Back to Journals » International Journal of General Medicine » Volume 19
Mendelian Randomization and Single-Cell RNA Sequencing Reveal CKAP4 and PFDN5 as Tumor Cell-Specific Causal Genes for Glioblastoma
Authors Huang C ![]() , Dong M, Yu Y, Wang C, Huang C, Li T, Su X, Shi F, Feng D
, Dong M, Yu Y, Wang C, Huang C, Li T, Su X, Shi F, Feng D
Received 24 February 2026
Accepted for publication 10 June 2026
Published 22 June 2026 Volume 2026:19 605071
DOI https://doi.org/10.2147/IJGM.S605071
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ching-Hsien Chen
Chong Huang, Minhai Dong, Yongjia Yu, Chunxi Wang, Chaojue Huang, Tang Li, Xiangsheng Su, Fengqiang Shi, Daqin Feng
Department of Neurosurgery, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, 530021, People’s Republic of China
Correspondence: Daqin Feng, Email [email protected]
Background: Glioblastoma (GBM) is an aggressive primary brain tumor with unclear etiology. We aimed to identify causal risk genes by integrating single-cell RNA sequencing (scRNA-seq) and Mendelian randomization (MR).
Methods: Differentially expressed genes (DEGs) from public databases were overlapped and analyzed via MR to screen for causal genes. A prognostic model was built using these genes and validated through immune infiltration analysis, single-cell mapping, and experimental assays (RT-qPCR, Western blot).
Results: A 6-gene prognostic signature (TMEM158, HOXB2, CKAP4, PEPD, PFDN5, NPC2) was established, where higher risk scores correlated with poorer overall survival and distinct immune profiles. scRNA-seq confirmed tumor cell-specific expression, validated experimentally. Multivariate MR highlighted CKAP4 and PFDN5 as having direct causal links to GBM.
Conclusion: The 6-gene signature predicts GBM prognosis, and CKAP4/PFDN5 are promising causal biomarkers and therapeutic targets. This integrated approach provides novel molecular insights and supports personalized therapy development in GBM.
Keywords: glioblastoma, single-cell analysis, Mendelian randomization analysis, differentially expressed genes, gene set enrichment analysis, drug sensitivity analysis
Introduction
Glioblastoma (GBM) represents the most prevalent and aggressive primary malignant neoplasm of the adult central nervous system.1 Despite the current standard therapeutic regimen—comprising maximal safe surgical resection followed by concurrent temozolomide (TMZ) administration with radiotherapy and subsequent maintenance TMZ therapy for 6–12 months2—clinical outcomes remain dismal, with median survival typically not exceeding 15 months. Consequently, the identification of robust molecular biomarkers for early diagnosis and the discovery of novel therapeutic targets are both urgently needed and critically important for improving patient prognosis. In recent years, multi-omics integration analysis has emerged as a powerful and transformative approach for deciphering the intricate molecular architecture of tumors.3 By synergistically combining genomic, transcriptomic, proteomic, and epigenomic data, multi-omics strategies substantially enhance our understanding of spatial and temporal heterogeneity in cancer biology and pave the way for innovative precision medicine approaches to target and modulate tumor progression.4
Single-cell RNA sequencing enables gene expression analysis at the single-cell level. This technique could provide a detailed understanding of GBM by elucidating heterogeneity, drug resistance mechanisms, and signaling pathways.5
Mendelian randomization (MR) has emerged as a powerful tool to identify causal risk genes for complex diseases and cancers, avoiding confounding and reverse causation.6,7 Recent applications in glioblastoma have further validated its ability to prioritize actionable therapeutic targets.8,9 It was usually the factors of concern, such as genes, that were taken as exposure factors, and diseases were regarded as outcome. This method is fundamentally grounded in Mendel’s law of independent assortment, which posits that genetic variations are randomly distributed within a population and are independent of potential confounding factors. In practice, Mendelian Randomization estimates the causal effect of an exposure on an outcome by identifying genetic variations—particularly single nucleotide polymorphisms (SNPs) that are significantly associated with the exposure factors under investigation. By utilizing these genetic variants as instrumental variables, MR effectively mitigates confounding bias and addresses the challenges of reverse causation that are prevalent in traditional observational studies.6 The process of conducting MR involves several critical steps. Firstly, researchers must select genetic variations that exhibit a strong association with the exposure factors of interest. Secondly, it is essential to validate that these genetic variations remain independent of any confounding factors. Finally, the causal effect is estimated by analyzing the relationship between the identified genetic variations and the corresponding outcomes. This systematic approach enhances the reliability of causal inference in epidemiological research.7 The IVs for MR analysis was required to meet three assumptions: 1. Relevance assumption: The SNPs were assumed to be strongly correlated with the exposure factors. 2. Independence assumption: The SNPs were assumed to be independent of the confounding factors. 3. Exclusion restriction assumption: The SNPs were assumed to affect the outcome only through the exposure factors. MR leverages data from genome-wide association studies (GWAS) to identify genetic variations associated with GBM. By selecting relevant genetic variants, MR assesses their causal effects on GBM risk, thereby enhancing our understanding of how specific genetic factors contribute to the development of this aggressive cancer. Consequently, this approach holds significant implications for advancing personalized medicine in the management of GBM, ultimately improving outcomes for patients.8,9
To date, no research has integrated MR analysis with single-cell analysis to explore how cell-type-specific gene expressions might differentially impact GBM risk. The innovation of our study lies in its identification of differentially expressed genes (DEGs) in specific cell types within GBM. Then, MR analysis with three major hypotheses was conducted to explore the causal relationship between the intersection genes of these DEGs and GBM. Moreover, the further screening of candidate core genes closely related to GBM prognosis, culminating in the construction of a risk prediction model. Moreover, our study comprehensively evaluates candidate biomarkers through gene set enrichment analysis, immune infiltration analysis, drug prediction and MR analysis. Finally, these biomarkers were validated using reverse transcription quantitative PCR and Western blot experiments. These findings offer new perspectives for understanding the complex mechanisms of GBM, and the identified biomarkers hold potential for transitioning from theoretical research to becoming instrumental tools in the clinical management of GBM.
Materials and Methods
Data Extraction
RNA-seq data for TCGA-GBM was downloaded from the TCGA Hub on the UCSC Xena website (http://xena.ucsc.edu/). The dataset comprised a total of 172 samples, including 5 healthy control samples and 167 GBM samples (140 primary and IDH-wildtype samples were selected for this study).10 The RNA-seq data from these 140 selected GBM tissue samples and 5 adjacent non-cancerous tissue samples were utilized for differential gene identification and prognostic analysis. Clinical information on patients with TCGA-GBM was shown in Supplementary Table 1. The GSE68848 dataset (comprising 228 GBM tissue samples and 28 normal samples) was used to validate the expression of prognostic genes. The CGGA-325 dataset, along with a single-cell RNA-seq (scRNA-seq) dataset, were downloaded from the CGGA database (http://www.cgga.org.cn/). The CGGA-325 dataset, which includes 74 GBM tissue samples with comprehensive survival information, was utilized to validate the prognostic model. In contrast, the scRNA-seq dataset contained 20 GBM tumor tissue samples and 8 samples from the tumor periphery, which were employed for single-cell analysis. The Genome-Wide Association Studies (GWAS) data of expression Quantitative Trait Loci (eQTL) of candidate genes was searched from Integrative Epidemiology Unit (IEU) open GWAS (https://gwas.mrcieu.ac.uk/). Additionally, the GWAS dataset for GBM (id: finn-b-C3_GBM) was acquired from the IEU OpenGWAS (https://gwas.mrcieu.ac.uk/). The dataset was selected based on the principle of having the most SNPs. This dataset comprises 218,792 samples, including 91 case samples and 218,701 control samples, along with 16,380,466 SNPs. The research design related to the MR data and other information about the potential population were described in Supplementary Table 2. This study also performed multi-omics integration. First, single-cell RNA-seq was used to identify cell-type-specific DEGs. Second, bulk RNA-seq was applied to identify prognosis-related DEGs. Third, GWAS-based Mendelian randomization was performed to prioritize causal genes overlapping from the above two sets. Finally, the causal genes were used to build a prognostic model, which was further interpreted by immune infiltration, functional enrichment, single-cell mapping, and experimental validation.
Single-Cell Analysis
Single-cell RNA sequencing (scRNA-seq) enables unbiased dissection of intratumoral heterogeneity and cell-type-specific gene expression at single-cell resolution, which is superior to traditional bulk RNA-seq that only provides average signals across millions of cells.5,11 In the CGGA-scRNA-seq samples, the scRNA-seq data were filtered using “Seurat” (version 4.0.5)11 (CreateSeuratObject), with min.cells = 3 and min.features = 10 as filtering criteria. Mitochondrial genes were quantified using “PercentageFeatureSet,” retaining cells with < 10% mitochondrial gene content. The scRNA-seq data were then standardized by “NormalizeData” and the 2,000 hypervariable genes were screened by “FindVariableFeatures” for subsequent cell type identification. To address batch effects between any two groups of samples, CCA was employed. The overall data was downscaled using PCA, and an elbow plot was generated to identify the optimal dimensions. Following dimensionality reduction, unsupervised cell clustering was conducted using “FindNeighbors” and “FindClusters,” visualized via “t-SNE.” Marker genes for each subpopulation were compared to known marker genes in the CellMarker database to determine cell subpopulation types, employing filtering parameters min.pct = 0.6, only.pos = TRUE, and logfc.threshold = 0.5. Identifying cell types were validated using the “SingleR” algorithm. Cells that play important roles in disease were selected as key cells.
