Back to Journals » Journal of Inflammation Research » Volume 15
Mechanistic Understanding of Lung Inflammation: Recent Advances and Emerging Techniques
Authors Keskinidou C ![]() , Vassiliou AG
, Vassiliou AG ![]() , Dimopoulou I, Kotanidou A, Orfanos SE
, Dimopoulou I, Kotanidou A, Orfanos SE
Received 20 December 2021
Accepted for publication 4 May 2022
Published 15 June 2022 Volume 2022:15 Pages 3501—3546
DOI https://doi.org/10.2147/JIR.S282695
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Chrysi Keskinidou, Alice G Vassiliou, Ioanna Dimopoulou, Anastasia Kotanidou, Stylianos E Orfanos
First Department of Critical Care Medicine and Pulmonary Services, School of Medicine, National and Kapodistrian University of Athens, “Evangelismos” Hospital, Athens, Greece
Correspondence: Alice G Vassiliou; Stylianos E Orfanos, First Department of Critical Care Medicine and Pulmonary Services, School of Medicine, National and Kapodistrian University of Athens, “Evangelismos” Hospital, Athens, Greece, Tel +30 210 7235521, Fax +30 210 7239127, Email [email protected]; [email protected]
Abstract: Acute respiratory distress syndrome (ARDS) is a life-threatening lung injury characterized by an acute inflammatory response in the lung parenchyma. Hence, it is considered as the most appropriate clinical syndrome to study pathogenic mechanisms of lung inflammation. ARDS is associated with increased morbidity and mortality in the intensive care unit (ICU), while no effective pharmacological treatment exists. It is very important therefore to fully characterize the underlying pathobiology and the related mechanisms, in order to develop novel therapeutic approaches. In vivo and in vitro models are important pre-clinical tools in biological and medical research in the mechanistic and pathological understanding of the majority of diseases. In this review, we will present data from selected experimental models of lung injury/acute lung inflammation, which have been based on clinical disorders that can lead to the development of ARDS and related inflammatory lung processes in humans, including ventilation-induced lung injury (VILI), sepsis, ischemia/reperfusion, smoke, acid aspiration, radiation, transfusion-related acute lung injury (TRALI), influenza, Streptococcus (S.) pneumoniae and coronaviruses infection. Data from the corresponding clinical conditions will also be presented. The mechanisms related to lung inflammation that will be covered are oxidative stress, neutrophil extracellular traps, mitogen-activated protein kinase (MAPK) pathways, surfactant, and water and ion channels. Finally, we will present a brief overview of emerging techniques in the field of omics research that have been applied to ARDS research, encompassing genomics, transcriptomics, proteomics, and metabolomics, which may recognize factors to help stratify ICU patients at risk, predict their prognosis, and possibly, serve as more specific therapeutic targets.
Keywords: acute respiratory distress syndrome, lung inflammation, mechanisms, biomarkers, omics
Introduction
In vivo and in vitro models have been important pre-clinical scientific tools in biological and medical research in the mechanistic and pathological understanding of the majority of diseases, as well as in their novel therapeutic approaches. Although in vitro methods are evolving, they cannot completely replace animal models. In vivo research using animal models can provide answers to the pathophysiology of the disease in a complex systemic manner, relevant to human responses.1 The progression of the physiological changes and the host’s responses in lung damage, especially in acute respiratory distress syndrome (ARDS), evolve over time, and therefore, they should be reproducible in the chosen animal model. The acute onset, disruption of the endothelial and epithelial barrier, and the prolonged hyper-inflammatory response in the lung, demonstrate the complexity of the experimental design and proper animal model establishment. Innate immune responses, including inflammatory cell infiltration and the “cytokine storm” are a hallmark of lung inflammation and ARDS. Cytokine storm is characterized by excessive inflammatory response to infectious and non-infectious diseases, in which pro-inflammatory cytokines are predominantly released. Cytokine storms in the systemic circulation and the lung alveolar environment can cause severe lung injury/ARDS.2 Since extensive reviews describing in detail the above-mentioned mechanisms have been previously published,3–6 we chose to focus on less commonly described lung injury mechanisms in experimental cell and animal models, and in related clinical acute lung inflammation, mainly ARDS. Although no existing animal model can reproduce every clinical aspect of ARDS and related inflammatory lung processes, they do provide important information about key elements of human response in lung pathologies.7
Experimental Lung Injury - Acute Lung Inflammatory Models
Ventilator-Induced Lung Injury (VILI) Model
The lung injury caused by the application of mechanical ventilation is called ventilator-induced lung injury (VILI).7 In this review we will only discuss VILI induced by mechanical forces, where the observed damage is the product of mechanical stretch and cellular mechanotransduction. The overstretching of the alveolar epithelium induces inflammatory responses, activation of capillary endothelial cells, irreversible opening of water channels across the epithelial barrier, causing endothelial disruption and induction of downstream signaling pathways.8,9 In VILI animal models, animal size is important since different animal models have different thresholds for VILI generation. Based on the animal model, different studies have applied different ventilator strategies, such as high tidal volumes,10 lung strain,11 and positive end-expiratory pressure (PEEP).12 Often researchers use a “two hit” animal model of VILI, were the injurious ventilation strategy is applied on pre-injured lungs, since uninjured lungs would require higher, and hence non-clinically relevant, tidal volumes to cause lung injury.13
Oxidative Stress
Under normal circumstances, oxygen metabolism releases reactive oxygen species (ROS) and reactive nitrogen species (RNS), both of which are known as pro-oxidants. These include superoxide radicals, hydrogen peroxide, hydroxyl radicals, nitric oxide, nitrogen dioxide, and peroxynitrite. The mitochondrial respiratory chain is the main source of ROS and nitric oxide synthases (NOS), namely, endothelial NOS (eNOS), inducible NOS (iNOS), and neuronal NOS (nNOS) for RNS.14 In addition, ROS-generating enzymatic systems, including xanthine oxidase, mitochondrial oxidases, and particularly protein nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) are important sources of ROS. ROS/RNS at low concentrations participate in cellular responses as regulators of signaling pathways and mediators of pathogen defense.15 In order to maintain the production of pro-oxidants under control, the cells produce endogenous anti-oxidant mediators. If the balance shifts in favor of the pro-oxidants for any reason, oxygen free radicals accumulate and oxidative damage occurs.16
The main mechanisms by which oxidative stress causes cellular damage are nucleic acid structural impairment, protein activity changes, inactivation of anti-oxidant enzymes, and alteration of transcription factors and gene expression.17–19 Oxidative stress causes permanent oxidation of several protein residues. ROS/RNS have the ability to modify histones at a post-translational level, affecting chromatin compaction and gene regulation. This occurs through histone methylation and acetylation, and also through post-translational modifications affected by the oxidative balance of the cell, and by epigenetic regulators. Notably, post translational modifications of the histones H3 and H4, as a result of disruptions in anti-oxidant response or direct interaction of oxygen radicals with the amino-terminal histone protrusions, have been described.20 The main function of the NOX family is to generate ROS through their NOX catalytic subunit, which aids the production of molecular oxygen by using NADPH as the electron donor. There are several NADPH oxidase isoforms, NOX1-5, which differ in the catalytic NOX subunit, as well as in cellular and tissue localization, ROS producing kinetics, and free radical type. NOX proteins are found in a variety of cell types, including the vasculature and blood cells. In the lung, NOX2 is highly expressed, in particular in alveolar macrophages and the airway epithelium, whereas NOX4 is highly expressed in the pulmonary smooth muscle cells. Their function, apart from participating in ROS production, includes defense and cellular responses, such as gene expression, signaling pathways, cell death induction, and response to mechanical stress.21,22 One of the main defense mechanisms against oxidative stress is the KEAP1-NRF2 [Kelch-like ECH-associated protein 1-nuclear factor (erythroid-derived 2)-like 2] regulatory pathway. NRF2 is a transcription factor responsible for the transcriptional regulation of antioxidant response elements (AREs), whereas KEAP1 is a NRF2 inhibitor and down regulator, suppressing the nuclear activation of antioxidant responsive elements.23,24 In normoxia, NRF2 binds to KEAP1 and then the complex is degraded. However, in the presence of ROS, the inhibitor is deactivated, and NRF2 is able to bind to AREs in the nucleus, enabling the transcription of antioxidant enzymes, such as heme oxygenase HO-1.25
It has been suggested that in both in vitro and in vivo VILI models, activation of NOX2 results in ROS production, hence worsening lung injury. In an in vitro VILI model, three different human and rat pulmonary cell lines were exposed to cycle mechanical stretch, in order to examine the involvement of the lung epithelium in ROS production. The authors suggested that after two hours of cycle stretching, NADPH oxidase activity increased, contributing to increased time- and magnitude-dependent ROS production.26 Moreover, NOX2 expression was shown elevated in a mouse model following injurious mechanical ventilation; it was proposed that the Toll-like receptor 4 (TLR4)/Tumor necrosis factor receptor–associated factor (TRAF)/NOX2 pathway was activated after ventilation with high tidal volumes, resulting in excessive ROS production, endoplasmic reticulum stress, and inflammation via the nuclear factor kappa B (NF-κB) pathway.27 On the other hand, it has been demonstrated that administration of NOX2 inhibitors or substances that prevent NOX2 activation prior to mechanical ventilation had a protective effect on mouse lung injury.27,28 The protective role of NRF2 has been suggested in many in vitro and in vivo VILI models. Specifically, the first study to demonstrate the protective role of NRF2 in VILI was in NRF2-deficient (NRF2−/−) mice. The NRF2−/− mice displayed higher vascular permeability levels and inflammatory responses, compared to wild-type mice, after two hours of injurious mechanical ventilation; administration of antioxidants reversed the VILI phenotype.29 Since then, several studies have also highlighted the protective effects of NRF2-dependent pathways in VILI through maintaining the oxidative balance. NRF2 activators could be used as potential therapeutic targets against VILI progression.30–35
Neutrophil Extracellular Traps (NETs)
Neutrophils eliminate pathogens through phagocytosis and degranulation, releasing several secreted products, including hormones, enzymes, and ROS.36 Another mechanism in which only activated neutrophils and not naive neutrophils engage in, is the release of neutrophil extracellular traps (NETs). The generated extracellular fibers are composed of granule proteins, such as neutrophil elastase and DNA, in particular histones H1, H2A, H2B, H3, and H4. NETs are released in the first ten minutes post-neutrophil activation, depending on the activator stimuli. NETs precede neutrophil phagocytosis and actively participate in the defense responses by eliminating pathogens, and halting pathogen spread.37 Even though NETs are very important components of the innate immune response, there is a fine line between their beneficial contribution and excess inflammatory response. Excessive NET formation in the lungs results in increased mucus viscosity, which is cytotoxic to lung epithelial and endothelial cells, promoting cell damage and disrupting the cellular matrix.38,39 In patients with severe non-thoracic blunt trauma, circulating histones are elevated and it has been suggested that they are able to induce distant organ damage, especially to the lungs.40
VILI is characterized, among others, by pulmonary edema, hyaline membrane formation, and neutrophil infiltration, as a result of the activation of pro-inflammatory and pro-fibrogenic pathways.41,42 During VILI, adhesion molecules, transforming growth factor (TGF)-β1, cytokines, and chemokines are upregulated and act as mediators for neutrophil recruitment in the ventilated lungs, inducing NET formation.38,42,43 A study by Rossaint et al reported the pathways through which NETs form and aid lung injury during injurious mechanical ventilation without the presence of infection. In a sterile, inflammatory VILI mouse model, they demonstrated that activated platelets triggered the binding of the chemokines CXCL4/CCL5 and β2-integrin to their respective receptors, G-protein-coupled receptors (GPCR) and MAC-1, hence leading to the release of NETs. Blocking any of these pathways resulted in reduced NET formation, ameliorating VILI severity.44 Moreover, in agreement with the above, another study demonstrated that mice ventilated with high tidal volumes had elevated NET markers and extracellular DNA in their lungs, the formation of which could be reduced by DNase I treatment. They also demonstrated that NET formation was partially regulated by TLR4.45 In an infectious VILI model, mice were intratracheally treated with LPS and were mechanically ventilated with high tidal volumes. Free DNA and citrullinated histones were detected in the bronchoalveolar lavage fluid (BALF) of the mice, indicating the presence of NETs in the alveolar space. Treatment with DNase reduced the NET markers in the BALF, and improved lung mechanics; however no other inflammatory-induced VILI parameters were altered.46 According to the findings of the above-mentioned studies, NETs are involved in inflammatory lung tissue damage during mechanical ventilation; thus, inhibiting NETs early in the development of VILI may be beneficial to lung compliance.
