")
Back to Journals » Journal of Experimental Pharmacology » Volume 12
MET Inhibitors for the Treatment of Gastric Cancer: What’s Their Potential?
Authors El Darsa H, El Sayed R , Abdel-Rahman O
Received 11 June 2020
Accepted for publication 9 September 2020
Published 6 October 2020 Volume 2020:12 Pages 349—361
DOI https://doi.org/10.2147/JEP.S242958
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Haidar El Darsa,1 Rola El Sayed,2 Omar Abdel-Rahman1
1Division of Medical Oncology, Department of Oncology, Cross Cancer Institute, University of Alberta, Edmonton, Alberta, Canada; 2Division of Hematology-Oncology, Department of Internal Medicine, American University of Beirut Medical Center, Beirut, Lebanon
Correspondence: Omar Abdel-Rahman
Division of Medical Oncology, Department of Oncology, Cross Cancer Institute, University of Alberta, Edmonton, Alberta, Canada
Email [email protected]
Abstract: Gastric cancer remains a disease with a dismal prognosis. Extensive efforts to find targetable disease drivers in gastric cancer were implemented to improve patient outcomes. Beyond anti-HER2 therapy, MET pathway seems to be culprit of cancer invasiveness with MET-overexpressing tumors having poorer prognosis. Tyrosine kinase inhibitors targeting the HGF/MET pathway were studied in MET-positive gastric cancer, but no substantial benefit was proven. Some patients responded in early phase trials but later developed resistance. Others failed to show any benefit at all. Etiologies of resistance may entail inappropriate patient selection with a lack of MET detection standardization, tumor alternative pathways, variable MET amplification, and genetic variation. Optimizing MET detection techniques and better understanding the MET pathway, as well as tumor bypass mechanisms, are an absolute need to devise means to overcome resistance using targeted therapy alone, or in combination with other synergistic agents to improve outcomes of patients with MET-positive GC.
Keywords: gastric cancer, MET over-expression, MET amplification, HGF, tyrosine kinase inhibitors, monoclonal antibodies
Introduction
Globally, gastric cancer (GC) represents a significant healthcare burden. It constitutes the fifth most common malignancy worldwide (5.7%), and the third most common cause of cancer-related mortality (8.2%).1 It is responsible for over 1,000,000 new cases and estimated 783,000 deaths in 2018. Its incidence rate is significantly elevated in Eastern Asia reaching (32/100,000), whereas in regions like Northern America and Europe, the incidence rate is generally low, reaching (5–6/100,000).1 Although rates of non-cardia gastric cancer have been steadily declining in western countries, the frequency of gastric cardia cancers has been soaring.2,3
GC is quite heterogeneous. It is categorized into several subtypes based on anatomy (cardia vs non-cardia), histology (diffuse vs intestinal) and molecular characteristics (microsatellite instability (MSI), Epstein–Barr virus (EBV) positivity, genomic stability (GS), and chromosomal instability (CIN)).4 Different tumor characteristics have different outcomes. Nevertheless, the prognosis of patients with advanced GC with standard therapy remains dismal worldwide, with a median overall survival (OS) of 8 to 12 months,5,6 and a 5-year OS reaching 5%.7,8 Therefore, alternative means were sought to improve outcomes.
The millennial advances in cell biology and genetic assays led scientists to scrutinize cell machinery responsible for oncogenesis, and attempt targeting driver mutations has shown to improve outcomes in a number of tumors. In GC, a variable genomic profile including variations in HER2, EGFR, FGFR2, KRAS and c-MET constitutes about 37% of cases that can be potentially targeted.9 For example, trastuzumab, an inhibitor of human epidermal growth factor receptor 2 (HER2), was shown to improve survival when added to chemotherapy in HER2 positive GC, becoming the first targeted treatment to be approved.10 However, as only 20% of cases of advanced GC overexpress HER2, it was imperative to look for alternative options.
Proto-oncogene c-Mesenchymal-Epithelial Transition (MET) signaling pathway plays an integral role in GC. An aberrant, over-activated MET pathway promotes disease progression, and serves as a common mechanism of resistance to HER targeted therapy.11 Therefore, the rationale for investigating c-MET targeting in GC was warranted. In this article, we will summarize the clinical significance of MET in GC onco-pathogenesis, elucidating the available results of trials including multiple MET inhibitors as single or combination therapy. We will also tackle the mechanisms of resistance to MET inhibitors, as well as the possible means to overcome it.
Role of MET in GC Onco-Pathogenesis
Mesenchymal-Epithelial Transition (MET) Pathway (Figure 1)
MET gene is located on chromosome 7q21-q31. It encodes the Hepatocyte Growth Factor (HGF) receptor, which is a member of the Receptor Tyrosine Kinase (RTK) family. RTKs are growth factors responsible for physiological responses such as embryogenesis, tissue regeneration, homeostasis, and wound healing.11 RTK activity is strictly regulated in normal cells while erratic activation in malignancy activates multiple downstream molecular signaling pathways,12 leading to tumorigenesis, cell survival, angiogenesis, metastasis, and resistance to anticancer agents.13,14
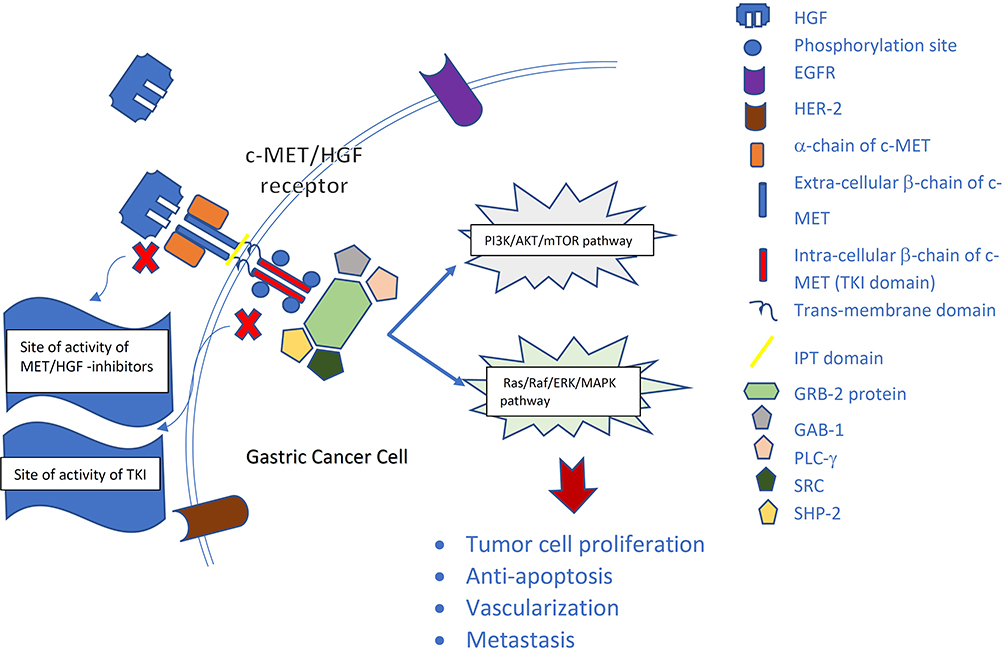 |
