Back to Journals » International Journal of Nanomedicine » Volume 18
Mesalazine–PAMAM Nanoparticles for Transporter-Independent Intracellular Drug Delivery: Cellular Uptake and Anti-Inflammatory Activity
Authors Gorzkiewicz M ![]() , Marcinkowska M, Studzian M, Karwaciak I, Pulaski L, Klajnert-Maculewicz B
, Marcinkowska M, Studzian M, Karwaciak I, Pulaski L, Klajnert-Maculewicz B ![]()
Received 22 September 2022
Accepted for publication 30 March 2023
Published 24 April 2023 Volume 2023:18 Pages 2109—2126
DOI https://doi.org/10.2147/IJN.S390763
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lijie Grace Zhang
Michal Gorzkiewicz,1,2 Monika Marcinkowska,1 Maciej Studzian,3,4 Iwona Karwaciak,4,5 Lukasz Pulaski,3,4 Barbara Klajnert-Maculewicz1
1Department of General Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, Lodz, Poland; 2Department of Molecular Medicine II, Medical Faculty, Heinrich Heine University Düsseldorf, Düsseldorf, Germany; 3Department of Oncobiology and Epigenetics, Faculty of Biology and Environmental Protection, University of Lodz, Lodz, Poland; 4Laboratory of Transcriptional Regulation, Institute of Medical Biology PAS, Lodz, Poland; 5Laboratory of Epigenetics, Institute of Medical Biology PAS, Lodz, Poland
Correspondence: Michal Gorzkiewicz, Department of General Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, 141/143 Pomorska Street, Lodz, 90-236, Poland, Tel +48 42 635 41 47, Email [email protected]
Background: Mesalazine is one of the main drugs used to treat inflammatory bowel diseases. However, its applicability is limited by its rapid inactivation and removal from the organism, as well as the need for its membrane transporter-dependent cellular uptake to exert therapeutic effect. The present study involved the development of an innovative nanocarrier, based on poly(amidoamine) (PAMAM) dendrimer of the 4th generation, to obtain higher concentrations of the drug in the intestinal epithelial cells, thus increasing its anti-inflammatory potential. The work involved synthesis and in vitro characterization of covalent PAMAM-mesalazine conjugate with succinic linker.
Results: PAMAM-mesalazine conjugate was synthesized and characterized by 1H NMR, 13C NMR, FTIR and MALDI-TOF MS. This allowed to confirm the purity of the obtained compound and intermediates. Based on the analyses, it was found that ~45 drug molecules were successfully attached to one molecule of PAMAM dendrimer. The conjugate was then characterized in terms of hydrodynamic diameter, zeta potential, spectral properties, drug release from the carrier, as well as cellular uptake and cytotoxicity in two in vitro models of gastrointestinal epithelium (CaCo-2 and HT-29 human cell lines). Analyzing cellular parameters related to the specific mechanism of action of mesalazine (inhibition of NF-κB signaling, decrease in interleukin and prostaglandin synthesis, and ROS scavenging), we showed that such a dendrimer-based carrier may enhance cellular uptake of the drug, which translated into its improved anti-inflammatory efficacy.
Conclusion: The use of PAMAM macromolecule as a carrier for mesalazine increases the bioavailability of the drug, ensuring enhanced cellular uptake and bypassing the need to utilize mesalazine-specific membrane transporters. All these characteristics translate into an improved anti-inflammatory activity of mesalazine in vitro. In conjunction with appropriately designed in vivo studies, such a compound may prove to be a promising alternative to the therapeutics currently used in inflammatory bowel diseases.
Keywords: PAMAM dendrimer, mesalazine, inflammatory bowel disease, drug delivery
Graphical Abstract:

Background
Inflammatory bowel diseases (IBDs) are complex conditions arising as a result of cross-talk between environmental and genetic factors, leading to altered immunological responses and inflammation in the intestine.1 IBDs are most commonly diagnosed in young patients aged between 15 and 30 years old.2 These disorders typically follow a relapsing–remitting course, with symptoms such as diarrhea and abdominal pain. IBDs may lead to extraintestinal complications, such as peripheral arthropathies of small and large joints, primary sclerosing cholangitis, or deep vein thrombosis. Extreme cases of IBD are linked to an increased risk of colorectal cancer.3,4 There are currently no therapies available that are fully effective in treating IBDs.
Mesalazine (5-aminosalicylic acid, 5-ASA) is one of the primary methods of treating IBDs. Its main activity involves inhibiting signaling through nuclear factor-κB (NF-κB), a transcription-regulating protein complex playing a key role in control of immunological processes such as cytokine and chemokine production. The drug is also a potent scavenger of free radicals, as well as an inhibitor of activity of cyclooxygenases and prostaglandin synthesis.5–7 However, the applicability of 5-ASA is notably limited due to its rapid inactivation and removal from the body. Moreover, the drug needs efficient, membrane transporter-dependent cellular uptake to reach its molecular targets and exert a therapeutic effect.7,8 Mesalazine treatment causes numerous side effects, such as myelosuppression and dysregulation of nervous, cardiovascular, digestive, and excretory systems, further limiting its efficacy.4
To increase intestinal concentrations and enhance the clinical effectiveness of mesalazine, several formulations and delivery vehicles have been developed. Current research is aimed at the application of innovative microcarrier and nanocarrier systems,9 including poly(lactic acid) and chitosan microspheres, polyurethane-chitosan graft copolymers, or silica- and polysaccharide-based nanoparticles.
Analyzing the data on currently studied drug nanocarriers, the dendrimers seem to be particularly promising. Due to their sphere-shaped, symmetrical architecture with external branches terminated with chemical moieties, these macromolecules have high solubility and biopermeability (the ability to cross various biological barriers), enabling efficient cellular uptake. Terminal functional groups are usually highly reactive and may be further modified to change the physicochemical features of a dendrimer, or to generate desirable activity, eg, therapeutic or catalytic activity. Drug–dendrimer conjugates improve the solubility of therapeutics, extend their systemic circulation time, and protect them against degradation.10 Such constructs can efficiently transport drugs directly to their target, thus helping to eliminate adverse effects. Conjugates that involve covalent bonding of the drug to the dendrimer may have higher stability than noncovalent complexes and also enable controlled, enzyme- or pH-triggered drug release at the site of action giving a chance to reduce the dose while maintaining the same therapeutic effect.11 All these features make dendrimers promising candidates for nanocarriers of mesalazine.
The present work aimed to increase the anti-inflammatory potential of mesalazine by developing an innovative nanocarrier of 5-ASA based on a 4th generation (G4) poly(amidoamine) (PAMAM) dendrimer to obtain higher concentrations of the drug in intestinal epithelial cells, which are the primary site of mesalazine action in IBDs. The project involved the synthesis and in vitro characterization of a PAMAM G4 macromolecule covalently conjugated to multiple particles of mesalazine with a succinic linker.
Materials and Methods
Synthesis and Characterization of the PAMAM-5-ASA Conjugate
The synthesis and chemical characterization of the PAMAM-5-ASA conjugate containing 45 5-ASA molecules chemically coupled to the PAMAM G4 scaffold (with 64 terminal amino groups) via a succinic linker is presented in Supplementary Material, Figures S1–S12. The conjugate and the individual components obtained during the synthesis were characterized by 1H NMR, 13C NMR, FTIR, and MALDI-TOF MS.
Size and Zeta Potential Measurements
Measurements of the hydrodynamic diameter and zeta potential were performed using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK). Water solutions containing studied compounds (free PAMAM G4 and PAMAM-5-ASA) at a final dendrimer concentration of 10 μM were placed in low-volume sized cuvettes (ZEN0112, Malvern Instruments Ltd., Malvern, UK) for size determination or in folded capillary cells (DTS 1070, Malvern Instruments Ltd., Malvern, UK) for zeta potential measurements and measured at 25°C. The data were analyzed using Malvern software.
Spectrofluorimetric Analysis
Fluorescence spectra were acquired using a PerkinElmer LS-50B spectrofluorometer. All measurements were performed in phosphate-buffered saline (PBS, pH 7.4) at room temperature (RT). The excitation wavelength was set at 350 nm, and spectra were collected at wavelengths from 380 to 600 nm. Excitation and emission slits were 5 and 7 nm, respectively. The measurements were carried out for tested compounds (free 5-ASA and PAMAM-5-ASA) at 5 μM concentration of mesalazine. Under the same conditions, the spectrum of free PAMAM G4 dendrimer was plotted at 111.1 nM concentration.
To analyze the spectral properties of compounds obtained after PAMAM-5-ASA hydrolysis, the conjugate was diluted in PBS and incubated at RT for 24 h. The mixture was then separated by centrifugation using an Amicon Ultra-4 centrifugal filter unit (3 kDa). The fractions collected from above and below the filter were diluted in PBS to match the initial volume and analyzed for spectral properties as described above.
Drug Release Studies
Solutions (200 µM) of free 5-ASA and PAMAM-5-ASA conjugate (with respect to mesalazine attached to the dendrimer) in PBS were enclosed in a dialysis membrane (SnakeSkin dialysis tubing, 3.5K MWCO, 22 mm, Thermo Fisher Scientific, Waltham, MA, USA) and immersed in PBS (pH 5 or 7.4). Samples from the inside of the tubing were collected at intervals (0.5, 1, 2, 4, 6, 8, and 24 h), and the fluorescence at 350 nm excitation and 430 nm emission (for PAMAM-5-ASA) or 490 nm (for free mesalazine) was measured on a PerkinElmer LS-50B spectrofluorometer. The results from these experiments are presented as the percentage decrease in fluorescence of the starting mesalazine or conjugate solution.
