Back to Journals » Breast Cancer: Targets and Therapy » Volume 17
Mendelian Randomization Integrating GWAS and eQTL Data Reveals DAAM1, a Potential Immune-Related Biomarker for Breast Cancer Prognosis
Authors Chen G, Zhang K, Wang Y, Zhang Z, Cao J, Gao G, Yu C, Dai Y, Qiao G, Cong Y ![]()
Received 21 September 2025
Accepted for publication 4 December 2025
Published 17 December 2025 Volume 2025:17 Pages 1247—1263
DOI https://doi.org/10.2147/BCTT.S569177
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Gang Chen,1,* Kun Zhang,1,* Yidan Wang,1,* Zhe Zhang,2,* Jianqiao Cao,1 Ge Gao,1 Chao Yu,1 Yuanping Dai,3 Guangdong Qiao,1 Yizi Cong1
1Department of Breast Surgery, The Affiliated Yantai Yuhuangding Hospital of Qingdao University, Yantai, People’s Republic of China; 2Department of Intensive Care Unit (Eastern), The Affiliated Yantai Yuhuangding Hospital of Qingdao University, Yantai, Shandong, People’s Republic of China; 3Department of Medical Genetics, Liuzhou Maternal and Child Health Hospital, Liuzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yizi Cong, Department of Breast Surgery, The Affiliated Yantai Yuhuangding Hospital of Qingdao University, Yantai, People’s Republic of China, Email [email protected] Guangdong Qiao, Department of Breast Surgery, The Affiliated Yantai Yuhuangding Hospital of Qingdao University, Yantai, People’s Republic of China, Email [email protected]
Background: The immune response plays a critical role in determining the prognosis of breast cancer (BC) patients. However, the underlying molecular mechanisms linking immune regulation to BC progression remain uncleared. This study aims to identify and functionally validate key immune-related genes that mechanistically impact on BC prognosis.
Methods: We used the Mendelian randomization (MR) integrating genome-wide association studies (GWAS) and expression quantitative trait loci (eQTL) data to prioritize candidate genes with a potential causal role in BC-related immune traits. This study was designed to establish a robust genetic rationale for candidate selection, minimizing false positives. Subsequent analyses focused on DAAM1, genes highlighted by the MR analysis, to explore their clinical relevance and biological functions. We validated their expression and association with immune infiltration levels using LASSO regression and patient tissue samples. Functional roles of DAAM1 were further investigated through in vitro assays based on cell proliferation, adhesion, invasion, and migration. The underlying mechanism was illustrated via Western blotting.
Results: Our integrated MR analysis identified DAAM1 as a top candidate with a genetically supported link to BC immune traits. DAAM1 expression was significantly elevated in BC tissues and inversely correlated with immune infiltration levels, suggesting a role in modulating the tumor immune microenvironment. Functional experiments demonstrated that DAAM1 knockdown effectively suppressed BC cell proliferation, adhesion, invasion, and migration. Mechanistically, Western blot analysis revealed that DAAM1 promotes these malignant phenotypes potentially through activating the epithelial-mesenchymal transition (EMT) pathway.
Conclusion: This study identified DAAM1 as a key immune-related prognostic biomarker in breast cancer, whose upregulation contributes to tumor progression and metastasis via EMT pathway. Our findings, elaborated in a causal inference framework, provide a mechanistic basis for DAAM1’s role in BC and underscore its potential as a therapeutic target.
Keywords: breast cancer, mendelian randomization, immune, prognostic, DAAM1
Introduction
Breast cancer (BC) poses a severe and multifaceted public health threat globally. It stands as the most frequently diagnosed cancer and the primary cause of cancer-related death in women worldwide.1 The burden is notably substantial in China, which harbors one of the world’s largest populations of BC patients.2 This trend is mirrored in other regions; for instance, in the United States, BC is the second leading cause of cancer mortality among women, with approximately 310,712 new cases estimated for 2024.3
The disease’s clinical heterogeneity is reflected in its major molecular subtypes—Luminal A, Luminal B, HER2-positive, and Triple-Negative Breast Cancer (TNBC).4 Notably, patients with similar tumor characteristics often exhibit divergent prognoses, a variation increasingly attributed to differences in individual immune responses and tumor immune microenvironment, which may be influenced by factors such as nodal (SLN, LN) or metastatic status.5 This underscores the critical need to identify robust immune-related biomarkers that can accurately predict patient outcomes.
Genome-wide association studies (GWAS) have successfully mapped numerous genetic variants associated with BC risk.6 However, a significant translational gap persists. The majority of these risk variants reside in non-coding genomic regions, implying they exert their effects by modulating gene expression, yet their specific functional roles in BC pathogenesis remain largely elusive. While previous integrative studies have combined GWAS with expression quantitative trait loci (eQTL) data from bulk breast tissue to nominate candidate risk genes,7,8 these efforts have often fallen short of establishing a causal link between gene expression and disease progression. Crucially, BC mortality is primarily driven by metastasis and therapy resistance, processes where the immune system and underlying mechanistic pathways, such as epithelial-mesenchymal transition (EMT), play pivotal roles. A focused investigation into how genetically influenced expression of specific genes drives these aggressive phenotypes is therefore warranted.
Mendelian randomization (MR) has emerged as a powerful statistical method to infer causality in observational data by using genetic variants (eg, eQTLs) as instrumental variables for a modifiable exposure (eg, gene expression).9,10 This approach can help bridge the gap between GWAS-identified risk loci and their functional consequences. To date, however, no MR study has systematically leveraged BC-specific eQTL data to dissect the causal role of gene expression in shaping the BC immune landscape and tumor progression.