Figure 1 c-MET/HGF pathway in gastric cancer pathogenesis. Hepatocyte growth factor (HGF) binds to c-MET, causing phosphorylation and activation of tyrosine kinase domain with consequent triggering of down-stream signaling via PI3K/AKT/mTOR pathway as well as RAS/RAF/ERK/MAPK pathway, eventually leading to tumor cell proliferation, tumor survival, angiogenesis and metastasis. |
MET is a disulfide heterodimer made of alpha and beta subunits. The alpha subunit is solely extracellular whereas the beta subunit contains a membrane-spanning segment, an intracellular cytoplasmic kinase domain, and a docking site in addition to the extracellular domain.15,16 HGF is a cytokine released by mesenchymal cells with a very high affinity to MET receptors. Binding of HGF to the extracellular domain of MET leads to receptor dimerization, tyrosine phosphorylation at the carboxy-terminal docking site, and finally kinase activation.16,17 This facilitates the binding of SRC HOMOLOGY-2 domain (SH2)-containing proteins and recruitment of proteins such as growth factor receptor-bound protein 2 (GRB2), GRB2-associated binding protein 1 (GAB1), Phospholipase C (PLC)- gamma, SRC, and SHP2. Ultimately, multiple downstream signaling pathways are activated, including Phosphatidyl-Inositol-3- Kinase (PI3K)/AKT, Extracellular signal-Regulated Kinase (ERK)/Mitogen-Activated Protein Kinase (MAPK) also known as (RAS-RAF-MEK-ERK) pathway, Signal Transducer and Activator of Transcription 3 (STAT3), and Nuclear Factor-κB (NF-κB).18,19 This pathway cascade activates CYCLIN D-CDK4/6 and phosphorylates Retinoblastoma Rb, releasing the E2F-1 transcription, which is essential in cell-cycle regulation. Moreover, several downstream genes mediating the phase G1-phase S transition are produced, enhancing cellular proliferation.20,21 In normal conditions, MET receptor is regulated through 26S proteasome-dependent ubiquitination, destruction, internalization, endocytosis, and eventual lysosomal degradation, all while retaining signaling capacity.22 However, aberrant MET signaling disrupts the process and promotes cell invasiveness, growth, angiogenesis, and metastasis, even in hypoxic conditions where excessive HGF is released.14,23
MET Alteration in GC (Table 1)
Aberrant c-MET pathway activation plays an important role in tumorigenesis. It can occur by protein overexpression, gene amplification, increased HGF ligand autocrine expression, enhanced paracrine ligand-mediated stimulation, inadequate c-MET degradation, ligand-independent activation and rarely gene mutation,19 in addition to the role of environmental conditions such as inflammation and hypoxia.24 The most common mechanism of MET pathway abnormal activation in GC is via protein overexpression with resultant excessive kinase activation. MET protein expression on immunohistochemistry (IHC) is predominantly detected in 50–65% of GC.25 Mainly, it is expressed in cancer cell cytoplasm, cell membrane and in stromal cells of tumors.26,27 Moreover, MET overexpression is mostly noted in dysplasia and precancerous intestinal metaplasia illustrating its critical role in the early phase of oncogenesis of GC.26,27 It is frequently encountered in well-differentiated tubular adenocarcinoma (67%), intestinal-type tumors (35%) and to a lesser extent in diffuse-type GC tumors (15–51%).25,28 MET overexpression has been linked to aggressiveness, tumor invasion depth, lymph node metastasis, distant metastasis, advanced tumor stage, recurrence, and poor survival.25–29
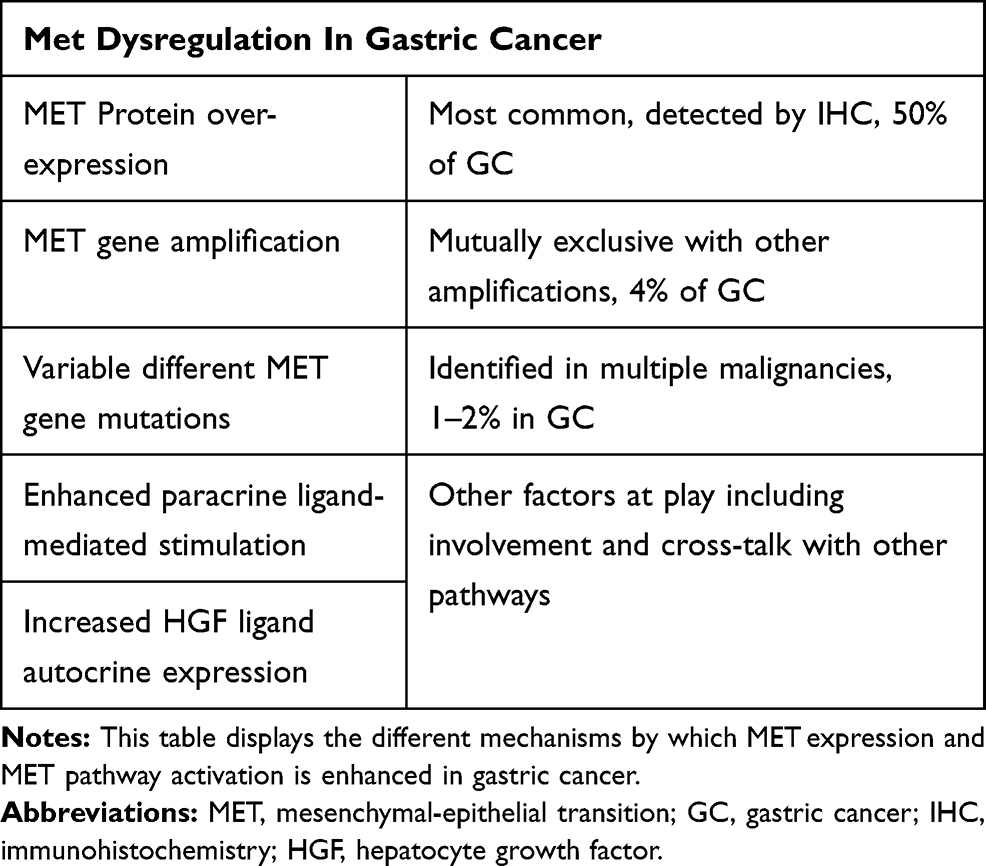 |
Table 1 MET Alteration in GC |
Another mechanism of aberrant MET pathway activation is through MET gene amplification, usually mutually exclusive with different other genes. Nevertheless, co-amplification can occur in around 4% (3.4%-7%) of GC, commonly intestinal sub-types,26,30 leading to de novo or secondary treatment resistance.9,31 Other activating genetic mutations of MET remain exceedingly rare in GC reaching only 1–2% of patients.4,32
Cross-Talk Between Pathways
MET co-expression and pathway activation exhibit significant cross talk with ERBB2 (HER2), Vascular Endothelial Growth Factor (VEGF), and its receptor (VEGFR) signaling pathways, which may cause resistance to targeted therapy and MET inhibitors.11,33,34 One example is the induction of HGF-independent c-MET activation in some cancer cellular models via Epidermal Growth Factor Receptor (EGFR) phosphorylation.35 RON (receptor originated from nantes) is another example of an RTK occasionally co-expressed with c-MET, where studies showed receptor interaction, one phosphorylating the other, and knockdown of one leading to compensation by the other.36
Moreover, c-MET expression in human GC specimens was found to be positively correlated to Jagged1 expression, responsible for activating the Notch1 signaling pathway that leads to COX-2 expression, elevation of prostaglandin E2, and resultant increased proliferation and migration ability of GC cells. However, constitutive activation of Notch1 limited HGF activity, repressed the c-MET oncogene, suppressed c-MET expression and decreased HGF sensitivity. It can be concluded that COX-2 and Notch knockdown or inhibitors may play a role in the therapeutic strategy against HGF/c-MET pathway.37,38
Targeted Therapy Against MET Pathway in GC
The development of inhibitors targeting MET/HGF or downstream signaling proteins has become an attractive goal for GC drug development, including variable tyrosine kinase inhibitors (TKIs), selective or multi-kinase inhibitors, and monoclonal antibodies.
