Back to Journals » Drug Design, Development and Therapy » Volume 14
Melatonin Alleviates Neuronal Damage After Intracerebral Hemorrhage in Hyperglycemic Rats
Authors Liang F, Wang J, Zhu X ![]() , Wang Z, Zheng J
, Wang Z, Zheng J ![]() , Sun Z, Xu S, Zhang J, Zhou J, Shi L
, Sun Z, Xu S, Zhang J, Zhou J, Shi L
Received 8 April 2020
Accepted for publication 16 June 2020
Published 2 July 2020 Volume 2020:14 Pages 2573—2584
DOI https://doi.org/10.2147/DDDT.S257333
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Georgios Panos
Feng Liang,1,* Jianli Wang,1,* Xiangyu Zhu,2,* Zhen Wang,1 Jingwei Zheng,1 Zeyu Sun,1 Shenbin Xu,1 Jianmin Zhang,1,3,4 Jingyi Zhou,1 Ligen Shi1
1Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China; 2Department of Neurology, Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 3Brain Research Institute, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China; 4Collaborative Innovation Center for Brain Science, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ligen Shi 88 Jiefang Road, Hangzhou City, Zhejiang Province 310009, People’s Republic of China
Tel +86-571-87784715
Fax +86-571-87784755
Email [email protected]
Jianmin Zhang 88 Jiefang Road, Hangzhou City, Zhejiang Province 310009, People’s Republic of China
Tel +86-571-87784715
Fax +86-571-87784755
Email [email protected]
Background: This study sought to investigate a novel effect of melatonin in reducing brain injury in an in vivo hyperglycemic intracerebral hemorrhage (ICH) model and further explore the mechanisms of protection.
Methods: Hyperglycemia ICH was induced in Sprague-Dawley rats by streptozocin injection followed by autologous blood injection into the striatum. A combined approach including RNA-specific depletion, electron microscopy, magnetic resonance, Western blots, and immunohistological staining was applied to quantify the brain injuries after ICH.
Results: Hyperglycemia resulted in enlarged hematoma volume, deteriorated brain edema, and aggravated neuronal mitochondria damage 3 days after ICH. Post-treatment with melatonin 2 hours after ICH dose-dependently improved neurological behavioral performance lasting out to 14 days after ICH. This improved neurological function was associated with enhanced structural and functional integrity of mitochondria. Mechanistic studies revealed that melatonin alleviated mitochondria damage in neurons via activating the PPARδ/PGC-1α pathway. Promisingly, melatonin treatment delayed until 6 hours after ICH still reduced brain edema and improved neurological functions. Melatonin supplementation reduces neuronal damage after hyperglycemic ICH by alleviating mitochondria damage in a PPARδ/PGC-1α-dependent manner.
Conclusion: Melatonin may represent a therapeutic strategy with a wide therapeutic window to reduce brain damage and improve long-term recovery after ICH.
Keywords: melatonin, intracerebral hemorrhage, hyperglycemia, mitochondria, apoptosis
Introduction
It is reported that more than 50% of stroke patients suffer from hyperglycemia.1 Hyperglycemia has been shown to be an independent determinant of hematoma expansion and contribute to worse functional outcomes after intracerebral hemorrhage (ICH).2 We previously reported that hyperglycemia exacerbated ICH-induced mitochondrial dysfunction, and accelerated perihematomal cell death in rodents.3 Further elucidating the mechanisms underlying hyperglycemia-aggravated mitochondrial dysfunction and brain injury might provide potential therapeutic targets to improve long-term outcomes after ICH.
Melatonin, which is mainly synthesized in the pineal gland, can penetrate subcellular compartments due to its amphipathic characteristic.4 Melatonin is a potent pharmacological antioxidant that alleviates mitochondrial dysfunction.5 In addition, melatonin can regulate glucose homeostasis.6 However, the effect of melatonin on ICH-induced brain injury in a context of hyperglycemia has not been evaluated. Peroxisome proliferator activated receptors (PPARs) are ligand-activated transcription factors that control glucose metabolism and energy production.7 Of the three PPAR family members, PPARδ is highly expressed in brain.8 Previous studies have indicated that PPARδ involved in regulating skeletal muscle mitochondrial biogenesis.9 PPARδ is at least two-fold more highly expressed in brain than in muscle,10 but studies regarding the function of PPARδ in brain are particularly rare. Peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α) is a co-activator of nuclear receptors and other transcription factors including PPARδ.11 Previous studies reported that PPARδ/PGC-1α pathway promoted the expression of genes that drive high-level energy production to ameliorate mitochondrial dysfunction and improve cell survival of neurons in Huntington’s disease.12 Interestingly, melatonin was reported to prevent mitochondrial damage via activating PPARδ pathway in mouse fibroblasts.13
The present study investigates the effect of melatonin post-treatment on neuronal mitochondria damage in a model of ICH with acute hyperglycemia. Furthermore, we elucidated a molecular mechanism involving PPARδ/PGC-1α activation for melatonin-afforded neuroprotection after hyperglycemic ICH.
Materials and Methods
Animal Studies
All surgical and experimental procedures were followed the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and Animal Research: Reporting of In vivo Experiments (ARRIVE) guidelines. This study was approved by the Animal Care Committee of Zhejiang University. Adult male Sprague-Dawley (SD) rats weighing 300–320g were housed on 12-h light/dark cycles under temperature- and humidity-controlled conditions.