Mitogen-Activated Protein Kinase (MAPK) Pathways
The ability of cells to sense extracellular stimuli, and convert this information into intracellular responses through signaling pathways, is fundamental for the cell’s survival and regulation. In mammals, mitogen-activated protein kinase (MAPK) families play a crucial role in important cellular functions, including cell cycle programming, proliferation, and apoptosis. The three MAPK cascades that have been extensively studied in mammalian cells are extracellular signal-regulated kinase (ERK), C-Jun N-terminal kinase (JNK), and p38 MAPK. Each MAPK pathway consists of a three-component module, including at least three enzymes that are necessary for MAPK activation. The MAPK kinase kinase (MKKK) is the first kinase of the module, and is responsible for the activation through phosphorylation of the sequential MAPK kinase (MKK), which in turn phosphorylates MAPK. Several isoforms of MKKKs, MKKs and MAPKs have been identified in mammals. The MAPK phosphorylation substrates can be either transcription factors, or enzymes and proteins.47–49
Growth factors and cytokines stimulate tyrosine kinase receptors (RTKs), and/or G protein-coupled receptors (GPCRs), which induce the cascade of the ERK MAPK pathway. The most representative ERK pathway consists of c-Raf-1 MKKK, which phosphorylates MEK1 and MEK2 MKK. MEKs then phosphorylate and activate the MAPKs ERK1 and ERK2. The ERKs translocate into the nucleus where they phosphorylate transcription factors that regulate cell proliferation and differentiation. The JNK pathway, also known as stress activated protein kinase (SAPK), is triggered by several stimuli, including stress, cytokines and growth factors. Several MKKKs and MKKs are activated in order to phosphorylate the three alternative forms of JNK, JNK1-3. JNKs can phosphorylate c-Jun on Ser-63 and Ser-73 by binding to its NH2-terminal activation domain, and can also phosphorylate JunB and JunD. The JNK pathway induces cellular responses that include apoptosis, inflammatory responses, and cell growth. Finally, the p38 pathway is also activated by cytokines, stress and mitogens. The four p38 isoforms α, β, γ, and δ are activated by different activated MKKK and MKK combinations. p38-induced cellular responses appear to have an important role in the cell’s fate, determining cell proliferation, survival, apoptosis, and stress responses.47–50
Cycle stretch of human bronchial epithelial, and pulmonary microvascular endothelial cells induced the activation of the MAPK signaling pathways, including p44/42, SAPK/JNK, and p38. In particular, p38 activation seemed to have an important role in interleukin (IL)-8 production, and therefore induction of the inflammatory response through the chemo-attraction of neutrophils.51,52 Many studies have used murine models of VILI exposed to high pressure or high tidal volume ventilation, and have demonstrated increased activation of MAP kinases, including p38, JNK, and ERK1/2, as well as NF-κB, and related transcription factors, such as c-Jun.53–55 Mice deficient in p38, MKK3, and JNK1 were less prone to VILI and cell death. Furthermore, JNK1−/− mice exhibited resistance to pulmonary edema development.53 In mice exposed to high-stretch mechanical ventilation, the quantification of MAP kinase markers demonstrated that within one minute of injurious ventilation, the endothelial cells exhibited significant phosphorylated levels of p38 and ERK1/2, suggesting that they are probably the first cell type to rapidly respond to stretch-induced injuries by activating MAP kinase pathways. Type I and II alveolar epithelial cells exhibited increased phosphorylation of p38 within the first minute, while alveolar macrophages showed signs of MAP kinase activation after five minutes of high stretch ventilation.56
Surfactant
Pulmonary surfactant is a phospholipid and protein layer that lines the alveolar wall. The main lipid component of surfactants is dipalmitoylphosphatidylcholine (DPPC), while the protein component consists of four associated surfactant proteins (SPs); two hydrophilic, SP-A and SP-D also termed collectins, and two hydrophobic, SP-B and SP-C. Surfactant synthesis occurs mainly in alveolar type II cells. One of the principal surfactant functions is the prevention of alveolar collapse through the reduction of surface tension.57 Surfactant can also modulate the host’s immune response, and facilitate in pathogen elimination.58,59 Several diseases have been associated with surfactant deficiency or overproduction. Lung surfactant integrity and activity is inhibited by injury-induced compounds, including plasma and blood proteins, ROS, RNS, and lytic enzymes.60
Mechanical ventilation can cause lung overdistention, edema formation, and decreased lung compliance.61 It has been demonstrated that mechanical ventilation without PEEP can impair surfactant layer composition and function, while ventilation with applied PEEP protects the lungs.62 Surfactant impairment increases alveolar wall tension and pressure parameters, increasing the susceptibility of lung collapse.63 In an adult rat VILI model, administration of exogenous surfactants restored the gas exchange imbalance, oxygenation, and lung mechanics.64 In another study, combining exogenous surfactants with PEEP in mechanically ventilated rats reduced TNF-decompartmentalization.65 Moreover, early surfactant administration resulted in a superior protective response in a surfactant deficient VILI rabbit model.66
Ion and Water Transport
Maintaining balanced water and ion transport in the lung is an important process to help maintain normal lung function. Pulmonary edema is characterized by impaired capillary and alveolar walls, and fluid collection in the alveolar compartment.67 Disturbed ion homeostasis has been associated with several lung pathologies.68 Aquaporins (AQPs) are a family of proteins that participate in water transportation as water channels. In mammals, there are more than ten homologous AQPs, and about one third are expressed in the lungs.69 AQPs participate actively in the trans-endothelial and trans-epithelial water flux in the lung. AQP1, the first characterized AQP, is expressed in alveolar epithelial cells and in microvascular endothelial cells. The expression of AQP4 is localized in the basolateral membranes of bronchial epithelium, and of AQP5 in the apical membranes of type I epithelial cells.70–73 AQP expression seems to differ between various types of lung injury, depending on the injury site each model induces. Apart from their role in soluble transportation, AQPs participate in different cellular processes, including cell proliferation and migration, and signaling transduction.74
Airway epithelial cells have the ability to control the transport of solutes and ions through ion channels that are distributed among their basal and apical membranes.68 Impaired edema clearance present in lung injuries results in altered ion transportation and fluid reabsorption.75 Sodium (Na+) transport from the apical membrane of the epithelium to the basolateral membrane, and then out to the interstitium and the circulation is the driving force for fluid clearance. In particular, sodium ions enter epithelial cells through the amiloride-sensitive epithelial Na+ channels (ENaC) present in the apical membrane of epithelial cells, and then exit through sodium-potassium-adenosine triphosphatase (Na, K-ATPase). ENaCs are expressed on the apical membranes of both type I and II epithelial cells, and have an important role in transcellular Na reabsorption. Na, K-ATPase is a transmembrane protein located on the basolateral surface of alveolar epithelial type II cells; with the conversion of ATP to ADP, three Na ions are pumped out and two K ions enter the cytoplasm. An osmotic gradient is generated that forces the passive water movement from the apical side with the help of aquaporins. The pathophysiology that occurs in the lungs during acute lung injury can alter the Na, K ATPase functions.68,75,76
AQPs could modulate the wet/dry lung ratio in VILI, thus acquiring a protective role in VILI.77 AQP1 expression in different VILI murine models has been reported to be either decreased or increased, depending on the experimental parameters. In mice subjected to high-stretch ventilation, AQP1 expression remained unaltered.72 In a rat VILI model using high tidal volumes, AQP1 expression decreased. However, in the same rat model using low tidal volume ventilation, AQP1 mRNA, and not protein, expression increased after two and four hours of ventilation.78,79 In a rat model, high volume ventilation decreased AQP1 expression, while AQP1 upregulation with a cyclooxygenase-2 inhibitor alleviated lung injury.80 As for AQP4, the mRNA and protein expression were found decreased in a mouse injurious ventilated model.72 High tidal volume ventilated rats exhibited reduced AQP5 expression. It was suggested that treatment with a p38 MAPK inhibitor could upregulate AQP5 and have a protective effect on lung injury.81 Moreover, in low tidal volume ventilation, the protein expression of AQP5 was found to be gradually increased, without, however, affecting permeability and edema formation.78
Based on the fact that Na, K-ATPase has been reported to be downregulated during injurious mechanical ventilation, a research team observed that overexpression of Na, K-ATPase in a VILI rat model of mild ventilation, increased Na, K-ATPase activity, and improved liquid clearance.82 The use of autologous transplantation of adipose-derived stromal cells (ADSCs) was tested as a therapeutic approach in order to ameliorate VILI in a rat model. Treatment with ADSCs in injurious ventilated mice increased Na, K-ATPase activity, and induced the gene and protein expression of Na+ channel subunits, improving alveolar fluid clearance.83 Another study suggested that intratracheal instillation of dopamine in a VILI rat model could activate the dopaminergic D2 receptors, resulting in rapid activation of Na, K-ATPase, positively affecting pulmonary edema clearance and survival.84
Septic Model