To address this gap, we hypothesized that a MR analysis integrating GWAS with BC-relevant eQTL data would reveal novel, causally implicated immune-related genes that mechanistically influence BC prognosis by regulating key oncogenic processes. Here, we present an analysis designed to test this hypothesis. We specifically aimed to identify genes with a causal role in BC immune traits and to functionally validate their impact on tumor cell behavior. Our integrated approach identified DAAM1 as a top candidate. Subsequent functional experiments confirmed that DAAM1 significantly promotes BC cell proliferation, adhesion, invasion, and migration, potentially through the activation of the EMT pathway. This study thus not only identifies DAAM1 as a potential prognostic biomarker and therapeutic target but also provides a causal and mechanistic framework for its role in BC progression. The structure of the study is shown in Supplement Figure S1.
Materials and Methods
Mendelian Randomization Analysis and Instrumental Variable Selection
To infer potential causal relationships between gene expression and BC risk, we performed a two-sample MR analysis. Genetic instruments for gene expression (exposure) were obtained from a breast tissue-specific expression quantitative trait locus (eQTL) dataset. Single nucleotide polymorphisms (SNPs) significantly associated with gene expression (P < 5×10−8) were selected as instrumental variables (IVs). To ensure independence of the IVs, we clumped SNPs based on linkage disequilibrium (LD) using a stringent threshold of r2 < 0.001 and a window size of 10,000 kb. Summary-level data for BC (outcome) were sourced from the Breast Cancer Association Consortium (BCAC) GWAS (ID: ieu-a-1130). The key MR assumptions were addressed as follows: the genetic variants were strongly associated with the exposure (relevance assumption); we verified that the selected SNPs were not directly associated with known major confounders of the exposure-outcome relationship (exchangeability assumption); and the instruments were assumed to influence BC risk only through their effect on gene expression (exclusion restriction assumption). The primary MR analysis was conducted using the inverse variance weighted (IVW) method for genes with multiple IVs. For genes with only one suitable SNP, the Wald ratio method was applied. Sensitivity analyses, including MR-Egger regression and the weighted median estimator, were performed to assess the potential impact of pleiotropy.
Analysis of Gene Expression Data and Identification of Differentially Expressed Genes (DEGs)
The gene expression profile of GSE45827 based on the GPL570 platform was downloaded from the GEO database (www.ncbi.nlm.nih.gov/geo/). It consisted of 41 basal and 30 Her2 positive, 29 luminal A 30 luminal B subtypes, and 11 normal tissues. The different genes were analyzed using GEO2R online tools. Differentially expressed genes with fold change (FC) ≥ 2.0, FC≤1/2.0 and adjusted p-values of < 0.05 were considered the cutoff criteria. Overlapping different expressed genes (DEGs) and Venn diagrams were obtained using online tools (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Gene Ontology (GO) and Anotation Kyoto Encyclopedia of Genes and Genomes (KEGG)
The overlap DEGs were analyzed using R software the packages of “clusterProfiler,” “org.Hs.eg.db”, enrichplot, and visualized by the package of “ggplot2” of R software.
Correlations Between DEGs and Immune
The correlation between overlapping DEGs and the abundance of tumor-infiltrating immune cells was assessed using the “linkET” package in R. The results were visualized with the “ggplot2” package.
Univariate and Multivariate Analysis
To identify immune-related prognostic factors, we applied the least absolute shrinkage and selection operator (LASSO) regression analysis. Patients were stratified into high- and low-risk groups based on the median risk score calculated using the “survminer” package in R. Subsequently, univariate and multivariate Cox regression analyses were performed, leading to the identification of five key immune-related genes. The predictive performance of this gene signature was evaluated using time-dependent receiver operating characteristic (ROC) curve analysis.
Construction and Verification of Nomogram
A nomogram was created using the R software “survival” and “rms” packages to predict the survival of BC patients based on factors such as diagnosis age, metastasis status, TNM stage, risk score, and stage. The accuracy of the nomogram in differentiating patients was assessed using a calibration curve.
Survival Analysis
The two DEGs were divided into two categories according to their median expression levels, and KM-plotter analysis was used to identify immune factors related to prognosis using the online tools, KM-plotter (https://kmplot.com/analysis/) and GSE45827 follow-up data. Statistical significance was set at or less than 0.05.
Validation of Immune Factors Using Immune Cluster Groups
ESTIMATE is a short name for the estimation of stromal and immune cells in malignant tumors using expression data; it is an effective method for inferring the fraction of stromal and immune cells in tumor samples.11 ESTIMATE arithmetic was used to take into account stromal, immune, and ESTIMATE scores and the tumor purity of the GSE45827 BC samples using R software, as previous reported.12 For assessing the diverse subtypes of immune infiltration cells, the “CIBERSORT” package of R software was used to analyze the 29 immune related cells in each BC patient according to the expression data.13 Finally, the different levels of gene expression were verified based on high and low immune infiltration groups via the “ggpubr” package in R software.
BC Patients
In our study, we selected five breast cancer patients who had been treated at the Department of Breast Surgery of the Affiliated Yantai Yuhuangding Hospital of Qingdao University. All patients signed informed consent forms approved by the Institutional Review Board of Yantai Yuhuangding Hospital (certificate number: 2024-139).
RNA Isolation and Q-PCR
RNA was extracted from BC patient tissue samples using TRIzol reagent, following the manufacturer’s instructions (Shandong Sparkjade Biotechnology Co., Ltd.) So, 1μg of RNA from each sample was converted to cDNA using the SPARK script II RT Plus Kit (Shandong Sparkjade Biotechnology Co., Ltd.) Subsequently, Q-PCR was performed using the SYBR Green qPCR Mix kit (with ROX; Shandong Sparkjade Biotechnology Co., Ltd.), according to the manufacturer’s protocol. Finally, the mRNA expression levels were determined using the 2−ΔΔCT formula.
