that has a significant impact on adaptive metabolic response as adipokine production, insulin sensitivity and FFAs release varies between the storage depots.124 Females have a higher percentage and a different pattern of body fat, with relatively more WAT in the hips and thighs. In contrast, males typically have the fat accumulation in the upper body and abdominal areas, the so-called android fat deposition.39 Multiple clinical and epidemiological studies have demonstrated the detrimental effect of visceral abdominal fat and protective effect of gluteal-femoral fat in T2DM, as a CV risk which eventually increases morbidity and mortality. However, this clear benefit ceases to exist after menopause indicating sex-related differences from the influence of sex hormones.125 Besides, sex differences in WAT are not limited to storage depots, as females have more BAT and an enhanced capacity to beige their WAT.126
The circulating levels of the metabolic hormones and their sensitivity also vary enormously.127 These hormones mediate some of the sex differences that are responsible for maintaining energy homeostasis in response to changing nutrient status.128 The complex interplay of genetics, epigenetics, and hormonal factors affect the structure, function, and physiology of above-mentioned tissue- and organ-system that impact metabolic flexibility.129 Thus, significant sex-based differences exist in the regulation of metabolism explains a varied development and progression of disease spectrum of DBCD.
Metabolic Inflexibility: Cellular and Metabolic Pathways
Calorie Excess and Reduced Physical Activity Causing Metabolic Inflexibility
Insulin and other glucoregulatory hormones, by virtue of their effects on various enzymes in the metabolic pathway, provide reducing equivalents to the mitochondria to maintain a dynamic utilization of glucose or lipid substrates in fed and fasting conditions, thus retaining a state of metabolic flexibility. The flexibility is compromised due to the continuous intake of calorie-dense foods with low nutrient values leading to the excess on the supply side of the supply-demand axis. Under normal circumstances, this is coupled to a sedentary lifestyle with a metabolic slowing down with age, leading to a state of metabolic inflexibility. The cells continuously adapt their metabolic pathways to meet their energy needs and respond to nutrient availability. At the same time, the mitochondria, which play a crucial role in maintaining cellular, tissue, and systemic flexibility, fine-tunes the redox reactions occurring at the ETC complexes in order to intake only required quantities of reducing equivalents to meet the ATP demand of the cell. Hence, excess fuel supply in the absence of a commensurate increase in demand results in the state of metabolic gridlock.24 Since the metabolic intermediates are prevented from entering the mitochondria, their accumulation ultimately results in feedback inhibition of uptake of both glucose and fat from circulation. The pancreas releases more insulin after sensing these excess circulating fuel substrates, resulting in the earliest manifestation of the insulin-resistant state.130 It has been observed that IR results subsequently in defects in the mobilization of fat and FAO, resulting in a vicious cycle of intracellular accumulation of FA, increased levels of TGs, and downregulation of insulin signaling.131 Altogether, these observations suggest that mitochondrial bioenergetics in the skeletal muscle and the liver plays a critical role in maintaining metabolic flexibility.23
In the WAT, surplus energy results in expanding the TG pool. Under normal physiological circumstances, the WAT can expand passively to accommodate excess nutrients. In response to excess glucose, amino acids, and FA, the triacylglycerol and glycogen depots are packed to the capacity and cannot accommodate more influx of the substrate. Intracellular accumulation of metabolites alters the morphology of WAT, and under normal circumstances, the influence of insulin, adipocytes undergo hypertrophy due to the activation of lipogenic genes.132 As a result of the enhanced FA uptake coupled with blunted FAO and lack of insulin-mediated inhibition of lipolysis, the net result is spilling excess into the circulation that is taken up by the non-WATs like the liver, muscle, heart, and pancreas leading to ectopic fat deposition.133 Data suggest that raised FFA levels drive IR in multiple organs.134,135 In this state, insulin-stimulated suppression of lipolysis is only partially achieved in WAT. While in the other organs, lipids are preferentially apportioned towards TGs’ synthesis and away from mitochondrial oxidation.136 However, tissues such as the pancreas and cardiomyocytes with limited compensatory FAO are predisposed to a high risk of steatosis.
On the other hand, the liver and muscle, which are better equipped for disposing of surplus FA through FAO, are affected much later. When stored TG exceeds the oxidative capacity of the cell, the excess feeds into non-oxidative pathways of FA metabolism, such as ceramide formation.137 The dysregulated release and storage of FA can further stimulate pro-inflammatory cytokines, which interfere with the local and systemic immune response. The lipid species and other mediators such as ROS, NO, diacylglycerol (DAG), impair insulin signaling through different mechanisms and enhance apoptosis.138 Therefore, excess lipid accretion induces lipotoxicity, which further contributes to metabolic inflexibility in different tissues.