Identification of Candidate Genes
In the CGGA-scRNA-seq samples of GBM and tumor peripheral tissue, differential genes for various cell types were identified using the “FindMarker” function, identifying DEGs1 with a P-value < 0.05. In the TCGA-GBM dataset, the “DESeq2” package (v1.36.1)12 was used to compare gene expression between GBM and normal samples, identifying DEGs2 with a P-value < 0.05 and |log2fold change (FC)| > 0.5. Additionally, candidate genes were identified by intersecting DEGs1 and DEGs2 using the “ggVenn” package (version 1.2.2). Furthermore, genes that were significantly upregulated or downregulated in GBM tumors were identified through single-cell RNA sequencing and differential expression analysis. And the roles of these genes in tumor progression were explored.
Mendelian Randomization Analysis
The two-sample Mendelian randomization (MR) analysis was performed independently of the single-cell RNA-seq expression matrices. While single-cell data were used to identify cell-type-specific differentially expressed genes (scRNA-seq DEGs, termed DEGs1), the MR analysis itself relied solely on independent genetic data: expression quantitative trait loci (eQTL) summary statistics for candidate genes and genome-wide association study (GWAS) summary statistics for GBM, both sourced from the IEU OpenGWAS database. Mendelian randomization (MR) is a genetically informed method that uses SNPs as instrumental variables to infer causal relationships, effectively reducing confounding and reverse causation compared with traditional observational correlation studies.6,7,13 Three basic premises underlie MR studies: (1) Relevance: a robust and significant correlation exists between instrumental variables (IVs) and exposure factor. (2) Independence: IVs are unrelated to confounding factors. (3) Exclusion restriction: IVs can only influence outcomes through exposure factors and not through other channels. The causal relationship between candidate genes (exposure factors) and GBM (outcome) was analyzed using the “TwoSampleMR” package (version 0.5.6).13 MR analysis was performed using the “mr” function, incorporating five algorithms: MR Egger,14 Weighted Median,15 Inverse Variance Weighted (IVW),16 Simple Mode,17 and Weighted Mode.18 The IVW method was deemed pivotal for result determination, with a significance threshold of P-value < 0.05, indicating that GBM potentially increases the risk of candidate genes, and an OR below 1 suggesting a protective effect. In order to obtain effective instrumental variables (IVs), the extract_instruments function in the TwoSampleMR (v 0.5.6) package19 was used to read the GWAS data of the exposure factors and outcome events. And the IVs were screened according to the following steps. Firstly, SNPs that were significantly associated with the exposure factors were selected (p < 5×10−8). Then, the SNPs with linkage disequilibrium were removed using the parameter “clump = TRUE” (R2 = 0.001, kb = 10,000). Based on the GWAS data of the outcome, the SNPs that were significantly associated with the outcome were removed (with “clump = TRUE” and “rsq = 0.8”). Meanwhile, the genes with a SNP count of at least 3 were retained, and the samples with complete SNP data were also kept. The strength of each IV was evaluated by calculating the F-statistic. The IVs with an F-statistic greater than 10 were considered to have no weak instrument bias and possess strong predictive ability. The IVs with an F-statistic less than 10 might generate bias and were therefore excluded. IVs with F statistics less than 10 are excluded. Among them, the formula for F was given as 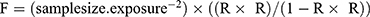 , R represents the degree of explanation of the exposure factor by the instrumental variable, and samplesize.exposure represents the number of instrumental variables. Through the above steps, high-quality IVs were obtained and used for the subsequent MR analysis to ensure the reliability of the results and the effectiveness of causal inference. Funnel, scatter, and forest plots were employed for result visualization.
, R represents the degree of explanation of the exposure factor by the instrumental variable, and samplesize.exposure represents the number of instrumental variables. Through the above steps, high-quality IVs were obtained and used for the subsequent MR analysis to ensure the reliability of the results and the effectiveness of causal inference. Funnel, scatter, and forest plots were employed for result visualization.
Sensitivity Analysis of MR
To validate the MR findings, a series of stringent sensitivity analyses were undertaken. The mr_heterogeneity function was initially applied to evaluate heterogeneity using Cochran’s Q test.20 The heterogeneity test was mainly conducted to check whether there was heterogeneity among the IVs, which could indicate pleiotropy or measurement errors. Among them, the fixed-effects IVW approach was applied if the p was more than 0.05; if not, the random-effects IVW method was applied. Subsequently, the mr_pleiotropy_test function was utilized to examine the possibility of horizontal pleiotropy and identify any potential confounders; a nonsignificant result (P- value > 0.05) indicated no significant pleiotropy.21 The stability of the causal estimates was further confirmed through Leave-One-Out (LOO) analysis, which systematically excluded individual SNPs to ensure the robustness of the results in the absence of any single variant.22 Pleiotropic Clustering for Mendelian Randomization (PCMR), an analytical framework for MR, was employed to more accurately estimate the causal effect of an exposure on an outcome by correcting for pleiotropic effects in the presence of horizontal pleiotropy. Specifically, the LD pruning (r2 < 0.1, kb = 10, p > 0.5) was performed using the ld_clump() function from the ieugwasr package (v 1.0.4) to obtain a background SNP set (X_clump1), while independent genetic variants significantly associated with the exposure were retained as the IVs set (X_clump). A two-stage analysis was then conducted via the PCMR method: potential pleiotropic clustering patterns were first identified based on X_clump1; subsequently, the causal effect of the exposure on the outcome was evaluated using X_clump while controlling for horizontal pleiotropy, and the robustness of the results was validated via the Bootstrap method. For GWAS and eQTL data, the p-values were converted into chi-squared statistics by using the qchisq function in the stats package (v 4.2.2). The lambda GC (λGC) was obtained through taking the ratio of the median chi-squared statistic to the theoretical median. Subsequently, a standardization process was conducted to adjust the λGC to the benchmark of 1000 samples. Eventually, a faceted bar plot was drawn by applying ggplot2 package (v 3.5.2) to present both the original and standardized λGC values. After that, based on the results of the MR analysis, the steiger test was adopted using the directionality_test function in the TwoSampleMR package (v 0.6.15) to evaluate the directionality between the exposure factors and the outcome. The conditions for passing the steiger test were that the correct causal direction was TRUE and p < 0.05. Finally, the genes that passed the MR analysis were named as key genes.
Enrichment Analysis of Key Genes
To elucidate the biological roles of the selected key genes, Gene Ontology (GO) contained cellular component (CC), molecular function (MF) and biological process (BP), were analyzed. Then, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses was conducted. GO and KEGG were employed the “ClusterProfiler” package (version 4.4.4) (P-value<0.05),23 based on Human Gene Annotation package “org.Hs.egdb” (version3.15.0). The enriched pathways were sorted by P-value score, visualizing through gapminder (version 1.0.0) (https://CRAN.R-project.org/package=gapminder) and GOplot (version 1.0.2).24
Construction and Validation of Risk Model
In the TCGA-GBM dataset, a risk model was constructed to investigate the prognostic impact of key genes on GBM patients. We constructed a risk model to investigate the prognostic impact of the key genes on GBM patients. The key genes were first subjected to univariate Cox analysis to identify those with greater prognostic significance (P-value < 0.02) via “survival” package (version 3.3–1). Next, the “glmnet” package (version 4.1–4) was used to perform least absolute shrinkage and selection operator (LASSO) analysis select biomarkers with highly correlated features. The risk coefficient for each selected biomarker was then calculated. To assess the prognostic utility of the risk model, GBM patients were assigned a risk score derived from the expression levels and associated risk coefficients of the identified genes. The median risk score was subsequently employed as a threshold to segregate GBM patients into high-risk and low-risk groups. The formula for risk score was:

n: the number of prognostic genes included in the model (n = 6 in this study). coef(gene_i): the regression coefficient of the i-th prognostic gene obtained from LASSO-Cox analysis. expr(gene_i): the normalized expression level of the i-th prognostic gene in the sample.
Regression coefficients for the six genes: HOXB2: coef = 0.13207; NPC2: coef = 0.28953; TMEM158: coef = −0.18435; PFDN5: coef = 0.12051; PEPD: coef = −0.30577; CKAP4: coef = 0.23731.
Additionally, survival curves were constructed for both the high-risk and low-risk groups in the TCGA-GBM dataset to assess the overall survival (OS) rates of patients with GBM. The accuracy of the risk model was validated using ROC analysis. Moreover, the findings were further validated using the CGGA-325 sample set to strengthen the reliability of the risk model.
Independent Prognostic Analysis
In the TCGA-GBM dataset, the distribution of patients among various clinical subtypes was analyzed using the χ2-test to determine any significant differences between the high-risk and low-risk groups. The “ggstatsplot” package (version 0.11.0)25 was utilized to illustrate the disparities in clinical factors between the two risk groups. Subsequently, to delve deeper into the prognostic significance of the clinical factors in conjunction with the risk model, a univariate Cox analysis was conducted, incorporating both the clinical factors and the risk score (P < 0.05). The clinical factors were also tested for the proportional hazards (PH) assumption, and those that satisfied this assumption were included in a multivariate Cox analysis to pinpoint independent prognostic factors (P < 0.05). To elucidate the connections between the clinical factors and risk model, we constructed a nomogram, integrating the identified biomarkers, to forecast the 1- to 3-year survival rates for GBM patients. Calibration curves and ROC were subsequently plotted to evaluate the validity and precision of the risk model in predicting GBM prognosis.