Tyrosine Kinase Inhibitors of MET Pathway (Table 2)
Selective Inhibitors
Tivantinib (ARQ197) is a non-adenosine triphosphate (ATP) competitive, selective MET inhibitor. It was first studied in a Phase I trial including 51 patients with GC, showing good tolerance with a recommended dose of 360 mg twice daily (BID). The best response was stable disease (SD) for ≥4 months in 14 patients. Tivantinib decreased intra-tumoral phosphorylated MET (p-MET), total c-MET levels, and phosphorylated Focal Adhesion Kinase (FAK). However, it increased terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick-end labeling (TUNEL) staining in tumor biopsy, which was correlated with apoptosis. Furthermore, c-MET blockade decreased circulating endothelial cells (CEC) in 58% (25 of 43) of patients.39
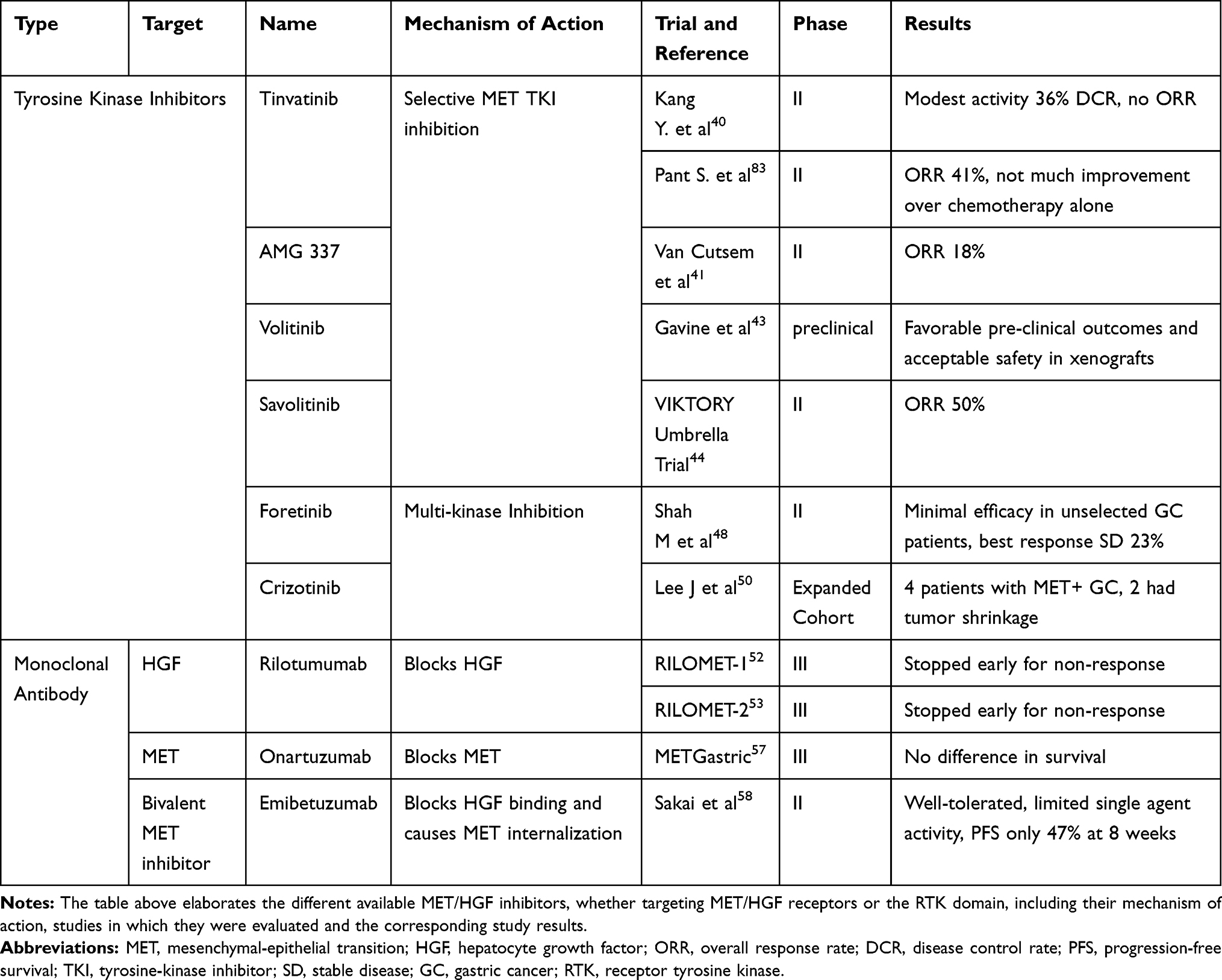 |
Table 2 MET Inhibitors |
In a Phase II study of 31 patients with advanced GC receiving tivantinib as second- or third-line therapy, modest activity with no objective response (OR) was observed. Disease control rate (DCR) was 36.7%, and median progression-free survival (PFS) was only 43 days. There was no clear correlation between efficacy and biomarkers including gene amplification of c-MET, c-MET, p-MET, and HGF expression. Concerning adverse effects (AEs), neutropenia and anemia were the most frequent (grade 3 or higher), each occurring in 13% of cases (4 of 30).40
AMG337 is a highly selective and potent small-molecule MET inhibitor. In one human phase I trial, 111 patients with solid tumors, 21% of gastro-esophageal origin, received AMG337 as second-line or later therapy, at a dose of 300 mg orally daily (QD). AMG337 showed a higher ORR of 29.6% among MET-amplified patients compared to 9.9% in all patients, regardless of MET-amplification status. The most common AEs were headache, fatigue, nausea, and vomiting.41 In a Phase II, single-arm study, 45 adults with MET-amplified GC and gastroesophageal junction (GEJ) adenocarcinoma, AMG337 showed promising anti-tumor activity with an ORR of 18% (8 partial responses). The median PFS and OS as well as the duration of response (DOR) were 3.4, 7.9, and 6.0 months, respectively. The most frequent AEs were headache (60%), nausea (38%), vomiting (38%), and abdominal pain (33%). However, 71% had grade ≥3 AEs and 59% had serious AEs.42
Volitinib is a potent, highly selective, ATP-competitive c-MET small-molecule TKI that showed favorable preclinical outcomes and an acceptable safety profile in xeno-graft models. In one Chinese study, volitinib was tested in 3 out of 34 GC models after proving MET gene amplification and c-MET overexpression, and it resulted in a significant reduction in tumor p-MET with tumor growth inhibition.43
Savolitinib is a first-in-class potent, highly selective MET inhibitor that showed clinical efficacy and safety in multiple tumor types as monotherapy or in combination in phase Ib/II trials. As part of the phase II VIKTORY trial (targeted agent eValuation In gastric cancer basKeT KORea studY), 25 patients with MET-amplified GC were recognized out of 772 patients (3.5%), 20 of whom received savolitinib as monotherapy, attaining the highest ORR reaching 50% (10/20, 95% CI: 28.0–71.9) and meeting the pre-specified PFS endpoint of 6 weeks.44
KRC-00715 is an exclusively selective c-MET inhibitor tested among 18 GC cell lines with c-MET overexpression. It significantly suppressed the growth of c-MET overexpressed cell lines, inducing G1/S arrest, reducing downstream signals, and impairing c-MET activity. In vivo, KRC-00715 proved activity in xenograft models with significant tumor size regression.45
Multi-Kinase Inhibitors
Foretinib (GSK1363089) is an oral multi-kinase inhibitor that targets MET, RON, AXL, TIE-2, and VEGFR2 receptors. In preclinical and xenograft GC models, foretinib exhibited significant c-MET inhibition, preventing cancer stemness,46 and strongly enhancing the antitumor effect of chemotherapy.47 In a phase II study enrolling 74 patients with metastatic GC, foretinib was given as intermittent dosing (240 mg/day for 5 consecutive days every 2 weeks) in 48 patients, and daily dosing (80 mg/day during 2-week cycles) in 26 patients. Minimal efficacy was noted in unselected patients, best response being SD in 23% (10 of 44) in those receiving intermittent dosing and 20% (5 of 25) in those on daily dosing. Only 4% (3 of 67) had MET amplification in tumor specimens, one of whom had SD. OS was 7.4 months with intermittent dosing and 4.3 months with daily dosing. Most treatment-related AEs were mild, encompassing fatigue, hypertension, nausea, and diarrhea, plus asymptomatic transaminase elevation. Grade 3 or higher treatment-related AEs occurred in 44% of patients on intermittent dosing and 35% of those on daily dosing.48
Crizotinib is an ATP-competitive, small-molecule TKI of MET and Anaplastic Lymphoma Kinase (ALK). In an expanded study cohort, four patients with MET-amplified GC received crizotinib, only two had tumor shrinkage (16% and 30%) with a PFS of 3.5 and 3.7 months, respectively.49 An ongoing pilot study is testing crizotinib in patients with c-MET-positive GC in third-line setting after chemotherapy failure; pending results.50
Monoclonal Antibodies Targeting HGF and c-MET
Rilotumumab (AMG 102)
Rilotumumab (AMG 102) is a fully humanized IgG2 monoclonal antibody that targets HGF, preventing receptor binding and consequently inhibiting c-MET activation. Its safety and efficacy were evaluated in a dose de-escalation phase Ib study and a double-blind randomized phase II study, in combination with epirubicin, cisplatin, and capecitabine (ECX) as first-line treatment. One hundred and twenty-one patients were randomized equally to receive rilotumumab at a dose of 15 mg/kg (n=40); a dose of 7.5 mg/kg (n=42), or placebo (n=39). Rilotumumab improved clinical outcomes with a median PFS in combined rilotumumab arms reaching 5.7 months versus 4.2 months with placebo. The ORR was 39%, and the DCR was 80% in the combined rilotumumab group. Subgroup analysis showed response in 50% of patients with high MET-expression, treated with rilotumumab. This sub-category had a statistically significant OS advantage with a median OS of 10.6 months compared to patients with low MET expression having a median OS of 5.7 months (Hazard Ratio (HR)=0.29; p = 0.012). In the MET negative subgroup, median OS was similar between rilotumumab and placebo groups. MET negative patients had better survival than those in the MET-positive group in the placebo arm with a median OS of 11.5 vs 5.7 months, respectively.51
Based on these results, two phase-III studies of rilotumumab were started including only IHC-selected MET over-expressers: RILOMET-1 and RILOMET-2. Both studies recruited patients with advanced untreated MET-positive (IHC ≥1+, ≥25% cells) GC or GEJ adenocarcinoma. In RILOMET-1 study, 690 patients were randomized to rilotumumab 15mg/kg or placebo in combination with ECX chemotherapy. In RILOMET-2, 450 Asian patients were randomized to rilotumumab 15mg/kg or placebo plus cisplatin and capecitabine (CX) chemotherapy.52,53 The primary endpoint in RILOMET-1 was OS, whereas in RILOMET-2 PFS and OS were co-primary endpoints. Unfortunately, the studies were stopped prematurely because of an increased death rate due to disease progression in the rilotumumab plus chemotherapy arm compared to chemotherapy alone (R vs P: 128 vs 107 deaths) in RILOMET-1. Rilotumumab was strikingly ineffective, with a median OS of 9.6 months compared to 11.5 months with chemotherapy alone (HR=1.37; p = 0.016). All clinical outcomes in all subgroups were statistically worse with rilotumumab.52 Therefore, RILOMET-2 trial was terminated shortly after.53 The most common AEs in the rilotumumab groups were neutropenia, anemia, peripheral edema, thromboembolism and fatigue.51,52