At the level of skeletal muscle, type I oxidative, and type II glycolytic muscle fibers respond differently to nutritional and physiological conditions.139 In response to excess fuel, there is a tendency for fiber shift from type I oxidative fiber containing a high supply of mitochondria that uses TG for fuel towards the type II glycolytic fiber with little mitochondrion and which uses glucose as the energy source.140,141 These findings are further supported by the observation that there is impairment in the balance between mitochondrial fusion and fission processes leading to abnormal mitochondrial fragmentation and degradation, thereby leading to a fiber shift to glycolytic type.142,143 Also, there is a downregulation of nuclear-encoded genes, such as PGCl-α, reducing mitochondrial biogenesis.144,145
Furthermore, overfed mitochondria continue to catabolize incoming metabolites leading to the competition between the substrates and enzyme, generating a feedforward inhibition in the network.146 As a consequence of the constant influx of FAs, the glucose oxidation is blunted by concurrent activation of PDK and inhibition of PDH.147 Intracellular accumulation of acetyl CoA, NADH, and ATP increased redox potential and inhibition of TCA enzymes. As a result of incomplete mitochondrial oxidation, there is ineffective switching between substrates causing a mitochondrial gridlock.24 Additionally, a high NADH/NAD+ ratio inhibits AMPK, reducing the expression of genes involved in ATP generation and retarding insulin signaling.148 Altogether, there are reduced levels of mitochondrial oxidative enzymes, decreased mitochondrial number, abnormal morphology, and lower ATP synthesis in the skeletal muscles of IR individuals.149,150 Besides, accumulated intracellular metabolites activate the protein kinase C pathway causing phosphorylation of IRS, inhibiting insulin signaling PI3K pathway, further reducing glucose uptake.151
Conversely, in the liver, there is increased hepatic mitochondrial oxidative capacity for both lipid and non-lipid substrates. This finding is supported by the gene expression study that revealed the upregulation of hepatic mRNAs related to OXPHOS, ROS, and gluconeogenesis.152 Further, there was a post-prandial increase in hepatic ATP by 6-fold in insulin-resistant subjects compared to lean control, suggesting augmented post-prandial adaptation of hepatic energy metabolism to calorie overload.153 The underlying mechanism is unclear; however, it is thought that due to increased release of FFA from visceral WAT, there is substrate competition between lipids and non-lipid fuel for oxidation. Interestingly, it has been shown that while β-oxidation and ketogenesis are dysfunctional in the diabetic liver, pyruvate carboxylase and TCA cycle flux appear to be elevated.154 This is in contrast to the mitochondria from skeletal muscle of diabetic rat models, where TCA cycle activity appears to be down-regulated. As such, inefficient hepatic mitochondrial functioning in conjunction with elevated FAs further compound oxidative stress from the ROS burden.155 The mitochondrial ROS activates the secretion of pro-inflammatory adipocytokines, eg, leptin, interleukins (Il-6, IL-8), Tumour Necrosis Factor (TNF- α), and monocyte chemoattractant protein-1 (MCP-1),156 further impairing insulin signaling. The perturbed ROS production affects the activities and functions of metabolic enzymes and signaling proteins compromising the flexibility of glucose and lipid metabolism in skeletal muscle and WAT.157
Pancreatic Beta-Cell Dysfunction
Chronic nutrient excess synergistically induces a deleterious effect on the β-cells’ mass and function. As a consequence of hyperglycemia coupled with high FFA, there is a compensatory increase in insulin production in the pancreatic β-cells. Despite the initial compensatory hyperinsulinemia, there is a relatively minor change in IR, maintaining normal glucose tolerance.158 Compensatory hyperinsulinemia helps maintain normal or near-normal levels of plasma glucose. As time progresses, β-cells increase cell mass to compensate for higher insulin requirements.159 It becomes even more challenging to control glycemic levels due to the inefficiency of insulin to inhibit glucagon secretion resulting in increased stress on β-cells. As a result, the overproduction of ROS damages β-cell’s mitochondrial DNA and cellular proteins.160 Inevitably, progressive dysfunction arises, resulting in loss of plasticity for insulin production and secretion, and eventual loss of β-cells.161 Likewise, lipotoxicity from the accumulation of FA in the β-cells further impairs the regulation of glucose metabolism in the skeletal muscle and the liver.162 The circulating cytokines also adversely affect β-cells by weakening their function and limiting the mass. Furthermore, impaired mitochondrial dynamics also play a vital role in nutrient-induced β-cell apoptosis, which may be a critical factor in pathogenesis.163 The additive effect of glucotoxicity and lipotoxicity compound the metabolic insult negatively impacting insulin sensitivity triggering pancreatic β-cells’ failure.164 The upregulation of mitochondrial fission causes overproduction of ROS that further contributes to mitochondrial dysfunction in hyperglycemia25,165 (Figure 7).
 |
Figure 7 Metabolic perturbation in different organ system due to mitochondrial dysfunction leading to metabolic inflexibility. |
Metabolic Inflexibility Associated with DBCD and the Spectrum of Micro- and Macro-Vascular Complications
Given the insulin’s central role in glucose and lipid metabolism, it is not surprising that multiple target organs are affected by its dysregulation manifesting as a continuum of the disease spectrum of DBCD. Typically, there is decreased sensitivity and/or response to insulin, affecting both metabolic and hemodynamic actions in different tissues and organs. It is likely that the IR originates within different tissues, and the degree of resistance varies between tissues due to considerable heterogeneity in insulin response.166,167 For instance, impaired glucose tolerance (IGT) is associated with skeletal muscle resistance and compensatory hyperinsulinemia compared to impaired fasting glucose, which is associated with hepatic IR and excessive endogenous glucose production.19 In contrast, combined IFG/IGT had a combination of increased gluconeogenesis, lack of suppression of hepatic glycogenolysis by insulin, and impaired glucose disposal in the peripheral tissues.168 As discussed in the previous section, metabolic inflexibility translates into these phenotypes of prediabetes resulting from the erratic selection of fuels in mitochondria, thereby interrupting the insulin-mediated PI3K pathway. In contrast, uninterrupted signaling of the MAPK pathway augments the mitogenic effect resulting in endothelial dysfunction and in endothelial cells and vascular smooth muscle cell proliferation.169,170 Thus, in the setting of IR due to the downregulation of PI3K and upregulation of the MAPK signaling pathway gives rise to prohypertensive, atherogenic, and thrombogenic background promoting atherosclerosis. Besides, advanced glycation end products formed from non-enzymatic glycation of proteins and lipids further induce endothelium damage, amplify inflammatory response, and increase the risk of microvascular dysfunction.171,172 However, the degree of damage and/or repair varies across different tissues or organs, since these mechanisms may be active preferentially affect some but not all organs, but generally they are associated with the development of microvascular complications.173
Overall, the evidence suggests that IR and β-cell dysfunction appears to be important initiating events for the development of the disease spectrum of diabetes and vascular complication.174 Besides hyperglycemia, other metabolic factors, including hypertension, obesity, and dyslipidemia, play a pathogenic role in the development of micro and macro-vascular complications. These abnormalities precede as an early event in the T2DM and associated micro- as well as macrovascular complications175,176 (Figure 7).
Unmet Need for Therapy Addressing Metabolic Inflexibility
Although the health benefits of lifestyle intervention to reduce body weight, including exercise and calorie restrictions, are well recognized in restoring metabolic flexibility in DBCD; however, these are difficult to sustain at a level that results in benefit in most patients.177,178 Also, currently available drugs such as sodium-glucose co-transporter-2 (SGLT2) inhibitors, glucagon-like peptide 1 (GLP-1) receptor agonists, dipeptidyl peptidase-4 (DPP-4) inhibitors, sulphonylureas, metformin, etc. have limitations. These therapies being wholly glucocentric, fail to control the disease progression and often fail to achieve accepted treatment goals. Besides, the treatment being symptomatic, is usually initiated often in a stage, when the other disease aspects such as risk factors (obesity, pre-hypertension, dyslipidemia, etc.) or complications (neuropathy, fatty liver disease, CVD, etc.) have progressed beyond control. Thus, there is a significant need for addressing metabolic inflexibility to prevent the progression along the DBCD disease spectrum from IR to prediabetes to frank diabetes and development of micro- and macro-vascular complications. There is no denying that there is a significant unmet need for safe and effective drugs that can prevent the progression of IR. An intensive and focused effort is needed to identify new targets for pharmacological interventions and also address dysglycemia, dyslipidemia, and hypertension collectively. Agents that either inhibit caloric intake or increase energy expenditure could result in reversing the process, and these could be approaches worth exploring.