Cell Culture
Two human colorectal adenocarcinoma cell lines, Caco-2 and HT-29, were purchased from ATCC (Manassas, VA, USA) and maintained under standard conditions in DMEM (Caco-2) or RPMI-1640 (HT-29) medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum, penicillin (100 U/mL) and streptomycin (100 µg/mL) (Sigma-Aldrich, St. Louis, MO, USA) at 37°C in an atmosphere of 5% CO2. Cells were subcultured twice a week.
Cytotoxicity Assay
To estimate the potential cytotoxic activity of the PAMAM-5-ASA conjugate, a resazurin assay was performed.12 Cells were seeded into 96-well black plates at a density of 1.5 × 104 cells per well 24 h before the experiment. The cells were subsequently treated with increasing concentrations of free mesalazine (0.1–10 mM), free PAMAM G4, or PAMAM-5-ASA conjugate (0.1–500 μM of the dendrimer) for 3 h and 24 h at 37°C in an atmosphere of 5% CO2. Following the incubation, resazurin was added to the culture medium at a final concentration of 10 µg/mL, and the plates were incubated at 37°C in the dark to allow conversion of resazurin to resorufin. The fluorescence of metabolized resazurin was measured after 30 and 90 min at 530 nm excitation and 590 nm emission using an EnVision plate reader (PerkinElmer, Waltham, MA, USA). Cell viability was calculated as the increase in resorufin fluorescence between 30 and 90 min and is presented as the percentage of untreated control.
Cellular Uptake Studies
To determine the cellular uptake of the tested compounds, Caco-2 and HT-29 cells were seeded into 12-well plates at a density of 3 × 105 cells per well 24 h prior to the experiment. The cells were then treated with 5-ASA and PAMAM-5-ASA at 2 mM final concentration of mesalazine for up to 4 h. To inhibit the transport of mesalazine across the cell membrane, cells were preincubated with inhibitors of organic anion-transporting polypeptides (OATPs) (cyclosporin A, rifampicin, budesonide, pravastatin, ritonavir) at a final concentration of 100 μM for 1 h.
Cells were then washed with PBS, lysed in 200 µL terbium solution (10 mM terbium(III) chloride, 20 mM EDTA (pH 8), 40 mM NaOH) with 0.1% Triton X-100, and transferred to black 96-well plates, and the fluorescence of terbium-salicylate complex was measured at 320 nm excitation and 550 nm emission using a PowerWave HT Microplate Spectrophotometer (BioTek Instruments Inc., Winooski, VT, USA). Standard curves of the terbium-salicylate fluorescence were prepared for free mesalazine and mesalazine conjugated with the PAMAM dendrimer under analogous conditions. The protein concentration in cell lysates was determined using the Pierce 660 nm Protein Assay Reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Results are presented as the quantity of mesalazine [pmol] per 1 µg of protein in the cell lysate.
Gene Expression Assay
The gene expression level was determined by quantitative real-time RT-PCR. Caco-2 and HT-29 cells were seeded into 12-well plates at a density of 3 × 105 cells per well 24 h before the experiment. Next, the cells were treated with increasing concentrations of 5-ASA and PAMAM-5-ASA for 15 min and then stimulated with tumor necrosis factor-α (TNF-α, final concentration: 5 ng/mL) or E. coli lipopolysaccharide (LPS, final concentration: 10 μg/mL) for an additional 3 h. To inhibit the transport of mesalazine across the cell membrane, in some experiments, cells were preincubated with 100 μM ritonavir for 1 h.
Cells were subsequently washed with PBS, and total cellular RNA was isolated using TRI Reagent (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s protocol. Complementary DNA (cDNA) was transcribed from mRNA using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA) and used for real-time PCR amplification with GoTaq qPCR Master Mix (Promega) according to the manufacturer’s protocol. Each 16 μL reaction volume contained 1 μL of cDNA and 0.25 μM of forward and reverse intron-spanning primers (for primer sequences, see Table 1). The reference genes (HPRT1, HMBS, and TBP) were selected according to the GeNorm procedure.13 PCR reactions were performed in 96-well microplates using the CFX96 Real-Time PCR Detection System (Bio-Rad). The expression level of assayed genes was calculated by the ΔΔCt method and expressed as the number of cognate mRNA copies per 1000 copies of geometric-averaged mRNA for the reference genes.
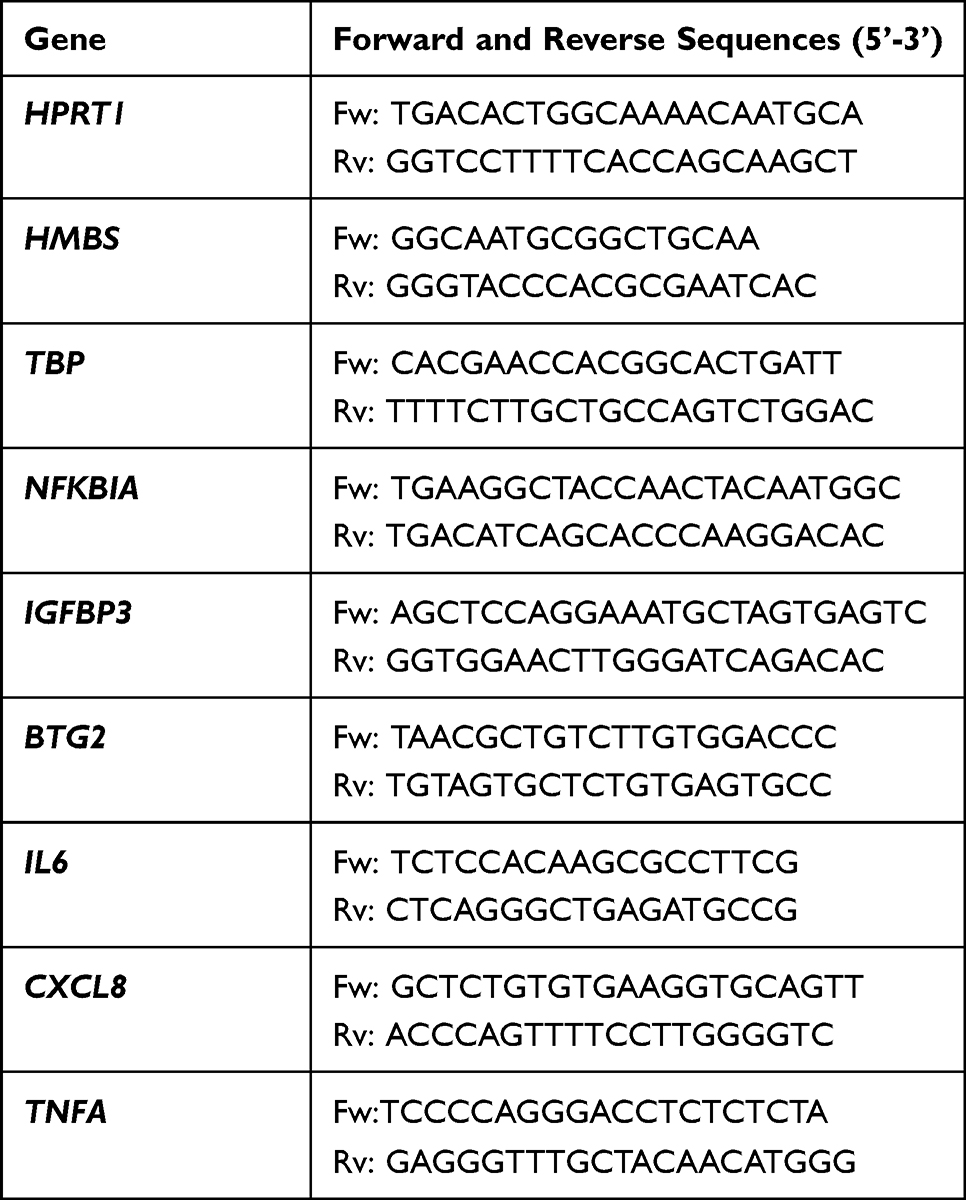 |
Table 1 Primer Sequences |
NF-κB Translocation Assay
Caco-2 and HT-29 cells were seeded into thin-bottom 96-well plates at a density of 1 × 104 cells per well 24 h before the experiment. Next, the cells were treated with 5-ASA and PAMAM-5-ASA at 2 mM final concentration of mesalazine for 15 min, then stimulated with TNF-α (5 ng/mL) for an additional 1 h. In some experiments, cells were preincubated with ritonavir (100 μM) and/or parthenolide (10 μM) for 1 h.