Sepsis is a complex syndrome characterized by a dysregulated host response to invaded pathogens. The secreted pro-inflammatory mediators, tumour necrosis factor (TNF)-α, IL-1, and IL-8, act as neutrophil recruitment mediators during the initial hyper-inflammatory phase.85 The mechanisms through which neutrophils kill pathogens are phagocytosis, degranulation, and NET formation, rendering a crucial role in infection elimination.86 Even though pre-clinical animal models of sepsis have provided important information on sepsis mechanisms, it should be noted that sepsis is a multifactorial syndrome and, therefore, animal models are not able to reproduce all clinical symptoms. The closer to the intensive care unit (ICU) environment the model is designed, the more clinically relevant answers the model could provide.87
One of the most applied methods to mimic human sepsis in an animal model is through endotoxins. The most common endotoxin used is lipopolysaccharide (LPS), a glycolipid found in the outer membrane of gram negative bacteria.88 The administration routes of LPS include intravenous, intraperitoneal, and intratracheal injection, and due to the simplicity of the method, it is very easy to achieve this septic model. However, one disadvantage of this model is that the systemic response to the endotoxin, does not replicate the one observed in human sepsis; this includes the time escalation and intensity of the cytokine storm observed, as well as the changes in hemodynamic equilibrium.89
Another animal model mimicking human sepsis induced lung injury is cecal ligation and puncture (CLP). This model induces acute lung injury secondary to peritonitis. In animals, peritonitis is experimentally generated through surgical ligation and perforation of the cecum with a needle.90 ARDS-like lung injury in CLP is promoted by hyperpermeability-induced pulmonary edema, neutrophil-mediated damage, and hypoxia.91,92 In contrast to the LPS-induced sepsis, the CLP effects develop within days with a milder onset, and therefore, is considered one of the proper animal models of sepsis. However, the requirement for surgical induction of injury is a disadvantage.93
Oxidative Stress
The role of NADPH oxidases has been investigated in many in vivo and in vitro septic models. In a mouse CLP-induced septic model and an in vitro LPS-induced septic model, NOX4 knockdown was associated with decreased mortality levels and ROS production, whereas NOX2 knockdown was linked to worse outcomes.94 It was suggested that NOX4 induction in LPS-induced endothelial cells is post-translationally mediated by the proteasome/ubiquitin pathway.95 In the same manner, NOX2 activity was increased in the alveolar epithelial cells and macrophages of LPS-induced septic mice, as well as in an in vitro model, leading to cell damage and loss of barrier integrity due to excess ROS production.96 NRF2 has been characterized as an important host component of the innate immune response in experimental models of sepsis. In LPS and CLP-induced septic mouse models, NRF2−/− deficient mice showed greater inflammatory response and mortality rates when compared to NRF2+/+ septic mice. Additionally, deregulated gene expression of key innate immunity components and antioxidant genes was established in NRF2−/− septic mice lungs, as early as 30 min post-infection, which later affected the severity of the inflammatory response.97 Furthermore, in a CLP mouse model, induction of NRF2 by depleting its inhibitor, KEAP1, in macrophages and neutrophils improved mouse outcomes, and protected against sepsis.98 Several studies, taking advantage of the protective function of NRF2 in sepsis, have examined several pharmacological and non-pharmacological components that are involved in NRF2-dependent pathways in order to alleviate lung injury caused by sepsis.99–101
NETs
NETs participate in sepsis progression. NETs aid in pathogen elimination, and also participate in organ dysfunction development. In CLP and LPS-induced septic mouse models, increased serum extracellular DNA was detected. Treatment with DNase decreased NETs, however increased pathogen burden and inflammation; on the other hand induction of NETs resulted in lung injury and increased mortality. Treatment with a combination of DNase and antibiotics ameliorated systemic inflammation and parameters of lung injury. It was concluded that there should be a dynamic balance between NET formation and pathogen elimination in order to achieve pathogen clearance and to avoid lung injury.102,103 In another study, in a LPS-induced mouse model, it was demonstrated that neutrophils interacting with activated platelets, through TLR4, induce NET production, promoting endothelial cell damage and organ dysfunction.104 These are in agreement with the results from a CLP mouse model, which showed that the interaction of thrombin-activated platelets with polymorphonuclear cells (PMNs) resulted in local NET formation, promoting subsequent immunothrombosis.105
MAPK Pathways
Increased phosphorylation of JNK and p38 MAPK in lung tissue of septic murine models after CLP has been reported, while disruption of MAPK signaling pathways through administration of JNK and p38 inhibitors have resulted in restored lung permeability, decreased leukocyte recruitment, modulation of systemic inflammatory response, and attenuation of the lung injury.106,107 Two different studies have examined the effects of the deficiency of important MAPK signaling pathway factors in intraperitoneally LPS-injected mice. In the first study, septic MKK3 deficient mice exhibited reduced inflammatory and oxidative stress markers, concluding that MKK3 deficiency has a protective role in endothelial cell damage.108 In the second study, deficiency of the MAPK phosphatase 5 (MKP5) in mice enhanced the phosphorylation of p38, JNK, and ERK in macrophages, resulting in induced neutrophil infiltration, edema formation, and inflammatory response.109 In an intratracheal LPS mouse model, alveolar macrophages exhibited increased activated levels of p38 and NF-κΒ only five minutes after LPS administration, while type I and II epithelial cells displayed signs of activation by that time. In contrast with the rapid response of the endothelial cells in the injurious mechanical ventilation mentioned priory, in the septic model, the endothelial cells showed signs of MAPK activation fifteen minutes after LPS administration.56
Surfactant
As previously mentioned, SP-A and SP-D are able to modulate immune responses. The C-terminal lectin domain of collectins binds to pathogens, and mediates their elimination through opsonization.58 Collectins can bind to Gram-negative bacteria by recognizing and binding to LPS, and therefore, can modulate cellular activation and responses following LPS exposure.110 In a study using a CLP adult sheep lung injury model, the protein expression of three out of four surfactant protein levels was decreased in the first 48 hours of lung injury and could, therefore, be used as a severity biomarker.111 Later, it was found that the changes in surfactant metabolism in septic lungs are the result of the decreased conversion of large surfactant aggregates to small ones, with the amount of large aggregates remaining unaltered.112 Under normal conditions, there is a balance between the large surfactants and the converted non-functioning small surfactants, however, in pathological conditions, this balance is disrupted.113
Ion and Water Transport
In LPS or CLP-induced lung injury models, the upregulation of AQP1 and AQP5 has been linked with a protective role in lung injury progression.114–117 In various murine models of LPS-induced lung injury, the expression of AQP1 and AQP5 was decreased. Reduced AQP expression levels have been linked with increased levels of inflammatory markers and apoptotic cells.72,118,119 The absence of AQP1 in mice given LPS intratracheally had no effect on the induced lung injury.120 Moreover, in a study examining the expression of AQP1 and AQP5 in an in vitro and an in vivo mouse model, exposure to LPS resulted in differential regulation of AQPs. In the in vitro model, LPS increased both the mRNA and protein expression of AQP1, however did not alter AQP5 expression. In the mouse model, intraperitoneal injection of LPS decreased the expression of AQP1, however not the mRNA expression of AQP5.121 Inhibition of AQP4 prior to LPS instillation has been shown to ameliorate lung injury and decrease mortality.122
Alveolar fluid clearance in a CLP-induced lung injury rat model was decreased, and when rats were treated with amiloride, the fluid clearance decreased significantly. Sepsis promoted the endocytosis of Na, K-ATPase proteins from the basolateral membrane into the cytoplasm of alveolar epithelial type II cells, impairing the active Na transport.123 The therapeutic benefit of ascorbic acid has been examined in a mouse model intraperitoneally injected with a fecal stem solution. Ascorbic acid could alleviate the pathology of lung injury and induce the activity of Na, K-ATPase.124 In a porcine model of sepsis-induced lung injury caused by fecal clot implantation, gene delivery of ENaC and Na, K-ATPase into alveolar cells, reduced edema formation, improved lung function, and decreased mortality.125
Ischemia/Reperfusion Model
Another type of lung injury extensively studied in animal models, due to its important clinical significance, is ischemia/reperfusion (I/R). Lung injury is generated through an ischemic period followed by a reperfusion period. Thoracic procedures, such as lung transplantation, pulmonary thromboendarterectomy and esophagectomy, and trauma, can cause ischemic/reperfusion periods, resulting in pulmonary complications, like ARDS. Apart from direct ischemia/reperfusion in the lungs, distant vascular bed and non-pulmonary sites, for example, gut ischemia/reperfusion, could also contribute to lung injury.126–128
In vivo models of ischemia/reperfusion include small animals, mainly mice, rats, and rabbits. They are chosen due to their easy handling, however they can provide limited clinical information. On the other hand, larger animals, like pigs, dogs, and sheep, approach the clinical symptoms in a more relevant manner, and have facilitated the pre-clinical research of new ARDS interventions. In order to achieve ischemia/reperfusion, major surgical intervention is required. Animals need to be sedated and mechanically supported. Access to the lungs is accomplished through thoracotomy, and then the clamping procedure with arterial forceps, ligature, and balloon occluder is performed. Ischemia can be performed either by clamping the pulmonary circulation and preserving the bronchial circulation, or by arresting bronchial circulation, through lung hilum clamping, which restricts both pulmonary and bronchial circulation. Air ventilation can also be stopped, inducing more severe lung damage.7,127 Apart from the chosen animal species, the extent of the ischemic area, the duration of ischemia, and the inflation state are parameters that determine the severity of the lung injury. The injury is characterized by increased alveolar epithelial permeability, edema formation, release of pro-inflammatory cytokines, and infiltration of polymorphonuclear cells. It should be noted that these damage responses develop at the ischemic/reperfused site, and also at the contralateral lung.7,129
