Back to Journals » Cancer Management and Research » Volume 12
Melatonin Regulates Cisplatin Resistance and Glucose Metabolism Through Hippo Signaling in Hepatocellular Carcinoma Cells
Received 10 September 2019
Accepted for publication 11 January 2020
Published 12 March 2020 Volume 2020:12 Pages 1863—1874
DOI https://doi.org/10.2147/CMAR.S230466
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Lina Mi,1 Hongyu Kuang2
1Department of Gastroenterology, The Fourth Affiliated Hospital of Harbin Medical University, Harbin 150001, Heilongjiang Province, People’s Republic of China; 2Department of Endocrinology, The First Affiliated Hospital of Harbin Medical University, Harbin 150001, Heilongjiang Province, People’s Republic of China
Correspondence: Hongyu Kuang
Department of Endocrinology, The First Affiliated Hospital of Harbin Medical University, No. 23, Youzhen Street, Nangang District, Harbin 150001, Heilongjiang Province, People’s Republic of China
Email [email protected]
Introduction and Aim: Hepatocellular carcinoma (HCC) is a primary malignancy that occurs in the liver. Clinical cases have been recorded worldwide, particularly in the Saharan area and Asia. In the present work, we aimed to probe the characteristics of melatonin involved in human HCC development, especially in cisplatin resistance and glucose metabolism.
Methods: Two HCC cells, HepG2 and Hep3B cells, were treated with melatonin. Cell cycle test was then used to define the role of melatonin in cell progression while Western blotting and qPCR assay were applied to determine the associated proteins in the treatment. Annexin V/PI staining and MTT assay was used to probe the involvement of melatonin in cisplatin-induced cell apoptosis process. Successively, we assessed glucose consumption in melatonin treated cells along with Western blotting for detection of GLUT-3 expression level. Yes-associated protein (YAP), a key regulator of Hippo signaling pathway, was further examined to characterize the function of melatonin on adjusting GLUT3 and Bcl-2 expression.
Results: Melatonin enabled inhibition of HepG2 and Hep3B proliferation and cell cycle progression via affecting the cell cycle-associated proteins. Annexin V/PI staining and MTT assay results demonstrated that melatonin assisted cisplatin-induced apoptosis accompanied with upregulated caspase-3 and poly ADP-ribose polymerase (PARP) cleavage, as well as Bcl-2 expression. It revealed that melatonin inhibits glucose uptake and ATP production via downregulation of Glucose transporter 3 (GLUT3). In addition, YAP was downregulated by melatonin treatment. The YAP depletion in HepG2 and Hep3B cells suppressed mRNA and protein expression of Bcl-2 and GLUT3, whereas overexpression of YAP in melatonin treated cells partly reversed the melatonin-induced inhibition on proliferation, cisplatin-induced apoptosis, and GLUT3 and Bcl-2 expression.
Conclusion: Melatonin hindered HCC proliferation and aided cisplatin resistance via regulating the Hippo signaling pathway.
Keywords: melatonin, hepatocellular carcinoma, proliferation, glucose metabolism, YAP, apoptosis
Introduction
Hepatocellular carcinoma has been considered as one common liver cancer due to vast increasing mortality among patients with chronic liver disease. Primary treatments including surgery and chemotherapy were supposed to prolong survival time. Chemotherapeutic drugs often undermine HCC patients’ life quality. A resurgence of interest on novel therapeutic approaches on HCC treatment occurs in recent studies. The development of bio-essential and non-toxic medicine for chemotherapy is suggestive of new alternative plan to overcome the limitations during the current chemotherapy and maintain the life expectancy among patients.
Melatonin (MEL) is a hormone secreted by the pineal gland and delivered light/dark cycle information to the circadian system.1–3 The characterized properties are known as circadian maintenance and oxidized cell damage reduction.4,5 Cisplatin is developed from platinum-based compounds and constitutively applied in many cancers as the first-line chemotherapeutic treatment with valid clinical activity against a wide spectrum of cancers, for instances, liver cancer, ovarian and small cell lung cancer.6 However, side effects can lead to severe cytotoxicity against normal cells and tissues as well as cause high incidence of chemoresistance.7 Combination studies of MEL and chemotherapy, likely cisplatin against cancer cells, reported previously indicating its ability to enhance chemotherapy-induced apoptosis in cancer cells, such as cisplatin-treated SK-OV-3 human ovarian cancer cells,8 doxorubicin-treated HepG2 and Bel-7402 human hepatoma cells,9 SK-N-MC human Ewing sarcoma cell line,10 and AR42J rat pancreatic tumor cell line.11 It revealed that MEL inhibited the proliferation, migration, and invasion of cancer cells.12–14 MEL were identified to involve in diverse signaling pathways including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), AKT, signal transducer, and activator of transcription 3 (STAT3) and hypoxia-inducible factors 1 (HIF1) pathways.15–18 In addition, conventional chemotherapy that accompanied with MEL treatment extended patients’ survival, indicating the potential beneficial effect in anti-cancer therapy.19
Hippo signaling pathway mediates tissue growth via suppressing cell proliferation and promoting apoptosis.20 Growing evidences revealed a dysregulation of Hippo signaling during human cancer development.21–23 Yes-associated protein (YAP), a key downstream effector of Hippo, has been widely investigated in human disease studies. Ectopic overexpression of YAP1 in the mouse liver leads to HCC development.24,25 This oncogenic function is further supported by its upstream regulators, which inhibit YAP1 activity by phosphorylation. In addition, knockout of Sav1 and Mst1/2 in the liver can cause HCC development as well.26,27 Meanwhile, Oroxylin A ameliorated angiogenesis in liver fibrosis by inhibiting Hippo-YAP signaling.28 Glypican-3 targeting HN3 and YAP was shown to significantly inhibit the growth of HCC cells and xenograft tumors in nude mice.29 A previous study described that MEL protects against lung fibrosis by regulating the Hippo/YAP pathway.30 Albeit closely related evidence, whether MEL was associated with Hippo signaling pathway in human cancers, in particular, HCC remains unclear. In order to address the question, two HCC cell lines were used in this study to investigate the role of MEL in HCC cell proliferation, glucose metabolism, and cisplatin-induced apoptosis.