Following gentle removal of the culture medium and a single wash with PBS, cells were immediately fixed for 20 min at RT with PBS-buffered 2% formaldehyde solution freshly prepared from paraformaldehyde. Subsequently, the cells were washed twice with blocking buffer (PBS, 1% BSA, 0.1% Triton X-100) and further incubated with the blocking buffer for 10 min at RT. NF-κB was then probed with Alexa Fluor 488-conjugated antibody (recombinant anti-NF-kB p65 antibody, EP2161Y, Alexa Fluor 488, Abcam, ab204263, 1:300) diluted in blocking buffer for 1 h at RT in the dark. Finally, after three wash cycles with the blocking buffer, nuclei were stained with 1 µg/mL Hoechst 33342 dye for 30 min at RT in the dark, then the cells were stored in PBS at 4°C and protected from light until imaging. Measurement of NF-κB p65 translocation from cytoplasm to nucleus was conducted with a high-content screening platform (ArrayScanVTi from Thermo Fisher) using the manufacturer’s proprietary software and protocol for measuring the ratio of nuclear to cytoplasmic staining.14 Measurements were made for >1000 cells per single well, and ratios were averaged within the well and taken as single biological replicates. Data are presented as the percentage of control (untreated) translocation ratio.
Cytokine and Prostaglandin Assays
Caco-2 and HT-29 cells were seeded into 6-well plates at a density of 5 × 105 cells per well 24 h before the experiment. Next, the cells were treated with 5-ASA and PAMAM-5-ASA at 2 mM final concentration of mesalazine for 15 min, then stimulated with TNF-α (5 ng/mL) or E. coli LPS (10 μg/mL) for an additional 1 h. In some experiments, cells were preincubated with ritonavir at a final concentration of 100 μM for 1 h. All the treatments were performed in FBS-free medium. The medium containing compounds was subsequently removed, the cells were washed twice with PBS, and fresh FBS-free medium was added to the cells for 4 h.
The protein concentration of interleukin-6 (IL-6), IL-8, and TNF-α was measured in the collected medium using ELISA Kits from Biorbyt Ltd. (orb50052, orb50054, and orb50111, respectively). The prostaglandin concentration was evaluated using Prostaglandin Screening ELISA Kit from Cayman Chemical (514012). The assays were performed according to the manufacturer’s protocol, and the absorbance was read using an EnVision plate reader (PerkinElmer, Waltham, USA) at the appropriate wavelengths.
ROS Generation Assay
Caco-2 and HT-29 cells were seeded into black 96-well plates at a density of 1 × 104 cells per well 24 h before the experiment. Next, the cells were treated with 5-ASA and PAMAM-5-ASA at 2 mM final concentration of mesalazine for 15 min, then stimulated with TNF-α (20 ng/mL) for an additional 1 h. In some experiments, cells were preincubated with ritonavir (100 μM) for 1 h. The cells were subsequently washed once with HBSS and incubated with dihydroethidium (DHE; 10 μM in HBSS) for 30 min in the dark, and the fluorescence was measured at 518 nm excitation and 605 nm emission using an EnVision plate reader (PerkinElmer, Waltham, USA).
Statistics
For statistical significance testing for single pairwise comparisons, the Student’s t-test was applied. For multiple comparisons, ANOVA followed by Tukey’s post-hoc test was applied. In all tests, p-values <0.05 were considered statistically significant. Data are presented as the arithmetic mean ± SD.
Results
Mesalazine was conjugated to a PAMAM G4 dendrimer to obtain drug delivery nanovehicles that would serve a dual purpose: facilitate the delivery of mesalazine to cells through the cellular membranes and ensure that the intracellular fate of mesalazine is commensurate with anti-inflammatory activity. A description of the synthesis procedure (using a succinic linkage) and the chemical properties of the resulting conjugates is contained in Supplementary Material. The obtained conjugates contained ca. 45 5-ASA molecules per one dendrimer molecule.
Biophysical characterization of the conjugate was directed at proving its suitability for drug delivery at the cellular level. We confirmed that derivatization of PAMAM G4 with 5-ASA significantly increased the hydrodynamic diameter of nanoparticles (from 3.91 ± 0.74 nm for PAMAM G4 to 4.95 ± 0.48 nm for PAMAM-5-ASA) and reduced the positive surface-charge density (zeta potential decreased from 25.47 ± 2.69 mV to 14.33 ± 2.75 mV). The decreased zeta potential value did not, however, lead to a higher propensity for aggregation; the proportion of aggregated material to monomers when sorted by size was lower for PAMAM-5-ASA than for PAMAM G4 (Figure S13).
To verify that the conjugate was a suitable drug-delivering nanoparticle, it was important to test if it could hydrolyze in physiological conditions, releasing unmodified 5-ASA molecules with potential pharmacological activity. This task was made easier by the discovery that conjugation of mesalazine to the dendrimer leads to a characteristic hypsochromic shift in the photometric absorption spectrum of mesalazine (Figure 1); the absorption maximum changes from ca. 490 nm to ca. 430 nm, most probably reflecting reduced solvent accessibility and higher structural rigidity of the bound state. We followed the spectral changes upon prolonged incubation (24 h) in solution, with subsequent ultrafiltration to separate the high-molecular-weight (dendrimer-bound) fraction from the low-molecular-weight (released mesalazine congener) fraction. From this experiment, we demonstrated that after hydrolysis, the low-molecular-weight fraction was spectroscopically identical to unmodified 5-ASA, while the remaining high-molecular-weight fraction was unchanged, and that the quantitative proportions were consistent with the occurrence of simple hydrolysis of the amide bond and release of the 5-ASA molecule (Figure 1). Moreover, the presence of a specific PAMAM-associated absorption peak (relatively weak absorption at ca. 400 nm) only in the unhydrolyzed fraction proved that the hydrolysis does not affect the dendrimer core and does not release its components.
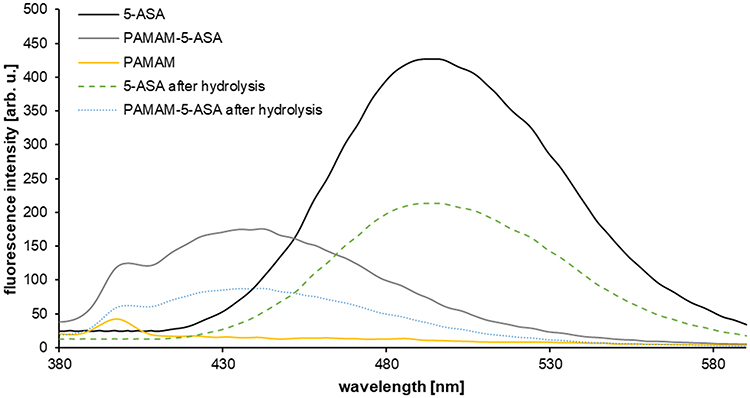 |
Figure 1 Emission spectra (exc. λ = 350) of mesalazine, PAMAM-5-ASA conjugate, free PAMAM G4, and compounds acquired after PAMAM-5-ASA hydrolysis. |
A similar approach was applied to verify the relative kinetics of the hydrolytic release process through a semi-permeable membrane, using dialysis as a separation technique. Spectrophotometric measurements (Figure 2) demonstrated that free 5-ASA molecules diffuse quickly; therefore, the slower kinetics observed upon incubation of PAMAM-5-ASA solution reflected hydrolysis itself, as the rate-limiting step. Regarding the intracellular fate of the conjugate molecules, an important additional result was the demonstration that the rate of hydrolysis does not vary with pH within the physiological range from quasi-neutral (cytoplasmic) pH 7.4 to acidic (lysosomal) pH 5.
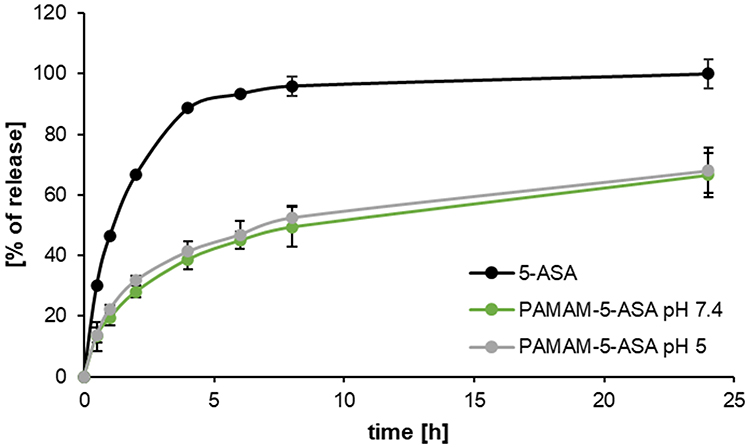 |
Figure 2 The release of 5-ASA from the PAMAM-5-ASA conjugate in different pH conditions, determined by equilibrium dialysis. Data are presented as mean ± SD, n = 6. |
A prerequisite for the potential clinical application of nanoparticles for drug delivery is their relative lack of extraneous biological activity, including acute cytotoxicity. This is especially important for PAMAM dendrimers, which are known to have strong membrane-disruptive properties and must be extensively modified for bioorthogonality. Fortuitously, the derivatization of the dendrimer with 5-ASA was sufficient to reduce extensive nonspecific and specific PAMAM cytotoxicity (Figure 3). Unmodified PAMAM molecules had a concentration- and time-dependent negative effect on the viability of colorectal cancer cells; the HT-29 cell line was more vulnerable to the effects of PAMAM than Caco-2 cells. In marked contrast, PAMAM-5-ASA was toxic to both cell lines only at supra-physiologically high concentrations (>100 µM) and upon prolonged incubation (24 h), making further experiments on its suitability for mesalazine delivery in vitro feasible.