Oxidative Stress
Endothelial cells have the ability to sense hemodynamic changes. Blocking blood flow results in changes in cell membrane polarization, and production of ROS through NOX2 activation. Additionally, a sudden restoration of blood flow induces NOX2 activation.130 In an ischemia/reperfusion mouse model, after one hour of ischemia followed by two hours of reperfusion, it was shown that NOX2 played an important role in invariant natural killer (NK) cell mediated IL-17 production, lung injury induction, edema formation, and neutrophil infiltration.131 Moreover, in a hilar clamp I/R mouse model, it was suggested that following I/R, a crosstalk between invariant NK cells, alveolar macrophages, and type II epithelial alveolar cells facilitates lung inflammation and dysfunction, via secretion of IL-17 and TNF-α in a NADPH oxidase-dependent mechanism.132 Inhibitors of NOX2 and NOX1/NOX4 have been shown to protect the lungs and alleviate lung injury induced by I/R. Hence they have been proposed as therapeutic approaches.133 The interruption of lung blood flow disrupts the physiological metabolic balance, causing toxic metabolic byproduct accumulation, hypoxia, and alteration of cellular pathways. The period following reperfusion does not restore the balance; on the contrary the ischemic lung injury worsens. One of the biggest mediators of I/R lung injury is ROS production.134 Hence, identifying major antioxidant components or activators/inhibitors of ROS producing pathways, and designing novel therapeutic strategies based on these cellular pathways, could provide new insights in I/R lung injury management. Several studies have demonstrated the NRF2-dependent protective pathways of lung injury are induced by I/R.135–139
NETs
Only a few studies have investigated the involvement of NETs in ischemia/reperfusion lung injury models. Until now, published studies have examined the role of NET formation in experimental lung transplantation. Sayah et al were the first to highlight the pathologic role of NETs in lung transplantation. In two experimental lung transplantation mouse models, hilar clamp and orthotopic lung transplantation after prolonged cold ischemia (OLT-PCI), the presence of rich NETs in the BALF of both models was detected, and increased platelet count in the latter model. The models displayed a platelet-dependent NET formation mechanism, emphasizing the therapeutic potential of DNase I treatment in primary lung graft dysfunction.140 A study by Scozzi et al, examined the impact of NET fragments after DNAase I treatment in a mouse orthotopic lung allograft damaged by I/R injury; even though at first DNase I treatment improved allograft lung function, the released NET fragments induced inflammatory cascade development, CD4+ T cell responses, and NET fragments eventually could jeopardize transplant lung acceptance.141
MAPK Pathways
Immunohistochemical analysis of the whole left lung of rats subjected to I/R, revealed that activated p38 and JNK are localized in alveolar macrophages, while ERK1/2 is found in endothelial and epithelial cells. The strategic site of expression of p38 and JNK is responsible for the protective effect of p38 and JNK inhibition in I/R induced lung injury.142 In rats subjected to I/R, inhibition of p38 attenuated lung injury and the induced inflammatory responses, specifically by decreasing the levels of IL-1β, IL-6, and cell adhesion molecules.143 These results are in agreement with two other studies examining lung injury induced by intestinal I/R in rats, which demonstrated that IL-1β expression levels were associated with p38.144,145 Although I/R physiologic and pathogenic parameters, such as aerodynamic and hemodynamic changes, as well as donor-recipient compatibility, cannot be introduced into cellular models, in vitro models provide important mechanistic information. Researchers using rat pulmonary microvascular endothelial cells and exposing them to conditions resembling lung transplantation procedures, including cold ischemia, reperfusion, and re-oxygenation, demonstrated the pivotal role of MAPK regulation in non-hypoxic I/R.146,147
Surfactant
Following I/R, the blood-air barrier is disturbed, alveolar and interstitial edema are formed, and the intra-alveolar surfactant is impaired.148 Pretreatment of lung transplants with exogenous surfactants prior to storage has been shown to alleviate lung injury, and improve lung function.149,150 In rat lungs subjected to I/R and cold storage, administration of exogenous surfactants prior to ischemia improved oxygenation, edema formation, and blood-air barrier impairment. Neither I/R nor exogenous surfactant altered alveolar epithelial type II cell parameters and, therefore, the suggested underlying mechanism of this improvement is that exogenous surfactants increase the total active endogenous intra-alveolar surfactants, and re-balance the ratio of large and small aggregates.151–153 Moreover, a study using a rat I/R model demonstrated that exogenous surfactant treatment improved histopathologic lung features, decreased apoptosis, and induced anti-inflammatory cytokine secretion levels.154
Ion and Water Transport
In rats subjected to I/R, inhibition of p38 attenuated lung injury and edema formation by reducing the expression of AQP1, a water channel expressed in lung endothelial cells.143 In a murine model, increased AQP1 expression was detected one and two weeks following I/R lung injury. In the same model, AQP1 deficient mice subjected to I/R exhibited impaired I/R resolution, negatively affected angiogenesis, and decreased survival. It was hence suggested that AQP1 could promote angiogenesis in I/R.155 In a lower limb I/R, the lung mRNA and protein expression of AQP1 and AQP5 was decreased, enhancing inflammation and pulmonary edema formation; pre-treatment with sodium hydrosulfide upregulated AQP1 and AQP5 expression, and reversed the inflammatory phenotype.156 Moreover, in a rat intestinal I/R-induced lung injury model, the expression of AQP4 was upregulated, while the injection of a p38 MAPK inhibitor resulted in downregulation of AQP4; this down-regulation decreased lung injury severity.144
Na, K-ATPase activity is impaired in I/R periods. In a porcine lung injury model of mesenteric artery I/R, gene delivery of ENaC and Na, K-ATPase into alveolar cells reduced edema formation, improved lung function, and decreased mortality.125 Several agents have been examined in order to evaluate whether they are effective in attenuating I/R-induced lung injury. In distal organ I/R animal models, iloprost (a prostanoid mainly used to treat pulmonary arterial hypertension), acetazolamide (a carbonic anhydrase inhibitor), and caffeic acid phenethyl ester (an antioxidant), have been found to upregulate and restore Na, K-ATPase activity, hence attenuating I/R-induced lung injury.157–159
Smoke Inhalation Model
Smoke inhalation is another type of lung injury reproduced in animal models. Following smoke inhalation, inflammatory mediators and cytokines are released, neutrophil accumulation is induced, and pulmonary edema is formed. Following this initial response, a fibrotic phase with hyaline formation and cellular hyperplasia occurs. The effects of smoke inhalation injuries affect the upper airway, the lower respiratory tract, as well as systemic physiological functions. The damage caused in the lower airway is characterized by injured type I alveolar epithelial cells, increased vascular permeability, secretion of cytokines, leading to protein fluid collection and edema formation in the alveoli space.160–162
Animal models of smoke inhalation-induced lung injury include both small and large animals. Studies use different types of smoke injury methods, such as cotton smoke, pine smoke, and wood shaving smoke. Animals are either directly exposed to the smoke and injury is induced through inhalation, or in larger animals, smoke is introduced through mechanical ventilation and the animal needs to be anesthetized. Larger animals could provide more accurate information due the pathophysiological similarities to human injury.160,163
Cigarette smoking is one of the main causes of morbidity and mortality worldwide, responsible for the development of chronic lung inflammation, including chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), pulmonary hypertension, and asthma. Tobacco smoke is a complex mixture of toxic components, carcinogens, and reactive oxygen species. It has been demonstrated that it alters vascular function, increases alveolar-capillary barrier permeability, and induces inflammation in smokers. Cigarette smoking has been recognized as a risk factor for ARDS development and poor prognosis.164–166 Since in this review we have focused on acute lung inflammation models, in the following section we will only provide a brief overview of published reviews relevant to the described mechanisms for chronic lung inflammation secondary to tobacco smoke.
Oxidative Stress
After exposure to wood smoke extract, ROS levels, intracellular mitogen and stress-activated signaling pathways, apoptosis, and IL-8 levels were increased in primary human bronchial epithelial and rat alveolar epithelial type II cells. Administration of NAPDH and ROS inhibitors and antioxidants reduced the above mentioned responses, emphasizing the important role of oxidative stress stimuli in smoke-induced injury.167,168 Moreover, exposure of human pulmonary artery endothelial cells to wood smoke extract induced upregulation of antioxidant enzymes, including HO-1, and intracellular ROS levels were also increased. The induced oxidative stress mediated cell apoptosis through mitochondrial release of apoptosis-inducing factor and endonuclease G.169 It should be noted, that several studies have examined the role of oxidative stress in smoke-induced lung injury in different animal models. They found increased levels of important oxidative stress markers, which in the context of the current review are not discussed.