Hyperglycemic ICH Model
Streptozotocin (STZ), as a highly selective pancreatic islet β-cells cytotoxic agent, is used to induce diabetes by causing complete β-cell necrosis. The hyperglycemic ICH model was established as follows. Briefly, adult male SD rats were housed in a comfortable environment with free access to food and water before experiments. The bodyweight of rats was recorded every day. At least 6–8 hours prior to STZ injection, all food and water will be removed. Immediately prior to injection, the STZ (Sigma-S0130, Sigma-Aldrich Trading Co., Ltd., St Louis, MO, USA) was dissolved in 50 mM sodium citrate buffer (pH 4.5) to a final concentration of 10 mg/mL. The rats received the STZ solution by intraperitoneal injection at 65 mg/kg for 3 days. During these 3 days, normal food and 10% sucrose water were accessed freely. Form the fourth day, the 10% sucrose water was switched to regular water. We used OneTouch Select Test Strips (Johnson & Johnson) to measure tail venous blood glucose every morning after overnight fasting. And blood glucose >250mg/dl was defined as hyperglycemia according to previous studies.14 These hyperglycemic rats were then employed to induce ICH models. The autologous blood injection model of ICH was established as previously reported with some modifications.15 In brief, rats were anesthetized with pentobarbital sodium and placed in a stereotaxic frame for aseptic surgery. A small burr hole (0.5mm) was drilled into the skull 3.0 mm right of and 0.2 mm posterior to bregma. Autologous blood was taken from the right femoral artery without anticoagulants. Within 2 minutes, 0.2mL autologous blood was infused into the striatum (6.0 mm deep below the surface of the skull) using a 26-G needle. The needle was slowly removed after an additional 10 mins delay to prevent backflow. The hole was sealed with bone wax. Body temperature was maintained during surgery with a heating blanket.
Drug and Small Interfering RNA (siRNA) Administration
Melatonin (N-acetyl-5-methoxytryptamine) was purchased from Sigma-Aldrich (St Louis, MO, USA) and dissolved in 1 mL saline containing 1% ethanol. Endogenous melatonin is secreted at night with a robust circadian rhythm and maximum plasma levels that occur around 3 to 4 am. Between 8am to 8pm, plasma melatonin is at a very low level.16 Exogenous melatonin was administrated in the daytime. Melatonin (5, 10, and 15 mg/kg) was injected intraperitoneally at 2, 24, and 48 h after ICH induction. Vehicle (saline containing 1% ethanol) was administered as control. The PPARδ siRNA, PGC1α siRNA, or scramble siRNA (Thermo Fisher Scientific) mixed with the transfection reagent (Engreen Biosystem Co., Ltd.; 500 pmol/10 μL) was injected into the right lateral ventricle at a rate of 2 μL/min 24 h before ICH.
Neurobehavioral Tests
Several neurobehavioral tests have been performed to detect the effect of melatonin on long-term recovery after ICH as previously described.17 Sensorimotor deficits were assessed by the corner test, foot fault, and adhesive removal tests. These neurobehavioral tests were carried out 1 d before and 1–14 d after ICH as described previously.18 In brief, the corner test was expressed as a relative proportion of right turn, and calculated as: right/(right+left)×100%. The foot fault test focused on each weight-bearing step, and calculated as: the number of fault foot/the total number of steps×100%. The adhesive removal test was expressed as the time to remove each adhesive tape (1×1mm2), with a maximum of 120s. All rats were trained daily for 3 days before ICH induction.
Hematoma Volume and Brain Edema Measurements
Hematoma volume and brain edema were measured by magnetic resonance imaging (MRI) as previously described.19 T2 weighted MRI was performed at 2h, 6h, 24h, day 3, day 5, and day 7 after ICH operation in a 3.0-Tesla GE Discovery MR750 scanner (General Electric Company, USA). Rats had T2 fast spin-echo sequences using a field of view of 60×60 mm, matrix of 256×256 mm and 18 coronal slices (1-mm-thick). All MRI data were analyzed by a blinded observer using Image J software (Rasband WS, ImageJ, NIH, http://rsb.info.nih.gov/ij). Hematoma volume was estimated by measuring the lesions with high sign in T2 mapping. Brain edema was estimated by measuring the volumes of the affected (VLES) and contralateral (VC) hemispheres and using the formula: brain edema (%) = 100 × (VLES - VC)/VC.
Mitochondrial Function Measurements
Intracellular adenosine triphosphate (ATP) level was measured by an ATP bioluminescent assay kit from Sigma (St. Louis) according to the manufacturer’s instruction. Mitochondrial DNA (mtDNA) was measured by a TIA-Namp Genomic DNA kit in accordance with the manufacturer’s instructions. Amount of mtDNA was determined using real-time PCR.
Mitochondrial Morphology Measurements
Electron microscopy was used to measure the number and morphology of mitochondria in neurons. Briefly, rats were sacrificed at 72h after ICH. Perihematomal brain tissues (1mm3) obtaining from right hemisphere were processed with glutaraldehyde (2.5%) at 4°C overnight. After several chemical processing steps as described previously,3 the samples were sliced into 100 nm sections and then stained with 4% uranyl acetate and 0.5% lead citrate. The transmission electron microscopy (Philips Tecnai 10) was used to scan the ultrastructure of neurons.