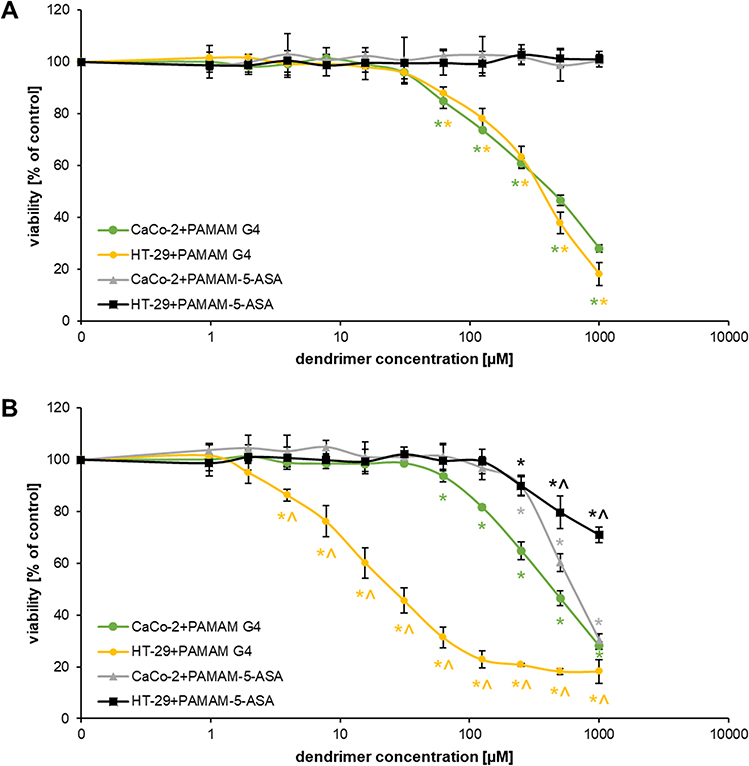 |
Figure 3 The cytotoxic effect of PAMAM and PAMAM-5-ASA on Caco-2 and HT-29 cells. Viability was determined by a resazurin assay after (A) 3 h and (B) 24 h of treatment. Data are presented as mean ± SD, n = 3. *p < 0.05, statistically significant difference compared with untreated control. ^p < 0.05, statistically significant difference between cell lines subjected to the same treatment. |
One of the main goals of designing conjugated nanoparticles is the provision of a cellular uptake route for pharmacologically active mesalazine that would be independent of permeation through dedicated organic ion channels/transporters (as downregulation of ion channels/transporters is a common route for the development of drug resistance). Therefore, experiments on the cellular uptake of free mesalazine or its dendrimer conjugate were designed around kinetic measurements without and with organic ion transport inhibitors. Interestingly, the time course of uptake of the free and nanoparticle-bound forms of mesalazine was highly similar in each of the tested cell lines (Caco-2 and HT-29, Figure 4A and B, respectively), with maximum intracellular concentrations reached after ca. 2 h of incubation. The extent of cell loading with mesalazine moieties was, however, remarkably higher for PAMAM 5-ASA than for free 5-ASA, although the incubation was performed at equimolar concentrations of the drug molecules. More importantly, the uptake of free 5-ASA was strongly inhibited by well-characterized inhibitors of organic anion transport (Figure 4C and D). The applied inhibitors had varying specificity for different transporter types; however, mesalazine does not have a single specific transporter and can permeate through several major facilitator superfamily (MFS) transporters. Interestingly, while budesonide was a potent inhibitor of 5-ASA uptake in Caco-2 cells, it was (as the only one of tested inhibitors) entirely inefficient in HT-29 cells, shedding some light on the molecular identity of at least the most important solute carriers responsible for 5-ASA uptake. In a key experiment, we showed that even the most potent inhibitors of direct 5-ASA uptake (cyclosporin A, pravastatin, and ritonavir) were completely ineffective at inhibiting the uptake of PAMAM-5-ASA in both cell lines (Figure 4E and F). This result proves that uptake of PAMAM-5-ASA occurs via a different route (most probably an endocytotic pathway), and that this mechanism accounts for all of the observed net flux of mesalazine between the extracellular and intracellular space. This finding means that the kinetics of conjugate uptake outcompete the potential sequential extracellular hydrolysis of the conjugate and transporter-mediated uptake of the released free drug.
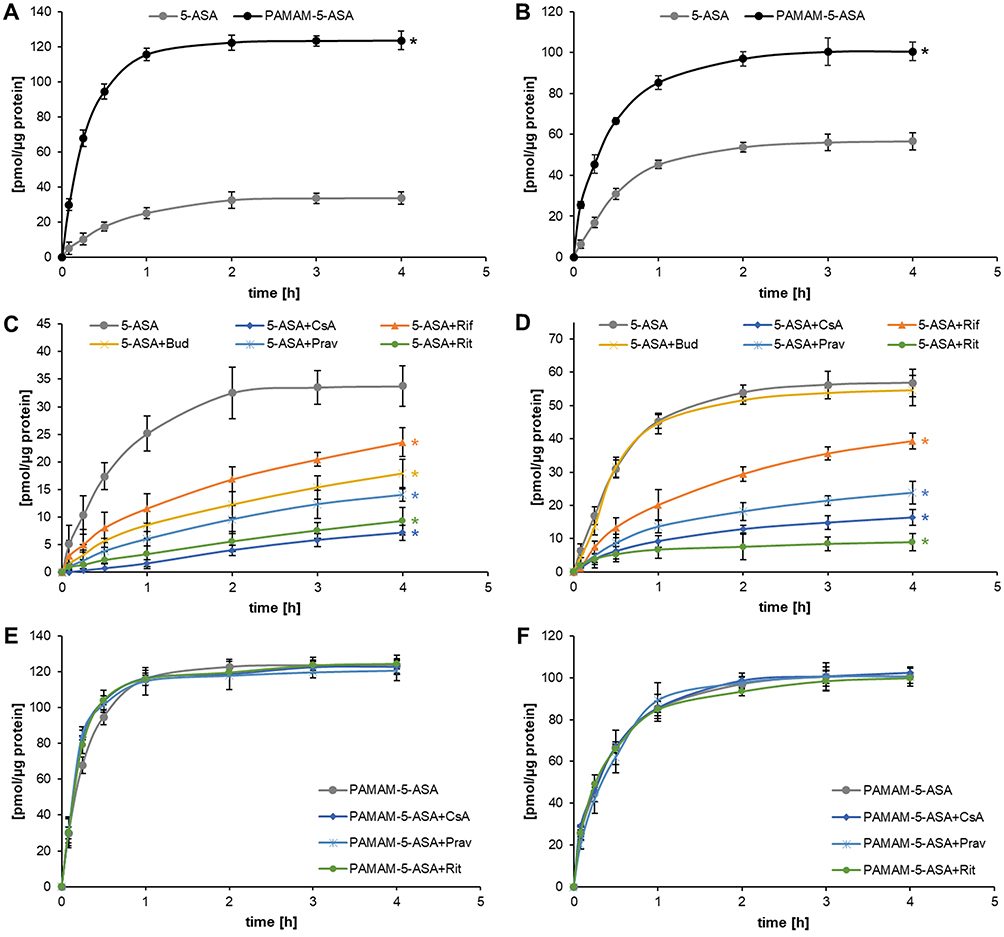 |
Figure 4 The cellular uptake of 5-ASA and PAMAM-5ASA in the absence (A and B) or presence (C–F) of organic anion-transporting polypeptide (OATP) inhibitors (cyclosporin A, CsA; rifampicin, Rif; budesonide, Bud; pravastatin, Prav; ritonavir, Rit) in Caco-2 (A, C and E) and HT-29 (B, D and F) cells. The uptake was measured using a spectrofluorimetric assay based on the formation of a ternary complex between mesalazine, terbium, and EDTA in an alkaline solution. Data are presented as mean ± SD, n = 5. *p < 0.05, statistically significant difference compared with 5-ASA uptake kinetics. |
The ability to inhibit certain signaling pathways is central to the anti-inflammatory action of mesalazine, especially in intestinal epithelium under generic conditions that mimic IBDs. The NF-κB pathway is particularly important for 5-ASA pharmacodynamics, and we performed several experiments to test the efficacy of PAMAM-5-ASA in modulating the NF-κB pathway. First, we tested the impact of various modalities of mesalazine on the expression level of marker genes known to be under direct transcriptional control by this pathway (NFKBIA, IGFBP3, BTG2) by real-time RT-PCR (Figure 5A and Table S1). The overall pattern of changes and, therefore, the experimental conclusions are similar for both tested cell lines and all three tested genes; Figure 5 shows only one gene in one cell line for clarity. TNF-α and lipopolysaccharide (LPS) from E. coli were chosen as a model stimuli that strongly activate the NF-κB pathway and consequently the expression of marker genes. Incubation of cells with either free 5-ASA or PAMAM-5-ASA concentration-dependently inhibited the expression of marker genes, mimicking the anti-inflammatory drug action in vivo. The level of inhibition by PAMAM-5-ASA was higher than that of free 5-ASA, reflecting the previously demonstrated potential of PAMAM to deliver higher intracellular loads of mesalazine. This result also showed that the delivered mesalazine is fully active. Most importantly, inhibiting 5-ASA uptake with ritonavir abolished the inhibitory effect of free 5-ASA, whereas ritonavir did not inhibit the effect of PAMAM-5-ASA. This result was fully in line with expectations derived from preceding experiments, strengthening the conclusion that dendrimer conjugation ensures cellular uptake of pharmacologically active mesalazine that circumvents organic ion membrane transporters.