Gene Set Enrichment Analysis (GSEA) Analysis
A comprehensive series of analyses was conducted to investigate the biological functions of the biomarkers. We investigated biological biomarkers by comprehensive series of analyses. Initially, we utilized log2FC to quantify the disparities between the high-risk and low-risk groups. Subsequently, the log2FC values were employed in GSEA (adjusted P < 0.05). We visualized the top-10 GSEA results by “GseaVis” package (version 0.0.5). Ultimately, we calculated the KEGG pathway scores in GBM samples using the “GSVA” package26 (version 1.38.2) and examined the disparities in KEGG pathways between the two groups via the “limma” package27 (version 3.46.0) (P-value < 0.01 log2|FC| > 0.015).
Immune-Related Analysis
Immune cell infiltration was quantified using the CIBERSORT algorithm to produce an immune score for each cell type. The TIDE score was used to evaluate immunotherapy response: a higher TIDE score indicates stronger immune exclusion and poorer predicted immunotherapy benefit.28,29 Immune infiltration scores were calculated by “CIBERSORT” algorithm for 22 immune cells from TCGA-GBM samples.29 We used the Wilcoxon test to compare the difference in immune infiltration scores between the high-risk and low-risk groups, and visualized the results with boxplot by “ggpubr” package (version 0.4.0)30 (P-value < 0.05). We conducted Spearman correlation coefficient (|cor| > 0.3, P-value < 0.05) to study the association between biomarkers and immune cell abundance. Additionally, based on 43 immune checkpoint genes obtained from reference,28 we employed Wilcoxon test to compare the expression levels of immune checkpoint genes across the two risk groups (P-value < 0.05). Furthermore, we obtained tumor immune dysfunction and exclusion (TIDE) scores and data on immune therapy responses for each GBM from the TIDE database. Subsequently, we conducted Wilcoxon test to compare the TIDE scores between the high-risk and low-risk groups. Ultimately, we analyzed the correlation between TIDE scores and the risk scores.
Drug Sensitivity Analysis
We calculated the half maximal inhibitory concentration (IC50) values for 138 common chemotherapeutic drugs from the GDSC database by the “pRRophetic” package (version 0.5).31 Subsequently, we employed the Wilcoxon test to compare the disparities of drug sensitivity across the low-risk and high-risk groups. Finally, we conducted correlation analysis between the top 10 sensitive drug for each group and risk score (P-value < 0.05, |cor| > 0.3).
Correlation Expression Analysis of Biomarkers
We generated boxplots by “ggpubr” package (version 0.4.0)30 from TCGA-GBM dataset to represent the variations in biomarker expression levels between GBM and normal samples. Biomarker expressions in GBM and normal samples from validation set GSE68848 were analyzed using the wilcoxon rank sum test with a threshold of P < 0.05. Additionally, the causal relationship between biomarkers and GBM was analyzed using the “TwoSampleMR” package (mv_harmonise_data function, version 0.5.6).13 Furthermore, biomarker expressions in various cell sub-populations were observed and visualized using t-SNE plots and violin plots.
Single-Cell Communication Analysis
To explore the communication network between different cells, the ligand-receptor interactions between different cell types in the disease and control groups of the single-cell dataset were analyzed using the CellChat package (v 1.6.1).32 The number and intensity of interactions between different cell types were visualized through network diagrams and heatmaps, which were plotted using the netVisual_circle and netVisual_heatmap functions, respectively. The key cells were subjected to dimensionality reduction and clustering analysis (resolution = 0.4) using the RunPCA, FindNeighbors, and FindClusters functions of the Seurat package (v 5.1.0), and were re-clustered into different subpopulations. Cell pseudotime trajectory analysis of key cell clusters was performed using the Monocle package (v 2.30.1).33 The trajectories of key cell clusters were segmented according to trajectory nodes. To detect the expression levels of biomarkers in the pseudotime of key cells, a dynamic heatmap was plotted to observe the expression of biomarkers. Functional enrichment analysis of each annotated cell cluster was conducted using the analyze_sc_clusters function of the ReactomeGSA package (v 1.16.1)34 and the enrichment results were extracted from different cells through the pathways function. Cell cycle stages for each cell were assigned using the CellCycleScoring function of the Seurat package (v 5.1.0) based on the built-in cell cycle-related gene set cc.genes. Subsequently, the PCA results were plotted using the DimPlot function, with coloring according to cell cycle stages.
Participants
In our study, we collected samples from GBM patients to facilitate RNA extraction and protein analysis. Specifically, 5 GBM tissue samples and 5 peripheral tumor tissue samples were obtained for RNA extraction, while an additional set comprising 3 GBM tissue samples and 3 peripheral tumor tissue samples was gathered for Western blot analysis. All patients involved in the study underwent surgical procedures in 2023. Eligible participants were adults aged 18 years or older with histologically confirmed supratentorial GBM. Inclusion criteria excluded individuals with known HIV or chronic hepatitis B or C infections, as well as those with any medical conditions that might affect the oral intake of medications. All subjects participating in the study were informed of consent; each participant provided written consent personally. The collection of specimens strictly followed the guidelines set forth by the Ethics Committee of The First Affiliated Hospital of Guangxi Medical University, in accordance with the principles of the Declaration of Helsinki, under the approval number 2024-E300-01. The sample sizes for RT-qPCR (n=5 pairs) and Western blot (n=3 pairs) were determined as a pilot validation based on resource availability and the primary aim of confirming the direction of expression changes observed in large-scale bioinformatic analyses.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated from 50 mg tissue samples using Trizol reagent. Subsequently, the RNA was reversed transcribed into complementary DNA utilizing the SureScript First-Strand cDNA Synthesis Kit (Servicebio, China). Quantitative PCR analysis was conducted utilizing the Fast SYBR Green qPCR Master Mix (Servicebio, China). The qPCR reactions were executed on a CFX96 real-time quantitative fluorescence PCR instrument (Bio-Rad, China), following this protocol: an initial pre-denaturation step at 95°C for 1 minute, followed by 40 cycles of denaturation at 95°C for 20 seconds, annealing at 55°C for 20 seconds, and extension at 72°C for 30 seconds. Subsequently, we quantified the relative expression levels of the genes by 2-ΔΔCt method (P<0.05). Each RT-qPCR analysis was replicated three times to ensure reliability. The primer sequences used are provided in Table 1.
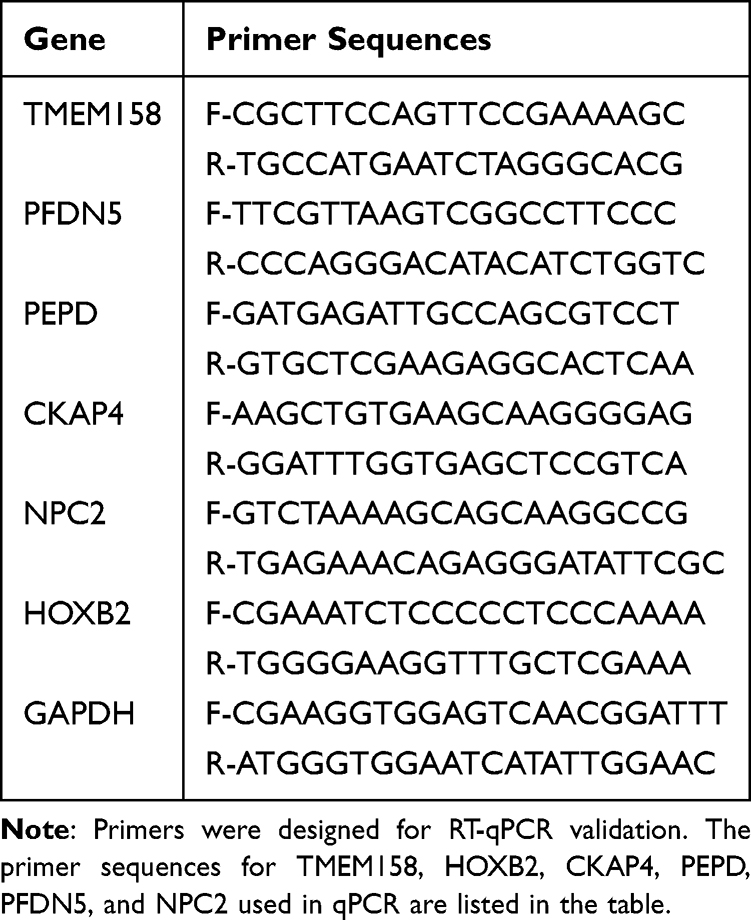 |
Table 1 Primer Sequences for qPCR |
Western Blot
Samples were lysed using RIPA buffer (HAT, China) supplemented with 1% phenylmethylsulfonyl fluoride (PMSF) and 1% protease inhibitor to ensure protein integrity. Equal amounts of total protein from each sample were resolved on 10% SDS-PAGE gradient gels, followed by transfer onto polyvinylidene difluoride (PVDF) membranes. To prevent non-specific binding, membranes were blocked with 5% skim milk. Subsequently, they were incubated with primary antibodies at 4°C overnight. The membranes were then exposed to secondary antibodies. Following incubation, the blots were thoroughly washed and analyzed using an image analysis system.
Statistical Analysis
Statistical analyses were conducted using R software (version 4.2.2). Statistical thresholds were defined as follows:
Single-cell DEGs: P < 0.05; Bulk RNA-seq DEGs: P < 0.05 and |log2FC| > 0.5; Survival & Cox regression: P < 0.2; LASSO selection: λ.min (optimal value); MR causal analysis: IVW P < 0.05; GO/KEGG enrichment: P < 0.05; GSEA: adjusted P (padj) < 0.05; Immune infiltration and drug sensitivity: P < 0.05; correlation: |cor| > 0.3 and P < 0.05; Box/violin plots: Wilcoxon rank-sum test P < 0.05.