Western Blotting
Perihematomal tissues were obtained for Western blotting detection at 72h following ICH induction. Western blotting was performed as previously described.20 The primary antibodies that were used contained peroxisome proliferator-activated receptor delta (PPARδ: Abcam, ab137724), peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC1α: Abcam, ab191838), mitochondrial transcription factor A (TFAM: Abcam, ab131607), nuclear respiratory factor 1 (NRF-1: Abcam, ab175932), cleaved Caspase 3 (Abcam, ab2302), and β-actin (Santa Cruz SC-47,778). Proteins were stained with species-specific HRP-linked antibodies (1:2000; Cell Signaling) and scanned by gel densitometry scanning.
Immunofluorescence Staining
The immunofluorescence staining was performed as previously described.21 In brief, the whole brain was obtained at 72h after ICH. After perfusing with PBS and 4% paraformaldehyde, the whole brain was immersed with 30% sucrose solution for dehydration. The fixed brains were cut coronally through the needle entry site, as well as 2 mm anterior and 2 mm posterior to bregma. Frozen slices (slice thickness: 8 μm) were obtained from a cryostat microtome (Leica CM3050S-3-1-1, Bannockburn, IL). The sections were incubated overnight at 4°C with primary antibodies, including NeuN (Abcam, ab177487 or ab104224), cleaved Caspase 3 (Abcam, ab2302), PPARδ (Abcam, ab137724), and PGC1α (Abcam, ab191838) followed by the appropriate fluorescence-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA). TUNEL staining was performed according to the manufacturer’s instruction (Roche Inc, Basel, Switzerland) as described in previous studies.22 The sections were visualized with a fluorescence microscope (LSM-710; Zeiss, Oberkochen, Germany). Expressions of the target proteins and apoptotic cells were quantified at the coronal level of the maximum hematoma diameter in three fields (each area 400×400 μm) immediately adjacent to the hematoma site. The percent of TUNEL-positive or cleaved Caspase-positive neurons was calculated in a blinded manner.
Statistical Analysis
All data were shown as the median with range. A Mann–Whitney U-test was used. Repeated measurement ANOVA with multivariate ANOVA followed by a Bonferroni test were performed for behavior tests. The analyses were performed using SPSS version 22.0 (SPSS Inc.). Statistical significance was defined as P < 0.05.
Results
Hyperglycemia Aggravated Brain Injury After ICH
T2 weight imaging was performed to detect the changing process of hematoma volume and brain edema from 2 hours to 7 days after ICH operation (Figure 1A). Hematoma volume showed no significant difference between hyperglycemic and normoglycemic ICH groups at baseline (P = 0.93; Figure 1B). Hyperglycemia delayed the shrinking process of hematoma volume from 3 days after ICH (P = 0.026; Figure 1B). In addition, hyperglycemia also raised the peak of brain edema at 3 days after ICH (P = 0.026; Figure 1C). In order to explore the brain injury in cellular level, we performed TUNEL staining at 3 days after ICH showing that both TUNEL-positive neurons and microglia were observed in perihematomal tissues in hyperglycemic rats (neurons: 24.02 ± 2.608%; microglia: 15.44 ± 1.588%; astrocyte: 3.903 ± 0.9600%; Figure 1D and E). Hyperglycemia increased the percentage of TUNEL positive and cleaved Caspase 3 positive neurons at 3 days after ICH (TUNEL: P = 0.026; cleaved Caspase 3: P = 0.041; Figure 1F and G). Subsequently, we examined the mitochondrial functions, including detecting mtDNA and ATP levels in perihematomal neurons at 3 days after ICH. The rats in normoglycemic ICH groups showed a significant decrease in neuronal ATP level compared with sham groups. Hyperglycemia exacerbated ICH-induced mitochondrial dysfunctions (mtDNA: P = 0.015; ATP: P = 0.015; Figure 1H and I). All these above data indicated that hyperglycemia aggravated brain injury after ICH.
 |
Figure 1 Hyperglycemia aggravated brain injury after ICH. (A) T2 weighted imaging was performed to detect the changing process of hematoma volume and brain edema from 2 hours to 7 days after ICH operation. (B) Hyperglycemia delayed the shrinking process of hematoma volume. (C) Hyperglycemia raised the peak of brain edema. (D) TUNEL staining was performed to explore the brain injury in cellular level. (E) Apoptosis of neuron and microglia were observed in perihematomal tissues after ICH. Hyperglycemia increased the percentage of (F) TUNEL positive and (G) cleaved Caspase 3 positive neurons. Hyperglycemia exacerbated this ICH-induced mitochondrial dysfunction, including (H) mtDNA and (I) ATP levels. Error bars represented mean ± standard deviation (SD). N = 6 per group. *P < 0.05 versus ICH group; ##P < 0.01 versus SHAM+DM group. Repeated measurement ANOVA with multivariate ANOVA followed by a Bonferroni test were performed for (B and C). Mann–Whitney U-tests were used for (F–I). Abbreviations: ICH, intracerebral hemorrhage; DM, diabetes mellitus; mtDNA, mitochondrial deoxyribonucleic acid; ATP, adenosine triphosphate. |
Higher Dose of Melatonin Obtained Better Neurological Functional Recovery
A large number of neuroprotectants have been developed to suppress apoptosis,23,24 while few studies focused on neuroprotection of hyperglycemic ICH-induced apoptosis. Our previous study reported that both low and high doses of melatonin (5 mg/kg and 10 mg/kg) showing significant improvements in neurological function in subarachnoid hemorrhage rats.21 However, only high dose of melatonin (10 mg/kg) alleviated brain edema.21 Hence, in the present study, we examined the effect of different doses of melatonin on neurological function including 5, 10, and 15 mg/kg. Medium and high doses of melatonin showed significant improvements in corner test from 3 days after ICH (difference among groups: P < 0.001; low dosage vs vehicle: P = 0.18; medium dosage vs vehicle: P = 0.002; high dosage vs vehicle: P = 0.01; Figure 2A). Similar results were observed in the fault foot (difference among groups: P < 0.001; low dosage vs vehicle: P = 0.058; medium dosage vs vehicle: P < 0.001; high dosage vs vehicle: P < 0.001; Figure 2B) and adhesive tests (difference among groups: P < 0.001; low dosage vs vehicle: P = 0.12; medium dosage vs vehicle: P = 0.01; high dosage vs vehicle: P = 0.006; Figure 2C). Higher doses of melatonin showed better functional recovery, but there are no significant differences among these treated groups. Furthermore, we detected the percentage of TUNEL-positive cells at 3 days after ICH which showed significant decrease of apoptotic neurons in medium and high dose of melatonin-treated groups, but not in the low dose of melatonin-treated groups (difference among groups: P < 0.001; low dosage vs vehicle: P = 0.093; medium dosage vs vehicle: P = 0.009; high dosage vs vehicle: P = 0.009; Figure 2D and E). Therefore, higher dose of melatonin (10 and 15 mg/kg) obtained better functional recovery after ICH compared with low dose of melatonin group (5 mg/kg). Higher dose of melatonin may cause some side effects, including headache, dizziness, and so on.25 Hence, the medium dose of melatonin (10 mg/kg) may be the best choice.