The impact of chronic exposure to cigarette smoke in the activation of NRF2 is thoroughly analyzed in a review by Müller et al, where the dual role of NRF2 activation is discussed.170 Another review worth mentioning examined the therapeutic potential of NRF2 in COPD animal models.171 Finally, a review by Kim et al has provided a comprehensive presentation of cigarette smoke models and has discussed the impact of NOX activation in cardiovascular diseases.172
NETs
To the best of our knowledge, no study has examined the effect of acute smoke exposure on NET formation. Most studies have explored the effects of cigarette smoke in animal smoke inhalation models. Mice exposed to cigarette smoke induced NET formation in the airways, which appeared to play an important role in the immune response. Moreover, treatment with aerosolized DNase I degraded NETs and ameliorated airway inflammation.173,174
MAPK Pathways
In two murine models of smoke-induced lung injury, fifteen minutes of cotton smoke exposure resulted in the activation of JNK. The activation of the JNK pathway has been shown to have a crucial role in airway epithelial cell apoptosis, as well as in mucous overproduction. Treatment with a JNK inhibitor alleviated these symptoms and increased animal survival. These results suggest that JNK can be used as a novel therapeutic target in smoke-induced lung injury.175,176
The reviews cited provide an in-depth analysis of the role and effect of the activation of the MAPK pathways in tobacco smoke-induced lung injury models.177–179
Surfactant
The normal surfactant intra-alveolar distribution is altered following smoke inhalation in mice. In a murine model exposed to smoke for 30 min, changes in surfactant metabolism appeared four hours after exposure and were sustained for twelve hours, including increased levels of newly secreted surfactants,180 whereas after eight hours the total phospholipid surfactant levels in the lung lavage was increased.181 Instilled porcine pulmonary surfactant in rats exposed to smoke inhalation attenuated the smoke-induced lung injury. In particular, porcine surfactant improved the histological damage, increased the endogenous SP-A levels, and inhibited pro-inflammatory cytokine secretion.182
The impact of tobacco smoke exposure on surfactant protein function, as well as the mechanistic effects of smoke on the pulmonary system have been thoroughly presented in the cited reviews.183,184
Ion and Water Transport
The effect of smoke on airway epithelial permeability was tested in murine tracheal epithelial monolayers. Two parameters of smoke inhalation injury were tested; thermal stress and acrolein exposure, one of the main fire smoke components. Inhibiting Na, K-ATPase activity by depleting Na ions or using ion channel inhibitors, and exposing cells to acrolein suppressed the short-circuit current, and activated transepithelial resistance. Smoke exposure seemed to damage the tight junctions and impair the airway epithelial barrier, which was mediated by disturbed transcellular Na, K-ATPase ion transport.185
Acid Aspiration Model
Aspirated fluids, including aspiration of gastric contents or chemical fluids with low pH, directly damage the airway epithelium, inducing a caustic insult. Following the initial response, a neutrophil-dependent inflammatory response occurs. It is characterized by impairment of the pulmonary vascular integrity. Fluid transportation, vascular leakage, alveolar hemorrhage, and edema formation comprise the pathological mechanisms of acid-induced lung injury.7,186 However, it should be noted that when acid enters the lower respiratory tract it is neutralized, and therefore the damage caused is not very extensive.186,187
In animal models, acid-induced lung injury is generated by instilling acidic fluids, most commonly hydrochloric acid (HCl), intratracheally, or directly to the bronchi. The severity of the lung injury depends on the acidity of the solution, and therefore, low pH solutions are used that approach the acidity of gastric fluids. Although HCl induces acid-induced lung injury, it does not completely replicate the injury caused by gastric aspiration. The gastric content, apart from HCl, consists of food particles, pathogens, and cytokines that further damage the lungs. The presence of pathogens in the gastric fluids could evolve the initial chemical pneumonitis to infectious aspiration pneumonia.7,186,188
Oxidative Stress
In NADPH oxidase-deficient (p47phox−/−) and NRF2-deficient (NRF2−/−) mice, intratracheally instilled with HCl, a protective role of NADPH oxidase and NRF2 in acid aspiration-induced lung injury was proposed. More specifically, in both models, increased markers of lung injury were detected. Induction of NRF2 activity in wild-type mice resulted in attenuation of the acid-induced lung injury, however did not alter neutrophil clearance, whereas NADPH oxidase limited lung injury by reducing neutrophil accumulation in the alveoli. These studies suggested that NADPH-generated ROS have a protective role in acid-induced lung injury in this setting, by modulating inflammatory and pulmonary damage responses.189,190 In a murine model of intratracheal instillation of HCl, NADPH oxidase subunits and mitochondrial oxidative stress levels were elevated. NADPH inhibitor treatment did not improve lung injury; however, the administration of a mitochondrial-targeted antioxidant factor reduced inflammation and protected against PMN infiltration.191
NETs
Mice challenged with intratracheal instillation of HCl exhibited increased NET levels in the BALF when compared to a sham group, while exogenous administration of NETs worsened injury. Treatment with alvelestat, a neutrophil elastase inhibitor, could act as a potential therapeutic strategy.192
MAPK Pathways
In an in vitro model using a human lung epithelial cell line exposed to HCl, the MAPK signaling pathways were activated in a time-dependent manner, promoting lung injury and apoptosis. Pre-treatment of the cells with p38 and JNK inhibitors decreased cell apoptosis, while pre-treatment with an ERK1/2 inhibitor did not affect apoptosis. This indicated that HCl effectively induced apoptosis via the JNK and p38 pathways, modulating epithelial cell injury and death.193 In another study using human epithelial lung cells exposed to hydrogen peroxide, p38 and ERK phosphorylated levels were upregulated, while pre-treatment with hydrogen sulfide decreased the phosphorylated levels.194
Surfactant
Rat models of intratracheal instillation of HCl and/or gastric particles developed severe lung injury, accompanied by surfactant dysfunction. The BAL retrieved from the rats revealed a great reduction in large surfactant aggregates, and their ability to lower surface tension was also impaired. Surfactant impairment strongly correlated with lung injury severity.195 In a study using adult rabbits treated intratracheally with HCl, the administration of different surfactants, natural, bovine, and synthetic recombinant, did not improve oxygenation. The authors attribute these results to extended protein leakage and surfactant inhibition.196 However, surfactant replacement seemed to be beneficial in ex vivo lung perfusion. In two porcine models exposed to gastric acid aspiration, surfactant administration either right before ex vivo lung perfusion or during, improved lung function and ameliorated inflammatory mediators.197,198 It appears that whether a surfactant-based therapeutic strategy will be successful, depends on many parameters, including the type of lung injury, the severity, and the surfactant composition.
Ion and Water Transport
In a murine model of intratracheal instillation of HCl, the mRNA and protein expression of AQP4 were found to be decreased, while the expression of AQP1 and AQP5 remained unaltered.72 In an early-stage oleic acid-induced lung injury rat model, expression of AQP4 increased and was dependent on MAPK signaling pathways.199 However, in a study investigating the effects of AQP depletion in different lung injury models, including HCl aspiration, the absence of AQPs did not affect physiological water clearance of the lung, or edema formation.200
In a murine model, both oleic acid and ouabain, a specific Na, K-ATPase inhibitor, induced lung injury and inhibited Na-K-ATPase for 24 hrs, indicating its important role in lung injury.201 Treatment of oleic acid-induced lung injury with propofol, restored the Na, K-ATPase activity, increased inducible nitric oxide synthase, and ameliorated the inflammatory response in rats.202
Radiation Model
Exposure to ionizing radiation for therapeutic purposes is one of the principal treatment strategies for thoracic malignancies. However, the lungs are relatively radiosensitive to ionizing radiation, restricting the use of lung radiotherapy, and inducing the most common complication of radiotherapy, radiation-induced lung injury. Following radiation exposure, an asymptomatic period occurs, which should not be mistakenly characterized as non-responsive, and has been defined as a “latent period” in which no major pathology is apparent. The biochemical modifications, however, that occur during this period are thought to be responsible for most of the effects of ionizing radiation in mammalian cells. The main mechanisms through which radiation results in pulmonary damage are through ROS and RNS, produced by water radiolysis, and through direct damage of macromolecules. Pathology manifests between 2 and 6 months following treatment, as acute pneumonitis occurs. The next pathological finding evolves between 6 and 24 months post-treatment, that is fibrosis.203,204
The use of animal models has been extremely beneficial in understanding the pathogenesis of radiation in humans and in improving therapeutic doses. A crucial factor that should be considered in the choice of the animal species is the similarity to human injury response in the model, as well as the use of similar radiation doses. Murine models are one of the main choices, as well as pigs and rats, and have provided useful information on the underlying mechanisms of radiation-induced lung injury.205
Oxidative Stress
Oxidative damage is responsible for the long-term toxicity following radiation exposure. The induced generation of free radicals impairs oxidative metabolism in the irradiated cells and neighboring cells, through cellular communication pathways.206,207 Among the enzymes that are unregulated by ROS and RNS production are NADPH oxidases. NOX4 levels are induced immediately after the lungs have been exposed to radiation, promoting tissue inflammation, and acting as a mediator of pulmonary fibrosis.208,209 Increased levels of NOX4 and oxidative stress in the pulmonary blood vessels and the epithelial cells, as well as increased apoptosis of type I pneumocytes was observed 6-weeks after radiation in the lungs of mice that had received one dose of radiation to the whole thorax. Treatment with a scavenger of ROS/RNS reduced both NOX4 and apoptosis, proposing an interplay between oxidative stress and apoptosis in radiation-induced lung injury.210
MAPK Pathways
In a murine model, high dose irradiation of the left lung, resulted in increased protein levels of the phosphorylated form of c-Raf, the upstream MKKK of the ERK1/2 pathway, even four weeks after radiation exposure, suggesting that c-Raf activation plays an important role in radiation-induced lung injury, and specifically in lung fibrosis.211 In another study, the activation of the ERK pathway was associated with cell proliferation after exposure to low dose ionizing radiation.212 As mentioned above, radiotherapy is a principal therapeutic intervention in cell malignancies, especially lung cancer. In an in vitro model of human lung cancer cells exposed to ionizing radiation, phosphorylation of p38 was elevated, reaching its maximum levels 3–6 hrs after exposure. Although the phosphorylation levels of the other two MAPKs were not altered, treatment with p38 and JNK inhibitors blocked the cells’ radiation-induced elongation and cell migration.213 It seems that the activation and the effects of the activated MAPK pathways are not brief after radiation exposure; they are sustained and regulate important cellular responses pertaining to the cell’s injury response and fate.