Results
Identification of Five Major Cell Clusters in GBM
The flowchart of our study is shown in Figure 1. Following quality control and normalization of the CGGA scRNA-seq data (Supplementary Figure 1), 21,021 genes and 2,000 highly variable genes were retained for analysis. Principal component analysis (PCA) using the first 30 significant components and subsequent clustering revealed nine initial cell clusters, which were annotated into five major cell types based on established marker genes: proliferating cells (high TOP2A), tumor cells, myeloid cells, lymphocytes, and normal cells (high MOG) (Figure 2A–C). Tumor and myeloid cells constituted the largest proportions of cells (Figure 2D–E), and tumor cells were designated as the key cellular population for subsequent analyses.
 |
Figure 1 Flowchart of the whole study. This study integrated single-cell RNA-seq (scRNA-seq), bulk RNA-seq, and two-sample Mendelian randomization (MR) to identify causal and prognostic genes in GBM. Differential expression analysis was performed using DESeq2 and Seurat; causal inference used the Inverse Variance Weighted (IVW) method; prognostic modeling used LASSO‑Cox regression;The predictive performance and generalizability of the model were rigorously validated using three independent external cohorts: GSE68848, and CGGA-325; experimental validation used RT‑qPCR and Western blot. |
 |
Figure 2 Cell clustering and annotation. (A) t‑SNE clustering based on scRNA‑seq data after quality control (min.cells = 3, min.features = 10, <10% mitochondrial genes). (B) Five cell types annotated using CellMarker and SingleR. (C) Dot plot of marker genes with filtering thresholds: min.pct = 0.6, logfc.threshold = 0.5. (D–E) Cell proportion statistics across clusters. |
Integration of single-cell and bulk RNA-seq identifies 41 causal GBM risk genes and their biological functions
Comparison of GBM versus peripheral tissue in the CGGA scRNA-seq dataset yielded 4,500 cell-type-specific differentially expressed genes (DEGs1). In the TCGA-GBM cohort, comparison of tumor versus normal tissue identified 9,793 DEGs2 (5,026 up, 4,767 down) (Figure 3A and B). Intersection of these two gene sets yielded 2,554 overlapping candidate genes (Figure 3C). The F-values of SNPs for 2,554 genes were calculated (Supplementary Table 3). Two-sample Mendelian randomization (MR) analysis, employing the inverse variance weighted (IVW) method as primary, identified 41 of these overlapping genes as having a significant causal relationship with GBM risk (Supplementary Table 4). Among these, 21 were risk factors (OR > 1) and 20 were protective factors (OR < 1). Using HOXB2as an example, MR analysis showed a significant causal effect (IVW OR > 1, p=0.0008), with sensitivity analyses (heterogeneity, pleiotropy, leave-one-out) confirming the robustness and reliability of the findings (Tables 2–4, Supplementary Table 5 and Figure 3D–H). The CHVP test p-values for all 41 genes were found to be >0.9e−07, indicating that no significant level of pleiotropic interference was detected in the instrumental variables (Supplementary Table 6). Genetic background standardization showed close alignment between datasets (Figure 3I). All SNPs had a variance explanation (R2) for the exposure that was greater than the variance explanation of SNPs for the outcome, and all P-values were less than 0.05. These results indicated that the direction was correct (Supplementary Table 7). To delve deeper into the biological roles of the 41 key genes, a comprehensive GO analysis was conducted, encompassing three key aspects: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC). In terms of biological processes, candidate genes are significantly associated with the regulation of viral entry into host cells, modulation by symbiont entry into host et al In terms of molecular function, candidate genes are significantly associated with amyloid-beta binding, 5′-3′ DNA helicase activity et al In terms of cellular component, candidate genes are significantly associated with trans-Golgi network membrane, trans-Golgi network et al (Supplementary Figure 2A) According to the KEGG analysis, the key genes exhibited a significant association with arginine and proline metabolism, as well as tuberculosis (Supplementary Figure 2B and Supplementary Table 8).
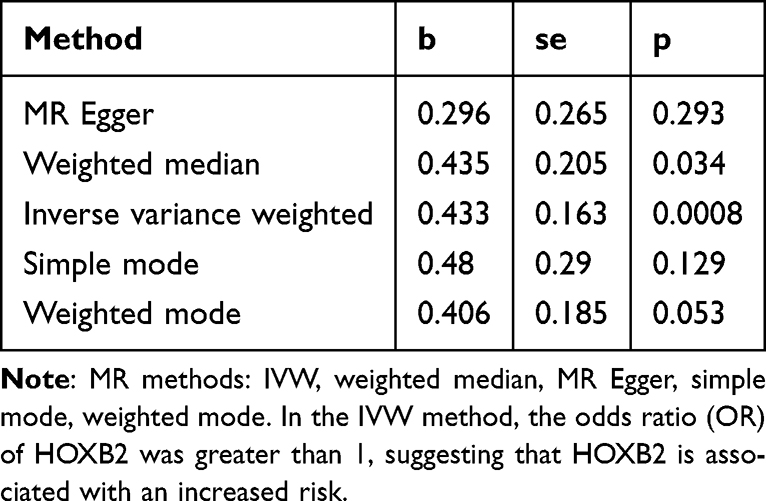 |
Table 2 MR Analysis of the HOXB2 Gene in Glioblastoma (GBM) |
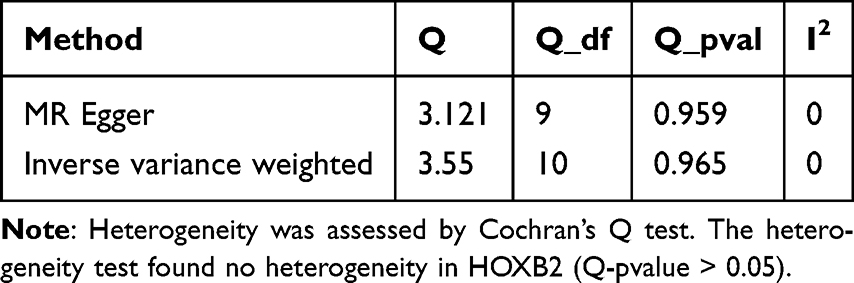 |
Table 3 Heterogeneity Test |
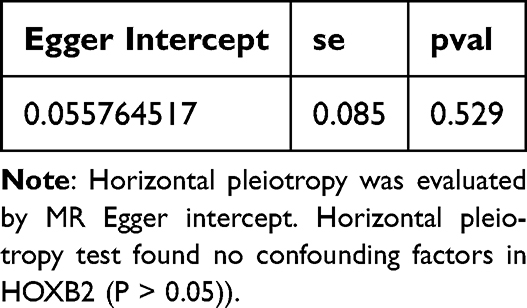 |
Table 4 Horizontal Pleiotropy |
 |
Figure 3 Screening for differentially expressed genes. (A) Volcano plot of DEGs in TCGA‑GBM using DESeq2; thresholds: P < 0.05 and |log2FC| > 0.5. (B) Heatmap of top 10 up/down DEGs. (C) Overlap of scRNA‑seq DEGs (DEGs1) and bulk DEGs (DEGs2). (D–H) Two‑sample MR using the IVW method (P < 0.05 as significance threshold), with leave‑one‑out (LOO) sensitivity analysis. (I) λGC before and after standardization. |
Construction and Validation of a Six-Gene Prognostic Risk Signature
Univariate Cox regression (P < 0.2)35 identified nine candidate prognostic genes from the 41 key genes (Figure 4A). Subsequent LASSO-Cox regression analysis selected an optimal 6-gene model (TMEM158, HOXB2, CKAP4, PEPD, PFDN5, NPC2) when the penalty parameter (λ) was minimized (Figure 4B and C). It is important to distinguish the aims of the different analyses: the multivariate MR identified CKAP4 and PFDN5 as having the strongest evidence for independent direct causal effects on GBM risk. In contrast, the LASSO-Cox prognostic model selected the combination of six genes that collectively provided the most accurate prediction of patient survival, irrespective of whether each gene’s influence is direct or mediated through other pathways. The risk score was calculated as: RiskScore = (0.13207 × HOXB2) + (0.28953 × NPC2) + (−0.18435 × TMEM158) + (0.12051 × PFDN5) + (−0.30577 × PEPD) + (0.23731 × CKAP4) (Table 5). In the TCGA-GBM cohort, patients stratified into high- and low-risk groups by the median risk score (cutoff=1.056) showed significantly different overall survival, with the high-risk group associated with poorer prognosis (Figure 4D–E). The model demonstrated predictive accuracy with 1-, 2-, and 3-year AUCs all > 0.6 (Figure 4F). These prognostic findings were successfully validated in the independent CGGA-325 dataset (Figure 4G–I).
 |
Table 5 The Risk Coefficients of the Biomarkers |
 |
Figure 4 Risk model construction, evaluation, and validation. (A) Univariate Cox regression (P < 0.2). (B and C) LASSO regression with λ.min to select 6 genes. (D) Risk score distribution. (E) Kaplan‑Meier survival curves compared by log‑rank test. (F) Time‑dependent ROC AUC. (G–I) External validation in CGGA‑325. Risk score was calculated as: RiskScore = Σ(coef_i × normalized expression_i). |
The Risk Score is an Independent Prognostic Factor and Delineates Distinct Functional Phenotypes
Clinical analysis revealed the high-risk group had a significantly higher proportion of deceased patients and of the mesenchymal molecular subtype (Table 6, Figure 5A and B). Both univariate and multivariate Cox analyses identified the risk score as the sole independent prognostic factor for GBM (Table 7, Figure 5C and D). A prognostic nomogram incorporating the risk score was constructed, and its calibration curves showed good agreement between predicted and observed 1-, 2-, and 3-year survival (Figure 5E–H). The nomogram’s predictive AUCs for 1- to 3-year all exceeded 0.6 (Figure 5I). Functional analysis revealed the high-risk group was enriched for pathways such as hematopoietic cell lineage and Staphylococcus aureus infection. Gene set variation analysis (GSVA) identified 28 differentially active pathways between risk groups, including activation of chemical carcinogenesis and cytochrome P450 drug metabolism, and inhibition of cell cycle and Notch signaling in the high-risk group (Supplementary Figure 3A–B).