 |
Figure 2 To explore the optimal dosage of melatonin in the therapy of hyperglycemic ICH. Behavior tests were performed to detect the effects of different doses of melatonin on brain recovery after hyperglycemic ICH, including (A) corner tests, (B) fault foot, and (C) adhesive removal tests. (D) TUNEL staining was performed to explore the anti-apoptotic effects of melatonin. (E) Significant decreases of apoptotic neurons were observed in 10 mg/kg and 15 mg/kg of melatonin treated groups, but not in the 5 mg/kg of melatonin treated groups. N = 6 per group. *P < 0.05 versus ICH+DM group. Repeated measurement ANOVA with multivariate ANOVA followed by a Bonferroni test were performed for (A–C). Mann–Whitney U-tests were used for (D). Abbreviations: ICH, intracerebral hemorrhage; DM, diabetes mellitus. |
Optimal Dose of Melatonin Alleviated Mitochondria Damage After Hyperglycemic ICH
We detected mitochondrial structure and function to evaluate the effect of melatonin on rescuing damaged mitochondria (Figure 3A). The optimal dose of melatonin (10 mg/kg) alleviated hyperglycemic ICH-induced mitochondria injury by preventing mitochondria loss (P = 0.026; Figure 3B) and reducing swollen mitochondria (P = 0.026; Figure 3C) at 3 days after ICH. Similar results were observed in rescuing mitochondrial dysfunction including ATP (P = 0.046; Figure 3D) and mtDNA levels (P = 0.045; Figure 3E) at 3 days after ICH.
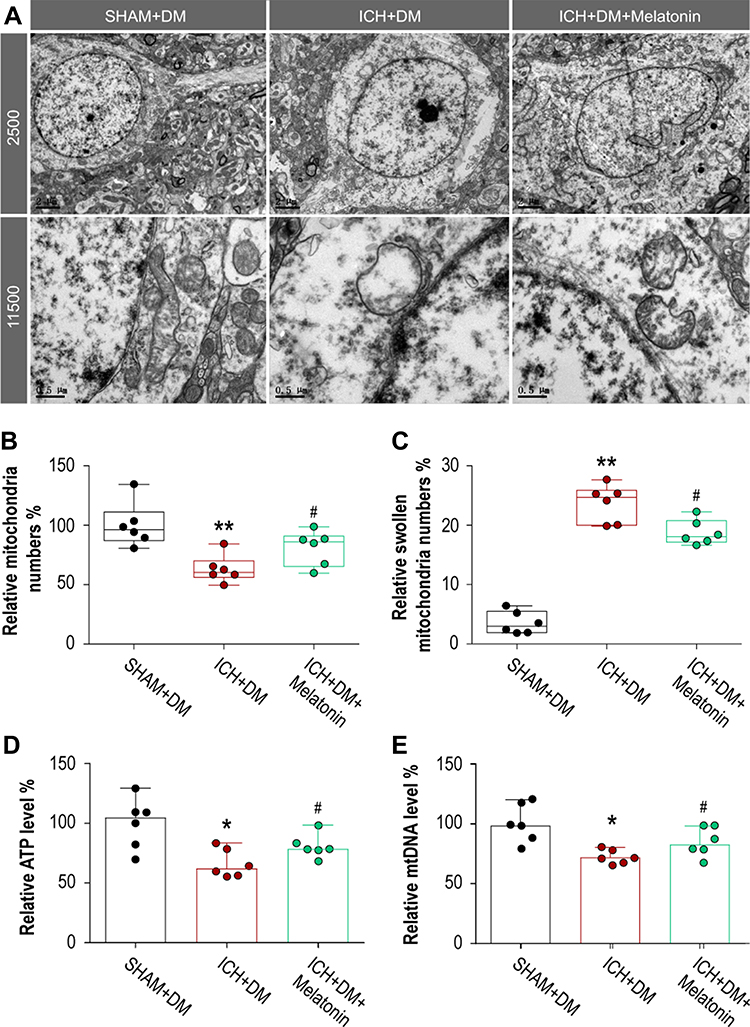 |
Figure 3 Optimal dose of melatonin alleviated mitochondria damage after hyperglycemic ICH. (A) Electron microscope was performed to mitochondrial structure and function. The optimal dose of melatonin (10 mg/kg) alleviated hyperglycemic ICH-induced mitochondria injury by preventing (B) mitochondria loss and reducing (C) swollen mitochondria. Similar results were observed in rescuing mitochondrial dysfunction including (D) ATP and (E) mtDNA levels. N = 6 per group. *P < 0.05 and **P < 0.01 versus SHAM+DM group; #P < 0.05 versus ICH+DM group. Mann–Whitney U-tests were used for (B-E). Abbreviations: ICH, intracerebral hemorrhage; DM, diabetes mellitus. |
Melatonin Activated the PPARδ/PGC-1α Pathway to Protect Neurons After ICH
To explore the potential mechanism of melatonin in protecting neuronal mitochondria after hyperglycemic ICH, we performed Western blotting (Figure 4A-D), immunofluorescence (Figure 4E), and siRNA techniques (Figure 5A-F). Hyperglycemic ICH rats showed a 14.3% decrease in the expression of PPARδ (P = 0.039, Figure 4A), a 16.7% decrease in the expression of PGC-1α (P = 0.002, Figure 4B), a 26.4% decrease in the expression of NRF-1 (P = 0.004, Figure 4C), and a 27.9% decrease in the expression of TFAM (P = 0.009, Figure 4D). Melatonin treatment diminished the reduction of these proteins induced by ICH, including PPARδ (P = 0.002, Figure 4A), PGC-1α (P = 0.002, Figure 4B), NRF-1 (P = 0.002, Figure 4C), and TFAM (P = 0.002, Figure 4D). In addition, immunofluorescence staining indicated that PPARδ and PGC-1α were mainly expressed in neurons (Figure 4E). We used PPARδ siRNA and PGC-1α siRNA to examine whether this pathway involved in the neuroprotection of melatonin. Both PPARδ siRNA and PGC-1α siRNA could reverse the effect of melatonin on NRF-1, TFAM, and cleaved-Caspase 3 (P < 0.05; Figure 5A-F). These results revealed that melatonin alleviates neuronal mitochondria damage by activating the PPARδ/PGC-1α pathway after ICH.
 |