Cell Culture
MDA-MB-231 and MCF7 cells were obtained from the Cell Resource Center at Peking Union Medical College and grown in Dulbecco’s modified Eagle’s medium (DMEM) supplied by Shandong Sparkjade Biotechnology Co., Ltd. The medium was supplemented with 10% fetal bovine serum (FBS) from PAN Seratech and 1% antibiotics from the same supplier. The cells were incubated at 37 °C in a 5% CO2 atmosphere at 95% humidity under standard conditions.
Cell Transfected
siRNAs specifically targeting DAAM1 (sense: 5′-GCCCUGAAAUCCUUCAACUTT-3′, antisense: 5′-AGUUGAAGGAUUUCAGGGCTT-3′) and negative control (NC, sense: 5′-UUCUCCGAACGUGUCACGUTT-3′, antisense: 5′-ACGUGACACGUUCGGAGAATT-3′) were synthesized by GenePharma. The transfection experiment was conducted using siRNA mate-plus, according to the manufacturer’s instructions.
Function Assay
In vitro Cell Proliferation Assay
The cells were detached and counted. Three thousand cells per 200 μL were seeded in 96-well plates. Following incubation, the culture medium was removed and 100 µL of 10% CCK-8 (Shandong Sparkjade Biotechnology Co., Ltd.) was added. After one hour’s incubation, the absorbance was measured at 450 nm.
In vitro Matrigel Adhesion Assay
To a 96-well plate, 100 µL of 1% (w/v) Matrigel was added to coat the artificial basement membrane overnight. Drying followed. Before seeding, 20,000 cells per 200 µL of culture medium were seeded into each well. The dried membrane was rehydrated in 100 µL of sterile distilled water. After incubation for 60 min, the medium was removed and cells washed with 200 µL of PBS, fixed in 4% formaldehyde for 30 min, and stained with 0.5% crystal for 30 min. Finally, the adherent cells were observed under a microscope after washing with distilled water and drying.
In vitro Trans Well Migration Assay
In vitro migration assay: A total of 20,000 cells were placed in Transwell chambers (8 μm pore size) containing 200 μL of medium. After a 48-hour incubation period, the cells were fixed with 4% formaldehyde, and those that had migrated through the pores were stained with crystal violet for microscopic examination.
Matrigel Invasion Assay
In the in vitro invasion assay, Matrigel-coated transwells (50 μg per well) were allowed to dry before use. To each filter, 20,000 cells were added. After 48 hours’ culture, the invading cells were fixed with 4% formaldehyde, stained with crystal violet, and visualized under a microscope.
Western Blot
Cell lysis was performed using RIPA buffer to ensure that equal protein concentrations were used for subsequent Western blot analysis. Protein samples were separated by SDS-PAGE (10% polyacrylamide gel) and transferred to nitrocellulose membranes. The membranes were then treated with a blocking solution containing 5% non-fat dry milk in TBS- T (Tris-buffered saline containing Tween-20) at room temperature for 60 min to prevent nonspecific binding. The membranes were then incubated with primary antibodies diluted in TBS-T: anti-E-cadherin (Santa Cruz Biotechnology), anti-GAPDH (Sangon Biotech Co., Ltd.), anti-N-cadherin (Santa Cruz Biotechnology), and anti-slug (Santa Cruz Biotechnology). After extensive washing with TBS-T, secondary antibodies conjugated to horseradish peroxidase (HRP) were used for detection. Protein bands were subsequently imaged using an imaging system equipped with ECL detection reagents.
Results
Overview of the Approach
Three gene expression profiles and GSE45827 gene chips were used in this study. They consisted of 11 normal tissues, 41 basal, 30 Her2 positive, 29 luminal A 30 luminal B BC tissues. Gene expression was analysed using GEO2R online tools, between normal and tumor samples and each subtype. A FC ≥ 2 or FC≤1/2 and adjusted P ≤ 0.05 were consider significant criteria. The volcano map for each subtype of up- and downregulated differently expressed genes (DEGs) are shown in Supplement Figure S2A–D. In total, 2820 up-regulated and 1005 down-regulated DEGs were selected from tumor and normal tissues using the vein diagram (Figure 1A and B). The DEGs heatmap was shown in Figure 1C.
 |
Figure 1 Identification of differentially expressed genes (DEGs) across breast cancer molecular subtypes. (A and B) Venn diagrams illustrating the overlap of significantly (A) up-regulated and (B) down-regulated DEGs identified across the four subtype-specific comparisons. (C) Hierarchical clustering heatmap of the consensus DEGs from (A and B) across all samples, demonstrating distinct gene expression patterns between normal tissues and BC subtypes. Rows represent genes, and columns represent individual samples grouped by tissue type (color bar). |
As previously reported,14–16 the eQTL P threshold was set to 5×10−8. If the linkage disequilibrium value was r2 < 0.001, related SNPs were defined. The outcome and exposure results were combined to maintain the same allele impact on exposure and outcome data. Data-weighted median and inverse variance weighting were used. Subsequently, the crossed exposure genes and DEGs were merged using an online Venn diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/), and 23 upregulated and 8 downregulated genes were selected (Figure 2).