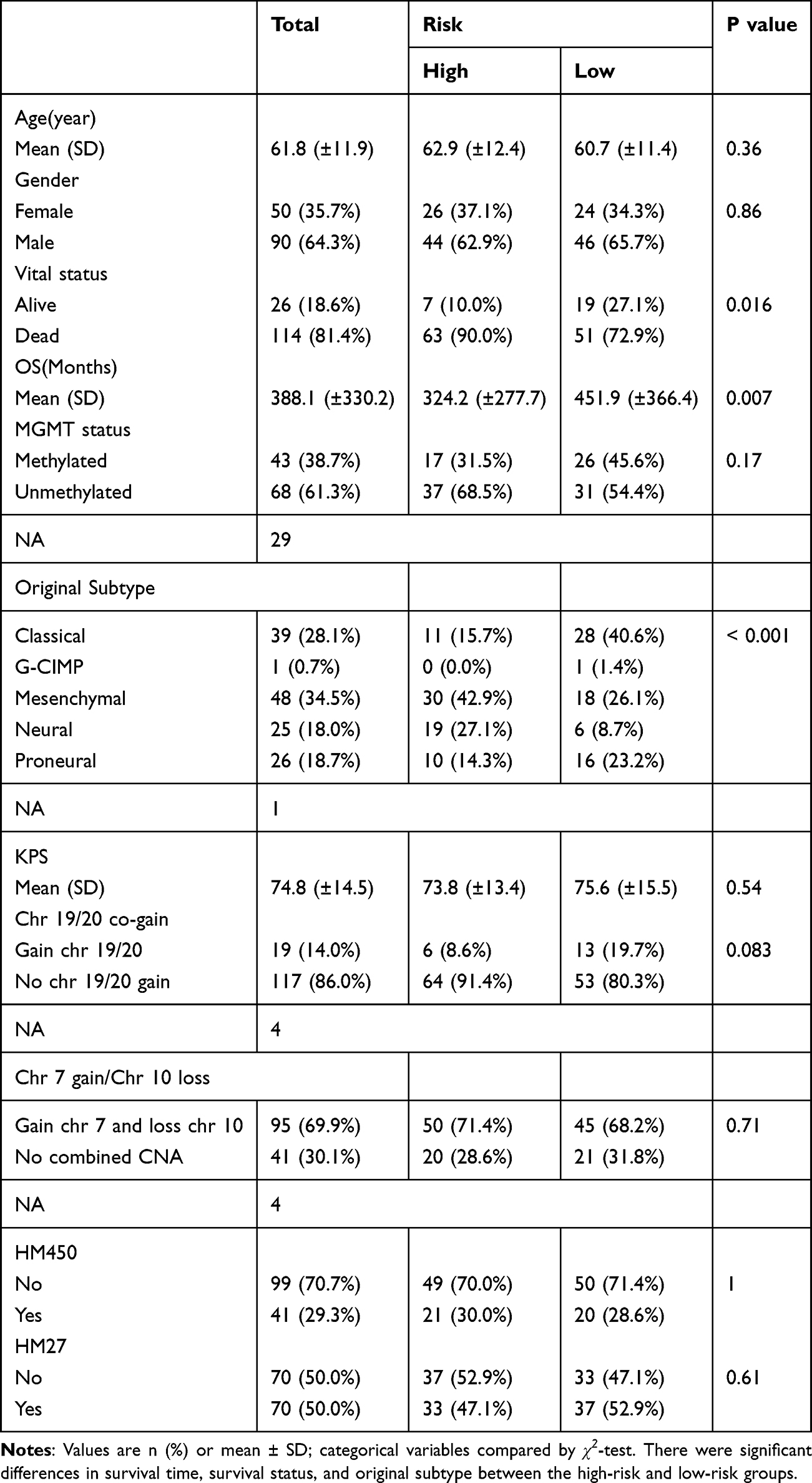 |
Table 6 Risk and Clinical Data in TCGA-GBM |
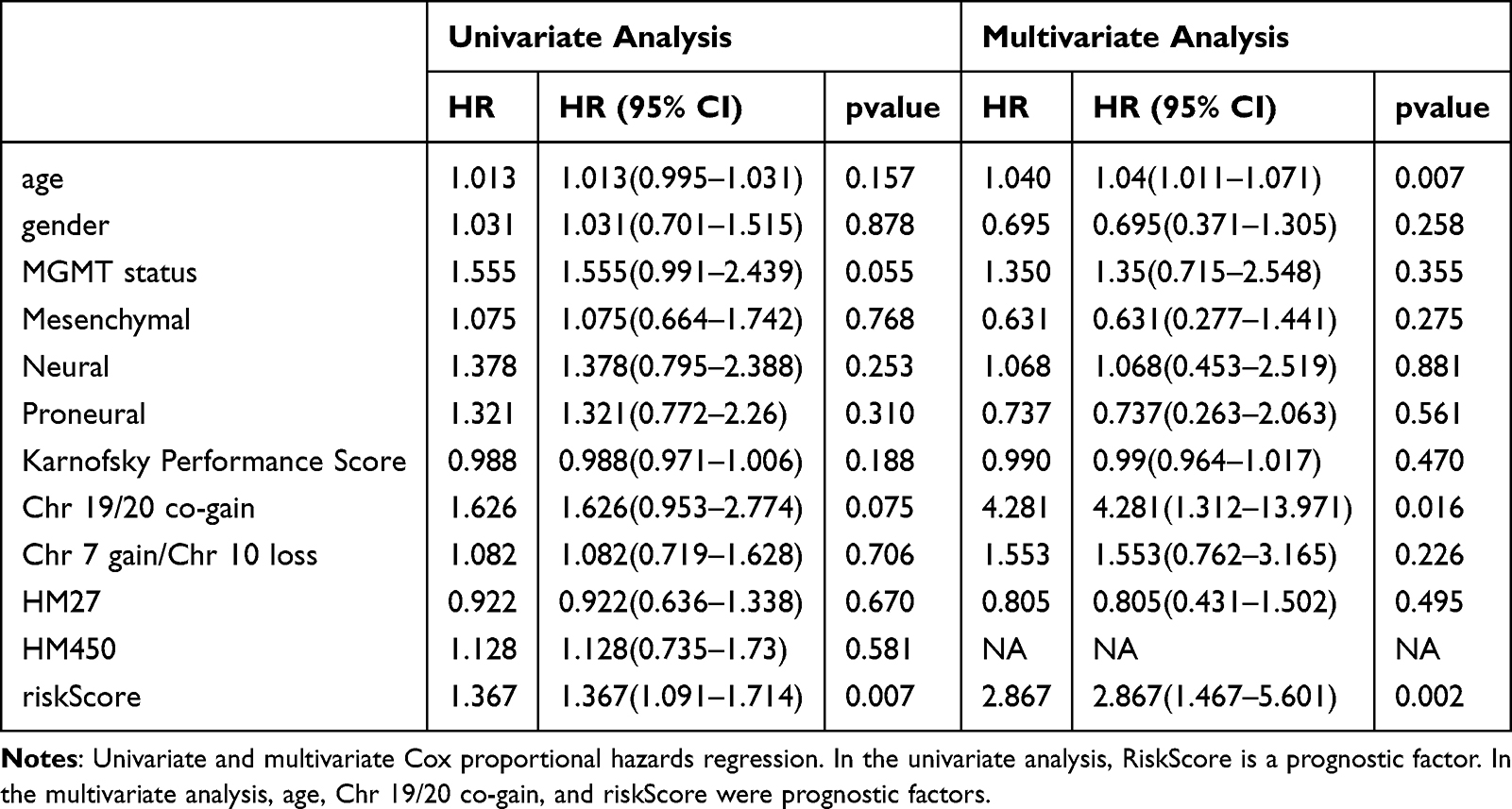 |
Table 7 Risk Score and Independent Prognostic Analysis of Clinical Factors |
 |
Figure 5 Correlation between prognostic model and clinical factors. (A and B) Categorical variables compared by χ2-test. (C and D) Univariate and multivariate Cox regression. (E) Nomogram for 1‑/2‑/3‑year survival. (F–H) Calibration curves. (I) AUC for nomogram prediction. |
Immune Microenvironment and Drug Sensitivity Associated with the Risk Signature
CIBERSORT analysis revealed significant differences in immune cell infiltration between risk groups, specifically in plasma cells, CD8+ T cells, and resting/activated memory CD4+ T cells (Figure 6A and B). Among the six biomarkers, NPC2 showed the strongest positive correlation with CD8+ T cell abundance and negative correlation with resting memory CD4+ T cells (Figure 6C). Expression of 27 immune checkpoint genes differed significantly between groups (Figure 6D). Analysis of Tumor Immune Dysfunction and Exclusion (TIDE) scores showed a negative correlation with risk scores, and the low-risk group had significantly higher TIDE scores, suggesting a potentially less favorable response to immunotherapy (Figure 6E and F). Drug sensitivity analysis predicted that low-risk patients were more sensitive to agents like AMG.706 and axitinib, whereas high-risk patients were more sensitive to drugs targeting metabolic and signaling pathways such as GDC0941 and DMOG (Supplementary Figure 4A). The sensitivity of several drugs (eg., AMG.706, DMOG) showed significant correlation with the risk score (Supplementary Figure 4B).