Figure 4 Melatonin activated the PPARδ/PGC-1α pathway. Western blotting was performed to detect the expression of (A) PPARδ, (B) PGC-1α, (C) NRF-1, and (D) TFAM. Immunofluorescence staining indicated that (E) PPARδ and (F) PGC-1α were mainly expressed in neurons. N = 6 per group. *P < 0.05 versus SHAM+DM group; #P < 0.05 versus ICH+DM group. Mann–Whitney U-tests were used for (A–D). Abbreviations: ICH, intracerebral hemorrhage; DM, diabetes mellitus. |
 |
Figure 5 To confirm whether melatonin alleviates neuronal mitochondria damage by activating the PPARδ/PGC-1α pathway. (A–C) We used PPARδ siRNA to block this pathway and detect the downstream proteins including (A) NRF-1, (B) TFAM, and (C) cleaved-Caspase 3. (D–F) We used PGC-1α siRNA to block this pathway and detect the downstream proteins including (D) NRF-1, (E) TFAM, and (F) cleaved-Caspase 3. N = 6 per group. *P < 0.05 versus ICH+DM group; #P < 0.05 versus ICH+DM+Melatonin+Scramble siRNA group. Mann–Whitney U-tests were for (A–F). Abbreviations: ICH, intracerebral hemorrhage; DM, diabetes mellitus. |
Delayed Melatonin Administration Showed Similar Effects on Improving Neurological Functional Recovery
In order to detect the potential application value of melatonin in treating ICH patients. We examined whether delayed melatonin administration (6 hours after ICH) still have neuroprotection. We observed that delayed melatonin administration alleviated brain edema at 3 days after hyperglycemic ICH (melatonin 6h vs melatonin 2h vs vehicle: P < 0.05, Figure 6A and B). In addition, delayed melatonin administration showed similar improvements of neurological behaviors including corner test (P < 0.05, Figure 6C) and fault foot (P < 0.05, Figure 6D).
 |
Figure 6 Delayed melatonin administration showed similar effects on improving neurological functional recovery. (A) T2 weight scanning was performed to examine whether delayed melatonin administration (6 hours after ICH) still have neuroprotection. (B) Delayed melatonin administration also alleviated brain edema at 3 days after hyperglycemic ICH. (C–D) Delayed melatonin administration showed similar improvements of neurological behaviors including (C) corner test and (D) fault foot. N = 6 per group. *P < 0.05 versus ICH+DM group. (E) The molecular mechanism might be involved in the role of melatonin in alleviating brain injury after hyperglycemic ICH. Hyperglycemic ICH aggravated mitochondria swelling leading to perihematomal neuron apoptosis. Melatonin activated PPARδ/PGC-1α pathway to increase the transcription of TFAM and NRF-1, which maintained the structure and function of mitochondria leading to reduction of neuron apoptosis. A Mann–Whitney U-test was used for (B). Repeated measurement ANOVA with multivariate ANOVA followed by a Bonferroni test were performed for (C and D). Abbreviations: ICH, intracerebral hemorrhage; DM, diabetes mellitus. |
Discussion
Melatonin alleviated brain injury and improved neurological functional recovery by protecting neuronal mitochondria after hyperglycemic ICH. Hyperglycemia not only delayed the shrinking process of hematoma volume but also raised the peak of brain edema after ICH. Melatonin showed its neuroprotective effects on alleviating hyperglycemia-induced brain injury after ICH. This inspiring property was associated with maintaining the homeostasis of neuronal mitochondria via activating the PPARδ/PGC-1α pathway. Moreover, the present study revealed that delayed melatonin administration still showed positive effects on improving neurological functional recovery after ICH. All these data indicated that melatonin may be a promising agent to treat those patients with hyperglycemic ICH.