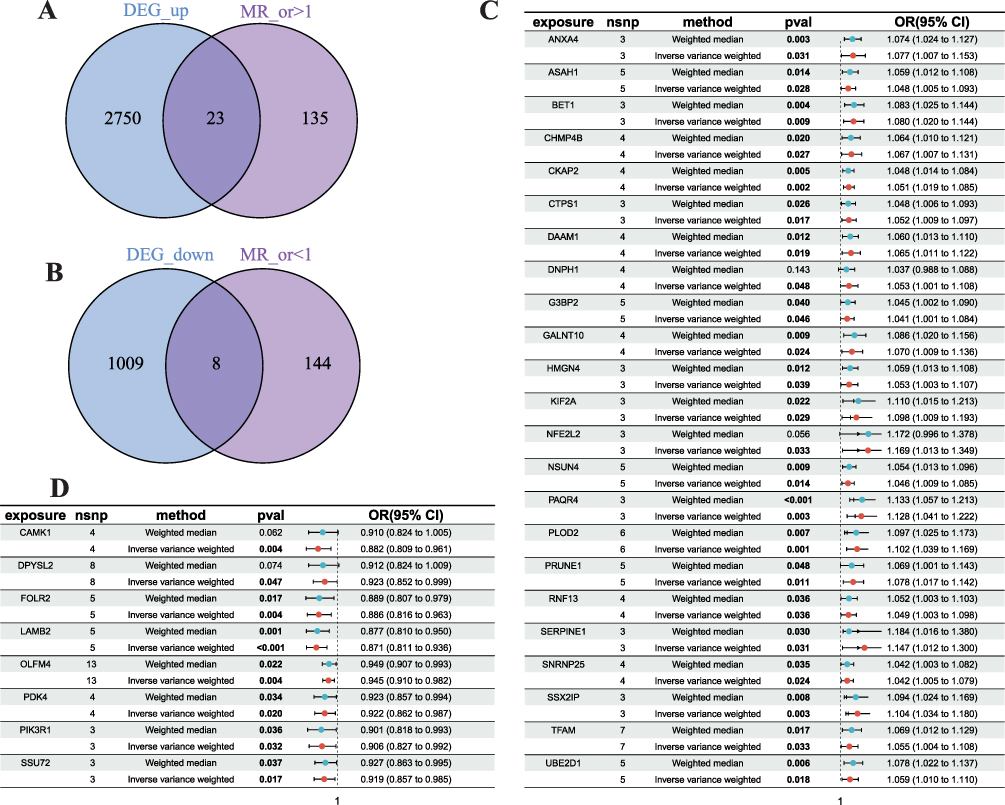 |
Figure 2 Integration of Mendelian Randomization (MR) and Transcriptomic Analysis. (A) Venn diagram identifying the overlap between upregulated differentially expressed genes (DEGs) and genes with a putative causal effect on BC risk (MR odds ratio > 1). (B) Venn diagram identifying the overlap between downregulated DEGs and potential protective genes (MR odds ratio < 1). (C and D) Forest plots of the MR analysis for the intersected (C) upregulated and (D) downregulated genes, showing the causal effect estimates (odds ratios) and 95% confidence intervals for each gene. |
GO and KEGG Enrichment Analysis of DEGs
In order to identify the DEGs’ potential mechanisms in BC, GO and KEGG analysis enrichment was performed, using the “clusterProfiler”, “org.Hs.eg.db”, and “ggplot2”. Upregulated DEGs were most enriched in “positive regulation of blood coagulation,” ‘spindle microtubule, “transcription coregulator binding” (Figure 3A). Downregulated DEGs were enriched in “positive regulation of nucleocytoplasmic transport”, “protein complex involved in cell-matrix adhesion”, and “phosphatidylinositol 3-kinase regulatory subunit binding” (Figure 3B). The KEGG pathway was enriched in the apelin signaling pathway and glioma, separately, in up- and downregulated genes (Figure 3C and D).
 |
Figure 3 Functional Enrichment Analysis of Consensus Differentially Expressed Genes (DEGs). (A and B) Gene Ontology (GO) enrichment analysis for the consensus (A) up-regulated and (B) down-regulated DEGs, showing the top significantly enriched biological processes. (C and D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for the consensus (C) up-regulated and (D) down-regulated DEGs, displaying the top significantly enriched pathways. |
Immuno Infiltration Analysis
The human leukocyte antigen (HLA) system encodes proteins and antigens to T cell immune response. In particular, the cells and tissues or the organ trace to the source and autoimmune diseases usually depend on the HLA system.17 Moreover, B and T cells usually function as a product of the long-lasting protective protein antibody and development of immune memory after the appearance of infection.18 Macrophages and monocytes are also phagocytic cells that are important for the innate immune response against a variety of infections.18 From these results, we can see that plasma cells were more numerous than normal tissues, but T cell regulatory and macrophage M0 levels were lower than those of normal tissues (Figure 4A and B). DEGs that regulate immune function are shown in Figure 4C.
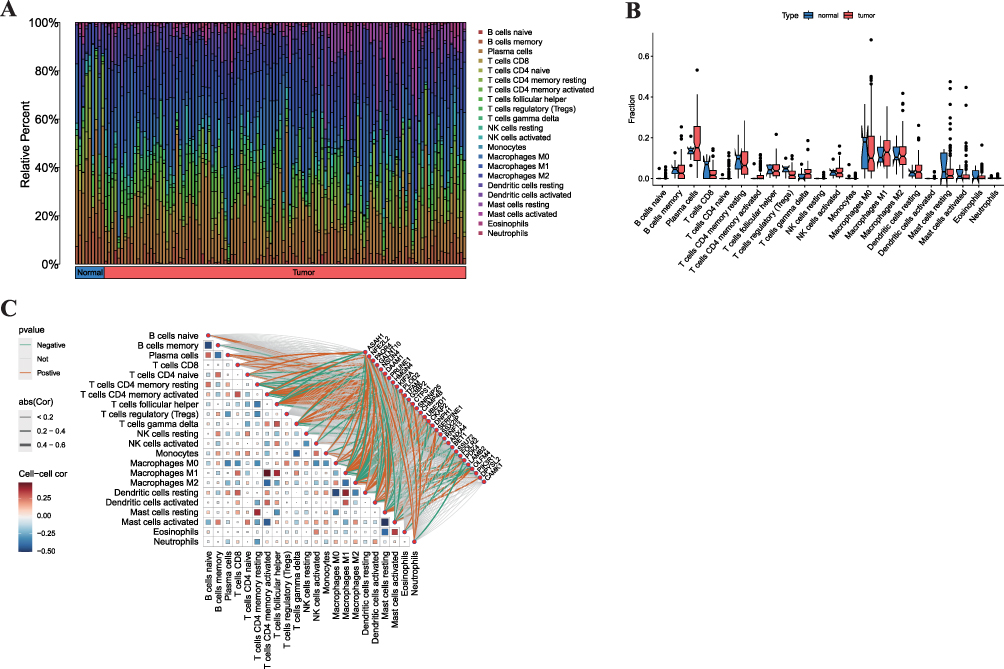 |
Figure 4 Characterization of the Tumor Immune Microenvironment. (A) Bar plot showing the relative abundance of 22 tumor-infiltrating immune cell types in normal breast tissues and breast cancer (BC) tissues, as estimated by CIBERSORT. (B) Box plot comparing the fractions of selected key immune cells between normal and BC tissues (**p < 0.01). (C) Correlation heatmap between the expression levels of the consensus DEGs and the abundance of immune cells. Red indicates positive correlation, blue indicates negative correlation. |
Univariate and Multivariate Analysis
From the LASSO regression analysis, the data were separated into high- and low-risk groups according to the median risk score using the Survminer package in R software. Univariate and multivariate Cox regression analyses were performed to test whether the DEGs could be independent prognostic indicators based on risk groups. The results showed that these five genes could be independent prognostic factors. The univariate and multivariate Cox regression analysis results are shown in Figure 5A and B. Risk factors, survival time, and the survival line are shown in Figure 5C, D and F, respectively. The heatmap also showed that the five selected factors were differentially expressed in the high and low immunoinfiltration groups (Figure 5E).