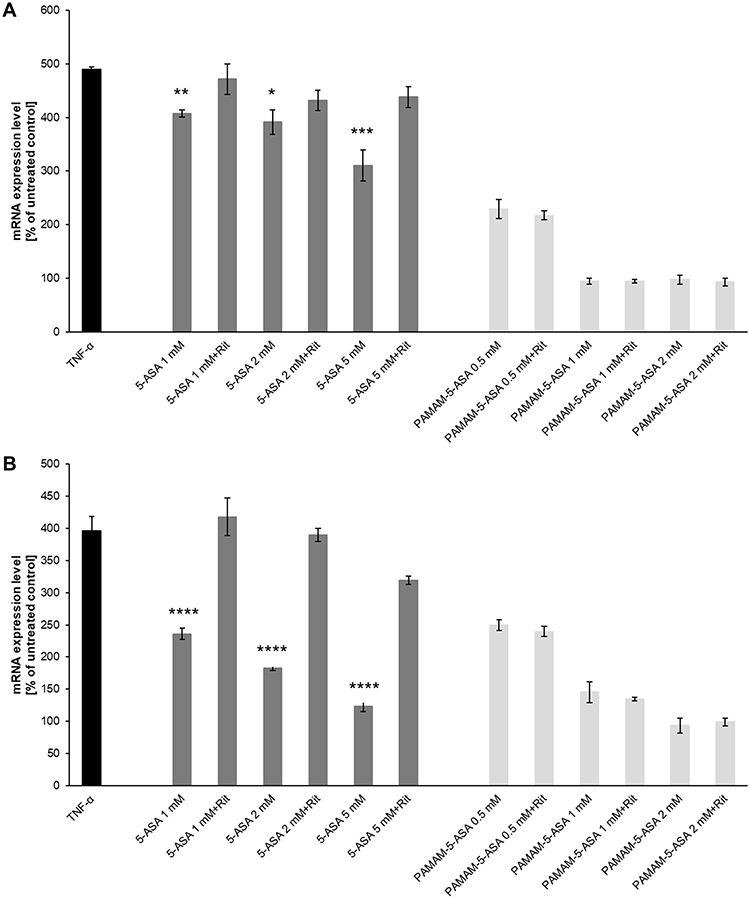 |
Figure 5 The effect of 5-ASA and PAMAM-5-ASA on (A) NFKBIA and (B) IL6 gene expression at the mRNA level in Caco-2 cells, in the presence or absence of ritonavir. The relative level of gene expression was evaluated using quantitative real-time RT-PCR. Data are presented as the percentage of cognate mRNA expression in control (untreated) cells (number of cognate mRNA copies per 1000 copies of geometric-averaged mRNA for reference genes), mean ± SD, n = 4. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, statistically significant difference compared with the corresponding sample pretreated with ritonavir. |
We further verified the direct impact of mesalazine on the NF-κB pathway. In a series of experiments that paralleled the treatments used to measure marker gene expression (Figures 6 and S14), we showed that both 5-ASA and PAMAM-5-ASA partially prevented TNF-α-induced nuclear translocation of activated NF-κB transcription factor (again, the level of inhibition induced by PAMAM-5-ASA was greater than that of free 5-ASA). Crucially, and similar to the results of marker gene expression, inhibition of organic ion transporters by ritonavir abolished the effect of free 5-ASA on TNF-α-induced nuclear translocation of activated NF-κB (restoring control levels of activation), whereas ritonavir did not affect the actions of the dendrimer conjugate. This result suggests a potential molecular target – namely NF-κB transcription factor – for dendrimer-delivered mesalazine in its clinically relevant anti-inflammatory action. Significantly, PAMAM-5-ASA (identical to the effect of free 5-ASA) acted only on cells that had been activated by the proinflammatory stimulus in all mechanistic assays and did not affect basal NF-κB signaling in unchallenged cells. This finding implies that PAMAM-5-ASA will not have unwanted off-target effects in vivo.
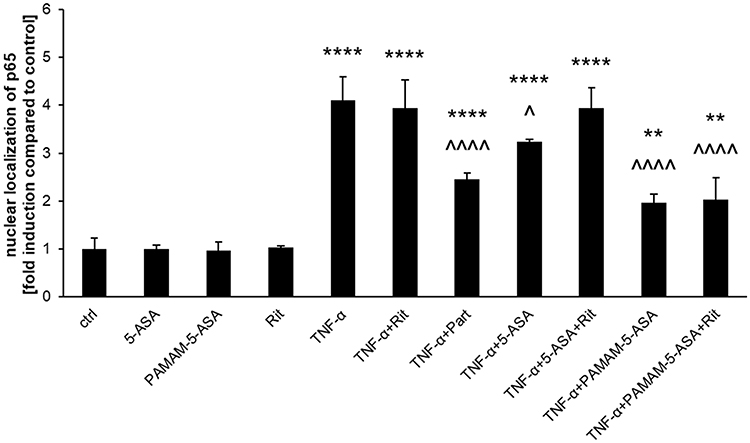 |
Figure 6 The effect of 5-ASA and PAMAM-5-ASA on the nuclear translocation of NF-κB in Caco-2 cells. The subcellular localization of NF-κB (both in the absence or presence of parthenolide, Part) was determined by high-content screening (immunofluorescence with an anti-p65 antibody and automated microscopy/image analysis). Data are presented as the fold-induction of nuclear to cytoplasmic staining ratio in control (untreated) cells, mean ± SD, n = 4. **p < 0.01, ****p < 0.0001, statistically significant difference compared with untreated control. ^p < 0.05, ^^^^p < 0.0001, statistically significant difference compared with TNF-α-treated cells. |
Inhibition of proinflammatory signaling by mesalazine must lead to detectable phenotypical changes in affected cells to effectively prevent inflammation-related symptoms at tissue level. We measured this property of dendrimer-delivered 5-ASA using several immunologically important endpoints that have a clear role in the pathology of IBDs: a) mRNA-level expression of proinflammatory cytokine genes; b) secretion of the active protein forms of these cytokines from cells; c) production of prostaglandins (another type of proinflammatory secondary mediator) by a series of biochemically activated enzymes; d) production of reactive oxygen species (ROS) by intracellular pro-oxidant enzymes that are targets for secondary messengers (such as prostaglandins). Gene expression was quantified by real-time RT-PCR, extracellular and intracellular concentrations of signaling mediators were determined by ELISA, and the ROS concentration was measured by fluorogenic substrate oxidation. We demonstrated a consistent pattern of results that supported our model of the mechanism of action of PAMAM-5-ASA. Specifically, mRNA and protein levels of biochemically stimulated proinflammatory cytokines IL-6, IL-8, and TNF-α were decreased by 5-ASA treatment, and PAMAM-5-ASA treatment decreased the level of these cytokines even more profoundly (Figures 5B, 7, S15–S17 and Table S1). Crucially, after ritonavir-mediated inhibition of membrane transporters, the potency of free 5-ASA was abolished, but the potency of the conjugate was not altered in all tested cases. Relevant control experiments proved that ritonavir has no direct off-target effects on the studied processes, and unstimulated cells were unaffected by any of the tested forms of mesalazine (Figure 7C). Similar results were obtained when the effects of free 5-ASA and PAMAM-5-ASA on prostaglandin concentrations were measured (Figures 8 and S18). This result strongly supports the notion that prostaglandin production is exclusively regulated (directly or indirectly) by the same mesalazine-sensitive signaling pathways that determine cytokine secretion. Inhibition of prostaglandin generation is clinically important, as prostaglandin generation is a central feature of pathological tissue-level inflammation and a common target of many nonsteroidal anti-inflammatory drugs. Finally, we determined the effect of free and dendrimer-bound mesalazine on the mechanistically distal (indirectly regulated) process of ROS generation. In experiments where ROS production was stimulated with physiologically relevant factors, and free and dendrimer-bound mesalazine inhibited ROS production, the latter was more pharmacologically potent (Figure 9). In this experimental setup, it was not possible to verify the effect of ritonavir-mediated transporter inhibition, as ritonavir is known to stimulate ROS production by an unrelated mechanism. However, free and dendrimer-bound mesalazine produced similar results in both tested cell lines, which attests to the likelihood of a common mechanism of action in both cell types, with ROS production located downstream of mediator (cytokine, prostaglandin) production in the proinflammatory pathway.