Cerebral microdialysis monitoring studies have reported that perihematomal tissue has long-lasting signs of disturbed metabolism.26 A metabolic penumbra theory has been proposed that there is a metabolic rather than ischemic penumbra in ICH.27 Glucose metabolism dysfunction has been reported in perihematomal tissues after ICH.28 Under normal physiologic conditions, the glucose content in the cerebrospinal fluid fluctuates between 0.82 and 2.4 mmol/L. When the glucose content reached 10 mmol/L, neuronal mitochondrial dysfunctions were observed in primary cortical neurons, including mitochondrial depolarization, compromised electron transport chain complexes, and intracellular ATP deprivation.29 If the glucose content kept going up to 30 mmol/L, those extremely disordered mitochondria would alter neuron morphology and promote neuron apoptosis.29 Our present study revealed that hyperglycemia exacerbated the ICH-induced mitochondrial dysfunctions including diminished ATP and mtDNA levels, leading to a mass of neuronal apoptosis. These findings were consistent with previous studies which also reported more neuronal apoptosis in hyperglycemic ICH rats than those in normoglycemic ICH rats.30
Melatonin acts as a regulator of mitochondrial bioenergetic function.31 Because of its amphipathic nature, melatonin can cross physiological barriers and penetrate into subcellular compartments. We observed that 10 and 15 mg/kg but not 5 mg/kg of melatonin could significantly improve neurological behavioral performance lasting out to 14 days after ICH with DM. Previous studies reported that 15,32 20,33 100, and 150 mg/kg34 of melatonin could suppress cell apoptosis after ICH. In addition, 5 mg/kg of melatonin was also reported to have effect on alleviating ICH-induced secondary brain injury in rats.5 However, there is a study indicating that 5 mg/kg of melatonin showed no effect on ameliorating basal ganglia edema after ICH.32 Our data indicated that the ICH+DM model has worse brain edema and neuron apoptosis compared with the ICH model. Hence, 5 mg/kg of melatonin may not enough to show its neuroprotection in this ICH+DM model. Melatonin dosage varied from 0.1 mg to 50 mg/kg and was administered orally in clinical studies.35 Melatonin (10 mg/kg) was used in 40 newborns with grade III or IV respiratory distress syndrome without major side effects.36 Both melatonin and its metabolites are powerful-free radical scavengers which can maintain the mitochondrial homeostasis.37 Our present study indicated that melatonin mitigated mitochondria swelling and prevented mitochondria loss after hyperglycemic ICH. This protective effect of melatonin on neuronal mitochondria leads to promote neuron survival and improve the long-term neurological function recovery. Interestingly, the present study revealed that melatonin activated PPARδ/PGC-1α pathway to prevent mitochondria damage by hyperglycemic ICH. PPARδ is a ligand-activated transcription factor that performs essential functions in glucose metabolism, energy production, and cellular differentiation.12 Because of its high expression in brain tissues, PPARδ was considered as a crucial target in therapy of central nervous system diseases. Previous studies have showed that increased PPARδ transactivation ameliorated mitochondrial dysfunction and improved neuron survival.12,38 The present study revealed that melatonin upregulated the expression of PPARδ, which was consistent with previous studies reporting that melatonin upregulated PPARγ to stimulate the biogenesis of mitochondria.13 In addition, melatonin could also upregulate PGC-1α that was a co-activator of PPARδ. PGC-1α controls a network of transcriptional programs that culminate in mitochondrial biogenesis and enhanced energy production.39 Our present study indicated that both PPARδ and PGC-1α show important positions in maintaining mitochondria homeostasis. Either PPARδ siRNA or PGC-1α siRNA could reverse the effects of melatonin on reducing neuron apoptosis after hyperglycemic ICH. PPARδ could activate PGC-1α to promote the expression of downstream proteins including TFAM and NRF-1.40 TFAM is a mitochondrial DNA-binding protein that controls mitochondrial transcription.41 And NRF-1 is a nuclear DNA-binding factor that also regulates mitochondrial transcription.41 These two proteins could maintain mitochondria homeostasis to prevent neuron apoptosis. In summary, hyperglycemic ICH aggravated mitochondria swelling leading to perihematomal neuron apoptosis. Melatonin activated PPARδ/PGC-1α pathway to increase the transcription of TFAM and NRF-1, which maintained the structure and function of mitochondria leading to reduction of neuron apoptosis (Figure 6E).
Several limitations should be noted in this present study. First, the present study only focused on the effect of melatonin on perihematomal neuronal apoptosis. In fact, we observed that perihematomal microglia is activated and involved in the pathophysiology of brain injury by hyperglycemic ICH. Our further study will explore the role of microglia in hyperglycemic ICH and whether melatonin could prevent microglia apoptosis. Second, we used STZ to induce T1DM rats, while most of the ICH patients combined with T2DM in clinical practice. Although the present study only focused on the neuroprotective effect of melatonin on acute hyperglycemic ICH, whether the brain tissues influenced by long-term diabetes will reduce the sensitivity to melatonin treatment should be considered. Third, the design of the present study is too simple to strictly prove that PPARδ/PGC-1α pathway was involved in alleviating mitochondria damage in neurons by melatonin. Other molecular pathways might also participate in this regulatory mechanism of melatonin. Mechanistic studies revealed that melatonin alleviated mitochondria damage in neurons via activating the PPARδ/PGC-1α pathway.