 |
Figure 5 Construction and Validation of an Immune-Related Prognostic Signature. (A and B) Forest plots from the (A) univariate and (B) multivariate Cox regression analyses, identifying independent prognostic factors among the candidate genes. (C-E) Distribution of patient risk scores (C), overall survival status of patients (D), and expression heatmap of the five-gene signature (E) across the cohort, ordered by increasing risk score. (F) Kaplan-Meier overall survival curves for patients stratified into high-risk and low-risk groups based on the median risk score (Log-rank p-value is shown). (G) ROC curves of five-gene prognostic signature for predicting 3-year, 5-year, and 10-year overall survival in BC patients. |
ROC curves were used to estimate the accuracy of survival time, 3-, 5-, and 10-year OS for BC patients, and the related area under curve (AUC) values were 0.728, 0.720, and 0.728, respectively (Figure 5G). These results indicated that 5 (CAMK1, SSX2IP, SERRPINE1, CTPS1, and DAAM1) may be independent prognostic factors for patients with BC.
Construction and Validation of Nomogram
To predict 3-, 5-, and 10-year overall survival for BC patients, a nomogram was established and features of the five DEGs were taken into account, such as diagnosis age, metastasis status, TNM stage, risk score, and survival rate (Figure 6A). The 3-, 5-, and 10-year calibration curves were compared to those of the nomogram, as shown in Figure 6B–D.
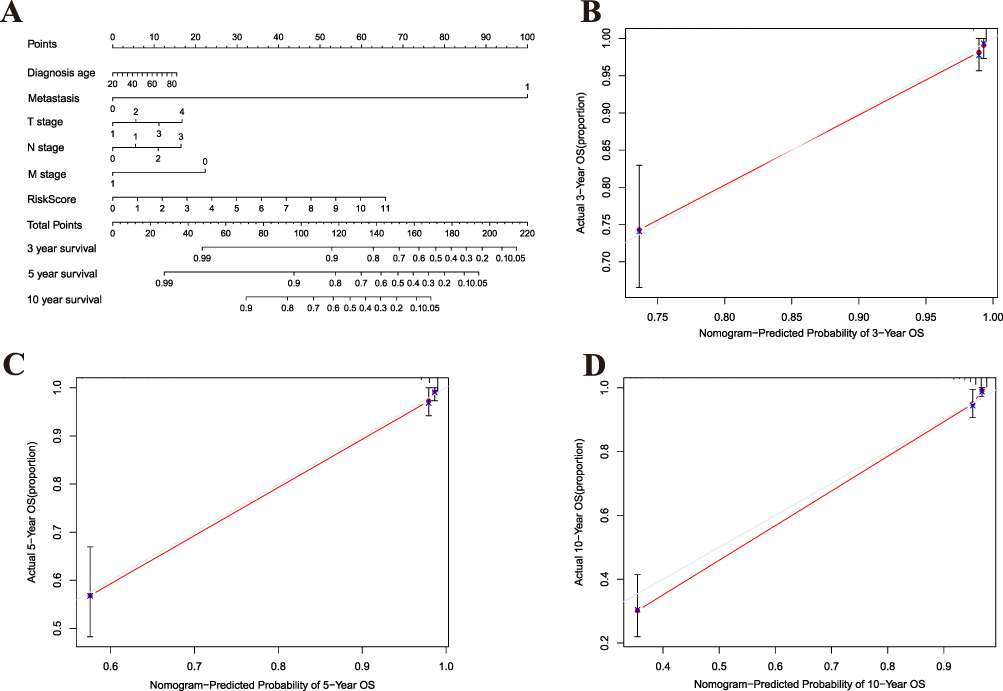 |
Figure 6 Development of a Prognostic Nomogram. (A) A nomogram integrating clinical factors (eg, age, TNM stage) and the molecular risk score to predict the probability of 3-, 5-, and 10-year overall survival for BC patients. (B–D) Calibration curves for the nomogram, comparing the predicted versus observed survival rates at (B) 3 years, (C) 5 years, and (D) 10 years. The dotted line represents perfect prediction. |
Prognostic Analysis
To identify the impact of the above 5 factors on the survival ability of BC patients, KM-plotter online tools (https://kmplot.com/analysis/) (Figure 7A–D) and GSE45827 were used Follow-up information was analysed (Figure 7E and F) and two (SERPINE1, DAAM1) factors were selected based on survival line.