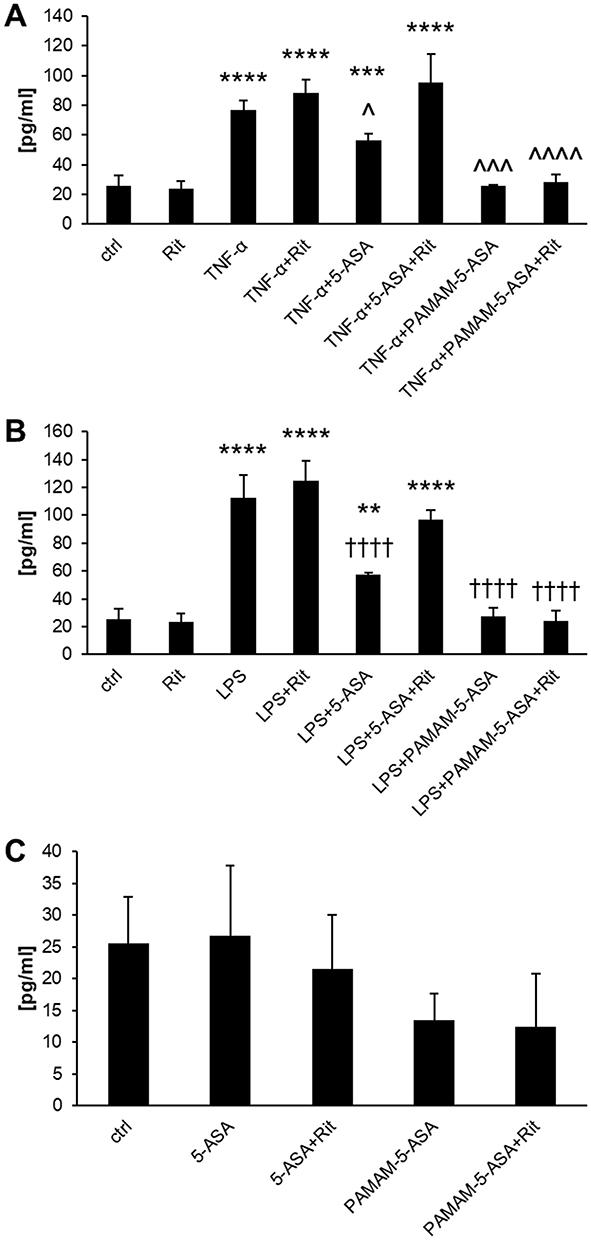 |
Figure 7 The effect of 5-ASA and PAMAM-5-ASA on the protein expression of IL-6 in Caco-2 cells stimulated with (A) TNF-α or (B) LPS, determined by ELISA in cellular supernatants. Panel C presents the cytokine concentration in control samples. Data are presented as the concentration of cytokine (pg/mL), mean ± SD, n = 4. **p < 0.01, ***p < 0.001, ****p < 0.0001, statistically significant difference compared with untreated control. ^p < 0.05, ^^^p < 0.001, ^^^^p < 0.0001, statistically significant difference compared with TNF-α-treated cells. ††††p < 0.0001, statistically significant difference compared with LPS-treated cells. |
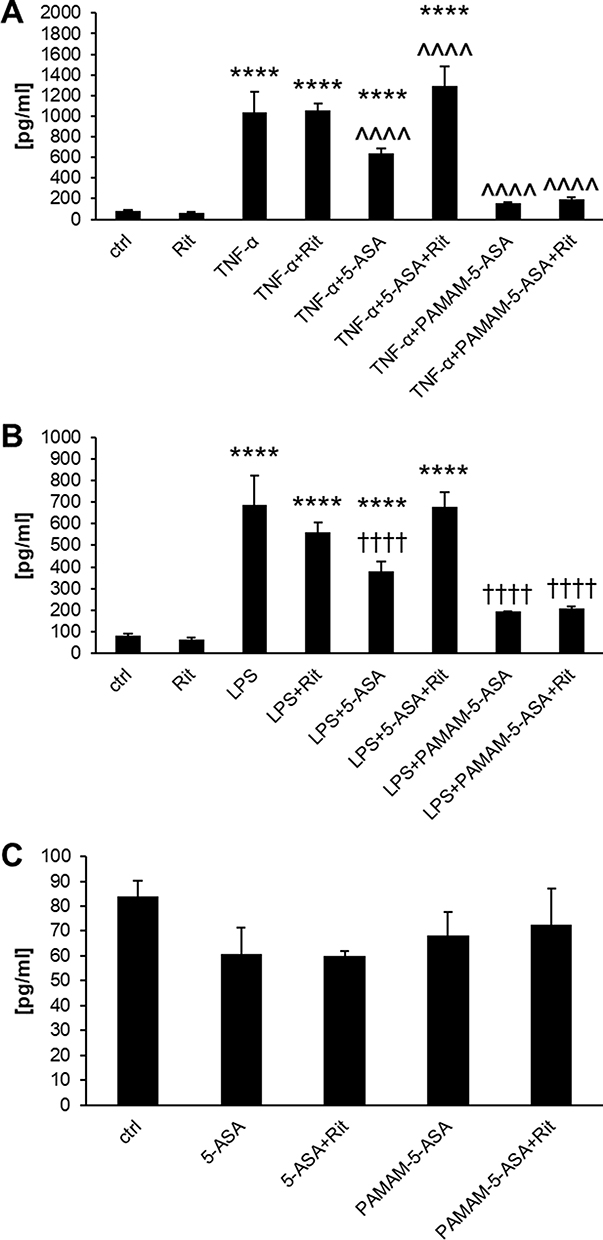 |
Figure 8 The effect of 5-ASA and PAMAM-5-ASA on the secretion of prostaglandins in Caco-2 cells stimulated with (A) TNF-α or (B) LPS, determined by ELISA in cellular supernatants. Panel C presents the secretion of prostaglandins in control samples. Data are presented as the concentration of prostaglandins (pg/mL), mean ± SD, n = 4. ****p < 0.0001, statistically significant difference compared with untreated control. ^^^^p < 0.0001, statistically significant difference compared with TNF-α-treated cells. ††††p < 0.0001, statistically significant difference compared with LPS-treated cells. |
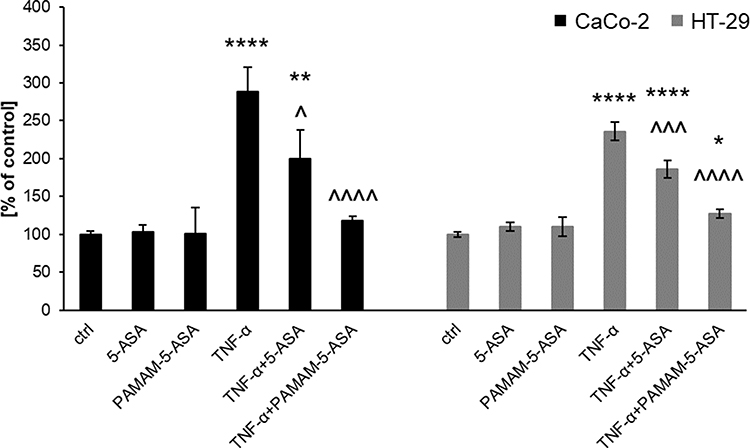 |
Figure 9 The effect of 5-ASA and PAMAM-5-ASA on intracellular ROS generation in Caco-2 and HT-29 cells stimulated with TNF-α, determined using a dihydroethidium probe. Data are presented as the percentage of ROS production in untreated control cells, mean ± SD, n = 3. *p < 0.05, **p < 0.01, ****p < 0.0001, statistically significant difference compared with untreated control. ^p < 0.05, ^^^p < 0.001, ^^^^p < 0.0001, statistically significant difference compared with TNF-α-treated cells. |
Discussion
Nanoparticle-mediated drug delivery is an active area of research to solve problems associated with drug pharmacokinetics and pharmacodynamics. Yet the majority of studies use a chemically unmodified drug that is associated with nanoparticles through physical forces and factors, namely encapsulation,15 entrapment,16 or adsorption.17 Drugs that are covalently conjugated to the chemical structure of the nanoparticle itself remain underused, even though they may have unique advantages with respect to preparation, formulation, standardization, and precise delivery to the site of action.18 The main reason for this reticence is the complex and difficult chemistry that is usually necessary for the reliable release of the biologically active compound from a conjugate, including optimal linker design, the need to maintain the structure of the pharmacophore, and the requirement to have a uniform coupling reaction on the nanoparticle surface.19,20 In this regard, nonsteroidal anti-inflammatory drugs (NSAIDs) are an especially convenient model due to their usually small and chemically simple molecules, as well as their well-researched pharmacokinetics.
Indeed, mesalazine was selected as the exemplar molecule in the present study because of its structural simplicity and known pharmacodynamics. Moreover, mesalazine is an attractive choice for studying novel drug delivery concepts due to its well-characterized but inadequately addressed practical problems with pharmacokinetics. Since mesalazine is too hydrophilic to enter cells by passive diffusion, cellular influx occurs via one of the several possible MFS membrane transporters, so inhibiting or reducing the expression of these transporters is a common source of drug resistance.21 Mesalazine is the drug of choice for treating several very common IBDs; therefore, emerging resistance is a broad clinical problem, and a novel drug delivery vehicle may have a high practical impact.
The efficient delivery of mesalazine for IBD treatment is hampered by its potential deactivation during gastrointestinal transit,7 and therefore drug delivery efforts have focused on enabling mesalazine to reach the large intestine. Applied approaches include rectal administration and application as large intestine-hydrolyzable congeners (eg, sulfasalazine).22 An additional approach uses polymeric delivery vehicles, such as gels,23 microparticles,24 and nanoparticles, that release the drug specifically in the colonic environment, for example, through the action of bacterial enzymes. However, nanoscale carriers are the only method that addresses the need to reach the intracellular site of action. This method was selected in the present study, building upon our experience with nanoparticle-mediated drug delivery25 and knowledge from available literature data on mesalazine.