In conclusion, melatonin alleviated brain injury and improved neurological functional recovery by protecting neuronal mitochondria after hyperglycemic ICH. The underlying mechanism might involve, at least in part, PPARδ/PGC-1α pathway. In addition, delayed melatonin administration still showed positive effects on improving neurological functional recovery after ICH.
Acknowledgments
We would like to thank all these rats sacrificed for science.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Luitse MJ, Biessels GJ, Rutten GE, Kappelle LJ. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012;11(3):261–271. doi:10.1016/S1474-4422(12)70005-4
2. Passero S, Ciacci G, Ulivelli M. The influence of diabetes and hyperglycemia on clinical course after intracerebral hemorrhage. Neurology. 2003;61(10):1351–1356. doi:10.1212/01.WNL.0000094326.30791.2D
3. Zheng JW, Shi LG, Liang F, et al. Sirt3 ameliorates oxidative stress and mitochondrial dysfunction after intracerebral hemorrhage in diabetic rats. Front Neurosci. 2018;12:414–428. doi:10.3389/fnins.2018.00414
4. Acuna-Castroviejo D, Escames G, Venegas C, et al. Extrapineal melatonin: sources, regulation, and potential functions. Cell Mol Life Sci. 2014;71(16):2997–3025. doi:10.1007/s00018-014-1579-2
5. Wang Z, Zhou F, Dou Y, et al. Melatonin alleviates intracerebral hemorrhage-induced secondary brain injury in rats via suppressing apoptosis, inflammation, oxidative stress, DNA damage, and mitochondria injury. Transl Stroke Res. 2018;9(1):74–91. doi:10.1007/s12975-017-0559-x
6. Li T, Ni L, Zhao Z, et al. Melatonin attenuates smoking-induced hyperglycemia via preserving insulin secretion and hepatic glycogen synthesis in rats. J Pineal Res. 2018;64(4):e12475. doi:10.1111/jpi.12475
7. Yessoufou A, Wahli W. Multifaceted roles of peroxisome proliferator-activated receptors (PPARs) at the cellular and whole organism levels. Swiss Med Wkly. 2010;140:w13071. doi:10.4414/smw.2010.13071
8. Kliewer SA, Forman BM, Blumberg B, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A. 1994;91(15):7355–7359. doi:10.1073/pnas.91.15.7355
9. Schuler M, Ali F, Chambon C, et al. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006;4(5):407–414. doi:10.1016/j.cmet.2006.10.003
10. Girroir EE, Hollingshead HE, He P, et al. Quantitative expression patterns of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) protein in mice. Biochem Biophys Res Commun. 2008;371(3):456–461. doi:10.1016/j.bbrc.2008.04.086
11. Shi Q, Gibson GE. Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis Assoc Disord. 2007;21(4):276–291. doi:10.1097/WAD.0b013e31815721c3
12. Dickey AS, Pineda VV, Tsunemi T, et al. PPAR-delta is repressed in Huntington’s disease, is required for normal neuronal function and can be targeted therapeutically. Nat Med. 2016;22(1):37–45. doi:10.1038/nm.4003
13. Guven C, Taskin E, Akcakaya H. Melatonin prevents mitochondrial damage induced by doxorubicin in mouse fibroblasts through Ampk-Ppar Gamma-dependent mechanisms. Med Sci Monit. 2016;22:438–446. doi:10.12659/MSM.897114
14. Liu J, Gao BB, Clermont AC, et al. Hyperglycemia-induced cerebral hematoma expansion is mediated by plasma kallikrein. Nat Med. 2011;17(2):206–210. doi:10.1038/nm.2295
15. Kitaoka T, Hua Y, Xi G, Hoff JT, Keep RF. Delayed argatroban treatment reduces edema in a rat model of intracerebral hemorrhage. Stroke. 2002;33(12):3012–3018. doi:10.1161/01.STR.0000037673.17260.1B
16. Zeitzer JM, Duffy JF, Lockley SW, Dijk DJ, Czeisler CA. Plasma melatonin rhythms in young and older humans during sleep, sleep deprivation, and wake. Sleep. 2007;30(11):1437–1443. doi:10.1093/sleep/30.11.1437
17. Freret T, Bouet V, Leconte C, et al. Behavioral deficits after distal focal cerebral ischemia in mice: usefulness of adhesive removal test. Behav Neurosci. 2009;123(1):224–230. doi:10.1037/a0014157
18. Wang J, Shi Y, Zhang L, et al. Omega-3 polyunsaturated fatty acids enhance cerebral angiogenesis and provide long-term protection after stroke. Neurobiol Dis. 2014;68:91–103. doi:10.1016/j.nbd.2014.04.014
19. Dang G, Yang Y, Wu G, et al. Early erythrolysis in the hematoma after experimental intracerebral hemorrhage. Transl Stroke Res. 2017;8(2):174–182. doi:10.1007/s12975-016-0505-3
20. Pariente R, Pariente JA, Rodriguez AB, Espino J. Melatonin sensitizes human cervical cancer HeLa cells to cisplatin-induced cytotoxicity and apoptosis: effects on oxidative stress and DNA fragmentation. J Pineal Res. 2016;60(1):55–64. doi:10.1111/jpi.12288