Materials and Methods
Cell Culture and Chemicals
HCC cell lines, HepG2 and Hep3B, were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in RPMI-1640 (Invitrogen; Thermo Fisher Scientific, Carlsbad, CA, USA) with 10% Fetal bovine serum (FBS, Invitrogen; Thermo Fisher Scientific, Carlsbad, CA, USA). The cultures were maintained at 37°C in a humidified incubator containing 5% (v/v) CO2 in air. Cells were seeded at a density of 1×106 cells/mL in six-well plates. MEL and cisplatin were purchased from Sigma–Aldrich (St. Louis, MO, USA). pcDNA3.1-hYAP plasmid was transfected into cells using Attractene Transfection Reagent. The sequence of YAP siRNA was GACAUCUUCUGGUCAGAGA and Dharmafect 1 was used for siRNA transfection. Trypsin, antibiotics, PBS, and propidium iodide were purchased from ThermoFisher. DMSO was obtained from Merck.
MTT Assay
Cells (5×103/well) were plated in 96-well plates and cultured overnight. MTT solution (volume, 20 μL; concentration, 5 mg/mL; Sigma, St. Louis, MO, USA) was added to each well and then incubated for another 4 h. MEL (in DMSO) was serially diluted to 1mM and 2mM and incubated for 24 or 48 hrs, cisplatin was serially diluted to 5 μM. The supernatant was removed and DMSO (150 μL) was added for proliferation and viability measurement preparation. Absorbance was measured at 490 nm. Data were obtained from triplicate wells per condition.
Cell viability (IC50) was determined for each agent by calculating the slope and intercept of different concentrations. The experiment was performed in triplicate, and repeated three times.
Flow Cytometry for Cell Cycle and Apoptosis Analysis
Cells in six-well plates were collected using 0.25% trypsin for 24 hrs after MEL treatment. After the washing protocol (PBS), cells were re-suspended in 250 μL of binding buffer and then fixed in 1% paraformaldehyde for 24 hrs. For staining and cell cycle analysis, 5 mg/mL propidium iodide was used. Annexin V/PI kit (ab14085, Abcam) was applied to cells for cell apoptosis analysis. Stained cells were analyzed by ACEA Flow cytometer (ACEA NovoCyte® 1000).
Western Blot
Total protein was extracted using lysis buffer (ThermoFisher, Rockford, IL) and Bradford method was used to quantify the protein. When SDS-PAGE assay was performed, 30 g of the protein was separated and then transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). For primary antibody incubation, cleaved caspase 3, cleaved YAP, Bcl-2, GLUT-3 (1:1000; Cell Signaling Technology, Danvers, MA, USA), and GAPDH (1:2000; Cell Signaling Technology, Danvers, MA, USA) were incubated overnight at 4°C. For secondary antibody incubation, peroxidase-coupled anti-mouse/rabbit IgG (1:1000, Cell Signaling Technology, USA) was incubated at 37°C for 2 hrs. The result was visualized using ECL (Thermo, Rockford, IL) and detected using a DNR BioImaging System (DNR, Jerusalem, Israel). The quantification of WB band density was performed by using Photoshop CS6 (Adobe).
Real-Time Quantitative PCR
Total RNA was isolated from HepG2 and Hep3B cells using TRIzol reagent (Life technology, St. Louis, MO, USA) following the manufacturer’s instructions. Then, reverse transcription of total RNA into cDNA using PrimerScript RT Master Mix kit (TakaRa, Dalian, China). Briefly, a total 20 μL of reverse-transcription reaction solution was prepared containing 4 μL of 5X RT Master Mix and 800 ng RNA and the mixture was reacted at 85°C for 2 mins and 37°C for 30 mins. PCR was performed using 7500 Realtime PCR System (Applied Biosystems) and SYBR Green master mix kit (Applied Biosystems). The fold change of target gene expression was assessed by the 2−ΔΔCt method. GAPDH was used as the reference gene. Experiments were repeated in triplicate. Primer sequences were listed as follows: CYCLIN D1 forward TGG AGG TCT GCG AGG AAC A, CYCLIN D1 reverse TTC ATC TTA GAG GCC ACG AAC AT; GAPDH forward AAG ATC ATC AGC AAT GCC TCC T, GAPDH reverse TGG TCA TGA GTC CTT CCA CGA T. Thermal cycling conditions were as follows: 95°C for 10 mins; followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Each PCR reaction was followed by continuous melt curve analysis.
ATP Production and Glucose Consumption
The ATP level in cell lines was determined using the ATP Bioluminescence Assay Kit. Harvested cultured cells were lysed and followed with a centrifugation at 10,000g for 2 min at 4°C. The measurement of ATP level was conducted via a mixture of 50 μL of the supernatant with 50 μL of luciferase reagent, which catalyzed the light production from ATP and luciferin. The emitted light correlated to the ATP concentration in a linear regression and measured using a microplate luminometer.
Glucose levels were assessed using glucose assay kit (Biovision). Assays were performed according to the manufacture instruction; cells were collected into centrifugal tube and collected supernatant after centrifugation. Glucose consumption was calculated as the difference in glucose concentration between the original supernatant and the supernatant from the cell cultures. Absorbance was measured at 563 nm using a Spectra Max M5 plate reader (Molecular Devices, LLC, Sunnyvale, CA, USA). Absorbance was measured at 490 nm. Data were acquired from triplicate wells per condition.