Conclusion
Insulin and other glucoregulatory hormones provide an integrated set of signals to maintain metabolic flexibility. Sex-based differences arising from genetics, epigenetics, and hormones influence metabolic flexibility. Disruption in metabolic flexibility is most commonly caused by excess food intake and sedentary lifestyle, contributing to mitochondrial dysfunction characterized by inefficient nutrient sensing and ineffective substrate switching deemed as “Metabolic inflexibility.” This condition alters metabolic and non-metabolic pathways leading to the development of IR and β-cell dysfunction. All these effects induce cellular events, including increase oxidative stress and inflammation, endothelial dysfunction, production and accumulation of advanced glycation end products, ectopic fat accumulation resulting in dyslipidemia, hypertension, and micro- and macro-vascular complications. These complications can be addressed through traditional lifestyle measures such as caloric restriction and exercise that are vital components for tackling metabolic inflexibility. However, these traditional lifestyle measures are challenging over long-term in the modern era of nutrient excess. Therefore, pharmacological interventions, in addition to lifestyle changes, can play a major role in the management of dysglycemia, dyslipidemia, and hypertension collectively, by addressing the underlying defect and help prevent progression to vascular and, consequently, end-organ damage. Targeting pathways that address mitochondrial dysfunction would exert a beneficial effect on metabolic inflexibility that may correct IR, β cell dysfunction, and endothelial dysfunction and, as a consequence, would be therapeutically effective across the entire continuum of DBCD.
Abbreviations
ACC, Acetyl CoA carboxylase; AMP, Adenosine Monophosphate; AMPK, AMP-activated kinase; ANG, Angiotensin II; AT1, Angiotensin-1; ATP, Adenosine TriPhosphate; BAT, Brown adipose tissue; BCAA, Branched Chain Amino Acid; BCKD, Branched Chain α Ketoacid Dehydrogenase; BMI, Body Mass Index; CoA, Coenzyme A; CPT-1, Carnitine PalmitoylTransferase-1; CR, Caloric Restriction; CVD, Cardiovascular Disease; DAG, Diacyl Glycerol; DBCD, Dysglycemia-Based Chronic Disease; ERK, Extracellular Signal-Regulated Kinases; ET-1, Endothelin-1; ETC, Electron Transport Chain; FA, Fatty Acid; FADH, Flavin Adenine Dinucleotide; FAO, Fatty Acid Oxidation; FATP, Fatty Acid Transport Protein; FAS, Fatty Acyl Synthase; FFA, Free Fatty Acid; FoxO-1, Forkhead-O1; GH, Growth Hormone; GK, Glucokinase; G6P, Glucose-6-Phosphate; GIP, Gastric Inhibitory Polypeptide; GLP-1, Glucagon-Like Peptide-1; GLUT, Glucose Transport; HDL, High Density Lipoprotein; 3-HB, 3-beta-hydroxybutyrate; ICAM, Intracellular Adhesion Molecule; ICMR, Indian Council of Medical Research; IDF, International Diabetes Federation; IL, Interleukin; IR, Insulin Resistance; IRS, Insulin Receptor Substrate; JAK/STAT, Janus Kinase-Signal Transducer and Activator; JNK, c-Jun N-terminal kinase; KB, Ketone Bodies; LDL, Low Density Lipoprotein; LDH, Lactate Dehydrogenase; LPL, Lipoprotein Lipase; MAPK, Mitogen-Activated Protein Kinase; MCP, Monocyte Chemoattractant Protein-1; NAD/NADH, Nicotinamide Adenine Dinucleotide; NEFA, Non-Esterified Fatty Acid; NO, Nitrous Oxide; OXPHOS, Oxidative Phosphorylation; PDH, Pyruvate Dehydrogenase; PDK, Pyruvate Dehydrogenase Kinase; PEPCK, Phosphoenolpyruvate Carboxykinase; PFK, Phosphofructokinase; PGC1-α, Peroxisome proliferator-activated receptor-gamma co-activator1-α; PI3K, Phosphatidylinositol 3-kinase; PKB, Protein Kinase B; PPAR, Peroxisome proliferator-activated receptor; RAAS, Renin Angiotensin Aldosterone System; REE, Resting Energy Expenditure; ROS, Reactive Oxygen Species; SIRT, Sirtuin; SNS, Sympathetic Nervous System; TCA, Tricarboxylic Acid Cycle; T2DM, Type II Diabetes Mellitus; TG, Triglyceride; TH, Thyroid hormone; TNF, Tumour Necrosis Factor; UCP1, Uncoupling Protein 1; VCAM, Vascular Cell Adhesion Molecule; VLDL, Very Low Density Lipoprotein; VSM, Vascular Smooth Muscle; WAT, White Adipose Tissue.
Acknowledgments
The authors are thankful to Dr Suchit Kumbhare and Dr. Deepa Joshi for editorial assistance.
Disclosure
Sanjay Kalra, Ambika Gopalakrishnan Unnikrishnan, Manash P Baruah, Rakesh Sahay, and Ganapathi Bantwal report no conflicts of interest in this work.
References
1. Saeedi P, Petersohn I, Salpea P, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843. doi:10.1016/j.diabres.2019.107843
2. Anjana RM, Deepa M, Pradeepa R, et al. ICMR INDIAB Collaborative Study Group. Prevalence of diabetes and prediabetes in 15 states of India: Results from the ICMR INDIAB population based cross sectional study. Lancet Diabetes Endocrinol. 2017;5:585–596.
3. Bhardwaj S, Misra A. Obesity, diabetes and the Asian phenotype. World Rev Nutr Diet. 2015;111:116–122.
4. Unnikrishnan R, Anjana RM, Mohan V. Diabetes in South Asians: is the phenotype different? Diabetes. 2014;63:53–55. doi:10.2337/db13-1592
5. Joshi SR, Anjana RM, Deepa M, et al. Prevalence of dyslipidemia in urban and rural India: the ICMR–INDIAB Study. PLoS One. 2014;9:e96808. doi:10.1371/journal.pone.0096808
6. Yadav D, Mishra M, Tiwari A, et al. Prevalence of dyslipidemia and hypertension in Indian Type 2 diabetic patients with metabolic syndrome and its clinical significance. Osong Public Health Res Perspect. 2014;5:169–175. doi:10.1016/j.phrp.2014.04.009
7. Diamond J. Medicine: diabetes in India. Nature. 2011;469:478–479. doi:10.1038/469478a
8. McKeigue PM, Shah B, Marmot MG. Relation of central obesity and insulin resistance with high diabetes prevalence and cardiovascular risk in South Asians. Lancet. 1991;337:382–386.