Another phase II 3-arm trial studied mFOLFOX6 (oxaliplatin, folinic acid, and fluorouracil) monotherapy or in combination with panitumumab or rilotumumab in HER2-negative, MET-positive advanced GC. The MEGA French study revealed that the addition of panitumumab or rilotumumab was not effective. The 4-month PFS was 71% with chemotherapy alone, 57% with panitumumab, and 61% with rilotumumab. There were more side effects in combination arms; grade 3 or more AEs occurring in 62% with chemotherapy alone, 83% with panitumumab and 89% with rilotumumab.54
Onartuzumab
Onartuzumab is a recombinant, fully humanized, monovalent monoclonal antibody that binds the extracellular domain of MET; prevents binding with HGF and blocks subsequent MET pathway signaling. In a Phase 2 study, onartuzumab was combined with erlotinib in the second or third-line setting, resulting in improved PFS and OS compared to placebo plus erlotinib.55 Another phase 2 trial also recruited HER2 negative advanced GC patients to test onartuzumab-mFOLFOX6 combination. There was no survival advantage over chemotherapy alone; neither in the general study population nor in MET-positive patients, only more toxicity.56 Hereafter, a Phase III MetGastric study investigating FOLFOX6 ± onartuzumab in HER2 negative, MET-positive advanced GC patients was prematurely halted. The addition of onartuzumab to first-line mFOLFOX6 did not improve OS, PFS, or ORR, irrespective of MET expression status. Grade 3 and above AEs were more frequently observed with onartuzumab including neutropenia, hypoalbuminemia, peripheral edema, thrombocytopenia, pulmonary embolism, and gastric perforation.57
Emibetuzumab
Emibetuzumab is a humanized immunoglobulin monoclonal bivalent anti-MET antibody. It blocks HGF-MET receptor interaction, and causes receptor internalization and degradation; therefore, suppressing ligand-independent MET activation. It was evaluated in a non-randomized, single-arm, phase 2 study including 15 Asian patients with MET-positive advanced GC, defined by IHC as ≥60% tumor-cell staining at >2 + intensity, having received ≥2 prior lines of chemotherapy. Emibetuzumab proved to be well tolerated with limited single-agent activity. PFS reached 47% at the 8-week landmark. Grade ≥ 3 AEs were mainly electrolyte imbalances: hyperkalemia, hyponatremia, and hyperuricemia.58
Agents Under Investigation
Several other monoclonal antibodies targeting HGF or MET in GC are being developed. For example, ficlatuzumab and TAK-701 are humanized monoclonal antibodies that specifically target soluble HGF, blocking its binding to c-MET. Phase I/II clinical studies are ongoing to evaluate their tolerability, safety, pharmacodynamics, and pharmacokinetics.59,60 ABT-700 is another humanized anti-c-MET monoclonal antibody which has interestingly revealed strong single-agent activity in MET-amplified GC patients based on results of a phase I study.61
Resistance to MET Targeted Therapy (Figure 2)
Unfortunately, preclinical and clinical data support the development of acquired resistance to HGF/c-MET inhibitors. Numerous mechanisms of resistance to anti-HGF/c-MET therapies need to be overcome to improve anticancer effects including: poor MET-status recognition, alternative signaling pathways, emergence of new mutations, and heterogeneity of MET expression.
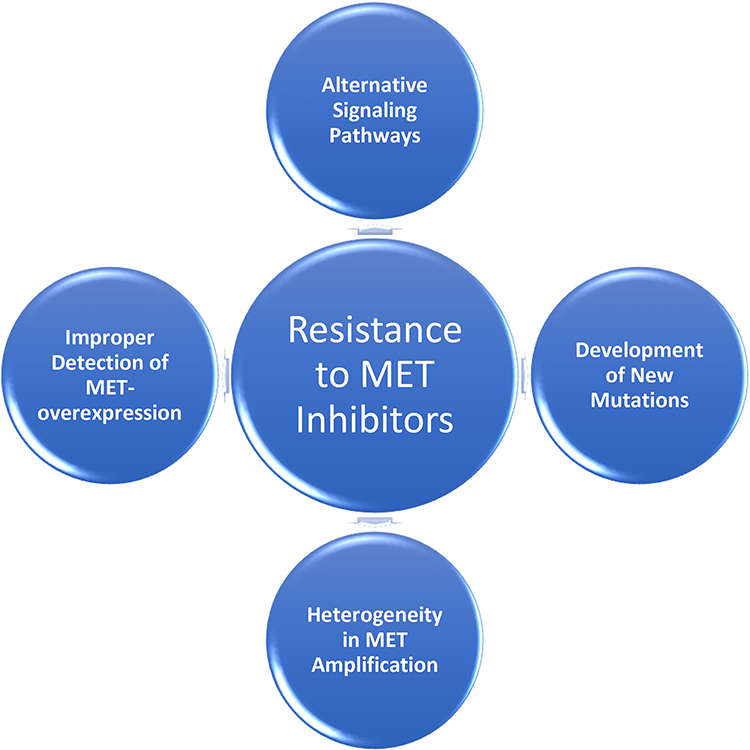 |
Figure 2 Mechanisms of Resistance to MET inhibitors. |
Poor MET-Status Recognition
Therapeutic decisions regarding MET targeted therapy use require reliable MET-status identification. Over many years, techniques for MET-status recognition varied from MET protein expression on IHC (protein level), to MET amplification via Fluorescent/Silver In-situ Hybridization (FISH/SISH), or even genome sequencing assays (gene level). Major discrepancies were noted and no consensus was reached. Moreover, IHC overexpression may not truly reflect gene/pathway driver status, as it does not always correlate with gene amplification, transcriptional activation, or hypoxia. Given this variability in MET-status determination with different diagnostic criteria, MET targeting may become somewhat challenging.62,63
Alternative Signaling Pathways
Alternative signaling pathways may be a rescue mechanism for cancer cells to overcome the effect of MET inhibitors. Interestingly, in one study, 40–50% of patients having MET-amplified GC were characterized to have HER2 and/or EGFR co-amplification that compromised c-MET inhibitor anti-tumoral effects.31 This predominantly occurs in CIN subtypes characterized by frequent somatic copy number alterations.4
Furthermore, pathway crosstalk and MET’s ability to heterodimerize with HER family members, including EGFR and HER2-3, with their subsequent activation, driven by overexpressed ligands (TGFα/EGF, heregulin), leads to reactivation of downstream PI3K/AKT/MEK/MAPK pathways;31,64 thereby overcoming MET inhibition, and enhancing tumor aggressiveness.65,66 Such co-amplification of MET and HER2 was translated into a reduction of the antitumor capacity with monotherapies such as lapatinib in HER2 amplified GC.11
Emergence of New Mutations
Acquired resistance can be driven by the emergence of new mutations within or outside the MET gene. Point mutations within the c-MET activation loop, which constitutes the drug target, destabilize the receptor, decrease its binding capacity and cause resistance to MET inhibitors while maintaining downstream MEK-ERK and PI3K-AKT signaling.66,67 In the VIKTORY trial, three patients treated with Savolitinib developed resistance through emerging mutations, particularly MET D1228H, MET D1228N, MET D1228V and MET Y1230C.67
Resistant mutations include point mutations, increased copy number; skip mutations or alterations in exon 14 splicing site.68,69 Examples include: MET Y1248H and MET D1246N.70
Other mutations such as KRAS and RON mutations have been noted to bypass pathway suppression by MET inhibitors.31
Heterogeneity in MET Amplification
Another mechanism of resistance to MET inhibitors is the dramatic heterogeneity in MET-amplification, which may differ within the same tumor as well as between different metastatic lesions and the primary tumor in GC. This leads to mixed responses to MET inhibition, and treatment failure due to the outgrowth of non-MET-amplified clones.9 Even upregulation of MET gene amplification may confer resistance to MET Inhibitors through association with E-cadherin and epithelial to mesenchymal transition (EMT).65
Microenvironment Interference and Immune Regulation