 |
Figure 6 Immune-related analysis. (A) Immune cell scores inferred by CIBERSORT. (B) Differences between risk groups by Wilcoxon test (P < 0.05) *P < 0.05, **P < 0.01. (C) Spearman correlation (**|cor| > 0.3, P < 0.05**). (D) Immune checkpoint gene expression. *P < 0.05, **P < 0.01. (E–F) TIDE score and correlation with RiskScore. **P < 0.01. |
Single-Cell Communication and Dynamics of Biomarker Expression
Cell-cell communication analysis revealed enhanced interaction networks, particularly between tumor and proliferating cells, in GBM compared to normal tissue (Figure 7A–F). The five major cell types were co-enriched in 1,709 metabolic pathways (Figure 7G and Supplementary Table 9). Sub-clustering of tumor cells identified 8 subpopulations (Figure 7H). Pseudotime trajectory analysis ordered these cells into 5 differentiation states, with subclusters 0 and 1 at the earliest stage (Figure 7I–K). Dynamic expression analysis along the pseudotime trajectory showed that HOXB2 and PEPD decreased gradually, NPC2, TMEM158 and PFDN5 increased then decreased, and CKAP4 increased progressively (Figure 7L). Cell cycle analysis revealed no pronounced heterogeneity across phases (Figure 7M).
 |
Figure 7 Tumor cell-related analyses between GBM and normal groups. (A–F) Cell–cell communication analyzed by CellChat. (G) Metabolic pathway activity. (H) UMAP clustering (resolution = 0.4). (I–K) Pseudotime trajectory by Monocle. (L) Biomarker dynamics. (M) Cell cycle scoring using Seurat. |
Validation of Biomarker Expression and Identification of CKAP4 and PFDN5 as Potential Causal Drivers
Expression of all six biomarkers (TMEM158, HOXB2, CKAP4, PEPD, PFDN5, NPC2) was significantly upregulated in GBM tumors in the TCGA dataset (Figure 8A). While univariate MR analysis indicated all six genes were significantly associated with GBM, multivariate MR analysis, accounting for pleiotropy, identified only CKAP4 and PFDN5 as retaining significant independent causal associations (Table 8 and Figure 8B). Single-cell expression analysis confirmed that these biomarkers were predominantly expressed in tumor and proliferating cells (Figure 8C). Experimental validation using RT-qPCR and Western blot on clinical GBM samples confirmed the significant upregulation of both mRNA and protein levels for all six genes compared to adjacent non-tumor tissue (Figure 8D–F). These expression findings were consistent in the external validation dataset GSE68848 (Supplementary Figure 5).
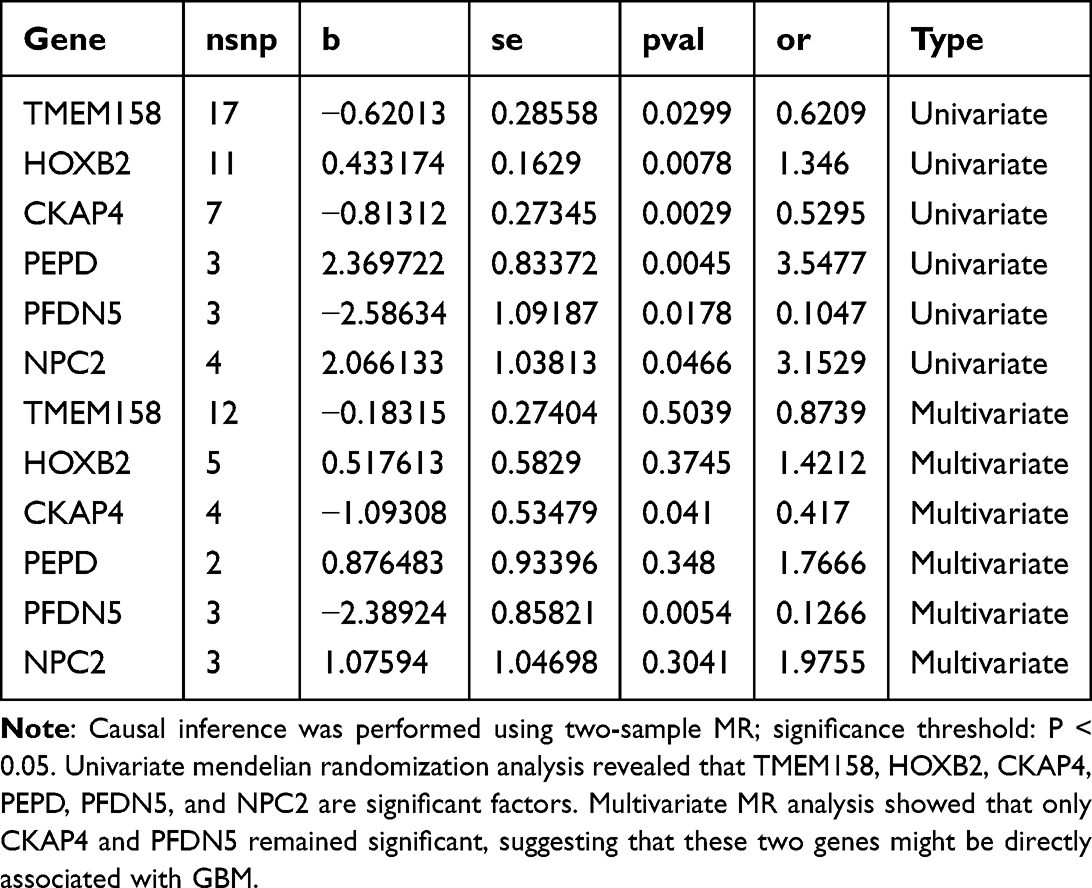 |
Table 8 Mendelian Randomization Analysis of Biomarkers |
 |
Figure 8 Expression of Prognostic Model Genes. (A) Expression in GBM vs normal using log2‑transformed counts. *P < 0.05, **P < 0.01, ***P < 0.001 (B) Multivariate Mendelian randomization to identify independent causal genes. (C) scRNA‑seq violin plots. (D) RT‑qPCR using 2−ΔΔCt method. (E and F) Western blot quantification. *p<0.05 **p<0.01. |
Discussion
Glioblastoma (GBM) is the most aggressive primary malignant tumor of the central nervous system.1 Current research predominantly relies on correlative associations, while systematic approaches that prioritize causal genetic factors and integrate multi-omics data for prediction remain limited. In this study, we integrated single-cell transcriptomics with Mendelian randomization (MR) to identify genetically implicated genes, subsequently developing and validating a prognostic signature for GBM. Through single-cell analysis, we characterized five distinct cell populations: tumor cells, proliferating cells, lymphocytes, myeloid cells, and normal cells. Furthermore, MR analysis identified 41 candidate key genes. We constructed a six-gene risk model comprising TMEM158, HOXB2, CKAP4, PEPD, PFDN5, and NPC2. The model demonstrated that elevated risk scores were significantly associated with poorer survival outcomes in GBM patients. Additionally, multivariate MR analysis suggested a potential direct causal association of CKAP4 and PFDN5 with GBM. RT-qPCR and Western blot analyses confirmed significantly upregulated mRNA and protein expression levels of these six genes in GBM tissues compared to controls. Collectively, these six genes constitute a functionally interconnected module that drives GBM progression by modulating proliferation, differentiation, protein homeostasis, immune infiltration, and oncogenic signaling pathways, thereby providing a mechanistic basis for their prognostic value.
Our single-cell analysis delineated the cellular architecture of GBM, identifying nine clusters consolidated into five major types: tumor cells, proliferating cells, myeloid cells, lymphocytes, and normal cells. The predominance of tumor and myeloid populations highlights a tumor–immune ecosystem central to GBM pathology.8,10 Annotation was validated by marker genes (TOP2A in proliferating cells, MOG in normal cells). By intersecting differentially expressed genes from single-cell (CGGA) and bulk (TCGA) datasets, we identified 2,554 consistently dysregulated genes. MR analysis established a causal relationship for 41 of these genes with GBM risk, moving beyond correlation to suggest potential drivers and protectors. Enrichment analyses (GO, KEGG) of these 41 genes revealed their involvement in viral entry/symbiosis pathways, amyloid-beta binding, helicase activity, and MAPK scaffolding, with cellular localization to the trans-Golgi network, linking them to immune evasion, proteostasis, and oncogenic signaling. Their association with arginine and proline metabolism further ties them to metabolic reprogramming and immunosuppression.