9. Enas EA, Singh V, Munjal YP, et al. Reducing the burden of coronary artery disease in India: challenges and opportunities. Indian Heart J. 2008;60:161–175.
10. Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities — the role of insulin resistance and the sympathoadrenal system. N Engl J Med. 2009. doi:10.1056/NEJM199602083340607
11. El-Atat F, Aneja A, Mcfarlane S, Sowers J. Obesity and hypertension. Endocrinol Metab Clin North Am. 2003;32:823–854. doi:10.1016/S0889-8529(03)00070-7
12. Levitan EB, Song Y, Ford ES, Liu S. Is nondiabetic hyperglycemia a risk factor for cardiovascular disease? A meta-analysis of prospective studies. Arch Intern Med. 2004;164:2147–2155. doi:10.1001/archinte.164.19.2147
13. Nguyen TT, Wang JJ, Wong TY. Retinal vascular changes in pre-diabetes and prehypertension: new findings and their research and clinical implications. Diabetes Care. 2007;30:2708–2715. doi:10.2337/dc07-0732
14. Gabir MM, Hanson RL, Dabelea D, et al. Plasma glucose and prediction of microvascular disease and mortality: evaluation of 1997 American Diabetes Association and 1999 World Health Organization criteria for diagnosis of diabetes. Diabetes Care. 2000;23:1113–1118. doi:10.2337/diacare.23.8.1113
15. Sarwar N, Gao P, Seshasai SR, et al. Emerging Risk Factors Collaboration. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215–2222. doi:10.1016/S0140-6736(10)60484-9
16. Barr ELM, Zimmet PZ, Welborn TA, et al. Risk of cardiovascular and all-cause mortality in individuals with diabetes mellitus, impaired fasting glucose, and impaired glucose tolerance: the Australian Diabetes, Obesity, and Lifestyle Study (AusDiab). Circulation. 2007;116:151–157. doi:10.1161/CIRCULATIONAHA.106.685628
17. Flier JS, Kahn CR, Roth J. Receptors, antireceptor antibodies and mechanisms of insulin resistance. N Engl J Med. 1979;300:413–419. doi:10.1056/NEJM197902223000808
18. Frayn KN, Coppack SW. Insulin resistance, adipose tissue and coronary heart disease. Clin Sci. 1992;82:1–8.
19. Abdul-Ghani MA, Tripathy D, DeFronzo RA. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 2006;29:1130–1139. doi:10.2337/dc05-2179
20. Mechanick JI, Garber AJ, Grunberger G, Handelsman Y, Garvey WT. Dysglycemia-based chronic disease: an American Association of Clinical Endocrinologists position statement. Endocr Pract. 2018;24:995–1011. doi:10.4158/PS-2018-0139
21. Kelley DE, Goodpaster B, Wing RR, Simoneau J-A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol Endocrinol Metab. 1999;277:E1130–E1141. doi:10.1152/ajpendo.1999.277.6.E1130
22. Neel JV. Diabetes mellitus: a “Thrifty” genotype rendered detrimental by “Progress”? Am J Hum Genet. 1962;14:353–362.
23. Smith RL, Soeters MR, Wüst RCI, Houtkooper RH. Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr Rev. 2018;39:489–517.
24. Muoio DM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159:1253–1262. doi:10.1016/j.cell.2014.11.034
25. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi:10.2337/diabetes.54.6.1615
26. Galgani JE, Moro C, Ravussin E. Metabolic flexibility and insulin resistance. Am J Physiol Endocrinol Metab. 2008;295:E1009–E1017. doi:10.1152/ajpendo.90558.2008
27. Bloom SR, Edwards AV. The role of the parasympathetic system in the control of insulin release in the conscious calf. J Physiol. 1981;314:37–46. doi:10.1113/jphysiol.1981.sp013688
28. Dimitriadis G, Mitrou P, Lambadiari V, Maratou E, Raptis SA. Insulin effects in muscle and adipose tissue. Diabetes Res Clin Pract. 2011;93(Suppl 1):S52–S59. doi:10.1016/S0168-8227(11)70014-6
29. Postic C, Dentin R, Girard J. Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab. 2004;30:398–408. doi:10.1016/S1262-3636(07)70133-7
30. Han H-S, Kang G, Kim JS, Choi BH, Koo S-H. Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med. 2016;48:e218. doi:10.1038/emm.2015.122
31. Adeva-Andany M, López-Ojén M, Funcasta-Calderón R, et al. Comprehensive review on lactate metabolism in human health. Mitochondrion. 2014;17:76–100. doi:10.1016/j.mito.2014.05.007
32. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13:572–587. doi:10.1038/nrendo.2017.80
33. Wakil SJ, Abu-Elheiga LA. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res. 2009;50:S138–S143. doi:10.1194/jlr.R800079-JLR200
34. Frayn KN, Arner P, Yki-Järvinen H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem. 2006;42:89–103. doi:10.1042/bse0420089
35. Shepherd PR, Kahn BB. Glucose transporters and insulin action – implications for insulin resistance and diabetes mellitus. N Engl J Med. 1999;341:248–257. doi:10.1056/NEJM199907223410406
36. Kelley DE. Skeletal muscle fat oxidation: timing and flexibility are everything. J Clin Invest. 2005;115:1699–1702. doi:10.1172/JCI25758
37. Jensen J, Rustad PI, Kolnes AJ, Lai Y-C. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front Physiol. 2011;2. doi:10.3389/fphys.2011.00112
38. Frayn KN, Shadid S, Hamlani R, et al. Regulation of fatty acid movement in human adipose tissue in the postabsorptive-to-postprandial transition. Am J Physiol Endocrinol Metab. 1994;266:E308–E317. doi:10.1152/ajpendo.1994.266.3.E308
39. Karastergiou K, Smith SR, Greenberg AS, Fried SK. Sex differences in human adipose tissues – the biology of pear shape. Biol Sex Differ. 2012;3:13. doi:10.1186/2042-6410-3-13
40. Nguyen TT, Hernández Mijares A, Johnson CM, Jensen MD. Postprandial leg and splanchnic fatty acid metabolism in nonobese men and women. Am J Physiol. 1996;271:E965–E972. doi:10.1152/ajpendo.1996.271.6.E965
41. Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol. 2017;8:1–8. doi:10.1002/cphy.c170012