Previous studies have associated mesalazine with various nanoparticles using several methods, for example, by coprecipitation with polymers (eg, with polyurethane-chitosan graft copolymer),26 by encapsulation inside polymeric nanoparticles (eg, composed of poly-lactic-co-glycolic acid),27 by surface adhesion (eg, to silicon dioxide nanoparticles),28 or by direct covalent conjugation (eg, to poly-caprolactone),29 which we also chose to apply for our study. The mode by which mesalazine is associated with the nanoparticle is crucial for determining the location of delivery and the kinetics of its release from the nanoparticle. However, in the case of intracellular delivery, the size and surface properties of the nanoparticle itself are of equal importance, as they determine the fate of the nanoparticle upon encountering the cell (the mechanism of internalization, eg, by endocytosis) and inside the cell (including drug release).20
Dendrimers are unique single-molecule, highly uniform nanoparticles that are attractive for covalent drug conjugation because of their homogenous and controllable surface chemistry.20 They have previously been tested in several studies as carriers for mesalazine, but the originality of our approach relies on a choice of coupling chemistry combined with careful investigation of the biophysical properties and the in vitro physiological action using several cell-based assays. Wiwattanapatapee et al conjugated mesalazine to a PAMAM dendrimeric scaffold using a bacterial enzyme-hydrolyzable azo bond to facilitate release. This linker is known to undergo intestine-specific cleavage, so, as expected, this method did not enhance the cellular uptake of mesalazine.30 A lysine-based dendrimer with a silsesquioxane core and a hydrolyzable Schiff base linker was also used as a carrier for mesalazine. However, this construct was evaluated in mice models only for the treatment of retinal degeneration, without any intracellular delivery experiments or biological studies of its anti-inflammatory action.31 Other studies aimed only to chemically characterize drug–dendrimer conjugates or noncovalent complexes32,33 using mesalazine as a model molecule and did not study biological aspects. Our design of a mesalazine–PAMAM conjugate is specifically aimed at increasing the intracellular concentration of mesalazine in the intestinal epithelium independently of membrane transporter activity, and our experiments were geared toward this goal. We sequentially analyzed mechanisms of release, intracellular delivery, and biological function. It is important to note that upon potential clinical application of the conjugate, it would be delivered directly to the intestinal lumen (either by rectal administration or by oral capsules designed to dissolve in the large intestine); in this study, measured release kinetics (on the order of several hours) and rate of cellular uptake (virtually saturated after one hour of exposure) were commensurate with efficient active drug targeting for intracellular site of action. Thus, our work is a pioneering effort regarding facilitating the delivery of this drug to its molecular targets.
The structural design of the conjugate in this study was adapted to the above goals; our experimental workflow: selection of PAMAM generation and surface modification was aimed at efficient internalization, as demonstrated in previous studies,34 while the chemical coupling method was chosen to ensure the intracellular release of unchanged mesalazine, as achieved in studies with other drugs.35 We obtained satisfactory results at each stage: biophysical characterization showed that kinetics and conditions of release are commensurate with intracellular delivery; we subsequently proved that the kinetics of cellular delivery are those expected for endocytosis-mediated internalization.36 The mechanism of cellular uptake of PAMAM dendrimers and PAMAM-drug conjugates that were analogous to the mesalazine conjugate used in this study have been extensively researched and documented to consist mainly of clathrin-mediated endocytosis,37–40 including in the same cell lines as used in the present study,41,42 yielding further support to our conclusion that this is indeed the drug delivery route we are observing. Moreover, the main goals of drug delivery from the physiological and clinical perspective were also achieved: intracellularly released mesalazine was active in anti-inflammatory assays that measured immune-related cellular activity (gene expression, signaling, mediator synthesis and release, redox homeostasis) to ensure a reliable and holistic view of mesalazine function based on extensive literature.7 More crucially, delivery of mesalazine as a nanoparticle conjugate circumvented the effect of inhibited transporter function; this finding is proof that it is possible to overcome natural resistance to free drug that is mediated by repression of transporter expression.21 Our results are comparable with other successful efforts that delivered anti-inflammatory compounds, namely tocopherol43 and dexamethasone,44 into cells by conjugating them to PAMAM dendrimers via succinate linkers; however, these studies tested the biological activity of the delivered compound only by individual methods at a superficial phenomenological level. Moreover, since the problem of escaping membrane transporter-mediated delivery is unique to mesalazine, our proof of concept showing that transporter-mediated resistance can be circumvented is unique and ground-breaking.
Conclusions
The results of our in vitro study are highly promising as we took care to document drug delivery at the level of accessibility of the molecular targets (release and intracellular concentration of active drug) and pharmacodynamics (inhibition of proinflammatory events by the released drug in physiologically relevant cell models of intestinal epithelium). The results now need to be tested in an extensive in vivo proof of concept study, for which relevant animal models of IBDs will need to be carefully selected if the integral advantages of our nanoparticle-based approach are to be salient. Such a study will be inherently much more complicated due to the additional confounding effects of whole-organism pharmacokinetics, which have previously derailed similar studies of promising nanoparticles.45 Our group intends to pursue an in vivo proof of concept study in the future; meanwhile, the results from the current in vitro research significantly add to our knowledge of the possibilities and perspectives of nanoparticle drug delivery.
Acknowledgments
Graphical abstract was prepared in part using image vectors from Servier Medical Art (www.servier.com) licensed under the Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Author Contributions
Conceptualization, MG, MM; synthesis and characterization of conjugate, MG, MM; methodology, MG, MM, MS, IK, LP; formal analysis, MG, MM, MS, IK; investigation, MG, MM, MS, IK; data curation, MG, MM, MS, IK; writing—original draft preparation, MG, LP; writing—review and editing, MG, MM, MS, IK, LP, BKM; visualization, MG, MM, LP; supervision, BKM; funding acquisition, MG. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Science Centre, Poland (Project 2018/31/N/NZ7/00374 “PAMAM dendrimer as carrier for mesalazine to enhance the efficacy of inflammatory bowel disease therapy - in vitro studies”).
Disclosure
Dr Michal Gorzkiewicz reports grants from National Science Centre Poland, during the conduct of the study; In addition, Dr Michal Gorzkiewicz has a patent P.432894 pending to University of Lodz. Dr Monika Marcinkowska reports grants from National Science Centre, during the conduct of the study; In addition, Dr Monika Marcinkowska has a patent P.432894 pending to University of Lodz. Prof. Dr. Barbara Klajnert-Maculewicz reports grants from National Science Centre, during the conduct of the study; In addition, Prof. Dr. Barbara Klajnert-Maculewicz has a patent P.432894 pending to University of Lodz. The authors report no other conflicts of interest in this work.
References
1. Cohen RD. Review article: evolutionary advances in the delivery of aminosalicylates for the treatment of ulcerative colitis. Aliment Pharmacol Ther. 2006;24(3):465–474. doi:10.1111/j.1365-2036.2006.03010.x
2. Loftus EV. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504–1517. doi:10.1053/j.gastro.2004.01.063
3. Kornbluth A, Sachar DB. Ulcerative colitis practice guidelines in adults: American College Of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2010;105(3):501–523. doi:10.1038/ajg.2009.727
4. Ham M, Moss AC. Mesalamine in the treatment and maintenance of remission of ulcerative colitis. Expert Rev Clin Pharmacol. 2012;5(2):113–123. doi:10.1586/ecp.12.2
5. Caprilli R, Cesarini M, Angelucci E, Frieri G. The long journey of salicylates in ulcerative colitis: the past and the future. J Crohns Colitis. 2009;3(3):149–156. doi:10.1016/j.crohns.2009.05.001
6. MacDermott RP. Progress in understanding the mechanisms of action of 5-aminosalicylic acid. Am J Gastroenterol. 2000;95(12):3343–3345. doi:10.1111/j.1572-0241.2000.03342.x
7. Williams C, Panaccione R, Ghosh S, Rioux K. Optimizing clinical use of mesalazine (5-aminosalicylic acid) in inflammatory bowel disease. Therap Adv Gastroenterol. 2011;4(4):23748. doi:10.1177/1756283X11405250
8. König J, Glaeser H, Keiser M, Mandery K, Klotz U, Fromm MF. Role of organic anion-transporting polypeptides for cellular mesalazine (5-aminosalicylic acid) uptake. Drug Metab Dispos. 2011;39(6):1097–1102. doi:10.1124/dmd.110.034991
9. Brakmane G, Winslet M, Seifalian AM. Systematic review: the applications of nanotechnology in gastroenterology. Aliment Pharmacol Ther. 2012;36(3):213–221. doi:10.1111/j.1365-2036.2012.05179.x
10. Mintzer MA, Grinstaff MW. Biomedical applications of dendrimers: a tutorial. Chem Soc Rev. 2011;40(1):173–190. doi:10.1039/B901839P
11. Menjoge AR, Kannan RM, Tomalia DA. Dendrimer-based drug and imaging conjugates: design considerations for nanomedical applications. Drug Discov Today. 2010;15(5–6):171–185. doi:10.1016/j.drudis.2010.01.009
12. O’brien I, Wilson T, Orton F, Pognan F. Investigation of the alamar blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267(17):5421–5426. doi:10.1046/j.1432-1327.2000.01606.x
13. Hellemans J, Vandesompele J. Selection of reliable reference genes for RT-qPCR analysis. Methods Mol Biol. 2014;1160:19–26.