From these causal candidates, Cox and LASSO regression distilled a robust 6-gene prognostic signature (TMEM158, HOXB2, CKAP4, PEPD, PFDN5, NPC2). This signature integrates multiple pathogenic axes: transcriptional regulation (HOXB2), lipid trafficking and immune modulation (NPC2), protein folding and proteostasis (PFDN5), proteolytic metabolism (PEPD), and membrane dynamics (CKAP4, TMEM158). The six-gene prognostic signature (TMEM158, HOXB2, CKAP4, PEPD, PFDN5, NPC2) encompasses multiple layers of GBM pathogenesis. HOXB2, a homeobox transcription factor, acts as a canonical oncogene in GBM by promoting stemness and mesenchymal transition.36,37 Our observation that HOXB2 was consistently identified as a risk factor in both MR and Cox regression supports its causal role in driving poor survival. CKAP4 exerts oncogenic functions primarily by activating the PI3K/AKT pathway and inhibiting the Hippo signaling pathway, thereby accelerating proliferation, migration, and angiogenesis in high-grade gliomas.38,39 This mechanistically explains why CKAP4 emerged as an independent causal gene in multivariate MR and predicts poor prognosis.40–42 PFDN5 acts as a co-factor of c-Myc and regulates protein folding and proteostasis.43,44 Deregulation of PFDN5 disrupts Myc-driven transcriptional programs and autophagy, leading to uncontrolled proliferation and metabolic reprogramming.45,46 Our study is the first to establish a causal link between PFDN5 and GBM, likely via Myc-mediated oncogenic signaling. PEPD (prolidase) contributes to collagen metabolism and cell survival;47,48 its dysregulation promotes invasiveness and treatment resistance.49,50 NPC2 is involved in lipid trafficking and antigen presentation;51,52 its strong correlation with CD8+ T-cell infiltration suggests a critical role in shaping the immune microenvironment of GBM.53 TMEM158 is a transmembrane protein that modulates cell-matrix adhesion and signaling,54 sustaining tumor cell survival and stemness.55 Collectively, these genes form a regulatory network that drives hallmark capabilities of GBM, including uncontrolled proliferation, immune evasion, metabolic reprogramming, and therapeutic resistance. The risk score, calculated from these genes, was an independent prognostic factor. Patients stratified into high- and low-risk groups showed significant survival differences, with the high-risk group enriched for the aggressive mesenchymal subtype. The signature’s prognostic value was confirmed in an independent cohort (CGGA-325) and was successfully incorporated into a clinically applicable nomogram.
Functionally, the high-risk phenotype was characterized by an altered immune landscape and metabolic adaptation. GSVA indicated activation of chemical carcinogenesis and cytochrome P450 drug metabolism pathways, alongside inhibition of cell cycle and Notch signaling, suggesting a shift towards therapy resistance. Immune deconvolution revealed significant differences in plasma cells, CD8+ T cells, and memory CD4+ T cells between risk groups. Notably, NPC2 expression strongly correlated with CD8+ T cell infiltration. Analysis of 27 immune checkpoints and TIDE scores suggested the high-risk group might be less immune-evasive and potentially more responsive to immunotherapy, indicating the model’s utility in predicting therapeutic vulnerability. This immune stratification can be attributed to the causal genes in our signature: NPC2 modulates lymphocyte infiltration, CKAP4 promotes an immunosuppressive microenvironment, and HOXB2 drives inflammatory signaling, collectively leading to distinct immune landscapes between risk groups. Correspondingly, drug sensitivity analysis predicted risk-dependent vulnerabilities: low-risk patients to anti-angiogenics (eg., AMG.706, axitinib) and high-risk patients to metabolic/PI3K-mTOR inhibitors (eg., GDC0941, DMOG), aligning with their distinct biological states.56,57
Single-cell communication analysis revealed enhanced interactions, particularly between tumor and proliferating cells in GBM. Pseudotime trajectory analysis of tumor subpopulations showed dynamic expression of the six biomarkers during differentiation, underscoring their roles in tumor progression. Cell cycle analysis showed no pronounced heterogeneity, indicating a uniformly proliferative state.
Validation across datasets (TCGA, GSE68848) and via RT-qPCR/Western blot confirmed consistent upregulation of all six biomarkers in GBM tissue, with enrichment in tumor and proliferating cells. Multivariate MR analysis refined these associations, identifying CKAP4 and PFDN5 as having the strongest evidence for independent, direct causal effects on GBM risk, distinguishing them as prime mechanistic candidates.
The roles of CKAP4 and PFDN5 warrant specific emphasis.
CKAP4 has been validated as a driver of glioma malignancy by inhibiting Hippo signaling and activating PI3K/AKT cascades,58,59 supporting our result that CKAP4 is an independent causal gene for GBM.41 Its upregulation correlates with poor survival, and our MR analysis provides novel genetic epidemiological evidence supporting a causal role in GBM, beyond prior functional studies.42,58,59 While prior studies have established CKAP4’s functional roles in glioma biology, our work provides novel genetic epidemiological evidence supporting a causal relationship between CKAP4 and GBM risk, moving beyond correlative expression associations. This causal inference, derived from Mendelian randomization, strengthens the rationale for targeting CKAP4 in GBM and distinguishes our contribution from previous descriptive or functional studies.58,59
PFDN5 regulates protein homeostasis and interacts with c-Myc to control transcription and proliferation,43,44 representing a novel causal dependency in GBM that warrants further functional validation.45,46 To our knowledge, this is the first study to propose a direct causal link between PFDN5 and GBM, identifying it as a novel dependency.
Collectively, we propose a model where this gene module drives GBM: CKAP4 via Hippo/PI3K signaling; PFDN5 via c-Myc/proteostasis; HOXB2 via stemness/mesenchymal transition; NPC2 via immune microenvironment remodeling; PEPD via invasiveness; and TMEM158 via cell survival. This integrated signature offers a composite biomarker for prognosis. Individually, these genes nominate distinct therapeutic avenues: CKAP4 and PFDN5 for targeted therapy;43,44,58,59 HOXB2 for epigenetic targeting; NPC2 and PEPD for metabolic/microenvironment strategies; and TMEM158 for adhesion-mediated signaling. Future diagnostic and therapeutic trials could prioritize validating these genes as blood- or tissue-based biomarkers and developing corresponding targeted agents, especially against CKAP4 and PFDN5.
Our study has limitations. The retrospective design may introduce bias, requiring validation in prospective, multi-center cohorts. Experimental validation, though consistent, was performed on a limited sample set. While MR suggests causality, the precise molecular mechanisms of CKAP4 and PFDN5 require functional validation in GBM models. Future work should bridge computational discovery with mechanistic studies and clinical translation to develop precision strategies for high-risk GBM patients.
Conclusion
In conclusion, by integrating single-cell RNA sequencing with two-sample Mendelian randomization, this study systematically identified both prognostic and genetically supported causal genes in glioblastoma. We established and validated a robust 6-gene prognostic signature (TMEM158, HOXB2, CKAP4, PEPD, PFDN5, NPC2), where a higher risk score was significantly associated with poorer overall survival, mesenchymal subtype enrichment, and distinct immune microenvironment profiles. Multivariate Mendelian randomization analysis further suggested that CKAP4 and PFDN5 may exert independent causal effects on GBM risk. These findings highlight the prognostic value of the six-gene signature for patient stratification and nominate CKAP4 and PFDN5 as priority candidates for further investigation as potential therapeutic targets in GBM. Collectively, the six genes provide a valuable resource for advancing diagnostic precision and targeted therapy development in GBM.
Abbreviations
TCGA, The Cancer Genome Atla; GEO, Gene Expression Omnibu; DEGs, Diferentially Expressed Genes; LASSO, Least Absolute Shrinkage and Selection Operator; OS, overall survival; ROC, receiver operating characteristic; GSEA, gene set enrichment analysis; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; GBM, glioblastoma.
Data Sharing Statement
The data underlying this article are available in the Cancer Genome Atlas (TCGA) at http://xena.ucsc.edu/, Chinese Glioma Genome Altas (CGGA) at http://www.cgga.org.cn/, and IEU OpenGWAS at https://gwas.mrcieu.ac.uk/.
Ethics Approval and Consent to Participate
The study was approved by the Ethics Committee of the Medical Ethics Committee of First Affiliated Hospital of Guangxi Medical University and conformed to the provisions of the Declaration of Helsinki. The approval number and date of approval are as follows: [2024-E300-01] and [2024]. Informed consent was obtained from all subjects involved in the study. All methods involved in human subjects were performed in accordance with the Declaration of Helsinki.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors declare no competing interests in this work.
References
1. Ostrom Q, Price M, Neff C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2016-2020. Neuro Oncology. 2023;25(Supplement_4):iv1–24. doi:10.1093/neuonc/noad149
2. Stupp R, Mason WP, Van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med. 2005;352:987–996. doi:10.1056/NEJMoa043330
3. Smita K, Rohan G, Rashmi K,A, Pravir K. Emerging trends in post-translational modification: shedding light on glioblastoma multiforme. Biochim Biophys Acta Rev Cancer. 2023;1878. doi:10.1016/j.bbcan.2023.188999
4. Liu Y, Sinjab A, Min J, et al. Conserved spatial subtypes and cellular neighborhoods of cancer-associated fibroblasts revealed by single-cell spatial multi-omics. Cancer Cell. 2025;43. doi:10.1016/j.ccell.2025.03.004
5. Johnson K, Anderson KJ, Courtois ET, et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nature Genetics. 2021;53(10):1456–1468. doi:10.1038/s41588-021-00926-8
6. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27.