42. Shao D, Tian R. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol. 2015;6:331–351.
43. Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19:181–192. doi:10.1016/j.cmet.2013.12.008
44. Wasserman DH, Spalding JA, Lacy DB, et al. Glucagon is a primary controller of hepatic glycogenolysis and gluconeogenesis during muscular work. Am J Physiol. 1989;257:E108–E117. doi:10.1152/ajpendo.1989.257.1.E108
45. Galbo H, Richter EAHilsted J, et al. Hormonal regulation during prolonged exercise. Ann NY Acad Sci. 1977;301:72–80. doi:10.1111/j.1749-6632.1977.tb38187.x
46. Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr. 2007;27:79–101. doi:10.1146/annurev.nutr.27.061406.093734
47. Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4:177–197. doi:10.1002/cphy.c130024
48. Pilkis SJ, Claus TH. Hepatic gluconeogenesis/glycolysis: regulation and structure/function relationships of substrate cycle enzymes. Annu Rev Nutr. 1991;11:465–515. doi:10.1146/annurev.nu.11.070191.002341
49. Schrauwen-Hinderling VB, Kooi ME, Schrauwen P. Mitochondrial function and diabetes: consequences for skeletal and cardiac muscle metabolism. Antioxid Redox Signal. 2016;24:39–51. doi:10.1089/ars.2015.6291
50. Cahill GF. Fuel metabolism in starvation. Annu Rev Nutr. 2006;26:1–22. doi:10.1146/annurev.nutr.26.061505.111258
51. Owen OE, Morgan AP, Kemp HG, et al. Brain metabolism during fasting. J Clin Invest. 1967;46:1589–1595. doi:10.1172/JCI105650
52. Shimomura Y, Honda T, Shiraki M, et al. Branched-chain amino acid catabolism in exercise and liver disease. J Nutr. 2006;136:250S–3S. doi:10.1093/jn/136.1.250S
53. Harris RA, Joshi M, Jeoung NH, Obayashi M. Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J Nutr. 2005;135:1527S–1530S. doi:10.1093/jn/135.6.1527S
54. Duffy PH, Feuers R, Leakey J, et al. Effect of chronic caloric restriction on physiological variables related to energy metabolism in the male Fischer 344 rat. Mech Ageing Dev. 1989;48:117–133. doi:10.1016/0047-6374(89)90044-4
55. Bruss MD, Khambatta CF, Ruby MA, Aggarwal I, Hellerstein MK. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am J Physiol Endocrinol Metab. 2009;298:E108–E116. doi:10.1152/ajpendo.00524.2009
56. Civitarese AE, Carling S, Heilbronn LK, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. doi:10.1371/journal.pmed.0040076
57. Guarente L. Mitochondria – a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi:10.1016/j.cell.2008.01.007
58. Gredilla R, Barja G. Minireview: the role of oxidative stress in relation to caloric restriction and longevity. Endocrinology. 2005;146:3713–3717. doi:10.1210/en.2005-0378
59. Li X. SIRT1 and energy metabolism. Acta Biochim Biophys Sin. 2013;45:51–60. doi:10.1093/abbs/gms108
60. Chang H-C, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2014;25:138–145. doi:10.1016/j.tem.2013.12.001
61. Romijn JA, Coyle EF, Sidossis LS, et al. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol. 1993;265:E380–E391. doi:10.1152/ajpendo.1993.265.3.E380
62. Hargreaves M, Spriet LL. Exercise metabolism: fuels for the fire. Cold Spring Harb Perspect Med. 2018;8:a029744. doi:10.1101/cshperspect.a029744
63. Spriet LL. New insights into the interaction of carbohydrate and fat metabolism during exercise. Sports Med. 2014;44(Suppl 1):S87–S96. doi:10.1007/s40279-014-0154-1
64. Messonnier LA, Emhoff C-AW, Fattor JA, et al. Lactate kinetics at the lactate threshold in trained and untrained men. J Appl Physiol. 2013;114:1593–1602. doi:10.1152/japplphysiol.00043.2013
65. Cantó C, Jiang LQ, Deshmukh AS, et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi:10.1016/j.cmet.2010.02.006
66. Arner P, Kriegholm E, Engfeldt P, Bolinder J. Adrenergic regulation of lipolysis in situ at rest and during exercise. J Clin Invest. 1990;85:893–898. doi:10.1172/JCI114516
67. Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi:10.2337/diabetes.49.11.1751
68. Henquin J-C, Ishiyama N, Nenquin M, Ravier MA, Jonas J-C. Signals and pools underlying biphasic insulin secretion. Diabetes. 2002;51:S60–S67. doi:10.2337/diabetes.51.2007.S60
69. González-Sánchez JL, Serrano-Ríos M. Molecular basis of insulin action. Drug News Perspect. 2007;20:527–531. doi:10.1358/dnp.2007.20.8.1157615
70. White MF. Insulin signaling in health and disease. Science. 2003;302:1710–1711. doi:10.1126/science.1092952
71. Baron AD. Hemodynamic actions of insulin. Am J Physiol. 1994;267:E187–E202. doi:10.1152/ajpendo.1994.267.2.E187
72. Steinberg HO, Baron AD. Vascular function, insulin resistance and fatty acids. Diabetologia. 2002;45:623–634. doi:10.1007/s00125-002-0800-2
73. Rattigan S, Wheatley C, Richards SM, Barrett EJ, Clark MG. Exercise and insulin-mediated capillary recruitment in muscle. Exerc Sport Sci Rev. 2005;33:43–48.
74. Potenza MA, Marasciulo FL, Chieppa DM, et al. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am J Physiol Heart Circ Physiol. 2005;289:H813–H822. doi:10.1152/ajpheart.00092.2005
75. Montagnani M, Golovchenko I, Kim I, et al. Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem. 2002;277:1794–1799. doi:10.1074/jbc.M103728200
76. Eringa EC, Stehouwer CD, Van Nieuw Amerongen GP, et al. Vasoconstrictor effects of insulin in skeletal muscle arterioles are mediated by ERK1/2 activation in endothelium. Am J Physiol Heart Circ Physiol. 2004;287:H2043–H2048. doi:10.1152/ajpheart.00067.2004
77. Rix I, Nexøe-Larsen C, Bergmann NC, et al. Glucagon physiology. In: Feingold KR, editor. Endotext. MDText.com, Inc.; 2000.