Surfactant
It is known that radiation causes alterations in the surfactant layer. Hence, a study tested the effect of SP-D deficient mice to γ-radiation. Their results showed that the absence of SP-D resulted in the induction of pro-inflammatory pathways, especially RNS generated by i-NOS.214 The increased secretion of alveolar surfactant within hours following lung radiation exposure was one of the first clinical manifestations of radiation-induced pneumonitis. In a rabbit radiation model, it was suggested that serum surfactant apoprotein levels could serve as a biomarker of mortality.215 In a murine model exposed to thoracic radiation, intranasal administration of a surfactant component one day post exposure secured lung function, reduced inflammation and oxidative stress.216
Ion and Water Transport
AQP1 and AQP5 expression in rats that survived acute pneumonitis following a single dose of thoracic irradiation, revealed that protein and mRNA levels were decreased after irradiation while, even though AQP5 was upregulated until the second week post irradiation, AQP5 levels decreased 4-weeks post radiation exposure. This study suggested that both AQP1 and AQP5 play an important role in the pathogenesis of radiation-induced lung injury.217 The role of AQP4 has also been examined; in a murine model the left lung was exposed to a single dose of radiation, and subsequently the mice were treated with an AQP4 inhibitor. Inhibition of AQP4 attenuated pneumonitis by reducing inflammatory and innate immunity mediators.218 In order to examine the effect of radiation on the activity of Na, K-ATPase, adenocarcinomic human alveolar basal epithelial cells were treated with ouabain, and then exposed to radiation. Na, K-ATPase activity was inhibited in an ouabain-dependent manner, and drug treatment impaired radiation-induced cell cycle arrest.219
Transfusion-Related Acute Lung Injury (TRALI) Model
Transfusion-related acute lung injury (TRALI) is a type of acute lung injury characterized by non-cardiogenic pulmonary edema and widespread leukocyte infiltration. The onset of TRALI is immediate, within the first 6-hrs following transfusion of blood or blood products; however, it can be misdiagnosed as volume overload.220 The passive transfer of granulocyte or lymphocytotoxic antibodies, or human leukocyte antigen (HLA)-specific antibodies may be responsible for the recipient’s complement activation and, subsequently, pulmonary injury.221 Antibodies are not the only cause of TRALI cases; the main theory explaining the development of TRALI is the “two-hit” hypothesis. This hypothesis considers as the first “hit” the patient’s medical condition and, as the second, transfusion of blood products, other than antibodies, that could modify the biological responses.204,222 Despite low mortality rates, TRALI is a leading cause of transfusion-related morbidity and mortality.223
Animal models of TRALI have expanded our understanding of the underlying pathological mechanisms. Once again, it should be noted that experimental animals cannot provide identical pathophysiological responses to TRALI as in critically ill patients, however these animal models have proven to be extremely valuable in such uncommon syndromes. Depending on the hypothesis studied, different stimuli have been held accountable for developing TRALI. Hence, TRALI animal models have facilitated the evaluation and confirmation of the different hypotheses proposed. Different experimental strategies based on in vitro, in vivo, and ex vivo models have been explored, using both antibody and non-antibody stimuli in the transfused blood. LPS or other agents can be used to mimic surgery, trauma, or infection as the “first hit”, and subsequently the transfusion of anti-leukocyte antibodies or bioactive lipids serve as the “second hit”.224 Several published reviews have presented in detail the different experimental TRALI animal models and have discussed the findings.222,225,226
Oxidative Stress
It has been demonstrated that stored blood components, and not fresh blood, can prime neutrophil NADPH oxidase in vitro. The NADPH oxidase was exclusively activated by outdated plasma.227 A study using an in vitro model of human microvascular endothelial cells demonstrated that anti-human neutrophil antigen-3a (HNA) antibodies mediated severe TRALI, leading to increased ROS generation and endothelial barrier disturbance. Moreover, in the same study, NOX2-deficient mice treated with anti–HNA-3a antibodies did not develop TRALI, indicating that endothelial cell–derived ROS may affect the endothelial barrier integrity.228
NETs
The involvement of NETs in TRALI has been examined in a limited number of studies. A study examined whether NETs formed in stored canine blood can act as mediators of TRALI incidences. The results indicated that NET markers were increased in stored red blood cells, and demonstrated that leukoreduced red blood cells prior to storage, reduced NET formation.229 Neutrophils play an important role in TRALI pathogenesis.230 In a neutrophil and platelet-dependent mouse model of TRALI, NETs were present in the lung microvasculature and plasma. Treatment with aspirin decreased NET formation and platelet deposition. Therefore, the authors suggested that NETs may promote lung injury in TRALI, and that targeting NET formation or platelet activation could be protective.231 Another study on a “two-hit” TRALI model showed that NETs were formed in the lungs of mice and that treatment with DNase I improved their condition.224
Ion and Water Transport
One of the main pathological pathways present in TRALI is the increased pulmonary capillary permeability. Fluid rich in protein collected in the alveolar space, caused pulmonary edema.232 Inflammatory stimuli activated the pulmonary endothelium and promoted the aggregation of neutrophils in the capillary space, leading to dysfunction of the lung alveolar-capillary permeability barrier.233 A murine TRALI model of passive transfusion of major histocompatibility complex (MHC) class I monoclonal antibodies exhibited increased lung vascular and epithelial permeability, decreased alveolar fluid clearance, and prominent neutrophil sequestration.234
Influenza-Induced Acute Lung Injury Model
The influenza virus is an infectious respiratory disease microbe that causes seasonal epidemics and pandemics. In mild infections, influenza affects the upper respiratory tract, while in more severe cases it affects the lower respiratory tract, and can even lead to death. The severity of infection is linked to viral replication in the lower respiratory tract, which is accompanied by significant inflammation caused by immune cell infiltration. Meanwhile, in more severe cases the influenza infection can progress to pneumonia, and eventually ARDS and death.235–237
In order to develop an animal model of virus-induced lung injury, the selected laboratory animal should be able to become infected by the pathogen, and replicate the clinical manifestations present in humans. Similarities in clinical signs, histopathologic changes, viral growth kinetics, and transmission should be manifested in the chosen animal model. If animal models are not naturally susceptible to infection, adaptation of the virus is performed in order to induce host susceptibility. Among the different animal models, only ferrets, guinea pigs, and, to a lesser extent, hamsters have exhibited efficient influenza virus transmission. However, due to cost and husbandry requirements, murine models are mostly used.236,238
Oxidative Stress
Mice lacking NRF2 showed impaired antioxidant regulation and induced lung inflammation when exposed to influenza and cigarette smoke. It seems that NRF2 plays an important role in infection susceptibility and cellular protection.239 In an in vitro influenza-induced acute lung injury model, the researchers used different influenza virus strains based on their pathogenicity to infect the human lung cell line, A549. Proteomic analysis revealed that greater proteomic changes were induced by the higher pathogenic influenza strains H5N1 and H7N9. More specifically, infection with the most pathogenic strain, H5N1, and to a lesser extent, H7N9, resulted in reduced nuclear localization of phosphorylated NRF2.240 Several agents have been shown to induce activation of the NRF2 pathway and exhibit anti-influenza cell protective properties, which could be considered as novel therapeutic strategies.241–245
NOX2-deficient mice exposed to influenza exhibited reduced inflammatory infiltrations and improved lung function when compared to control mice.246 In agreement with the above study, another study used two influenza strains with low and high pathogenicity, H3N2 and H1N1, respectively, and infected wild-type and NOX2-deficient mice. The absence of NOX2 reduced airway inflammation, oxidative stress, apoptosis, and viral titers, indicating that the combination of NOX2 inhibitors and antiviral therapies could be an effective therapy against influenza infections.247 In a murine model of influenza-induced lung injury, mice were challenged intranasally with inactivated H5N2 influenza virus triggering the generation of ROS. In another set of experiments, mice mutant for NCF1, a major component of the NADPH oxidase complex, were challenged with inactivated H5N2; this ameliorated oxidative stress and controlled the severity of lung injury.248 In in vitro and in vivo experiments, human pulmonary carcinoma cell lines and murine primary airway epithelial cells were exposed to H1N1. The expression of NOX2 was downregulated, while the expression of NOX4 was increased, acting as a major regulator of oxidative stress and viral replication.249 In contrast, NOX1 appeared to play the opposite role to NOX4 and NOX2 in a study of mice infected with influenza A. At the early stages of infection, NOX1 suppressed lung inflammation and reduced oxidative stress.250
NETs
Mice challenged with sublethal doses of influenza virus exhibited induced NET formation, particularly in the alveoli and airways, as well as in tissue injury sites. Moreover, when neutrophils isolated from influenza-infected mice were co-cultured with infected alveolar epithelial cells in vitro, NET formation was strongly up-regulated. The induced formation of NETs in the lung air space promoted cytotoxicity, microvascular thrombosis, and was associated with alveolar damage in influenza-induced pneumonitis.251,252 In another study, NETs were induced by the complement component C5a, and treatment with an inhibitor of complement C5 activation resulted in reduced NET formation and amelioration of lung inflammation.251,253 Furthermore, neutrophils isolated from healthy volunteers exposed to either seasonal H1N1 or the highly pathogenic H5N1, exhibited varying NET formations; NETs were present only in H1N1-infected neutrophils, while no NET formation was observed in H5N1-challenged neutrophils. However, in the H5N1-infected alveolar epithelium, a greater neutrophil permeability was noticed. The absence of NET formation in H5N1 infection could explain the varying pathogenesis of influenza infections.254
MAPK Pathways
Activation of the MAPK signaling pathways has been suggested to regulate host immune responses against influenza infections.255 In various in vitro and in vivo studies that used different strains of influenza virus to develop influenza-induced lung injury, MAPK signaling seemed to have an important role during infection. The levels of phosphorylated JNK, p38, and ERK were induced, and this was associated with lung injury, modulation of the inflammatory response, apoptosis, and viral replication.249,256–258 Different agents, including MAPK inhibitors, anti-oxidants, flavonoids, monoclonal antibodies, as well as drug repositioning, have been tested as therapeutic strategies in models of influenza-induced lung injury. All of these agents managed to reduce activation of the MAPK signaling pathways, which had a protective effect on improving influenza-induced lung injury.243,244,259–269
Surfactant
As mentioned above, pulmonary surfactants have a critical role in the lung’s normal function and pathological processes. Metabolomics analysis of serum, lung tissue, and BALF from influenza-infected mice revealed metabolome alterations during key phases of influenza-induced lung injury. Most of the altered metabolic pathways included pulmonary surfactants, indicating a possible implication of pulmonary surfactants in respiratory failure progression and following tissue restoration.270 Moreover, it was demonstrated that H1N1 infection changed the surfactant lipid metabolism of alveolar type II cells, which could in turn promote surfactant impairment and contribute to acute lung injury development.271 Therefore, several studies have examined the therapeutic potential of surfactant replacement in influenza infections. Administration of surfactant lipids, surfactant nano-emulsions, and artificial surfactants have been tested in different influenza-infected animal models. The results were positive, showing that surfactant administration prevented and disrupted influenza infection, and preserved lung function.272–274
Ion and Water Transport
In an in vitro study, influenza A virus inhibited ENaC in rat alveolar type II cells, while in an in vivo set of experiments using rat lungs, influenza reduced fluid transport across monolayers. It seems that the attachment of the virus to the alveolar epithelial cells can facilitate infection establishment and, when it becomes overwhelming, can lead to ARDS and even death.275 Influenza A virus impaired the function of Na,K-ATPase in the plasma membrane of human and murine alveolar epithelial cells, and the lung epithelium of infected mice.276 In an in vitro study, infection of human alveolar epithelial cells with the highly pathogenic strains H5N1 and H7N9 resulted in significant impairment of the alveolar fluid clearance, and protein permeability compared to the lower pathogenic seasonal strains H1N1 and H3N2. Moreover, H5N1 infection induced a greater down-regulation of Na,K-ATPase when compared to H1N1 infection. These differences were attributed to secreted factors from the alveolar epithelial cells rather than the virus strain.277 In a mouse model of influenza A virus-induced lung injury, flavonoid extracts from the Lamiaceae plant Mosla scabra enhanced the expression of AQP5. The induction of AQP5 expression was thought to be a mechanism for restoring water permeability in the mice lungs, thereby reducing edema, inflammation, and apoptosis.261
Streptococcus (S.) pneumoniae-Induced Acute Lung Injury Model