7. Susanna C,L, Adam S,B, Stephen B. Mendelian randomization for cardiovascular diseases: principles and applications. Eur Heart J. 2023;44. doi:10.1093/eurheartj/ehad736
8. Zhao S, Xie Y, Ding X, et al. Exploring the causal relationship between antihypertensive drugs and glioblastoma by combining drug target Mendelian randomization study, eQTL colocalization, and single-cell RNA sequencing. Environ Toxicol. 2024;39. doi:10.1002/tox.24210
9. Xiang L, Wei G, Chen H, Minghua W, Xiaoling S. Causal relationship between inflammatory proteins and glioblastoma: a two-sample bi‑directional mendelian randomization study. Front Genet. 2024;15. doi:10.3389/fgene.2024.1391921
10. Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: state of the art and future directions. CA Cancer J Clin. 2020;70. doi:10.3322/caac.21613
11. Yu L, Shen N, Shi Y, et al. Characterization of cancer-related fibroblasts (CAF) in hepatocellular carcinoma and construction of CAF-based risk signature based on single-cell RNA-seq and bulk RNA-seq data. Front Immunol. 2022;13. doi:10.3389/fimmu.2022.1009789
12. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15. doi:10.1186/s13059-014-0550-8
13. Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7. doi:10.7554/eLife.34408
14. Xian W, Wu D, Liu B, et al. Graves disease and inflammatory bowel disease: a bidirectional mendelian randomization. J Clin Endocrinol Metab. 2022;108. doi:10.1210/clinem/dgac683
15. Liu Z, Wang H, Yang Z, Lu Y, Zou C. Causal associations between type 1 diabetes mellitus and cardiovascular diseases: a Mendelian randomization study. Cardiovasc Diabetol. 2023;22. doi:10.1186/s12933-023-01974-6
16. Mounier N, Kutalik Z. Bias correction for inverse variance weighting Mendelian randomization. Genet Epidemiol. 2023;47. doi:10.1002/gepi.22522
17. Dai Z, Xu W, Ding R, et al. Two-sample Mendelian randomization analysis evaluates causal associations between inflammatory bowel disease and osteoporosis. Front Public Health. 2023;11. doi:10.3389/fpubh.2023.1151837
18. Xu J, Zhang S, Tian Y, et al. Genetic causal association between iron status and osteoarthritis: a two-sample mendelian randomization. Nutrients. 2022;14. doi:10.3390/nu14183683
19. Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7. doi:10.7554/eLife.34408
20. Qin Q, Zhao L, Ren A, et al. Systemic lupus erythematosus is causally associated with hypothyroidism, but not hyperthyroidism: a Mendelian randomization study. Front Immunol. 2023;14. doi:10.3389/fimmu.2023.1125415
21. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23. doi:10.1093/hmg/ddu328
22. Cui Z, Feng H, He B, He J, Tian Y. Relationship between serum amino acid levels and bone mineral density: a mendelian randomization study. Front Endocrinol. 2021;12. doi:10.3389/fendo.2021.763538
23. Yu G, Wang L, Han Y, He Q. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–287. doi:10.1089/omi.2011.0118
24. Walter W, Sánchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31. doi:10.1093/bioinformatics/btv300
25. Shi Y, Wang Y, Dong H, et al. Crosstalk of ferroptosis regulators and tumor immunity in pancreatic adenocarcinoma: novel perspective to mRNA vaccines and personalized immunotherapy. Apoptosis. 2023;28(9–10):1423–1435. doi:10.1007/s10495-023-01868-8
26. Liang L, Yu J, Li J, et al. Integration of scRNA-Seq and bulk RNA-Seq to analyse the heterogeneity of ovarian cancer immune cells and establish a molecular risk model. Front Oncol. 2021;11:711020. doi:10.3389/fonc.2021.711020
27. Ritchie M, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
28. Zhang J, Han X, Lin L, et al. Unraveling the expression patterns of immune checkpoints identifies new subtypes and emerging therapeutic indicators in lung adenocarcinoma. Oxidative Med Cell Longev. 2022;2022:3583985. doi:10.1155/2022/3583985
29. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12. doi:10.1038/nmeth.3337
30. Cheng Q, Chen X, Wu H, Du Y. Three hematologic/immune system-specific expressed genes are considered as the potential biomarkers for the diagnosis of early rheumatoid arthritis through bioinformatics analysis. J Translat Med. 2021;19(1):18. doi:10.1186/s12967-020-02689-y
31. Geeleher P, Cox N, Huang R. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One. 2014;9:e107468. doi:10.1371/journal.pone.0107468
32. Jin S, Guerrero-Juarez CF, Zhang L, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12. doi:10.1038/s41467-021-21246-9
33. Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32. doi:10.1038/nbt.2859
34. Griss J, Viteri G, Sidiropoulos K, Nguyen V, Fabregat A, Hermjakob H. ReactomeGSA - efficient multi-omics comparative pathway analysis. Mol Cell Proteomics. 2020;19. doi:10.1074/mcp.TIR120.002155
35. McTavish H. A demonstration of photosynthetic state transitions in nature: shading by photosynthetic tissue causes conversion to state 1. Photosynthesis Res. 1988;17:247–254. doi:10.1007/bf00035451
36. Miyazaki Y, Hamada J-I, Tada M, et al. HOXD3 enhances motility and invasiveness through the TGF-beta-dependent and -independent pathways in A549 cells. Oncogene. 2002;21:798–808. doi:10.1038/sj.onc.1205126
37. Bi Y, Mao Y, Su Z, et al. HOXB-AS1 accelerates the tumorigenesis of glioblastoma via modulation of HOBX2 and HOBX3 at transcriptional and posttranscriptional levels. J Cell Physiol. 2021;236(1):93–106. doi:10.1002/jcp.29499
38. Osugi Y, Fumoto K, Kikuchi A. CKAP4 regulates cell migration via the interaction with and recycling of integrin. Mol Cell Biol. 2019;39. doi:10.1128/mcb.00073-19
39. Gao L, Wang Q, Ren W, et al. The RBP1-CKAP4 axis activates oncogenic autophagy and promotes cancer progression in oral squamous cell carcinoma. Cell Death Dis. 2020;11. doi:10.1038/s41419-020-2693-8
40. Kimura H, Yamamoto H, Harada T, et al. CKAP4, a DKK1 receptor, is a biomarker in exosomes derived from pancreatic cancer and a molecular target for therapy. Clin Cancer Res. 2019;25. doi:10.1158/1078-0432.ccr-18-2124
41. Pan YB, Wang S, Yang B, et al. Transcriptome analyses reveal molecular mechanisms underlying phenotypic differences among transcriptional subtypes of glioblastoma. J Cell Mol Med. 2020;24. doi:10.1111/jcmm.14976
42. Lu GF, You CY, Chen YS, et al. MicroRNA-671-3p promotes proliferation and migration of glioma cells via targeting CKAP4. Onco Targets Ther. 2018;11. doi:10.2147/ott.s177325
43. Rothman S. How is the balance between protein synthesis and degradation achieved? Theor Biol Med Model. 2010;7. doi:10.1186/1742-4682-7-25
44. Ariga H. Common mechanisms of onset of cancer and neurodegenerative diseases. Biol Pharm Bull. 2015;38. doi:10.1248/bpb.b15-00125
45. Zhong Y, Yang L, Xiong F, et al. Long non-coding RNA AFAP1-AS1 accelerates lung cancer cells migration and invasion by interacting with SNIP1 to upregulate c-Myc. Signal Transduct Target Ther. 2021;6. doi:10.1038/s41392-021-00562-y
46. Annunziata I, Van de Vlekkert D, Wolf E, et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat Commun. 2019;10. doi:10.1038/s41467-019-11568-0
47. Yang L, Li Y, Bhattacharya A, Zhang Y. A recombinant human protein targeting HER2 overcomes drug resistance in HER2-positive breast cancer. Sci Transl Med. 2019;11. doi:10.1126/scitranslmed.aav1620
48. Surazynski A, Donald SP, Cooper SK, et al. Extracellular matrix and HIF-1 signaling: the role of prolidase. Int J Cancer. 2007;122. doi:10.1002/ijc.23263
49. Yang L, Li Y, Bhattacharya A, Zhang Y. PEPD is a pivotal regulator of p53 tumor suppressor. Nat Commun. 2017;8. doi:10.1038/s41467-017-02097-9
50. Lubick KJ, Robertson SJ, McNally KL, et al. Flavivirus antagonism of type i interferon signaling reveals prolidase as a regulator of IFNAR1 surface expression. Cell Host Microbe. 2015;18. doi:10.1016/j.chom.2015.06.007
51. Torres S, Balboa E, Zanlungo S, et al. Lysosomal and mitochondrial liaisons in niemann-pick disease. Front Physiol. 2017;8:982. doi:10.3389/fphys.2017.00982
52. Wei D, Shen S, Lin K, et al. NPC2 as a prognostic biomarker for glioblastoma based on integrated bioinformatics analysis and cytological experiments. Front Genetics. 2021;12:611442. doi:10.3389/fgene.2021.611442
53. Wei D, Shen S, Lin K, et al. NPC2 as a prognostic biomarker for glioblastoma based on integrated bioinformatics analysis and cytological experiments. Front Genet. 2021;12. doi:10.3389/fgene.2021.611442
54. Marx S, Dal Maso T, Chen J-W, et al. Transmembrane (TMEM) protein family members: poorly characterized even if essential for the metastatic process. Seminars Cancer Biol. 2020;60:96–106. doi:10.1016/j.semcancer.2019.08.018
55. Barradas M, Gonos ES, Zebedee Z, et al. Identification of a candidate tumor-suppressor gene specifically activated during Ras-induced senescence. Experi Cell Res. 2002;273(2):127–137. doi:10.1006/excr.2001.5434
56. Zhou Q. BMS-536924, an ATP-competitive IGF-1R/IR inhibitor, decreases viability and migration of temozolomide-resistant glioma cells in vitro and suppresses tumor growth in vivo. Onco Targets Ther. 2015;8. doi:10.2147/ott.s80047
57. Halvorson KG, Barton KL, Schroeder K, et al. A high-throughput in vitro drug screen in a genetically engineered mouse model of diffuse intrinsic pontine glioma identifies BMS-754807 as a promising therapeutic agent. PLoS One. 2015;10. doi:10.1371/journal.pone.0118926
58. Luo T, Ding K, Ji J, et al. Cytoskeleton-associated protein 4 (CKAP4) promotes malignant progression of human gliomas through inhibition of the Hippo signaling pathway. J Neurooncol. 2021;154. doi:10.1007/s11060-021-03831-6
59. Xu K, Zhang K, Ma J, et al. CKAP4-mediated activation of FOXM1 via phosphorylation pathways regulates malignant behavior of glioblastoma cells. Transl Oncol. 2023;29. doi:10.1016/j.tranon.2023.101628
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.