Immunofluorescence Assay
HepG2 and Hep3B were seeded into 30 mm dishes containing 13 mm, collagen-coated coverslips (Hurst Scientific, WA). After 24 h cells were fixed by incubation with PBS containing 4% (w/v) paraformaldehyde (Merck, #104005100) for 15 min at room temperature. After washing twice with PBS for 5 min, fixed cells were blocked and permeabilized by incubation with PBS containing 5% (w/v) BSA and 0.3% (v/v) Triton X-100 for 1 h at room temperature. Primary antibodies and secondary antibodies were diluted in 1% (w/v) BSA/0.3% (v/v) Triton X-100/PBS and applied to coverslips and incubated at 4°C overnight. Cells were washed twice with PBS, before 0.3 μM Hoechst stain diluted in PBS was applied for 5 mins to stain nuclei. Cells were washed a further three times before coverslips were mounted onto glass slides using Gelvatol mounting medium (10.5% (w/v) polyvinyl alcohol, 21% (v/v) glycerol, 0.106 M Tris pH 8.5, sodium azide). Slides were viewed using a Leica SP8 microscope, and images were captured using LasX Controller software (Leica microsystem, 3.7.0).
Statistical Analysis
Statistical analyses were performed using SPSS version 16 for Windows. The Student’s t-test and one-way analysis of variance (ANOVA) with tukey post hoc test was used to compare differences between two groups or among multiple groups. Data was expressed as Mean ± SD. p<0.05 was considered to indicate statistical significance.
Results
MEL Inhibited HCC Proliferation and Cell Cycle Progression
HepG2 and Hep3B cells were treated with MEL at 1mM and 2mM concentration as referred to previous study.31 MTT assay showed that proliferation rates of HepG2 and Hep3B cells were decreased significantly by MEL in a concentration related property at 72 and 96 hrs post treatment (Figure 1A). The IC50 values of MEL for cell viability inhibition in HepG2 and Hep3B cells were determined as 2.3 and 2.9 mM, respectively. Cell cycle analysis indicated that HCC cells G1 percentage enhanced and S phase declined after MEL (2mM) treatment (Figure 1B), implying MEL arrested HCC cells at G1 phase.
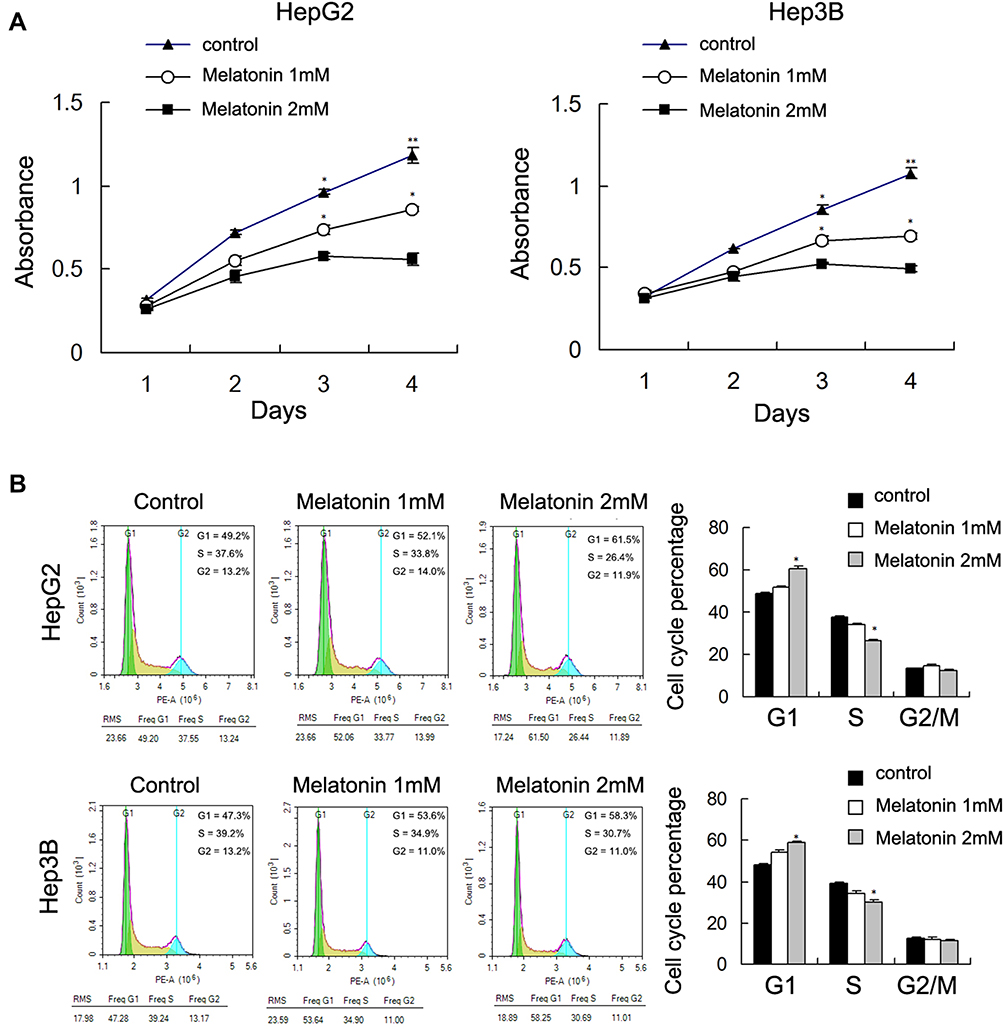 |
Figure 1 MEL inhibited hepatocellular carcinoma cell proliferation and cell cycle progression. (A) HepG2 and Hep3B cells were treated with MEL at the concentration of 1mM and 2mM. MTT assay showed that proliferation rates of the two cells were decreased significantly when treated with two concentrations of MEL. (B) Cell cycle analysis indicated that G1 percentage increased while S phase decreased after MEL treatment for 48 h. N = 3, *p < 0.05, **p < 0.01 comparing with control group. |
MEL Regulated Cell Cycle-Related Proteins
As Cyclin D1 were proposed as essential cycle-related protein in cells32–36. Western blot and qPCR were used to determine the role of MEL on Cyclin D1 expression. Cells were treated with 1mM or 2mM MEL. Western blotting analysis demonstrated that MEL incubation observably inhibited the expression of Cyclin D1 in both HepG2 and Hep3B cell lines. qPCR results confirmed similar phenomenon that MEL exposure led to a reduction of Cyclin D1 associated mRNA, in both HepG2 and Hep3B cells (Figure 2A and B).