The tumor microenvironment (TME) is an important factor in resistance to targeted therapies. In MET-positive GC models, tumor-associated fibroblasts within the microenvironment oversecrete HGF, activating downstream signals, promoting cancer colony formation, and causing tumor resistance. HGF secretion is enhanced by lactate within the TME, in addition to paracrine HGF provided by the extra-cellular matrix. Hypoxia, another characteristic of the TME, significantly reduces MET phosphorylation, while maintaining downstream signaling.69
Furthermore, HGF/MET signaling affects immune cells within the TME, such as mast cell activation by MET-α2β1 integrin,71 and dendritic cell (DC) impairment by diminished antigen presentation through matrix-metalloproteinase MMP2-MMP9.72 MET inhibition also limits the activity of anti-tumor neutrophils, aiding in tumor growth.73 More recent studies showed that MET inhibition, in general, upregulated Programmed death-ligand 1 (PDL-1) expression, compromising the killing effect of MET inhibitors.74,75
V-Combination Therapy and Future Perspectives to Bypass Resistance
Synergism with Other Targeted Therapies
Combinatorial targeting of multiple pathways such as EGFR, HER2, and HGF/c-MET axis could potentially maximize the anti-tumorigenic effect for certain MET-addicted GC patients.11,31,55,76
Combination with VEGF inhibitors is another possibility given the proven benefit of ramucirumab in advanced GC.77,78 In colorectal models, volitinib, a selective MET inhibitor, plus apatinib, a VEGF inhibitor, have shown synergy with significant tumor suppression and apoptosis.79
Use of Autophagy Inhibitors
After the use of MET-TKIs, some cells were noted to resort to protective autophagy through the MET/mTOR/ULK1 cascade, where cancer cells become less sensitive to further therapies.80,81 Autophagy inhibitors such as the immunomodulatory hydroxychloroquine, or mammalian target of rapamycin (mTOR) inhibitor everolimus can be used in addition to MET inhibitors to overcome resistance. Preclinical models in MET-amplified tumors showed that autophagy blockade helped MET-TKIs better control tumor growth, making the combination a promising therapeutic option to explore.82
Combination with Chemotherapy
Multiple studies tested chemotherapy combined with MET inhibitors. One phase I/II study included 32 patients with previously untreated advanced GC and GEJ cancers, to receive FOLFOX (day 1) plus Tivantinib (360 mg PO BID, days 1–14 in 2-week cycles) for a median of eight cycles. ORR was 41%. Treatment-related toxicities were mainly hematological, gastrointestinal, and peripheral neuropathy. Median PFS and OS were 6.1 and 9.6 months, respectively, similar to FOLFOX alone. Patients with high c-MET expression had inferior PFS and OS, proving the correlation of c-MET expression with poorer outcomes.83 Another ongoing Phase I–II trial is testing the combination of AMG-337 with mFOLFOX6 in c-MET-positive advanced-stage GC patients.84 Volitinib was also tested in combination with docetaxel, and results showed good tolerance and superior anti-tumor efficacy than either agent alone.43 Furthermore, MET-targeting monoclonal antibodies: rilotumumab, onartuzumab and emibetuzumab, were tested in combination with chemotherapy.52,53,56–58 However, results were disappointing.
Combination with Radiation Therapy
Radiation therapy has been found the culprit of up-regulation of c-MET expression and activity, mainly through the ATM and NF-κB signaling pathways,85 where levels of c-MET increased with radiation time and dose.86 In GBM models, radiotherapy enhanced HGF secretion, and radio-resistance was related to high HGF levels.87 Irradiation also showed to increase c-MET phosphorylation leading to HGF- independent increased downstream signaling.88
Moreover, the role c-MET/HGF pathway has proven to play in DNA repair with consequent radiation resistance,89 inclined researchers to test c-MET inhibitors with radiation therapy, with proof of capability to sensitize cancer cells to radiation in vitro and in vivo.90 For example, crizotinib sensitized cetuximab-resistant KRAS mutant colorectal cancer cells to radiation and improved outcomes in patients undergoing chemo-radiation.91 Similarly in breast cancer, a study of 208 pre-menopausal patients with breast cancer suggested that adding a MET inhibitor to radiotherapy might be an option for patients with c-MET overexpression.92 In esophageal cancer, foretinib increased radio-sensitivity via c-MET modulation, where it prompted cell apoptosis, and induced cell-cycle arrest by irradiation, thus diminishing the tumor burden, and improving patient outcomes.93
Use of Immunotherapy
Future perspectives should better explore combining HGF/MET inhibitors with immunotherapy. HGF/MET signaling pathway is intertwined with tumor immunity affecting DCs, mast cells, and neutrophils plus causing up-regulation of PD-1/PDL-1 expression. Researchers have attempted MET inhibition as an immunologic stimulant, and a bi-specific MET/PD-1 dual-acting monoclonal antibody was created and tested with good pre-clinical results.94 Moreover, NK1-targeted chimeric antigen receptors (CAR-T cell immunotherapy) were also developed to mediate MET-dependent T-cell activation against malignant cells.95
MET Knockdown and Molecular Therapy
Knockdown of c-MET not only significantly diminished tumor cell growth, migration and invasion, but also induced apoptosis, and enhanced activity of chemotherapy.96 At the level of mRNA, lentivirus-mediated RNA silencing of c-MET markedly suppressed the peritoneal dissemination of gastric cancer in vitro and in vivo.97
A number of non-coding RNAs (ncRNAs) were discovered, including microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) that have been involved in HGF/MET pathway, responsible for tumor aggressiveness and metastasis.98 Some lncRNAs, such as lncRNA-TUG1, are overexpressed in GC, particularly in diffuse-type, and cause significant proliferation of GC through indirect activation of c-MET, whose knockdown remarkably impairs migration, invasion and metastasis both in vitro and in vivo.99 Other miRNAs such as miR-206, miR-1 and miR-34a on the other hand, act as tumor suppressors, down-regulating c-MET in vitro as well as in xenograft models.100,101 More studies should be performed to better characterize the relationships among lncRNAs, miRNAs, and c-MET, and perhaps devise new therapies.
Finally, knockdown of N-acetylgalactosaminyltransferase 2 (GALNT2), an enzyme that mediates glycosylation and suppresses malignant phenotypes in GCs through MET/HGF activity, enhanced MET phosphorylation and decreased MET expression, thereby suppressing aggressive tumor phenotypes.102
Conclusion
Deciphering the underlying molecular heterogeneity of cancer could have significant clinical utilization by developing specified targeted therapies. Despite the success of targeting HER2 in advanced GC, research failed to find other targeted therapies with substantial proven benefit. Low 5-year GC survival rates emphasize the need for novel techniques to develop effective targeted therapies. The role of HGF/MET pathway in tumor prognosis has driven attention towards this entity as a target of inhibition to improve outcomes. Several selective/non-selective c-MET TKIs and monoclonal antibodies were tested; yet, none proved substantial clinical benefit.