21. Di Liberto G, Pantelyushin S, Kreutzfeldt M, et al. Neurons under T cell attack coordinate phagocyte-mediated synaptic stripping. Cell. 2018;175(2):458–471.e419. doi:10.1016/j.cell.2018.07.049
22. Ying GY, Jing CH, Li JR, et al. Neuroprotective effects of valproic acid on blood-brain barrier disruption and apoptosis-related early brain injury in rats subjected to subarachnoid hemorrhage are modulated by heat shock protein 70/matrix metalloproteinases and heat shock protein 70/AKT pathways. Neurosurgery. 2016;79(2):286–295. doi:10.1227/NEU.0000000000001264
23. Kinarivala N, Patel R, Boustany RM, Al-Ahmad A, Trippier PC. Discovery of aromatic carbamates that confer neuroprotective activity by enhancing autophagy and inducing the anti-apoptotic protein B-Cell lymphoma 2 (Bcl-2). J Med Chem. 2017;60(23):9739–9756. doi:10.1021/acs.jmedchem.7b01199
24. Makoukji J, Saadeh F, Mansour KA, et al. Flupirtine derivatives as potential treatment for the neuronal ceroid lipofuscinoses. Ann Clin Transl Neurol. 2018;5(9):1089–1103. doi:10.1002/acn3.625
25. Besag FMC, Vasey MJ, Lao KSJ, Wong ICK. Adverse events associated with melatonin for the treatment of primary or secondary sleep disorders: a systematic review. CNS Drugs. 2019;33(12):1167–1186. doi:10.1007/s40263-019-00680-w
26. Miller CM, Vespa PM, McArthur DL, Hirt D, Etchepare M. Frameless stereotactic aspiration and thrombolysis of deep intracerebral hemorrhage is associated with reduced levels of extracellular cerebral glutamate and unchanged lactate pyruvate ratios. Neurocrit Care. 2007;6(1):22–29. doi:10.1385/NCC:6:1:22
27. Vespa PM. Metabolic penumbra in intracerebral hemorrhage. Stroke. 2009;40(5):1547–1548. doi:10.1161/strokeaha.108.542803
28. Zazulia AR, Videen TO, Powers WJ. Transient focal increase in perihematomal glucose metabolism after acute human intracerebral hemorrhage. Stroke. 2009;40(5):1638–1643. doi:10.1161/strokeaha.108.536037
29. Peng Y, Liu J, Shi L, et al. Mitochondrial dysfunction precedes depression of AMPK/AKT signaling in insulin resistance induced by high glucose in primary cortical neurons. J Neurochem. 2016;137(5):701–713. doi:10.1111/jnc.13563
30. Chiu CD, Chen CC, Shen CC, et al. Hyperglycemia exacerbates intracerebral hemorrhage via the downregulation of aquaporin-4: temporal assessment with magnetic resonance imaging. Stroke. 2013;44(6):1682–1689. doi:10.1161/STROKEAHA.113.675983
31. Ganie SA, Dar TA, Bhat AH, et al. Melatonin: a potential anti-oxidant therapeutic agent for mitochondrial dysfunctions and related disorders. Rejuvenation Res. 2016;19(1):21–40. doi:10.1089/rej.2015.1704
32. Lekic T, Hartman R, Rojas H, et al. Protective effect of melatonin upon neuropathology, striatal function, and memory ability after intracerebral hemorrhage in rats. J Neurotrauma. 2010;27(3):627–637. doi:10.1089/neu.2009.1163
33. Lu J, Sun Z, Fang Y, et al. Melatonin suppresses microglial necroptosis by regulating deubiquitinating enzyme A20 after intracerebral hemorrhage. Front Immunol. 2019;10:1360. doi:10.3389/fimmu.2019.01360
34. Xu W, Lu X, Zheng J, et al. Melatonin protects against neuronal apoptosis via suppression of the ATF6/CHOP pathway in a rat model of intracerebral hemorrhage. Front Neurosci. 2018;12:638. doi:10.3389/fnins.2018.00638
35. Vural EM, van Munster BC, de Rooij SE. Optimal dosages for melatonin supplementation therapy in older adults: a systematic review of current literature. Drugs Aging. 2014;31(6):441–451. doi:10.1007/s40266-014-0178-0
36. Gitto E, Reiter RJ, Cordaro SP, et al. Oxidative and inflammatory parameters in respiratory distress syndrome of preterm newborns: beneficial effects of melatonin. Am J Perinatol. 2004;21(4):209–216. doi:10.1055/s-2004-828610
37. Acuna Castroviejo D, Lopez LC, Escames G, et al. Melatonin-mitochondria interplay in health and disease. Curr Top Med Chem. 2011;11(2):221–240. doi:10.2174/156802611794863517
38. Kinarivala N, Suh JH, Botros M, Webb P, Trippier PC. Pharmacophore elucidation of phosphoiodyn A - Potent and selective peroxisome proliferator-activated receptor beta/delta agonists with neuroprotective activity. Bioorg Med Chem Lett. 2016;26(8):1889–1893. doi:10.1016/j.bmcl.2016.03.028
39. Cui L, Jeong H, Borovecki F, et al. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127(1):59–69. doi:10.1016/j.cell.2006.09.015
40. Hondares E, Pineda-Torra I, Iglesias R, et al. PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem Biophys Res Commun. 2007;354(4):1021–1027. doi:10.1016/j.bbrc.2007.01.092
41. Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203. doi:10.1146/annurev.physiol.010908.163119
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.