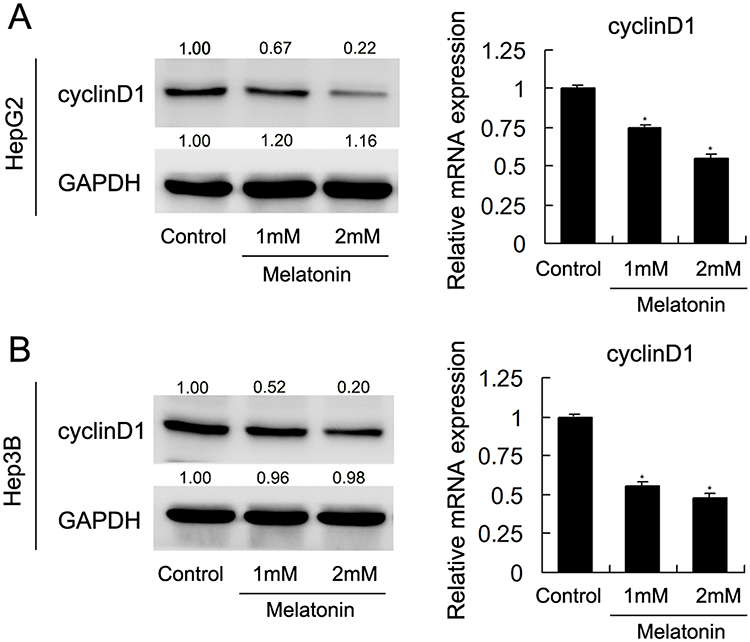 |
Figure 2 MEL regulated cell cycle-related proteins. (A) Western blot and qPCR results showed that MEL inhibited Cyclin D1 at protein and mRNA level in HepG2 and Hep3B cells. (B) Western blot and qPCR results showed that MEL suppressed protein and mRNA expression of Cyclin D1 in HepG2 and Hep3B cells. N = 3, *p < 0.05, **p < 0.01 comparing with control group. |
MEL Regulated Cisplatin-Induced Cell Apoptosis and Related Proteins
To explore how MEL affects cisplatin-mediated apoptosis of HCC cells, HepG2 and Hep3B cells were treated with cisplatin (5μM) with MEL (1, 2mM) and then examined by MTT assay. Cell viability was markedly decreased after treated with MEL in both two cell lines (Figure 3A). The IC50 values of cisplatin in HepG2 and Hep3B cells were determined as 13.6 and 11.0 μM, respectively, while co-treatment of MEL reduced IC50 to 3.6 μM and 1.8 μM in HepG2 and Hep3B cell, respectively. Thus, we examined apoptosis rate of HCC cells using Annexin V/PI kit. As exhibited in Figure 3A and B, MEL promoted apoptosis in dose-related manner in cisplatin-treated HCC cells (Figure 3B).
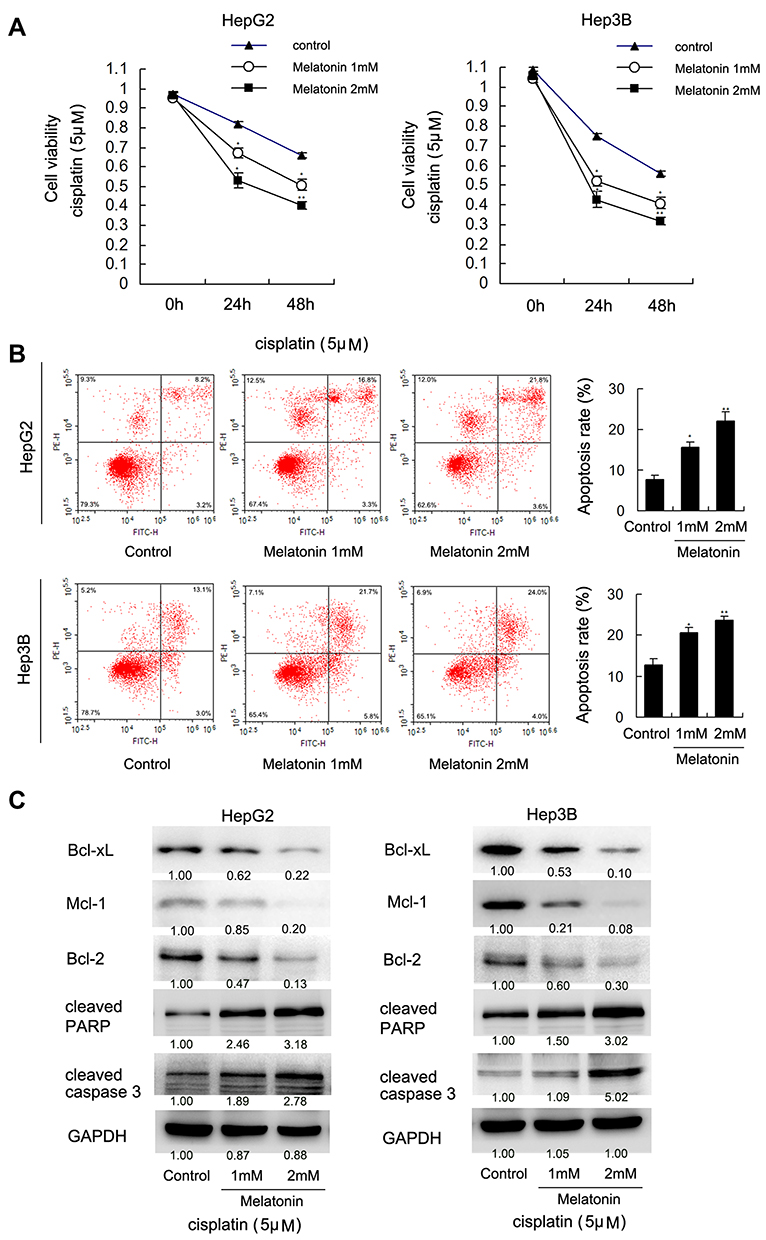 |
Figure 3 MEL promoted cisplatin-induced apoptosis. (A) HepG2 and Hep3B cells were treated with cisplatin (5 μM) together with MEL (1, 2 mM). MTT assay showed that cell viability was decreased significantly after treated with MEL in both two cell lines. (B) Annexin V/PI staining result demonstrated that MEL significantly increased apoptosis rate in cisplatin-treated hepatocellular carcinoma cells. The upper right quadrant of each plot depicts early apoptotic population of cells. (C) western-blot result showed that MEL upregulated cleaved caspase 3, PARP, Bcl-xL, Mcl-1, and Bcl-2 level. N = 3, *p < 0.05, **p < 0.01 comparing with control group. |