The complexity of MET signaling pathway, the lack of consensus and poor biomarker determination as well as the diverse resistance mechanisms (cross-talk, new mutations, upregulated gene amplification), all resulted in the limitation of clinical efficacy of MET inhibition. Scientists have attempted multiple means to overcome resistance and render these tumors more sensitive to treatment via combination with other targeted treatment, chemotherapy, radiation therapy, immunotherapy or molecular approaches.
As a conclusion, it is vital to standardize MET-status determination, so as to properly select patients for trials. Furthermore, a profound understanding of the coexistence of genetic alterations, the complex cross-talk between pathways, and resistance mechanisms will provide guidance for the innovation and validation of effective combination strategies that may improve patient outcomes in GC.
Author Contributions
All authors, HE, RS, and OA, contributed equally to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no potential conflicts of interest for this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
2. Howson CP, Hiyama T, Wynder EL. THE DECLINE IN GASTRIC CANCER: EPIDEMIOLOGY OF AN UNPLANNED TRIUMPH. Epidemiol Rev. 1986;8(1):1–27. doi:10.1093/oxfordjournals.epirev.a036288
3. Bosetti C, Bertuccio P, Levi F, et al. Cancer mortality in the European Union, 1970-2003, with a joinpoint analysis. Ann Oncol. 2008;19(4):631–640. doi:10.1093/annonc/mdm597
4. Bass AJ, Thorsson V, Shmulevich I, et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209.
5. Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label Phase 3 trial. Lancet Oncol. 2013;14(6):481–489. doi:10.1016/S1470-2045(13)70096-2
6. Guimbaud R, Louvet C, Ries P, et al. Prospective, Randomized, Multicenter, Phase III Study of Fluorouracil, Leucovorin, and Irinotecan Versus Epirubicin, Cisplatin, and Capecitabine in Advanced Gastric Adenocarcinoma: A French Intergroup (Fédération Francophone de Cancérologie Digestive, Fédération Nationale des Centres de Lutte Contre le Cancer, and Groupe Coopérateur Multidisciplinaire en Oncologie) Study. J clin oncol. 2014;32:3520–3526.
7. Le DT, Ott PA, Korytowsky B, et al. Real-world Treatment Patterns and Clinical Outcomes Across Lines of Therapy in Patients With Advanced/Metastatic Gastric or Gastroesophageal Junction Cancer. Clin Colorectal Cancer. 2020;19(32–38.e3):32–38.e3. doi:10.1016/j.clcc.2019.09.001
8. Cancer Stat Facts: Stomach Cancer. Available at: https://seer.cancer.gov/statfacts/html/stomach.html. Acessed May 13, 2020.
9. Deng N, Goh LK, Wang H, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61:673–684.
10. Bang Y, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697. doi:10.1016/S0140-6736(10)61121-X
11. Chen C, Kim H, Liska D, et al. MET Activation Mediates Resistance to Lapatinib Inhibition of HER2-Amplified Gastric Cancer Cells. Mol Cancer Ther. 2012;11(3):660–669. doi:10.1158/1535-7163.MCT-11-0754
12. Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. United States: Elsevier Inc; 2000.
13. Zhang Y, Vande Woude GF. HGF/SF‐met signaling in the control of branching morphogenesis and invasion. J Cell Biochem. 2003;88(2):408–417. doi:10.1002/jcb.10358
14. Appleman LJ. MET Signaling Pathway: A Rational Target for Cancer Therapy. J clin oncol. 2011;29(36):4837–4838. doi:10.1200/JCO.2011.37.7929
15. Komada M, Hatsuzawa K, Shibamoto S, et al. Proteolytic processing of the hepatocyte growth factor/scatter factor receptor by furin. FEBS Lett. 1993;328(1–2):25–29. doi:10.1016/0014-5793(93)80958-W
16. Vande Woude GF, Birchmeier C, Gherardi E, Birchmeier W. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915–925. doi:10.1038/nrm1261
17. Liu X, Newton RC, Scherle PA. Developing c-MET pathway inhibitors for cancer therapy: progress and challenges. Trends Mol Med. 2009;16(1):37–45. doi:10.1016/j.molmed.2009.11.005
18. Bradley CA, Salto-Tellez M, Laurent-Puig P, et al. Targeting c-MET in gastrointestinal tumours: rationale, opportunities and challenges. Nat Rev Clin Oncol. 2017;14:562–576.
19. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103.
20. Petrocca F, Visone R, Onelli MR, et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13(3):272. doi:10.1016/j.ccr.2008.02.013
21. Wiggan O, Taniguchi-Sidle A, Hamel PA. Interaction of the pRB-family proteins with factors containing paired-like homeodomains. Oncogene. 1998;16(2):227–236. doi:10.1038/sj.onc.1201534
22. Hammond DE, Urbé S, Vande Woude GF, Clague MJ. Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene. 2001;20(22):2761–2770. doi:10.1038/sj.onc.1204475
23. Pennacchietti S, Michieli P, Galluzzo M, et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3(4):347–361. doi:10.1016/S1535-6108(03)00085-0
24. Anestis A, Zoi I, Karamouzis MV. Current advances of targeting HGF/c-Met pathway in gastric cancer. Ann Translat Med. 2018;6(12):247. doi:10.21037/atm.2018.04.42
25. Janjigian YY, Tang LH, Coit DG, et al. MET Expression and Amplification in Patients with Localized Gastric Cancer. Cancer Epidemiol Biomarkers Prevent. 2011;20(5):1021–1027. doi:10.1158/1055-9965.EPI-10-1080
26. Lee HE, Kim MA, Lee HS, et al. MET in gastric carcinomas: comparison between protein expression and gene copy number and impact on clinical outcome. Br J Cancer. 2012;107(2):325–333. doi:10.1038/bjc.2012.237
27. Sun Y, Tian -M-M, Zhou L-X, You W-C, Li J-Y. Value of c-Met for Predicting Progression of Precancerous Gastric Lesions in Rural Chinese Population. Chin J Cancer Res. 2012;24(1):18–22. doi:10.1007/s11670-012-0018-x
28. Retterspitz MF, Mönig SP, Schreckenberg S, et al. Expression of {beta}-catenin, MUC1 and c-met in diffuse-type gastric carcinomas: correlations with tumour progression and prognosis. Anticancer Res. 2010;30:4635.
29. Guo T, Yang J, Yao J, et al. Expression of MACC1 and c-Met in human gastric cancer and its clinical significance. Cancer Cell Int. 2013;13(1):121. doi:10.1186/1475-2867-13-121
30. Liu YJ, Shen D, Yin X, et al. HER2, MET and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br J Cancer. 2014;110(5):1169–1178. doi:10.1038/bjc.2014.61
31. Kwak EL, Ahronian LG, Siravegna G, et al. Molecular Heterogeneity and Receptor Coamplification Drive Resistance to Targeted Therapy in MET-Amplified Esophagogastric Cancer. Cancer Discov. 2015;5(12):1271–1281. doi:10.1158/2159-8290.CD-15-0748
32. Dulak AM, Stojanov P, Peng S, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–486.
33. Kim J, Fox C, Peng S, et al. Preexisting oncogenic events impact trastuzumab sensitivity in ERBB2-amplified gastroesophageal adenocarcinoma. J Clin Invest. 2014;124(12):5145–5158. doi:10.1172/JCI75200
34. Zhang J, Jiang X, Jiang Y, et al. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. European Journal of Medicinal Chemistry. 2016; 108:495-504.
35. Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between Epidermal Growth Factor Receptor and c-Met Signal Pathways in Transformed Cells. J Biol Chem. 2000;275:8806–8811
36. Zhang Y, Xia M, Jin K, et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018;17:45.
37. Huang K, Sung I, Fang W, et al. Correlation between HGF/c-Met and Notch1 signaling pathways in human gastric cancer cells. Oncol Rep. 2018;40:294–302.