 |
Figure 7 Prognostic Value of DAAM1 and SERPINE1. (A–D) Kaplan-Meier survival analysis from the KM-plotter database, evaluating the association of (A and B) DAAM1 and (C and D) SERPINE1 expression with (A and C) overall survival (OS) and (B and D) relapse-free survival (RFS) in BC patients. (E and F) Validation of the prognostic value of (E) DAAM1 and (F) SERPINE1 using overall survival data from the GSE45827 cohort. |
Immuno Infiltration Verification
Based on the above high and low immune infiltration groups, immune infiltration was verified using ESTIMATE score, immune score, stromal score, and tumor purity (Figure 8A–D). Immune-related prognostic factors were also assessed, but only one (DAAM1) factor was significantly different between the high and low immune infiltration groups (Figure 8E and F). The sample relevance of immune-related cells in the high and low immune infiltration groups and the ESTIMATE score, stromal score, immune score, and tumor purity of each patient are shown in Figure 8G.
 |
Figure 8 Association of Candidate Genes with the Immune Contexture. (A–D) Comparison of the (A) ESTIMATE score, (B) Stromal score, (C) Immune score, and (D) Tumor purity between the high and low immune infiltration groups (***p < 0.001). (E and F) Expression levels of (E) DAAM1 and (F) SERPINE1 in the high versus low immune infiltration groups (**p < 0.01, ns: not significant). (G) Comprehensive heatmap displaying the enrichment levels of 29 immune-related cells, alongside the ESTIMATE-derived scores and tumor purity, for each patient in the high and low immune infiltration groups. |
Expression Validation
The above immune-related prognostic factors were verified using five paired BC patient tissues from Yantai Yuhuangding Hospital. DAAM1 expression was significantly higher in BC tissues than in the adjacent normal tissues (Figure 9A). In contrast, the CPTAC database (https://ualcan.path.uab.edu/cgi-bin/CPTAC-Result.pl?genenam=DAAM1&ctype=Breast) revealed that DAAM1 protein levels were higher in BC tissues than in normal tissues (Figure 9B).
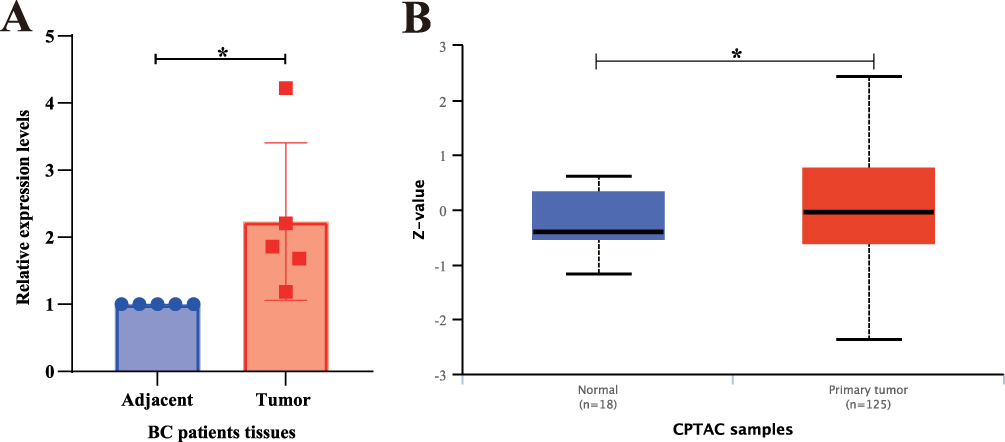 |
Figure 9 Validation of DAAM1 Overexpression in Breast Cancer Tissues. (A) Relative mRNA expression level of DAAM1 in paired breast cancer (BC) and adjacent normal tissues from our patient cohort (*p < 0.05). (B) Validation of DAAM1 protein expression level in BC versus normal breast tissues using data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) database (*p < 0.05). |
DAAM1 Promotes BC Proliferation and Adhesion, Invasion, and Migration
To determine the effect of DAAM1 on BC, DAAM1 was knocked down by transient siRNA transfection in MCF7 and MDA-MB-231 cells. DAAM1 expression was verified by Q-PCR (forward primer: 5′-CAGGAAGATCTGCCCAAGGA-3′, reverse primer: 5′-GTGATTAATTCGGCTCATCTCAAA-3′) and Western blotting (Figure 10A and B). Functional assay showed that, after DAAM1 knockdown, the proliferation (Figure 10C and D), adhesion (Figure 10E and F), invasion (Figure 10G and H), and migration (Figure 10I and J) of MDA-MB-231 and MCF7 cells were suppressed.