Next, apoptosis-related protein, cleaved PARP and caspase 3, as well as Bcl-xL, Mcl-1, and Bcl-2 was examined by immunoblotting, it demonstrated that MEL upregulated cleaved PARP and caspase 3, nonetheless downregulated Bcl-xL, Mcl-1, and Bcl-2 expression in both two cell lines with cisplatin treated (Figure 3C).
MEL Involved in ATP Production, Glucose Metabolism, and YAP Expression
Glucose metabolism and ATP production associated in cancer cell progression and chemoresistance. Glucose uptake is a vital step during glucose metabolism, MEL treatment (1 mM) reduced glucose consumption in HCC cells, compared with control group at 72 hrs post treatment (Figure 4A). Western blot data indicated that MEL reduced GLUT-3 expression (Figure 4B). Aerobic glycolysis of glucose supplied energy in cancer cells. ATP production is likely to influence on aerobic glycolysis and our data demonstrated that MEL significantly reduced ATP production (Figure 4C).
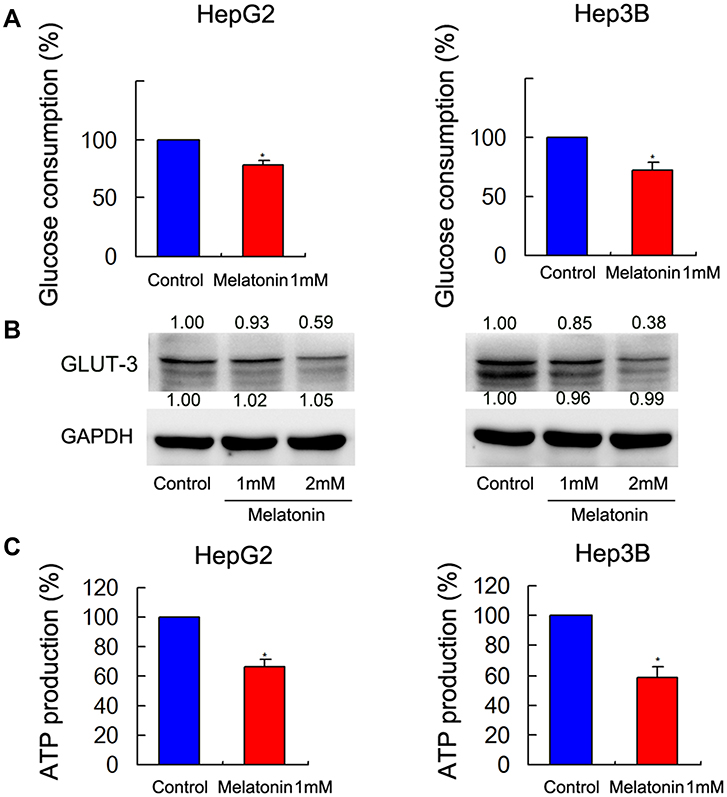 |
Figure 4 MEL regulated ATP production, GLUT-3 expression, and glucose metabolism. (A) MEL treatment (1 mM, 24 hrs) reduced glucose consumption compared in the two cell lines. (B) Western blot analysis revealed that MEL treatment reduced expression of Bcl-2 and GLUT-3 in the two cell lines. (C) MEL (1 mM, 24 h) reduced cancer cell ATP production in both two cell lines. N = 3, *p < 0.05 comparing with control group. |
YAP Regulated Proteins Associated with Glucose Metabolism and Chemoresistance
Furthermore, it demonstrated that MEL enabled to decrease YAP expression, while increasing Ser127 phosphorylation of YAP in HepG2 and Hep3B cell lines (Figure 5A and B). Immunofluorescence assay showed that MEL treatment increased cytoplasmic distribution of YAP in the in HepG2 and Hep3B cell lines, while YAP majorly located in nucleus in the control group (Figure 5C and D), suggesting MEL regulated in Hippo pathway.