Streptococcus pneumoniae is one of the primary causes of community-acquired pneumonia (CAP) worldwide, and also has a role in Hospital-acquired pneumonia (HAP). Streptococcus (S.) pneumoniae, or pneumococcus, is a highly invasive gram-positive bacterium responsible for high mortality rates worldwide. At least 97 S. pneumoniae serotypes have been characterized, highlighting its high adaptability. Children, the elderly, and adults with comorbidities are at higher risk of infection. Pneumococci first invade the host’s upper respiratory tract, where they colonize the nasopharyngeal epithelial cells asymptomatically. If the pathogens are not cleared by the host immune defences, bacteria migrate to sterile tissues and organs, causing pneumococcal diseases such as meningitis, bacteremia, and pneumonia.278,279 Pneumococcal pneumonia is the main type of pneumococcal diseases. Bacteria migrating through the lower respiratory tract escape the mucous defences and adhere to alveolar epithelial cells. Activation of immune responses, acute inflammation, disruption of the alveolar epithelium, and fluid accumulation in the alveoli are some of the early stages of pneumonia establishment.278,280,281
Several animal models of S. pneumoniae infection have been used to study infection progression, pathogenesis and to explore novel vaccine and drug candidates. Although different animal models have been used in experimental S. pneumoniae infection protocols, murine models are the most frequently encountered models, and have displaced other animal models. Different S. pneumoniae strains result in different outcomes and, therefore, different aspects of the infection can be studied.282 In order to achieve lung infection, different infection routes can be used, including intratracheal or direct intrabronchial instillation, aerosol or intranasal aspiration, and intraperitoneal or intravenous injection. Intranasal or aerosol exposure mimic the natural route of S. pneumoniae infection in humans. While procedures that deliver the bacterial inoculum directly into the respiratory tree require more invasive techniques, they are more effective for less virulent serotypes. Intranasal bacterial inhalation causes bronchopneumonia, while intratracheal instillation leads to lobar pneumonia. In addition, high-virulent serotypes are able to cause pneumonia and bacteremia in healthy mice. However, if a low-virulent serotype is used, immune-deficient animals are preferred.282,283
Oxidative Stress
In in vivo, ex vivo and in vitro models, S. pneumoniae infection induced oxidative stress and NRF2 was activated. Moreover, treatment with a NRF2 inducer restored the oxidative balance in the airway epithelial cells, suggesting a novel therapeutic strategy in pneumococcal pneumonia.284 The role of NRF2 in host response was examined in a murine pneumonia model. NRF2-deficient and wild-type mice were intratracheally instilled with S. pneumoniae, and afterwards gene profiling analysis of the whole lungs and neutrophils was performed. Six hours post-instillation, NRF2-deficient mice exhibited greater bacterial clearance and lower neutrophil aggregation; however, 24-hrs post-instillation, the accumulation of lung neutrophils was greater in the NRF2-null mice, possibly due to the extent of lung injury and the absence of the cytoprotective effects of the NRF2 gene.285 Moreover, in a pneumococcal model, mice deficient for the GP91phox subunit of the NADPH oxidase were intratracheally challenged with S. pneumoniae. When compared to the wild-type mice, the GP91phox-null mice exhibited no defect in bacterial clearance. The activation and accumulation of neutrophils was increased, however their presence did not increase lung injury. These findings indicated that in pneumococcal pneumonia, NADPH oxidase can regulate the inflammatory response and is not responsible for bacterial killing.286
NETs
Exposure of murine neutrophils to different serotypes of S. pneumoniae resulted in NET formation; the extent of NET formation was dependent on capsule thickness, and correlated with disease severity.287 The pneumococcal protein α-enolase could also induce NET formation.288 NET formation was also examined in in vitro and in vivo models of secondary S. pneumoniae infection after primary influenza infection. NET formation was induced, however NETs did not protect against the secondary infection. NET formation correlated with excessive inflammatory response and alveolar-capillary barrier dysfunction, determining disease severity.287,289 The elimination of bacterial infection by NETs seems to be controversial. There are several studies reporting that proteins present on the S. pneumoniae surface enable bacteria to escape from NET capture, allowing pneumonococcal migration to sterile sites.290,291
MAPK Pathways
In different pulmonary models of S. pneumoniae infection, pneumococcal challenge induced activation of MAPK signaling, indicating that the host’s immune response after infection is at least in part MAPK-dependent. Several studies have demonstrated that p38 and JNK are rapidly activated after pneumococcal infection, modulating lung cell activation, inflammatory response, and caspase-dependent cell apoptosis. Treatment with MAPK inhibitors reduced lung inflammation, apoptosis, and pneumococci-dependent gene transcription.292–296 Moreover, in an in vivo model, young, mature, and aged mice were intratracheally infected with S. pneumoniae. Alveolar macrophage JNK and p38 decreased with age, while alveolar macrophage ERK activation increased with age following infection. These findings were attributed to age-related TLR dysfunction in alveolar macrophages, which might explain the enhanced susceptibility to bacterial pneumonia in the elderly.297
Surfactant
The role of the surfactant layer has been examined in various animal models of S. pneumoniae infection. Surfactant proteins have the ability to bind and agglutinate pneumococci. In an in vitro model using recombinant SP-D, SP-D was capable of binding to most pneumococci, however different serotypes aggregated to a different extent.298 Intranasal exposure of S. pneumoniae to SP-D-deficient mice resulted in persistent bacterial inoculation, inflammatory response in the airways, early onset of bacteraemia, and dysfunctional bacterial clearance.299 Increased levels of SP-D were found in the BALF of infected mice, while morphologic injuries were detected only in type II alveolar cells and not type I cells in the first three days following infection.300 Moreover, the regeneration of the alveolar epithelium was investigated in mice recovering from S. pneumoniae infection. The alveolar epithelial type II cells expressing SP-C exhibited increased activity during infection, resulting in regeneration of the alveolar epithelium. Increased alveolar epithelial type II cell proliferation was observed within 7-days post-infection, while the differentiation of alveolar epithelial type II cells to type I occurred during the resolution of lung inflammation.301
Ion and Water Transport
In a rat model of S. pneumoniae infection, the expression of AQP5 was decreased by 70% 24-hrs post-infection in lung tissue. AQP5 levels increased three days after infection, without exceeding the control levels.300 In a study using ventilated and blood-free perfused murine lungs challenged with a virulent S. pneumoniae factor, an early onset of pulmonary microvascular barrier function impairment and severe pulmonary hypertension was observed.302 In an ex vivo model of perfused human lungs exposed to a high dose of S. pneumoniae, either intravenously in order to reproduce a bacteremia model, or through direct bacterial instillation into the distal airspaces, mimicking a pneumonia model, the injury induced to the alveolar epithelium was examined. In contrast to the pneumonia model, in the bacteremia model the alveolar epithelial permeability remained intact, no alveolar edema was detected, while alveolar fluid clearance was not impaired. Based on these results, it was proposed that the lung and alveolar epithelium is resistant to bacteremia, and has the ability to rapidly clear bacteria from the lungs, protecting them from lung injury development.303 Finally, in an in vitro model using human lung microvascular endothelial cells, induction of cells with a S. pneumoniae virulence factor revealed a protective role of ENaC in restoring capillary barrier function, presenting a novel therapeutic approach for improving barrier function during pneumonia.304
Coronavirus-Induced Acute Lung Injury Model
Prior to the ongoing coronavirus infectious disease (COVID-19), caused by the severe acute respiratory coronavirus 2 (SARS-CoV-2), two other coronavirus outbreaks, the severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002, and the Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012, had emerged. All of these viruses belong to the Coronaviridae family, β-coronaviruses genus, and result from zoonotic coronaviruses. The infections share some clinical manifestation similarities and dissimilarities. The SARS-CoV outbreak caused the most severe disease with the highest mortality rate. The outbreak was quickly restricted, mainly due to insufficient transmission. Afterwards, the MERS-CoV outbreak had an extremely high mortality rate (around 34%), however the outbreak was also quickly contained.305 Several genetic variants of SARS-CoV-2 have emerged since the beginning of the pandemic. Increased virus transmissibility, advanced host immunity, and vaccine-induced immunity escape viral mechanisms, highlight the importance of vigilance.306
Coronavirus animal models should provide information about virus transmission, prevention, pathogenesis, and treatment. One of the main parameters that should be considered when developing a coronavirus animal model is that the animals should express the same receptors the virus uses to invade humans. Moreover, basic clinical manifestations and disease severity should be reproducible in the chosen animal model. Transgenic animals (primarily murine models), virus-adapted animals, and immune-deficient animals can be used to mimic clinical manifestations as closely as possible. Small animals, including murine models and ferrets are suitable for therapeutic and pathogenesis studies. Non-human primates are closer to the immune and genetic profile of humans and, therefore exhibit similar pathophysiological responses. However, non-human primates are harder to maintain and are not cost-efficient. For SARS-CoV, ferrets and non-human primate models reproduced better the clinical symptoms and immunohistochemistry observed in infected humans. Transgenic mice, rhesus macaques, and marmosets are the most frequently used animal models for MERS-CoV. Murine models have been used to examine molecular mechanisms, pathogenesis, and vaccine development induced by SARS-CoV-2. Furthermore, in vitro models, primary cell cultures, and organoids have been an important tool in understanding the SARS-CoV-2 infection progression.307,308
NETs
In intranasally SARS-CoV-2-infected hamsters, induced NET formation in the lungs was observed 3-days post-infection, which remained increased until day 6 post-infection. NET formation was associated with vascular pathology, including endotheliitis and vasculitis.309 In SARS-CoV-2-infected hamsters, treatment with disulfiram, a drug used in alcohol use disorder, reduced NETs, neutrophil infiltration in the lungs, and perivascular fibrosis, however did not affect viral clearance. Disulfiram improved the histology of the infected lungs, implying a potential therapeutic benefit in COVID-19 treatment.310
MAPK Pathways
SARS-CoV can activate the p38 signaling pathways.311–313 The entry of SARS-CoV into the cells resulted in the cytoplasm transportation of the protein syntenin, which binds to the E envelope protein of SARS-CoV, leading to overexpression of inflammatory cytokines, through the activation of the p38 pathway. Both silencing of syntenin and infection with a recombinant SARS-CoV lacking the E envelope protein decreased p38 activation and the inflammatory response. Moreover, treatment with a p38 inhibitor increased the survival rates of the SARS-CoV-infected mice.311 The involvement of p38 in SARS-CoV infection has also been demonstrated by an in vitro and in vivo study, which showed that p38 was implicated in the pro-fibrotic response.314 In a COVID-19-mimic mouse model, treatment of mice with p38 and ERK inhibitors reduced the BALF cytokine levels and neutrophil infiltration.315
Surfactant
Among the differentially expressed genes that contribute to acute lung inflammation following lethal and sublethal doses of SARS-CoV, decreased expression of surfactant proteins was observed in infected mice.316 Proteomic analysis of hamster lung tissues infected with SARS-CoV-2 revealed altered expression of surfactant proteins at early stages of infection (4-days post-infection), when compared to mock controls. This indicates dysregulated surfactant metabolism following viral infection.317 An in vitro study using a recombinant fragment of human SP-D examined its therapeutic potential in SARS-CoV-2 infection. The recombinant fragment of human SP-D had the ability to restrict the interaction of the viral S1 spike protein with cells overexpressing the human angiotensin converting enzyme 2 (hACE2). This suggested that recombinant SP-D had a protective role against SARS-CoV-2, acting as a viral entry inhibitor.318
Ion and Water Transport
An in vivo murine model of SARS-CoV infection examined the pathogenesis differences between young and adult infected mice. Infected adult mice exhibited early and exacerbated acute pro-inflammatory responses in the lungs, which were accompanied by severe pulmonary edema and diffuse alveolar damage, leading to lethal respiratory illness. On the contrary, young mice were relatively resistant to the virus, and did not develop a severe respiratory illness.319 Moreover, pulmonary edema formation was observed in the lungs of MERS-CoV-infected non-human primate animal models. However, the degree of the pulmonary edema, as well as the disease severity, was different between the two models. In the lungs of infected rhesus macaques, focal interstitial pneumonia and pulmonary edema developed, mimicking the pathological processes in mild MERS-CoV infections. On the other hand, the edema that developed in the common marmoset lung tissue was widespread with diffused neutrophil infiltration and fibrinous exudates, rendering it a more acute and severe infection model.320 Finally, the role of aquaporins in the pathophysiology of SARS-CoV-2 infection was examined in an in vivo study. Immunohistochemical staining of AQP1 in hamster lung samples infected with SARS-CoV-2 revealed that perivascular edema formation was associated with a decline in AQP1 expression. Three days post-infection, AQP1 expression was completely reduced in vessels with edema. AQP1 seemed to have a key role in paracellular leakage and edema formation in SARS-CoV-2 infections.321
Table 1 lists the major findings of the studies on the lung injury mechanisms discussed in the models presented.