78. Boden G, Rezvani I, Owen OE. Effects of glucagon on plasma amino acids. J Clin Invest. 1984;73:785–793. doi:10.1172/JCI111272
79. Liljenquist JE, Bomboy JD, Lewis SB, et al. Effects of glucagon on lipolysis and ketogenesis in normal and diabetic men. J Clin Invest. 1974;53:190–197. doi:10.1172/JCI107537
80. Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60:2441–2449. doi:10.2337/db11-0425
81. Muscogiuri G, Chavez A, Gastaldelli A, et al. The crosstalk between insulin and renin-angiotensin-aldosterone signaling systems and its effect on glucose metabolism and diabetes prevention. Curr Vasc Pharmacol. 2008;6:301–312. doi:10.2174/157016108785909715
82. Beard KM, Lu H, Ho K, Fantus IG. Bradykinin augments insulin-stimulated glucose transport in rat adipocytes via endothelial nitric oxide synthase–mediated inhibition of Jun NH2-terminal kinase. Diabetes. 2006;55:2678–2687. doi:10.2337/db05-1538
83. Marrero MB, Schieffer B, Paxton WG, et al. Direct stimulation of Jak/STAT pathway by the angiotensin II AT 1 receptor. Nature. 1995;375:247–250. doi:10.1038/375247a0
84. Francesco A, Emanuela L, Angela S, Francesco P, Giorgio S. Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ Res. 2004;94:1211–1218. doi:10.1161/01.RES.0000126501.34994.96
85. Pendergrass KD, Gwathmey TM, Michalek RD, Grayson JM, Chappell MC. The angiotensin II - AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem Biophys Res Commun. 2009;384:149–154. doi:10.1016/j.bbrc.2009.04.126
86. Lightfoot YL, Chen J, Mathews CE. Oxidative stress and beta cell dysfunction. Methods Mol Biol. 2012;900:347–362.
87. Schindler C, Bramlage P, Kirch W, Ferrario CM. Role of the vasodilator peptide angiotensin-(1–7) in cardiovascular drug therapy. Vasc Health Risk Manag. 2007;3:125–137.
88. Gironacci Mariela M, Valera María S, Irene Y, Clara P. Angiotensin-(1–7) inhibitory mechanism of norepinephrine release in hypertensive rats. Hypertension. 2004;44:783–787. doi:10.1161/01.HYP.0000143850.73831.9d
89. Luther JM, Luo P, Kreger MT, et al. Aldosterone decreases glucose-stimulated insulin secretion in vivo in mice and in murine islets. Diabetologia. 2011;54:2152–2163. doi:10.1007/s00125-011-2158-9
90. Favre GA, Esnault VLM, Van Obberghen E. Modulation of glucose metabolism by the renin-angiotensin-aldosterone system. Am J Physiol Endocrinol Metab. 2015;308:E435–E449. doi:10.1152/ajpendo.00391.2014
91. Keller U, Schnell H, Girard J, Stauffacher W. Effect of physiological elevation of plasma growth hormone levels on ketone body kinetics and lipolysis in normal and acutely insulin-deficient man. Diabetologia. 1984;26:103–108. doi:10.1007/BF00281115
92. Macfarlane DP, Forbes S, Walker BR. Glucocorticoids and fatty acid metabolism in humans: fuelling fat redistribution in the metabolic syndrome. J Endocrinol. 2008;197:189–204. doi:10.1677/JOE-08-0054
93. Kuo T, McQueen A, Chen T-C, Wang J-C. Regulation of glucose homeostasis by glucocorticoids. Adv Exp Med Biol. 2015;872:99–126.
94. Wise JK, Hendler R, Felig P. Influence of glucocorticoids on glucagon secretion and plasma amino acid concentrations in man. J Clin Invest. 1973;52:2774–2782. doi:10.1172/JCI107473
95. Ortiga-Carvalho TM, Sidhaye AR, Wondisford FE. Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat Rev Endocrinol. 2014;10:582–591. doi:10.1038/nrendo.2014.143
96. Cordeiro A, Souza LL, Einicker-Lamas M, Pazos-Moura CC. Non-classic thyroid hormone signalling involved in hepatic lipid metabolism. J Endocrinol. 2013;216:R47–R57. doi:10.1530/JOE-12-0542
97. Bianco AC, Dumitrescu A, Gereben B, et al. Paradigms of dynamic control of thyroid hormone signaling. Endocr Rev. 2019;40:1000–1047.
98. Shu L, Hoo RL, Wu X, et al. A-FABP mediates adaptive thermogenesis by promoting intracellular activation of thyroid hormones in brown adipocytes. Nat Commun. 2017;8:14147. doi:10.1038/ncomms14147
99. Cypess AM, Lehman S, Williams G, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi:10.1056/NEJMoa0810780
100. Virtanen KA, Lidell ME, Orava J, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi:10.1056/NEJMoa0808949
101. Cicatiello AG, Di Girolamo D, Dentice M. Metabolic effects of the intracellular regulation of thyroid hormone: old players, new concepts. Front Endocrinol. 2018;9. doi:10.3389/fendo.2018.00474