 |
Figure 5 MEL associated with YAP. (A, B) Western blot showed that MEL treatment reduced expression of YAP and increased Ser127 phosphorylation of YAP in both cell lines. (C, D) Immunofluorescence assay observed the cellular localization of YAP after MEL treatment in HepG2 and Hep3B cells. |
Previous studies suggested a complex correlation existed among Hippo signaling, glucose metabolism, and cancer cell apoptosis. Bcl-2 and GLUT-3 were identified as target genes of Hippo signaling pathway.21–23 We examined the expression of Bcl-2 and GLUT-3 upon YAP silencing. After we depleted YAP by transfection of HCC cell with YAP siRNA, it revealed that YAP depletion downregulated Bcl-2 and GLUT-3 (Figure 6A and B), suggesting these two proteins served as downstream targets of YAP in HepG2 and Hep3B cells.
 |
Figure 6 YAP silencing regulated GLUT-3 and Bcl-2 level. (A, B) Western blot showed that YAP depletion downregulated Bcl-2 and GLUT-3 in both cell lines. |
Overexpression of YAP Reverses MEL Effect on Cells
To validate whether MEL inhibited cell growth and chemoresistance through regulation of YAP, we overexpressed YAP using transfection and compared with control cells. YAP overexpression can partially restore cell viability and reduced apoptosis rate that was influenced by MEL in two cell lines (Figure 7A). YAP overexpression also rescues the phenotype of cell cycle which was arrested by MEL, as evidenced by reduced G1 phase percentage and restored S phase percentage (Figure 7B). In addition, YAP overexpression rescues the decrease of GLUT-3, cyclin D1 as well as Bcl-2 expression (Figure 7C). These data revealed that MEL regulated HCC cell proliferation to assist chemosensitivity via modulation of Hippo signaling pathway.
 |
Figure 7 Overexpression of YAP reversed the role of MEL. (A) YAP overexpression partly reduced apoptosis rate induced by cisplatin in both cell lines. (B) Cell cycle analysis indicated that G1 percentage decreased while S phase increased after YAP overexpression for 48 h. (C) Western blot showed that YAP overexpression restored Cyclin D1, Bcl-2, and GLUT-3 in both cell lines. N = 3, *p < 0.05 comparing with control group. |
Discussion
In the work, MEL inhibited the proliferation of HepG2 and Hep3B cells. Cell cycle progression was arrested at G0/G1 point with downregulation of Cyclin D1 by MEL. Cyclin D1 overexpression has been proved to associate with early cancer onset and tumor progression33 and led to oncogenesis by an enhancement of anchorage-independent growth and VEGF-dependent angiogenesis.33,37 The overexpression enabled to result in down-regulation of Fas, which, in turn, induced an increased chemotherapeutic resistance and a protection from apoptosis.
MEL treatment promoted cisplatin-induced apoptosis and downregulated Bcl-2 expression.38 It also hindered glucose metabolism of HCC cells via a decline of GLUT-3 expression. It was appealing that MEL downregulated Hippo inhibitor YAP, a central effector in Hippo signaling pathway.39,40 Both Bcl-2 and GLUT-3 were regulated by YAP. The restoration of YAP partially undermined the inhibitive effect of MEL on HCC cells, suggesting MEL involved in Hippo signaling. Accumulating studies also implied that MEL enhances cisplatin anti-cancer effect in many cancer cells by stimulating mitochondrial ROS generation,41 this aspect requires further investigation.
The treatment of MEL reduced HCC cell growth and mediated cell cycle-related protein expression. Similar feature has been defined in various cancer cell lines. In previous studies, detailed linkage of MEL to Hippo signaling was not investigated. Hippo signaling pathway maintained tissue growth balance by cell proliferation inhibition and promoting apoptosis.42–44 Dysregulation of Hippo signaling were identified during malignancy formation,32,45 including HCC.33 YAP is a crucial effector in Hippo signaling. Overexpression of YAP was detected in several cancers46 and enabled to aid cell cycle progression via inducing cyclin proteins' expression and inhibiting p21.47,48 In the present study, YAP silencing caused a reduced expression of Bcl-2 and GLUT-3, while its overexpression counteracted the MEL suppressive properties on HCC cells, indicating that MEL inhibited HCC growth partly through its inhibiting effect on YAP.
In addition, MEL promoted apoptosis accompanied with downregulation of Bcl-2, and upregulated cleaved caspase 3 and PARP. Bcl-2 was identified as a YAP downstream protein.47 In our work, YAP depletion downregulated Bcl-2 protein expression as a similar effect as MEL treatment mediated. Notably, YAP overexpression partially restored cell viability and reduced cisplatin-induced apoptosis. These results directed that MEL was capable of maintaining chemosensitivity through inhibition of YAP/Bcl-2 signaling.
We also determined that MEL suppressed glucose metabolism with downregulation of GLUT-3. One typical feature of cancer metabolism is likely to alter energy production from oxidative phosphorylation to glycolysis. Elevated glucose uptake, glucose consumption, and lactate production were probed in various cancers, these biological events were defined as an essential process in cancer cell survival and resistance to chemotherapy. Upregulation of rate-limiting glucose metabolic enzymes including GLUT family protein associated with elevated glucose consumption and ATP production of cancer cells.49 In addition, the previous study revealed that GLUT-3 is a target of YAP.50 Thus, our results undoubtedly confirmed that MEL treatment suppressed HCC glucose metabolism via YAP/GLUT-3 signaling pathway.