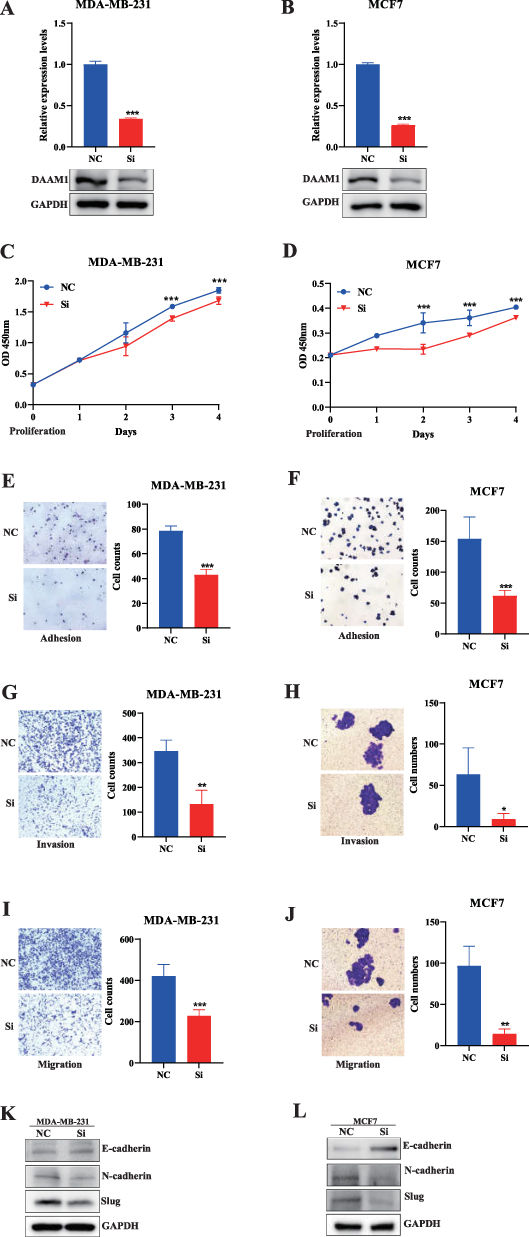 |
Figure 10 DAAM1 Promotes Malignant Phenotypes in Breast Cancer Cells via EMT. (A and B) Validation of DAAM1 knockdown efficiency at the (A) mRNA (Q-PCR) and (B) protein (Western blot) levels in MDA-MB-231 and MCF7 cells (***p < 0.001). (C–J) Functional consequences of DAAM1 knockdown on (C and D) cell proliferation (CCK-8 assay), (E and F) cell adhesion, (G and H) Matrigel invasion, and (I and J) cell migration (Transwell assay) (***p < 0.001, **p < 0.01, *p < 0.05). (K and L) Western blot analysis (K) and corresponding quantification (L) of epithelial-mesenchymal transition (EMT) markers (E-cadherin, N-cadherin, Slug) following DAAM1 knockdown, suggesting DAAM1 promotes invasion by activating the EMT pathway. |
DAAM1 Promotes BC Cell Proliferation and Adhesion, Invasion, and Migration via EMT Pathways
The EMT pathway, which facilitates cancer cell metastasis, is associated with E-cadherin and N-cadherin.19 Western blotting showed that DAAM1 might participate in the EMT pathway to promote BC cell proliferation, adhesion, invasion, and migration (Figure 10K and L).
Discussion
Our study establishes a genetically informed and functionally validated role for DAAM1 as a novel immune-related prognostic biomarker in breast cancer. By employing a MR framework, we moved beyond correlative associations to nominate DAAM1 as a gene with a potential causal influence on BC-related immune traits. This integrative approach, culminating in experimental validation, provides a compelling narrative that DAAM1 drives tumor progression by promoting aggressive cellular phenotypes via the EMT pathway, all within a context of immune-suppressed microenvironment.
The central finding of our MR analysis—that genetically predisposed expression of DAAM1 is linked to BC risk—addresses a critical gap in translating GWAS-identified loci into functional mechanisms. While previous studies have largely been descriptive, associating DAAM1 overexpression with metastasis in various cancers,20–22 our work provides a causal inference grounded in genetic instrumentation. This significantly strengthens the rationale for focusing on DAAM1, mitigating concerns of reverse causation or confounding that often plague observational studies.
Functionally, demonstrated that DAAM1 knockdown robustly inhibits BC cell proliferation, adhesion, invasion, and migration. More importantly, we bridged this phenotype to a specific molecular mechanism: the activation of the EMT program. The downregulation of E-cadherin coupled with the upregulation of N-cadherin and Slug upon DAAM1 silencing offers a plausible mechanistic explanation for its pro-metastatic role. This aligns with DAAM1’s established biochemical function as a formin protein responsible for orchestrating actin cytoskeleton dynamics.23–25 We therefore posit that DAAM1 facilitates EMT and subsequent metastasis not merely as a passive marker, but as an active regulator of the cytoskeletal remodeling that is fundamental to this process. This hypothesis provides a direct link from genetic variation to molecular function and ultimately to cellular behavior.
A particularly intriguing and novel finding is the inverse correlation between DAAM1 expression and immune infiltration. While the role of DAAM1 in cell motility is recognized, its function in the tumor immune microenvironment (TIME) of BC is largely unexplored. Our data suggest that high DAAM1 expression characterizes an “immune-cold” tumor phenotype. We speculate that this may occur through two non-mutually exclusive mechanisms: first, that the DAAM1-EMT axis directly enables tumor cells to evade immune surveillance, a known consequence of EMT;26 and second, that DAAM1 may influence the secretion of immunomodulatory factors or alter the physical structure of the tumor to impede immune cell recruitment. This finding positions DAAM1 at the nexus of tumor cell intrinsic motility and extrinsic immune modulation, a duality that merits intense future investigation.
It is important to acknowledge the limitations of our study. Firstly, while the MR analysis suggests causality, the precise molecular pathways connecting the genetic instruments to DAAM1 expression require further fine-mapping and functional genomic studies. Secondly, our functional validation, though robust, was conducted in vitro. The complexity of the TIME necessitates future validation using immune-competent in vivo models to directly confirm DAAM1’s role in immune evasion and to explore its potential as an immunotherapeutic target. Lastly, the observational nature of the immune infiltration correlation means that other unexplored factors could contribute to this phenotype.
In conclusion, this study provides a multi-layered justification for DAAM1 as a key player in BC progression. We have established a genetic rationale through MR, demonstrated its essential role in driving metastasis via EMT, and uncovered its association with an immunosuppressive microenvironment. These findings not only deepen our understanding of breast cancer biology but also highlight DAAM1 as a promising candidate for developing novel therapeutic strategies, particularly for aggressive, immune-resistant breast cancer subtypes.