102. Sayre NL, Lechleiter JD. Fatty acid metabolism and thyroid hormones. Curr Trends Endocrinol. 2012;6:65–76.
103. Aguayo-Mazzucato C, Zavacki AM, Marinelarena A, et al. Thyroid hormone promotes postnatal rat pancreatic β-cell development and glucose-responsive insulin secretion through MAFA. Diabetes. 2013;62:1569–1580. doi:10.2337/db12-0849
104. Simonides WS, van Hardeveld C. Thyroid hormone as a determinant of metabolic and contractile phenotype of skeletal muscle. Thyroid. 2008;18:205–216. doi:10.1089/thy.2007.0256
105. Seino Y, Fukushima M, Yabe D. GIP and GLP-1, the two incretin hormones: similarities and differences. J Diabetes Investig. 2010;1:8–23. doi:10.1111/j.2040-1124.2010.00022.x
106. Yabe D, Seino Y. Two incretin hormones GLP-1 and GIP: comparison of their actions in insulin secretion and β cell preservation. Prog Biophys Mol Biol. 2011;107:248–256. doi:10.1016/j.pbiomolbio.2011.07.010
107. Shimabukuro M, Koyama K, Chen G, et al. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc Natl Acad Sci USA. 1997;94:4637–4641. doi:10.1073/pnas.94.9.4637
108. Lihn AS, Pedersen SB, Richelsen B. Adiponectin: action, regulation and association to insulin sensitivity. Obes Rev. 2005;6:13–21. doi:10.1111/j.1467-789X.2005.00159.x
109. Arner P. Control of lipolysis and its relevance to development of obesity in man. Diabetes Metab Rev. 1988;4:507–515. doi:10.1002/dmr.5610040507
110. McCurley A, Pires PW, Bender SB, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18:1429–1433. doi:10.1038/nm.2891
111. Pruthi D, McCurley A, Aronovitz M, et al. Aldosterone promotes vascular remodeling by direct effects on smooth muscle cell mineralocorticoid receptors. Arterioscler Thromb Vasc Biol. 2014;34:355–364. doi:10.1161/ATVBAHA.113.302854
112. Waterland RA. Epigenetic mechanisms affecting regulation of energy balance: many questions, few answers. Annu Rev Nutr. 2014;34:337–355. doi:10.1146/annurev-nutr-071813-105315
113. Kral JG, Biron S, Simard S, et al. Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics. 2006;118:e1644–e1649. doi:10.1542/peds.2006-1379
114. Smith J, Cianflone K, Biron S, et al. Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J Clin Endocrinol Metab. 2009;94:4275–4283. doi:10.1210/jc.2009-0709
115. Nathanielsz PW, Poston L, Taylor PD. In utero exposure to maternal obesity and diabetes: animal models that identify and characterize implications for future health. Obstet Gynecol Clin. 2007;34:
116. Nicholas LM, Rattanatray L, MacLaughlin SM, et al. Differential effects of maternal obesity and weight loss in the periconceptional period on the epigenetic regulation of hepatic insulin-signaling pathways in the offspring. FASEB J. 2013;27:3786–3796. doi:10.1096/fj.13-227918
117. Borengasser SJ, Zhong Y, Kang P, et al. Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. Endocrinology. 2013;154:4113–4125. doi:10.1210/en.2012-2255
118. Liu H-W, Mahmood S, Srinivasan M, Smiraglia DJ, Patel MS. Developmental programming in skeletal muscle in response to overnourishment in the immediate postnatal life in rats. J Nutr Biochem. 2013;24:1859–1869. doi:10.1016/j.jnutbio.2013.05.002
119. Stöger R. Epigenetics and obesity. Pharmacogenomics. 2008;9:1851–1860. doi:10.2217/14622416.9.12.1851
120. Baker MS, Li G, Kohorst JJ, Waterland RA. Fetal growth restriction promotes physical inactivity and obesity in female mice. Int J Obes. 2015;39:98–104. doi:10.1038/ijo.2013.146
121. Li G, Kohorst JJ, Zhang W, et al. Early postnatal nutrition determines adult physical activity and energy expenditure in female mice. Diabetes. 2013;62:2773–2783. doi:10.2337/db12-1306
122. Bauman AE, Reis RS, Sallis JF, et al. Correlates of physical activity: why are some people physically active and others not? Lancet. 2012;380:258–271. doi:10.1016/S0140-6736(12)60735-1
123. de Vilhena e Santos DM, Katzmarzyk PT, Seabra AFT, Maia JAR. Genetics of physical activity and physical inactivity in humans. Behav Genet. 2012;42:559–578. doi:10.1007/s10519-012-9534-1
124. Fuente-Martín E, Argente-Arizón P, Ros P, Argente J, Chowen JA. Sex differences in adipose tissue: it is not only a question of quantity and distribution. Adipocyte. 2013;2:128–134. doi:10.4161/adip.24075
125. Santos RL, da Silva FB, Ribeiro RF, Stefanon I. Sex hormones in the cardiovascular system. Horm Mol Biol Clin Investig. 2014;18:89–103. doi:10.1515/hmbci-2013-0048
126. Frank AP, Palmer BF, Clegg DJ. Do estrogens enhance activation of brown and beiging of adipose tissues? Physiol Behav. 2018;187:24–31. doi:10.1016/j.physbeh.2017.09.026
127. Clegg DJ, Riedy CA, Smith KAB, Benoit SC, Woods SC. Differential sensitivity to central leptin and insulin in male and female rats. Diabetes. 2003;52:682–687. doi:10.2337/diabetes.52.3.682
128. Bouret SG. Role of early hormonal and nutritional experiences in shaping feeding behavior and hypothalamic development. J Nutr. 2010;140:653–657. doi:10.3945/jn.109.112433
129. Dearden L, Bouret SG, Ozanne SE. Sex and gender differences in developmental programming of metabolism. Mol Metab. 2018;15:8–19. doi:10.1016/j.molmet.2018.04.007
130. Ormazabal V, Nair S, Elfeky O, et al. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol. 2018;17:122. doi:10.1186/s12933-018-0762-4
131. Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi:10.1126/science.1082889
132. McLaughlin T, Craig C, Liu L-F, et al. Adipose cell size and regional fat deposition as predictors of metabolic response to overfeeding in insulin-resistant and insulin-sensitive humans. Diabetes. 2016;65:1245–1254. doi:10.2337/db15-1213
133. Snel M, Jonker JT, Schoones J, et al. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol. 2012;2012. doi:10.1155/2012/983814
134. Azzu V, Vacca M, Virtue S, Allison M, Vidal-Puig A. Adipose tissue-liver cross talk in the control of whole-body metabolism: implications in nonalcoholic fatty liver disease. Gastroenterology. 2020;158:1899–1912. doi:10.1053/j.gastro.2019.12.054
135. Sobczak IS, Blindauer A, Stewart A. Changes in plasma free fatty acids associated with type-2 diabetes. Nutrients. 2019;11:2022. doi:10.3390/nu11092022
136. Ademowo OS, Dias HKI, Burton DGA, Griffiths HR. Lipid (per) oxidation in mitochondria: an emerging target in the ageing process? Biogerontology. 2017;18:859–879. doi:10.1007/s10522-017-9710-z
137. Unger RH, Orci L. Lipotoxic diseases of nonadipose tissues in obesity. Int J Obes. 2000;24:S28–S32. doi:10.1038/sj.ijo.0801498
138. Metcalfe LK, Smith GC, Turner N. Defining lipid mediators of insulin resistance: controversies and challenges. J Mol Endocrinol. 2019;62:R65–R82.