Taken together, MEL inhibited HCC cell proliferation and glucose metabolism, promoted cisplatin-induced apoptosis. It suppressed YAP expression and the downstream targets, Bcl-2 and GLUT-3. Overexpression of YAP reversed the biological effect of MEL, suggesting that MEL possessed a promising potential as non-toxic drug candidate for HCC therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Anisimov VN. Effects of exogenous MEL—a review. Toxicol Pathol. 2003;31(6):589–603. doi:10.1080/01926230390257885
2. Karasek M, Pawlikowski M. Pineal gland, melatonin and cancer. NEL review. Neuro Endocrinol Lett. 1999;20(3–4):139–144.
3. Patel J. Melatonin. Pineal gland hormone--a brief review. Indian J Med Sci. 1998;52(12):567–568.
4. Okatani Y, Wakatsuki A, Watanabe K, et al. Melatonin inhibits vasospastic action of oxidized low-density lipoprotein in human umbilical arteries. J Pineal Res. 2000;29(2):74–80. doi:10.1111/jpi.2000.29.issue-2
5. Wakatsuki A, Okatani Y, Ikenoue N, et al. Melatonin protects against oxidized low-density lipoprotein-induced inhibition of nitric oxide production in human umbilical artery. J Pineal Res. 2001;31(3):281–288. doi:10.1111/jpi.2001.31.issue-3
6. Ishikawa T, Higuchi K, Kubota T, et al. Prevention of intrahepatic distant recurrence by transcatheter arterial infusion chemotherapy with platinum agents for stage I/II hepatocellular carcinoma. Cancer. 2011;117(17):4018–4025. doi:10.1002/cncr.v117.17
7. Huang T-H, Chiu Y-H, Chan Y-L, et al. Antrodia cinnamomea alleviates cisplatin-induced hepatotoxicity and enhances chemo-sensitivity of line-1 lung carcinoma xenografted in BALB/cByJ mice. Oncotarget. 2015;6(28):25741. doi:10.18632/oncotarget.v6i28
8. Kim J-H, Jeong S-J, Kim B, et al. Melatonin synergistically enhances cisplatin-induced apoptosis via the dephosphorylation of ERK/p90 ribosomal S6 kinase/heat shock protein 27 in SK-OV-3 cells. J Pineal Res. 2012;52(2):244–252. doi:10.1111/jpi.2012.52.issue-2
9. Fan LL, Sun GP, Wei W, et al. Melatonin and Doxorubicin synergistically induce cell apoptosis in human hepatoma cell lines. World J Gastroenterol. 2010;16(12):1473. doi:10.3748/wjg.v16.i12.1473
10. Casado-Zapico S, Rodriguez-Blanco J, GarcÃ-a-Santos G, et al. Synergistic antitumor effect of melatonin with several chemotherapeutic drugs on human Ewing sarcoma cancer cells: potentiation of the extrinsic apoptotic pathway. J Pineal Res. 2010;48(1):72–80. doi:10.1111/jpi.2009.48.issue-1
11. Uguz AC, Cig B, Espino J, et al. Melatonin potentiates chemotherapy-induced cytotoxicity and apoptosis in rat pancreatic tumor cells. J Pineal Res. 2012;53(1):91–98. doi:10.1111/jpi.2012.53.issue-1
12. Cos S, Sanchez-Barcelo E. Differences between pulsatile or continuous exposure to melatonin on MCF-7 human breast cancer cell proliferation. Cancer Lett. 1994;85(1):105–109. doi:10.1016/0304-3835(94)90245-3
13. Cos S, Sanchez-Barcelo E. Melatonin inhibition of MCF-7 human breast-cancer cells growth: influence of cell proliferation rate. Cancer Lett. 1995;93(2):207–212. doi:10.1016/0304-3835(95)03811-A
14. Liburdy R, Sloma TR, Sokolic R, et al. ELF magnetic fields, breast cancer, and melatonin: 60 Hz fields block melatonin’s oncostatic action on ER + breast cancer cell proliferation. J Pineal Res. 1993;14(2):89–97. doi:10.1111/jpi.1993.14.issue-2
15. Carbajo-Pescador S, Ordoñez R, Benet M, et al. Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br J Cancer. 2013;109(1):83. doi:10.1038/bjc.2013.285
16. Gao Y, Xiao X, Zhang C, et al. Melatonin synergizes the chemotherapeutic effect of 5-fluorouracil in colon cancer by suppressing PI3K/AKT and NF-κB/iNOS signaling pathways. J Pineal Res. 2017;62(2):e12380. doi:10.1111/jpi.2017.62.issue-2
17. León J, Casado J, Jiménez Ruiz SM, et al. Melatonin reduces endothelin-1 expression and secretion in colon cancer cells through the inactivation of FoxO-1 and NF-κβ. J Pineal Res. 2014;56(4):415–426. doi:10.1111/jpi.2014.56.issue-4
18. Shen Y-Q, Guerra-Librero A, Fernandez-Gil BI, et al. Combination of melatonin and rapamycin for head and neck cancer therapy: suppression of AKT/mTOR pathway activation, and activation of mitophagy and apoptosis via mitochondrial function regulation. J Pineal Res. 2018;64(3):e12461. doi:10.1111/jpi.2018.64.issue-3
19. Lissoni P, Chilelli M, Villa S, et al. Five years survival in metastatic non-small cell lung cancer patients treated with chemotherapy alone or chemotherapy and melatonin: a randomized trial. J Pineal Res. 2003;35(1):12–15. doi:10.1034/j.1600-079X.2003.00032.x
20. Deel MD, Li JJ, Crose LES, et al. A review: molecular aberrations within hippo signaling in bone and soft-tissue sarcomas. Front Oncol. 2015;5:190. doi:10.3389/fonc.2015.00190
21. Hao J, Zhang Y, Jing D, et al. Role of Hippo signaling in cancer stem cells. J Cell Physiol. 2014;229(3):266–270. doi:10.1002/jcp.24455
22. Wang Y, Ding W, Chen C, et al. Roles of Hippo signaling in lung cancer. Indian J Cancer. 2015;52(5):1. doi:10.4103/0019-509X.168949
23. Ramos A, Camargo FD. The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 2012;22(7):339–346. doi:10.1016/j.tcb.2012.04.006
24. Dong J, Feldmann G, Huang J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–1133. doi:10.1016/j.cell.2007.07.019
25. Camargo FD, Gokhale S, Johnnidis JB, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17(23):2054–2060. doi:10.1016/j.cub.2007.10.039
26. Lu L, Li Y, Kim SM, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A. 2010;107(4):1437–1442. doi:10.1073/pnas.0911427107
27. Lee K-P, Lee J-H, Kim T-S, et al. The Hippo–Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(18):8248–8253. doi:10.1073/pnas.0912203107
28. Zhang C, Bian M, Chen X, et al. Oroxylin A prevents angiogenesis of LSECs in liver fibrosis via inhibition of YAP/HIF‐1α signaling. J Cell Biochem. 2018;119(2):2258–2268. doi:10.1002/jcb.26388
29. Kim KM, Choi YJ, Hwang JH, et al. Shear stress induced by an interstitial level of slow flow increases the osteogenic differentiation of mesenchymal stem cells through TAZ activation. PLoS One. 2014;9(3):e92427. doi:10.1177/1556264614544277
30. Zhao X, Sun J, Su W, et al. MEL protects against lung fibrosis by regulating the hippo/YAP pathway. PLoS One. 2018;19(4):1118. doi:10.1186/s12881-017-0516-2
31. Carbajo-Pescador S, Steinmetz C, Kashyap A, et al. MEL induces transcriptional regulation of Bim by FoxO3a in HepG2 cells. Br J Cancer. 2013;108(2):442.