Code Availability
The underlying R software code for this study is not publicly available but may be made available to qualified researchers upon reasonable request from the corresponding author.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are available at https://gwas.mrcieu.ac.uk/ and https://www.ncbi.nlm.nih.gov/geo/.
Ethics Approval and Consent to Participate
This study complies with the Declaration of Helsinki.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the Natural Science Foundation of Shan Dong Province (ZR2021MH340), Special Fund for Breast Disease Research of Shandong Medical Association (YXH2021ZX053), and Yantai Science and Technology Innovation Development Plan (2024YT06000852).
Disclosure
All authors claim that there exists no competing interest.
References
1. Siegel RL, Kratzer TB, Giaquinto AN, Sung H, Jemal A. Cancer statistics, 2025. CA Cancer J Clin. 2025;75(1):10–45. doi:10.3322/caac.21871
2. Sun K, Zhang B, Lei S, et al. Incidence, mortality, and disability-adjusted life years of female breast cancer in China, 2022. Chin Med J. 2024;137(20):2429–2436. doi:10.1097/CM9.0000000000003278
3. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12–49. doi:10.3322/caac.21820
4. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi:10.1200/JCO.2008.18.1370
5. Escala-Garcia M, Guo Q, Dork T, et al. Genome-wide association study of germline variants and breast cancer-specific mortality. Br J Cancer. 2019;120(6):647–657. doi:10.1038/s41416-019-0393-x
6. Cai X, Liang C, Zhang M, Dong Z, Weng Y, Yu W. Mitochondrial DNA copy number and cancer risks: a comprehensive Mendelian randomization analysis. Int J Cancer. 2024;154(8):1504–1513. doi:10.1002/ijc.34833
7. Head ST, Dezem F, Todor A, et al. Cis- and trans-eQTL TWAS of breast and ovarian cancer identify more than 100 risk associated genes in the BCAC and OCAC consortia. bioRxiv. 2023. doi:10.1101/2023.11.09.566218
8. Zhang Z, Fang T, Chen L, Ji F, Chen J. Multi-omics Mendelian randomization integrating GWAS, eQTL, and mQTL data identified genes associated with breast cancer. Am J Cancer Res. 2024;14(3):1433–1445. doi:10.62347/BCZW1355
9. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi:10.1038/ng.3538
10. Dang X, Zhang Z, Luo XJ. Mendelian randomization study using dopaminergic neuron-specific eQTL nominates potential causal genes for Parkinson’s disease. Mov Disord. 2022;37(12):2451–2456. doi:10.1002/mds.29239
11. Yoshihara K, Shahmoradgoli M, Martinez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi:10.1038/ncomms3612
12. Chen G, Cao J, Zhao H, Cong Y, Qiao G. Identification and verification of immune-related gene prognostic signature based on ssGSEA for breast cancer. Cent Eur J Immunol. 2022;47(2):139–150. doi:10.5114/ceji.2022.118081
13. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi:10.1038/nmeth.3337
14. Wang F, Liu D, Zhuang Y, et al. Mendelian randomization analysis identified genes potentially pleiotropically associated with periodontitis. Saudi J Biol Sci. 2021;28(7):4089–4095. doi:10.1016/j.sjbs.2021.04.028
15. Yang Z, Yang J, Liu D, Yu W. Mendelian randomization analysis identified genes pleiotropically associated with central corneal thickness. BMC Genomics. 2021;22(1):517. doi:10.1186/s12864-021-07860-3
16. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–665. doi:10.1002/gepi.21758
17. Hurley CK. Naming HLA diversity: a review of HLA nomenclature. Hum Immunol. 2021;82(7):457–465. doi:10.1016/j.humimm.2020.03.005
18. Crooke SN, Ovsyannikova IG, Poland GA, Kennedy RB. Immunosenescence: a systems-level overview of immune cell biology and strategies for improving vaccine responses. Exp Gerontol. 2019;124:110632. doi:10.1016/j.exger.2019.110632
19. Zhu L, Tian Q, Gao H, et al. PROX1 promotes breast cancer invasion and metastasis through WNT/beta-catenin pathway via interacting with hnRNPK. Int J Biol Sci. 2022;18(5):2032–2046. doi:10.7150/ijbs.68960
20. Mei J, Xu B, Hao L, et al. Overexpressed DAAM1 correlates with metastasis and predicts poor prognosis in breast cancer. Pathol Res Pract. 2020;216(3):152736. doi:10.1016/j.prp.2019.152736
21. Mei J, Huang Y, Hao L, et al. DAAM1-mediated migration and invasion of ovarian cancer cells are suppressed by miR-208a-5p. Pathol Res Pract. 2019;215(7):152452. doi:10.1016/j.prp.2019.152452
22. Zhang Y, Bai X, Zhang Y, Li Y. Daam1 overexpression promotes gastric cancer progression and regulates ERK and AKT signaling pathways. Onco Targets Ther. 2021;14:4609–4619. doi:10.2147/OTT.S316157
23. Aspenstrom P, Richnau N, Johansson AS. The diaphanous-related formin DAAM1 collaborates with the Rho GTPases RhoA and Cdc42, CIP4 and Src in regulating cell morphogenesis and actin dynamics. Exp Cell Res. 2006;312(12):2180–2194. doi:10.1016/j.yexcr.2006.03.013
24. Habas R, Kato Y, He X. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell. 2001;107(7):843–854. doi:10.1016/s0092-8674(01)00614-6
25. Zhu Y, Tian Y, Du J, et al. Dvl2-dependent activation of Daam1 and RhoA regulates Wnt5a-induced breast cancer cell migration. PLoS One. 2012;7(5):e37823. doi:10.1371/journal.pone.0037823
26. Luond F, Sugiyama N, Bill R, et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev Cell. 2021;56(23):3203–3221e11. doi:10.1016/j.devcel.2021.11.006
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.