14. Chantong B, Kratschmar DV, Nashev LG, Balazs Z, Odermatt A. Mineralocorticoid and glucocorticoid receptors differentially regulate NF-kappab activity and pro-inflammatory cytokine production in murine BV-2 microglial cells. J Neuroinflammation. 2012;9(1):260. doi:10.1186/1742-2094-9-260
15. Kumari A, Singla R, Guliani A, Yadav SK. Nanoencapsulation for drug delivery. EXCLI J. 2014;13:265–286.
16. Spirescu VA, Chircov C, Grumezescu AM, Andronescu E. Polymeric nanoparticles for antimicrobial therapies: an up-to-date overview. Polymers. 2012;13(5):724. doi:10.3390/polym13050724
17. Chircov C, Spoială A, Păun C, et al. Mesoporous silica platforms with potential applications in release and adsorption of active agents. Molecules. 2020;25(17):3814. doi:10.3390/molecules25173814
18. Shrivastava P, Gautam L, Jain A, Vishwakarma N, Vyas S, Vyas SP. Lipid drug conjugates for improved therapeutic benefits. Curr Pharm Des. 2020;26(27):3187–3202. doi:10.2174/1381612826666200311124003
19. Natfji AA, Osborn HM, Greco F. Feasibility of polymer-drug conjugates for non-cancer applications. Curr Opin Colloid Interface Sci. 2017;31:51–66. doi:10.1016/j.cocis.2017.07.004
20. Dubey SK, Salunkhe S, Agrawal M, et al. Understanding the pharmaceutical aspects of dendrimers for the delivery of anticancer drugs. Curr Drug Targets. 2020;21(6):528–540. doi:10.2174/1389450120666191031092259
21. Liang E, Proudfoot J, Yazdanian M. Mechanisms of transport and structure-permeability relationship of sulfasalazine and its analogs in caco-2 cell monolayers. Pharm Res. 2000;17(10):1168–1174. doi:10.1023/A:1026450326712
22. Ye B, van Langenberg DR. Mesalazine preparations for the treatment of ulcerative colitis: are all created equal? World J Gastrointest Pharmacol Ther. 2015;6(4):137–144. doi:10.4292/wjgpt.v6.i4.137
23. Dhaneshwar SS. Colon-specific prodrugs of 4-aminosalicylic acid for inflammatory bowel disease. World J Gastroenterol. 2014;20(13):3564–3571. doi:10.3748/wjg.v20.i13.3564
24. Varshosaz J, Jaffarian Dehkordi A, Golafshan S. Colon-specific delivery of mesalazine chitosan microspheres. J Microencapsulation. 2006;23(3):329–339. doi:10.1080/02652040600612405
25. Gorzkiewicz M, Deriu MA, Studzian M, et al. Fludarabine-specific molecular interactions with maltose-modified poly (propyleneimine) dendrimer enable effective cell entry of the active drug form: comparison with clofarabine. Biomacromolecules. 2019;20(3):1429–1442. doi:10.1021/acs.biomac.9b00010
26. Mirabbasi F, Dorkoosh FA, Moghimi A, Shahsavari S, Babanejad N, Seifirad S. Preparation of mesalamine nanoparticles using a novel polyurethane-chitosan graft copolymer. Pharm Nanotechnol. 2017;5(3):230–239. doi:10.2174/2211738505666171103120026
27. Hallan SS, Hallan SS, Hallan SS, Mishra N, Mishra N, Mishra N. Microparticles as controlled drug delivery carrier for the treatment of ulcerative colitis: a brief review. Saudi Pharm J. 2016;24(4):458–472. doi:10.1016/j.jsps.2014.10.001
28. Tang H, Xiang D, Wang F, Mao J, Tan X, Wang Y. 5-ASA-loaded sio2 nanoparticles-a novel drug delivery system targeting therapy on ulcerative colitis in mice. Mol Med Rep. 2017;15(3):1117–1122. doi:10.3892/mmr.2017.6153
29. Pertuit D, Moulari B, Betz T, et al. 5-amino salicylic acid bound nanoparticles for the therapy of inflammatory bowel disease. J Control Release. 2007;123(3):211–218. doi:10.1016/j.jconrel.2007.08.008
30. Wiwattanapatapee R, Lomlim L, Saramunee K. Dendrimers conjugates for colonic delivery of 5-aminosalicylic acid. J Control Release. 2003;88(1):1–9. doi:10.1016/S0168-3659(02)00461-3
31. Wu X, Yu G, Luo C, et al. Synthesis and evaluation of a nanoglobular dendrimer 5-aminosalicylic acid conjugate with a hydrolyzable Schiff base spacer for treating retinal degeneration. ACS Nano. 2014;8(1):153–161. doi:10.1021/nn4054107
32. Namazi H, Adeli M. Dendrimers of citric acid and poly (ethylene glycol) as the new drug-delivery agents. Biomaterials. 2005;26(10):1175–1183. doi:10.1016/j.biomaterials.2004.04.014
33. Saboktakin MR, Tabatabaie RM, Maharramov A, Ramazanov MA. An aromatic macromolecule as a nano drug carrier: polyether dendrimer/poly (2-hydroxyethyl methacrylate). Polym Polym Compos. 2010;18(8):429–434.
34. Gorzkiewicz M, Janaszewska A, Ficker M, Svenningsen SW, Christensen JB, Klajnert-Maculewicz B. Pyrrolidone-modified PAMAM dendrimers enhance anti-inflammatory potential of indomethacin in vitro. Colloids Surf B. 2019;181:959–962. doi:10.1016/j.colsurfb.2019.06.056
35. Zolotarskaya OY, Xu L, Valerie K, Yang H. Click synthesis of a polyamidoamine dendrimer-based camptothecin prodrug. RSC Adv. 2015;5(72):58600–58608. doi:10.1039/C5RA07987J
36. Studzian M, Szulc A, Janaszewska A, Appelhans D, Pułaski Ł, Klajnert-Maculewicz B. Mechanisms of internalization of maltose-modified poly (propyleneimine) glycodendrimers into leukemic cell lines. Biomacromolecules. 2017;18(5):1509–1520. doi:10.1021/acs.biomac.7b00046
37. Oddone N, Zambrana AI, Tassano M, Porcal W, Cabral P, Benech JC. Cell uptake mechanisms of PAMAM G4-FITC dendrimer in human myometrial cells. J Nanopart Res. 2013;15(7):1–14. doi:10.1007/s11051-013-1776-1
38. Mohammadpour R, Safarian S, Buckway B, Ghandehari H. Comparative endocytosis mechanisms and anticancer effect of HPMA copolymer‐and PAMAM dendrimer‐MTCP conjugates for photodynamic therapy. Macromol Biosci. 2017;17(4):1600333. doi:10.1002/mabi.201600333
39. Zhu S, Hong M, Zhang L, Tang G, Jiang Y, Pei Y. Pegylated PAMAM dendrimer-doxorubicin conjugates: in vitro evaluation and in vivo tumor accumulation. Pharm Res. 2010;27(1):161–174. doi:10.1007/s11095-009-9992-1
40. Bastien E, Schneider R, Hackbarth S, et al. PAMAM G4.5-chlorin E6 dendrimeric nanoparticles for enhanced photodynamic effects. Photochem Photobiol Sci. 2015;14(12):2203–2212. doi:10.1039/c5pp00274e
41. Kitchens KM, Kolhatkar RB, Swaan PW, Ghandehari H. Endocytosis inhibitors prevent poly (amidoamine) dendrimer internalization and permeability across Caco-2 cells. Mol Pharm. 2008;5(2):364–369. doi:10.1021/mp700089s
42. Goldberg DS, Ghandehari H, Swaan PW. Cellular entry of G3.5 poly (amido amine) dendrimers by clathrin-and dynamin-dependent endocytosis promotes tight junctional opening in intestinal epithelia. Pharm Res. 2010;27(8):1547–1557. doi:10.1007/s11095-010-0153-3
43. Wang Y, Shen W, Shi X, et al. Alpha‐tocopheryl succinate‐conjugated G5 PAMAM dendrimer enables effective inhibition of ulcerative colitis. Adv Healthc Mater. 2017;6(14):1700276. doi:10.1002/adhm.201700276
44. Choksi A, Sarojini KVL, Vadnal P, Dias C, Suresh PK, Khandare J. Comparative anti-inflammatory activity of poly (Amidoamine)(PAMAM) dendrimer–dexamethasone conjugates with dexamethasone-liposomes. Int J Pharm. 2013;449(1–2):28–36. doi:10.1016/j.ijpharm.2013.03.056
45. Zhang M, Du Y, Wang S, Chen B. A review of biomimetic nanoparticle drug delivery systems based on cell membranes. Drug Des Dev Ther. 2020;14:5495–5503. doi:10.2147/DDDT.S282368
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Classification of Nanomaterial Drug Delivery Systems for Inflammatory Bowel Disease
Wang H, Zhou F, Shen M, Ma R, Yu Q
International Journal of Nanomedicine 2025, 20:1383-1399
Published Date: 3 February 2025