32. LaQuaglia MJ, Grijalva JL, Mueller KA, et al. Yap subcellular localization and hippo pathway transcriptome analysis in pediatric hepatocellular carcinoma. Sci Rep. 2016;6:30238. doi:10.1038/srep30238
33. Sohn BH, Shim -J-J, Kim S-B, et al. Inactivation of Hippo pathway is significantly associated with poor prognosis in hepatocellular carcinoma. Clin Cancer Res. 2016;22(5):1256–1264. doi:10.1158/1078-0432.CCR-15-1447
34. Xiong Y, Hannon GJ, Zhang H, et al. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366(6456):701. doi:10.1038/366701a0
35. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366(6456):704. doi:10.1038/366704a0
36. Wang X, Yan Y, Chen X, et al. The antitumor activities of marsdenia tenacissima. Front Oncol. 2018;8:473. doi:10.3389/fonc.2018.00473
37. Fernández JG, Rodríguez DA, Valenzuela M, et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced β-catenin/Tcf-Lef dependent transcription. Mol Cancer. 2014;13(1):209. doi:10.1186/1476-4598-13-209
38. Bennukul K, Numkliang S, Leardkamolkarn V. Melatonin attenuates cisplatin-induced HepG2 cell death via the regulation of mTOR and ERCC1 expressions. World J Hepatol. 2014;6(4):230. doi:10.4254/wjh.v6.i4.230
39. Sardo FL, Muti P, Blandino G, et al. Melatonin and Hippo pathway: is there existing cross-talk? Int J Mol Sci. 2017;18(9):1913. doi:10.3390/ijms18091913
40. Zhao X, Sun J, Su W, et al. Melatonin protects against lung fibrosis by regulating the Hippo/YAP pathway. Int J Mol Sci. 2018;19(4):1118. doi:10.3390/ijms19041118
41. Fujita H, Hirose K, Sato M, et al. Metformin attenuates hypoxia-induced resistance to cisplatin in the HepG2 cell line. Oncol Lett. 2019;17(2):2431–2440. doi:10.3892/ol.2018.9869
42. Huang J, Wu S, Barrera J, et al. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122(3):421–434. doi:10.1016/j.cell.2005.06.007
43. Guerrant W, Kota S, Troutman S, et al. YAP mediates tumorigenesis in neurofibromatosis type 2 by promoting cell survival and proliferation through a COX-2–EGFR signaling axis. Cancer Res. 2016;76(12):3507–3519. doi:10.1158/0008-5472.CAN-15-1144
44. Muramatsu T, Imoto I, Matsui T, et al. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis. 2010;32(3):389–398. doi:10.1093/carcin/bgq254
45. Kim W, Khan SK, Liu Y, et al. Hepatic Hippo signaling inhibits protumoural microenvironment to suppress hepatocellular carcinoma. Gut. 2018;67(9):1692–1703. doi:10.1136/gutjnl-2017-314061
46. Zhang Y, Li K, Ying Y, et al. C21 steroid-enriched fraction refined from Marsdenia tenacissima inhibits hepatocellular carcinoma through the coordination of Hippo-Yap and PTEN-PI3K/AKT signaling pathways. Oncotarget. 2017;8(66):110576.
47. Chen X, Gu W, Wang Q, et al. C-MYC and BCL-2 mediate YAP-regulated tumorigenesis in OSCC. Oncotarget. 2018;9(1):668.
48. Zhang HT, Gui T, Sang Y, et al. The BET bromodomain inhibitor JQ1 suppresses chondrosarcoma cell growth via regulation of YAP/p21/c-Myc signaling. J Cell Biochem. 2017;118(8):2182–2192. doi:10.1002/jcb.25863
49. Liu J, Wen D, Fang X, et al. p38MAPK signaling enhances glycolysis through the up-regulation of the glucose transporter GLUT-4 in gastric cancer cells. Cell Physiol Biochem. 2015;36(1):155–165. doi:10.1159/000374060
50. Wang W, Xiao Z-D, Li X, et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17(4):490. doi:10.1038/ncb3113
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.