38. Wang H, Rao B, Lou J, et al. The Function of the HGF/c-Met Axis in Hepatocellular Carcinoma. Fron Cell Dev Biol. 2020;8:55.
39. Timothy A, Olmos D, Andre T, et al. Phase I Trial of a Selective c-MET Inhibitor ARQ 197 Incorporating Proof of Mechanism Pharmacodynamic Studies. J Clin Oncol. 2011;29(10):1271–1279. doi:10.1200/JCO.2010.31.0367
40. Kang Y, Kang Y, Muro K, et al. A phase II trial of a selective c-Met inhibitor tivantinib (ARQ 197) monotherapy as a second- or third-line therapy in the patients with metastatic gastric cancer. Invest New Drugs. 2014;32(2):355–361. doi:10.1007/s10637-013-0057-2
41. Hong DS, LoRusso P, Hamid O, et al. Phase I Study of AMG 337, a Highly Selective Small-molecule MET Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2019;25(8):2403–2413. doi:10.1158/1078-0432.CCR-18-1341
42. Van Cutsem E, Karaszewska B, Kang Y, et al. A Multicenter Phase II Study of AMG 337 in Patients with MET -Amplified Gastric/Gastroesophageal Junction/Esophageal Adenocarcinoma and Other -Amplified Solid Tumors. Clin Cancer Res. 2019;25(8):2414–2423. doi:10.1158/1078-0432.CCR-18-1337
43. Gavine PR, Ren Y, Han L, et al. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol Oncol. 2015;9(1):323–333. doi:10.1016/j.molonc.2014.08.015
44. Lee J, Kim ST, Kim K, et al. Tumor Genomic Profiling Guides Patients with Metastatic Gastric Cancer to Targeted Treatment: the VIKTORY Umbrella Trial. Cancer Discov. 2019;9:1388–1405.
45. Park CH, Cho SY, Ha JD, et al. Novel c-Met inhibitor suppresses the growth of c-Met-addicted gastric cancer cells. BMC Cancer. 2016;16(1):35. doi:10.1186/s12885-016-2058-y
46. Sohn S, Kim B, Sul HJ, Choi BY, Kim HS, Zang DY. Foretinib Inhibits Cancer Stemness and Gastric Cancer Cell Proliferation by Decreasing CD44 and c-MET Signaling. Onco Targets Ther. 2020;13:1027–1035. doi:10.2147/OTT.S226951
47. Awasthi N, Grojean M, Monahan S, et al. Abstract 5807: the dual c-Met/VEGFR2 inhibitor foretinib augments chemotherapy response in preclinical models of gastric cancer. Cancer Res. 2018;78:5807.
48. Shah MA, Wainberg ZA, Catenacci DVT, et al. Phase II Study Evaluating 2 Dosing Schedules of Oral Foretinib (GSK1363089), cMET/VEGFR2 Inhibitor, in Patients with Metastatic Gastric Cancer. PLoS One. 2013;8(3):e54014. doi:10.1371/journal.pone.0054014
49. Jochen K, Eunice L, Ackerman A, et al. MET Amplification Identifies a Small and Aggressive Subgroup of Esophagogastric Adenocarcinoma With Evidence of Responsiveness to Crizotinib. J Clin Oncol. 2011;29(36):4803–4810. doi:10.1200/JCO.2011.35.4928
50. Lee J, Pilot A Study of Crizotinib in Patients With c-MET Positive Gastric Adenocarcinoma as a Third-line Chemotherapy. nct; 2015. Available from: https://www.openaire.eu/search/dataset?datasetId=opentrials__::e30647408d8fe4bc48c26469d3c9de88.
51. Iveson T, Donehower R, Davidenko I, et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation Phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 2014;15(9):1007–1018. doi:10.1016/S1470-2045(14)70023-3
52. Catenacci DVT, Tebbutt NC, Davidenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(11):1467–1482. doi:10.1016/S1470-2045(17)30566-1
53. A phase 3 study of rilotumumab (AMG 102) with cisplatin and capecitabine (CX) as first-line therapy in gastric cancer (RILOMET-2). Available at: https://www.clinicaltrials.gov/ct2/manage-recs/fdaaa. Accessed May, 15, 2020.
54. Malka D, François E, Penault-Llorca F, et al. FOLFOX alone or combined with rilotumumab or panitumumab as first-line treatment for patients with advanced gastroesophageal adenocarcinoma (PRODIGE 17-ACCORD 20-MEGA): a randomised, open-label, three-arm phase II trial. Eur J Cancer. 2019;115:97–106. doi:10.1016/j.ejca.2019.04.020
55. David R, Thomas J, Rodryg A, et al. Randomized Phase II Trial of Onartuzumab in Combination With Erlotinib in Patients With Advanced Non–Small-Cell Lung Cancer. J Clin Oncol. 2013;31:4105–4114. doi:10.1200/JCO.2012.47.4189
56. Shah MA, Cho J, Tan IB, et al. A Randomized Phase II Study of FOLFOX With or Without the MET Inhibitor Onartuzumab in Advanced Adenocarcinoma of the Stomach and Gastroesophageal Junction. Oncologist. 2016;21:1085–1090. doi:10.1634/theoncologist.2016-0038
57. Shah MA, Bang Y, Lordick F, et al. Effect of Fluorouracil, Leucovorin, and Oxaliplatin With or Without Onartuzumab in HER2-Negative, MET-Positive Gastroesophageal Adenocarcinoma: the METGastric Randomized Clinical Trial. JAMA Oncol. 2017;3(5):620–627. doi:10.1001/jamaoncol.2016.5580
58. Sakai D, Chung HC, Oh D, et al. A non-randomized, open-label, single-arm, Phase 2 study of emibetuzumab in Asian patients with MET diagnostic positive, advanced gastric cancer. Cancer Chemother Pharmacol. 2017;80(6):1197–1207. doi:10.1007/s00280-017-3445-z
59. Tabernero J, Elez ME, Herranz M, et al. A Pharmacodynamic/Pharmacokinetic Study of Ficlatuzumab in Patients with Advanced Solid Tumors and Liver Metastases. Clin Cancer Res. 2014;20:2793–2804.
60. Jones SF, Cohen RB, Bendell JC, et al. Safety, tolerability, and pharmacokinetics of TAK-701, a humanized anti-hepatocyte growth factor (HGF) monoclonal antibody, in patients with advanced nonhematologic malignancies: first-in-human phase I dose-escalation study. J Clin Oncol. 2010;28(15_suppl):3081. doi:10.1200/jco.2010.28.15_suppl.3081
61. Strickler JH, LoRusso P, Yen C, et al. Phase 1, open-label, dose-escalation, and expansion study of ABT-700, an anti-C-met antibody, in patients (pts) with advanced solid tumors. J Clin Oncol. 2014;32(15_suppl):2507. doi:10.1200/jco.2014.32.15_suppl.2507
62. Kim HS, Chon HJ, Kim H, et al. MET in gastric cancer with liver metastasis: the relationship between MET amplification and Met overexpression in primary stomach tumors and liver metastasis. J Surg Oncol. 2018;117(8):1679–1686. doi:10.1002/jso.25097
63. Marano L, Chiari R, Fabozzi A, et al. c-Met targeting in advanced gastric cancer: an open challenge. Cancer Lett. 2015;365(1):30–36. doi:10.1016/j.canlet.2015.05.028
64. Mark Y, Sun MY, Chen C-T, et al. HER kinase activation confers resistance to MET tyrosine kinase inhibition in MET oncogene-addicted gastric cancer cells. Mol Cancer Ther. 2008;7(11):3499–3508. doi:10.1158/1535-7163.MCT-08-0374
65. Khoury H, Naujokas MA, Zuo D, et al. HGF Converts ErbB2/Neu Epithelial Morphogenesis to Cell Invasion. Mol Biol Cell. 2005;16(2):550–561. doi:10.1091/mbc.e04-07-0567
66. Qi J, McTigue MA, Rogers A, et al. Multiple Mutations and Bypass Mechanisms Can Contribute to Development of Acquired Resistance to MET Inhibitors. Cancer Res. 2011;71(3):1081–1091. doi:10.1158/0008-5472.CAN-10-1623
67. Frigault MM, Markovets A, Nuttall B, et al. Mechanisms of Acquired Resistance to Savolitinib, a Selective MET Inhibitor in MET-Amplified Gastric Cancer. In: JCO Precision Oncology. 2020:222–232.