139. Szent-Györgyi AG. The early history of the biochemistry of muscle contraction. J Gen Physiol. 2004;123:631–641. doi:10.1085/jgp.200409091
140. Picard M, Hepple RT, Burelle Y. Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: tailoring the organelle for optimal function. Am J Physiol Cell Physiol. 2012;302:C629–C641. doi:10.1152/ajpcell.00368.2011
141. Wüst RCI, Jaspers RT, van Heijst AF, et al. Region-specific adaptations in determinants of rat skeletal muscle oxygenation to chronic hypoxia. Am J Physiol Heart Circ Physiol. 2009;297:H364–H374. doi:10.1152/ajpheart.00272.2009
142. Putti R, Sica R, Migliaccio V, Lionetti L. Diet impact on mitochondrial bioenergetics and dynamics. Front Physiol. 2015;6. doi:10.3389/fphys.2015.00109
143. Liu R, Jin P, Wang Y, et al. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS One. 2014;9:e92810. doi:10.1371/journal.pone.0092810
144. Kumari S, Anderson L, Farmer S, Mehta SL, Li PA. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl Stroke Res. 2012;3:296–304. doi:10.1007/s12975-012-0158-9
145. Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi:10.1073/pnas.1032913100
146. van Eunen K, Simons SMJ, Gerding A, et al. Biochemical competition makes fatty-acid β-oxidation vulnerable to substrate overload. PLoS Comput Biol. 2013;9:e1003186. doi:10.1371/journal.pcbi.1003186
147. Sugden MC, Holness MJ. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem. 2006;112:139–149. doi:10.1080/13813450600935263
148. Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8:92–103. doi:10.1038/nrendo.2011.138
149. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi:10.1056/NEJMoa031314
150. Mogensen M, Sahlin K, Fernstrom M, et al. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56:1592–1599. doi:10.2337/db06-0981
151. Cusi K, Maezono K, Osman A, et al. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. doi:10.1172/JCI7535
152. Misu H, Takamura T, Matsuzawa N, et al. Genes involved in oxidative phosphorylation are coordinately upregulated with fasting hyperglycaemia in livers of patients with type 2 diabetes. Diabetologia. 2007;50:268–277. doi:10.1007/s00125-006-0489-8
153. Koliaki C, Roden M. Alterations of mitochondrial function and insulin sensitivity in human obesity and diabetes mellitus. Annu Rev Nutr. 2016;36:337–367. doi:10.1146/annurev-nutr-071715-050656
154. Fletcher JA, Deja S, Satapati S, et al. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight. 2020;4.
155. Koliaki C, Szendroedi J, Kaul K, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi:10.1016/j.cmet.2015.04.004
156. Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. 2007;92:1023–1033. doi:10.1210/jc.2006-1055
157. Forrester Steven J, Kikuchi Daniel S, Hernandes Marina S, et al. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122:877–902. doi:10.1161/CIRCRESAHA.117.311401
158. Reaven G. Insulin resistance and coronary heart disease in nondiabetic individuals. Arterioscler Thromb Vasc Biol. 2012;32:1754–1759. doi:10.1161/ATVBAHA.111.241885
159. Cerf ME, Chapman CS, Louw J. High-fat programming of hyperglycemia, hyperinsulinemia, insulin resistance, hyperleptinemia, and altered islet architecture in 3-month-old wistar rats. ISRN Endocrinol. 2012;627270:2012.
160. Newsholme P, Cruzat VF, Keane KN, et al.Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J. 2016;473.
161. Boland BB, Rhodes CJ, Grimsby JS. The dynamic plasticity of insulin production in β-cells. Mol Metab. 2017;6:958–973. doi:10.1016/j.molmet.2017.04.010
162. Pinnick K, Neville M, Clark A, Fielding B. Reversibility of metabolic and morphological changes associated with chronic exposure of pancreatic islet beta-cells to fatty acids. J Cell Biochem. 2010;109:683–692. doi:10.1002/jcb.22445
163. Molina AJA, Wikstrom JD, Stiles L, et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–2315. doi:10.2337/db07-1781
164. Carpentier AC, Bourbonnais A, Frisch F, Giacca A, Lewis GF. Plasma nonesterified fatty acid intolerance and hyperglycemia are associated with intravenous lipid-induced impairment of insulin sensitivity and disposition index. J Clin Endocrinol Metab. 2010;95:1256–1264. doi:10.1210/jc.2009-1932
165. Yu T, Sheu -S-S, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. 2008;79:341–351. doi:10.1093/cvr/cvn104
166. Reaven GM, Olefsky JM. Relationship between heterogeneity of insulin responses and insulin resistance in normal subjects and patients with chemical diabetes. Diabetologia. 1977;13:201–206. doi:10.1007/BF01219700
167. Goodpaster BH, Sparks LM. Metabolic flexibility in health and disease. Cell Metab. 2017;25:1027–1036. doi:10.1016/j.cmet.2017.04.015
168. Basu R, Barosa C, Jones J, et al. Pathogenesis of prediabetes: role of the liver in isolated fasting hyperglycemia and combined fasting and postprandial hyperglycemia. J Clin Endocrinol Metab. 2013;98:E409–E417. doi:10.1210/jc.2012-3056
169. Wu G, Meininger CJ. Nitric oxide and vascular insulin resistance. BioFactors. 2009;35:21–27. doi:10.1002/biof.3
170. Wang CCL, Gurevich I, Draznin B. Insulin affects vascular smooth muscle cell phenotype and migration via distinct signaling pathways. Diabetes. 2003;52:2562–2569. doi:10.2337/diabetes.52.10.2562
171. Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic complications. Korean J Physiol Pharmacol. 2014;18:1–14. doi:10.4196/kjpp.2014.18.1.1
172. Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106:842–853. doi:10.1161/CIRCRESAHA.109.212217
173. Barrett EJ, Liu Z, Khamaisi M, et al. Diabetic microvascular disease: an endocrine society scientific statement. J Clin Endocrinol Metab. 2017;102:4343–4410.
174. Edwards CM, Cusi K. Prediabetes: a worldwide epidemic. Endocrinol Metab Clin North Am. 2016;45:751–764. doi:10.1016/j.ecl.2016.06.007
175. Ginsberg HN, Zhang Y-L, Hernandez-Ono A. Metabolic syndrome: focus on dyslipidemia. Obes Silver Spring Md. 2006;14(Suppl 1):41S–49S. doi:10.1038/oby.2006.281
176. Petrie JR, Guzik TJ, Touyz RM. Diabetes, hypertension, and cardiovascular disease: clinical insights and vascular mechanisms. Can J Cardiol. 2018;34:575–584. doi:10.1016/j.cjca.2017.12.005
177. Larson-Meyer DE, Heilbronn LK, Redman LM, et al. Effect of calorie restriction with or without exercise on insulin sensitivity, β-cell function, fat cell size, and ectopic lipid in overweight subjects. Diabetes Care. 2006;29:1337–1344. doi:10.2337/dc05-2565
178. Rynders CA, Blanc S, DeJong N, Bessesen DH, Bergouignan A. Sedentary behaviour is a key determinant of metabolic inflexibility: sedentary behaviour and metabolic flexibility. J Physiol. 2018;596:1319–1330. doi:10.1113/JP273282
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.