68. Baldacci S, Kherrouche Z, Descarpentries C, et al. MET exon 14 splicing sites mutations: A new therapeutic opportunity in lung cancer. Rev Mal Respir. 2018;35:796. doi:10.1016/j.rmr.2018.01.011
69. Huang X, Li E, Shen H, et al. Targeting the HGF/MET Axis in Cancer Therapy: challenges in Resistance and Opportunities for Improvement. Fron Cell Dev Biol. 2020;8:152. doi:10.3389/fcell.2020.00152
70. Li A, Yang J, Zhang X, et al. Acquired MET Y1248H and D1246N Mutations Mediate Resistance to MET Inhibitors in Non-Small Cell Lung Cancer. Clin Cancer Res. 2017;23:4929–4937. doi:10.1158/1078-0432.CCR-16-3273
71. McCall-Culbreath KD, Li Z, Zutter MM. Crosstalk between the alpha2beta1 integrin and c-met/HGF-R regulates innate immunity. Blood. 2008;111:3562.
72. Singhal E, Sen P. Hepatocyte growth factor-induced c-Src-phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin pathway inhibits dendritic cell activation by blocking IκB kinase activity. Int J Biochem Cell Biol. 2011;43(8):1134–1146. doi:10.1016/j.biocel.2011.04.006
73. Finisguerra V, Di Conza G, Di Matteo M, et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature. 2015;522(7556):349–353. doi:10.1038/nature14407
74. Martin V, Chiriaco C, Modica C, et al. Met inhibition revokes IFNγ-induction of PD-1 ligands in MET-amplified tumours. Br J Cancer. 2019;120(5):527–536. doi:10.1038/s41416-018-0315-3
75. Benkhoucha M, Molnarfi N, Dunand-Sauthier I, et al. Hepatocyte Growth Factor Limits Autoimmune Neuroinflammation via Glucocorticoid-Induced Leucine Zipper Expression in Dendritic Cells. J Immunol. 2014;193(6):2743–2752. doi:10.4049/jimmunol.1302338
76. Zucali PA, Ruiz MG, Giovannetti E, et al. Role of cMET expression in non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitors. Ann Oncol. 2008;19(9):1605–1612. doi:10.1093/annonc/mdn240
77. Fuchs, Charles S, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383(9911):31–39. doi:10.1016/S0140-6736(13)61719-5
78. Wilke H, Muro K, Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15:1224–1235.
79. Chen X, Guan Z, Lu J, et al. Synergistic antitumor effects of cMet inhibitor in combination with anti-VEGF in colorectal cancer patient-derived xenograft models. J Cancer. 2018;9(7):1207–1217. doi:10.7150/jca.20964
80. Humbert M, Medová M, Aebersold DM, et al. Protective autophagy is involved in resistance towards MET inhibitors in human gastric adenocarcinoma cells. Biochem Biophys Res Commun. 2013;431(2):264–269. doi:10.1016/j.bbrc.2012.12.120
81. Schroeder RD, Choi W, Hong DS, McConkey DJ. Autophagy is required for crizotinib-induced apoptosis in MET-amplified gastric cancer cells. Oncotarget. 2017;8(31):51675–51687. doi:10.18632/oncotarget.18386
82. Lin X, Peng Z, Wang X, et al. Targeting autophagy potentiates antitumor activity of Met-TKIs against Met-amplified gastric cancer. Cell Death Dis. 2019;10(2):139. doi:10.1038/s41419-019-1314-x
83. Pant S, Patel M, Kurkjian C, et al. A Phase II Study of the c-Met Inhibitor Tivantinib in Combination with FOLFOX for the Treatment of Patients with Previously Untreated Metastatic Adenocarcinoma of the Distal Esophagus, Gastroesophageal Junction, or Stomach. Cancer Invest. 2017;35(7):463–472. doi:10.1080/07357907.2017.1337782
84. Rajdev L, Phase A I and Randomized Phase II Double Blinded Placebo Controlled Study of mFOLFOX6 ± AMG 337 in the First Line Treatment of Patients With Her2/Neu Negative and High MET Expressing Advanced Gastric and Esophageal Adenocarcinoma. nct; 2015. Available from: https://www.openaire.eu/search/dataset?datasetId=opentrials__::bb9210e67017b0a3da1f9eef1cdfe740.
85. Tumors resistant to radiation therapy may be controlled by the MET oncogene. Biotech Week. 2011;20:1550.
86. De Bacco F, Luraghi P, Medico E, et al. Induction of MET by Ionizing Radiation and Its Role in Radioresistance and Invasive Growth of Cancer. JNCI. 2011;103:645–661.
87. Sheng-hua C, Yan-bin M, Zhi-an Z, et al. RETRACTED: radiation-enhanced hepatocyte growth factor secretion in malignant glioma cell lines. Surg Neurol. 2007;68:610–613.
88. Qian L, Mizumoto K, Inadome N, et al. Radiation stimulates HGF receptor/c‐Met expression that leads to amplifying cellular response to HGF stimulation via upregulated receptor tyrosine phosphorylation and MAP kinase activity in pancreatic cancer cells. Int J Cancer. 2003;104:542–549.
89. Todorova PK, Mukherjee B, Burma S. MET signaling promotes DNA repair and radiation resistance in glioblastoma stem-like cells. Ann Translat Med. 2017;5:61.
90. Bhardwaj V, Cascone T, Cortez MA, et al. Modulation of c-Met signaling and cellular sensitivity to radiation: potential implications for therapy. Cancer. 2013;119:1768–1775.
91. Cuneo KC, Mehta RK, Kurapati H, Thomas DG, Lawrence TS, Nyati MK. Enhancing the Radiation Response in KRAS Mutant Colorectal Cancers Using the c-Met Inhibitor Crizotinib. Transl Oncol. 2019;12:209–216.
92. Veenstra C, Pérez-Tenorio G, Fornander T, Skoog L, Nordenskjöld B, Abstract SO. 427: c-Met reduces response to radiation in breast cancer. Cancer Res. 2013;73:427.
93. Chen G, Dai W, Zhu H, et al. Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met. J Cancer. 2017;8:983–992.
94. Sun Z, Wu Y, Hou W, et al. A novel bispecific c-MET/PD-1 antibody with therapeutic potential in solid cancer. Oncotarget. 2017;8:29067–29079.
95. Thayaparan T, Petrovic RM, Achkova DY, et al. CAR T-cell immunotherapy of MET-expressing malignant mesothelioma. OncoImmunology. 2017;6:e1363137.
96. Avan A, Quint K, Nicolini F, et al. Enhancement of the Antiproliferative Activity of Gemcitabine by Modulation of c-Met Pathway in Pancreatic Cancer. Curr Pharm Des. 2013;19(5):940–950. doi:10.2174/138161213804547312
97. Chen J. Lentivirus-mediated RNA silencing of c-Met markedly suppresses peritoneal dissemination of gastric cancer in vitro and in vivo. Acta Pharmacol Sin. 2012;33:513–522.
98. Zhan H, Tu S, Zhang F, Shao A, Lin J. MicroRNAs and Long Non-coding RNAs in c-Met-Regulated Cancers. Fron Cell Dev Biol. 2020;8:145. doi:10.3389/fcell.2020.00145
99. Liu X, Sun R, Chen J, et al. Crosstalk Mechanisms Between HGF/c-Met Axis and ncRNAs in Malignancy. Fron Cell Dev Biol. 2020;8:23.
100. Zheng Z, Yan D, Chen X, et al. MicroRNA-206: effective Inhibition of Gastric Cancer Progression through the c-Met Pathway. PLoS One. 2015;10(7):e0128751. doi:10.1371/journal.pone.0128751
101. Zhang Z, Kong Y, Yang W, et al. Upregulation of microRNA-34a enhances the DDP sensitivity of gastric cancer cells by modulating proliferation and apoptosis via targeting MET. Oncol Rep. 2016;36(4):2391–2397. doi:10.3892/or.2016.5016
102. Liu S, Shun C, Hung K, et al. Mucin glycosylating enzyme GALNT2 suppresses malignancy in gastric adenocarcinoma by reducing MET phosphorylation. Oncotarget. 2016;7(10):11251–11262. doi:10.18632/oncotarget.7081
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.