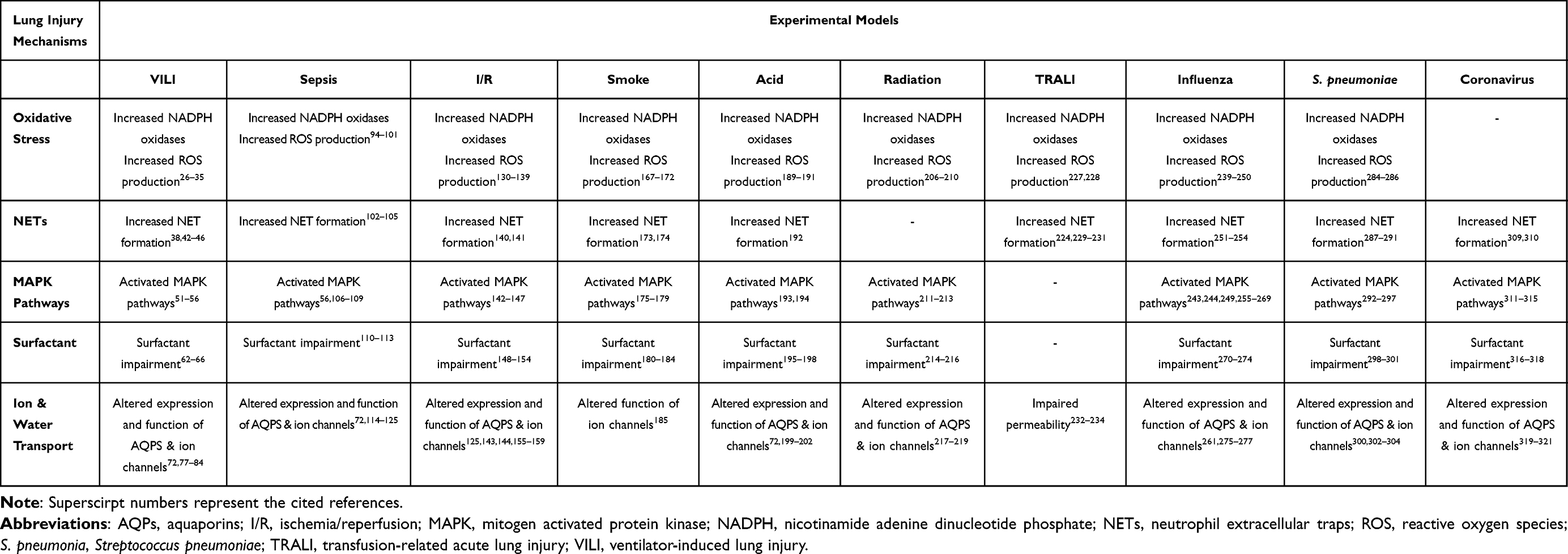 |
Table 1 Major Aspects of the Lung Injury Mechanisms in Experimental Models |
Clinical Acute Lung Inflammation - Acute Respiratory Distress Syndrome (ARDS)
Contrary to induced lung injury experimental models, the cause of ARDS in clinical studies is not easily recognizable. However, understanding the causative insults that lead to the clinical manifestations is a very useful tool in diagnosis and treatment strategies. Throughout this section of the review, we present clinical studies that have explored the selected mechanisms.
Figure 1 depicts the key mechanisms of lung injury discussed in this review that contribute to the progression of ARDS.
 |
Figure 1 Key mechanisms of lung injury that contribute to the progression of acute lung inflammation/acute respiratory distress syndrome (ARDS). Abbreviations: ERK, extracellular signal-regulated kinase; JNK, C-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MKK, MAPK kinase; MKKK, MAPK kinase kinase; NET, neutrophil extracellular trap; RNS, reactive nitrogen species; ROS, reactive oxygen species. Notes: Pulmonary and extrapulmonary stimuli causing lung injury induce physiological changes and related host’s responses, and when the response becomes overwhelming, ARDS is established. (I) Stimuli can shift the balance towards pro-oxidant release, leading to oxidative damage as a result of accumulation of oxygen free radical. (II) Activated neutrophils aggregate in the pulmonary microvasculature, interstitium, and the alveoli, releasing neutrophil extracellular traps (NETs). (III) These generated extracellular fibers consist of granule proteins and histones. Extracellular signals trigger the activation of mitogen-activated protein kinase (MAPK) signaling pathways, promoting cellular responses important for the cell’s fate. (IV) The maintenance of lung surfactant integrity is important for the prevention of alveolar collapse. Lung injury-induced compounds disrupt the integrity of the lung surfactant layer and impair its protective function. (V) Damaged capillary and alveolar walls exacerbate pulmonary edema formation by increasing the alveolar–capillary barrier permeability. Water and ion transportation homeostasis is disturbed. The expression and function of the water channels termed aquaporins (AQPs), is differentially regulated based on the type of injurious insult. Moreover, impaired edema clearance negatively affects ion transportation and fluid reabsorption. The image is adapted from Vassiliou AG, Kotanidou A, Dimopoulou I, Orfanos SE. Endothelial Damage in Acute Respiratory Distress Syndrome. Int J Mol Sci. 2020;21(22):8793. doi:10.3390/ijms21228793.67 © 2020 by the authors. Licensee MDPI, Basel, Switzerland. Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/). |
ARDS in the ICU
Mechanical ventilation is the most important therapeutic intervention in critically ill patients with respiratory failure admitted in the ICU. Although mechanical ventilation is a life-saving procedure, the mechanical pressures, gas volumes and concentrations, and the ventilator rates exceed the ones physiologically applied, causing lung injury. The common element of the pathophysiological mechanisms is the initial volutrauma and barotrauma in the alveolar units.322 This entity is clinically known as ventilator-associated lung injury (VALI), which resembles the experimental findings of ventilator-induced lung injury (VILI). However, as mentioned, in clinical practice it is difficult to determine the initial causative insult that leads to ARDS development, and clinical trials examining whether different mechanical ventilation strategies cause ARDS are not feasible. Therefore, the contribution of VALI to ARDS progression cannot be defined. There are no clinical manifestations in VALI diagnosis, and the relationship between ARDS and VALI is not clearly understood.323,324 A better understanding of ARDS mechanics could provide new insights in mechanical ventilation strategies that could help eliminate the risk of ventilator-associated complications.
ARDS is characterized by an acute lung inflammatory response. Therefore, it is considered the most appropriate clinical syndrome to study pathogenic mechanisms of lung inflammation. Based on the revised Berlin Definition, ARDS is a heterogeneous syndrome characterized by an acute onset, distinctive radiographic findings, and increased mortality. The categorization criteria use the level of patients’ hypoxemia to define the severity of ARDS as mild, moderate, or severe. Diffuse alveolar damage, which is pathophysiologically translated into edema formation, inflammation, and hyaline membrane formation, is one of the main morphological features of ARDS.325 Several direct and indirect lung injury etiologies increase the risk of ARDS development. Sepsis, ischemic periods followed by reperfusion, aspiration, toxic inhalation, and radiation are inciting events that lead to ARDS.326
Oxidative Stress
Excessive production of reactive oxygen intermediates contributes to endothelial cell damage, loss of endothelial barrier integrity, and pulmonary edema formation, which aid in ARDS progression.327 In ARDS, exogenous derived sources, such as mechanical ventilation, and endogenous, such as an injured endothelium, activated neutrophils in the lungs, parenchymal cells, and circulating oxidant-producing enzymes, are thought to be the primary causes of ROS-mediated lung injury.17 At the early stages of ARDS, NOX1 expression levels are increased in the alveolar epithelium of ARDS patients, and NOX1 is partially associated with lung epithelial cell death.328 Several genetic variants have been linked with ARDS susceptibility and mortality. In particular, a single nucleotide polymorphism (SNP) in the NRF2 gene (NFE2L2-rs6721961) has been associated with an increased 28-day mortality risk in ARDS patients.329
NETs
One of the major ARDS hallmarks is the accumulation of activated neutrophils in the pulmonary microvasculature, interstitium, and the alveoli. Neutrophils are the first line of the host’s defense against pathogens. In ARDS, the activation and migration of neutrophils to the sites of inflammation is achieved through chemotaxis, as a result of the secreted pro-inflammatory cytokines and chemokines.330 NETs have been associated with disease severity and outcome in several diseases, like pneumonia, sepsis and ARDS.331,332
MAPK Pathways
Transcriptional profile analysis of neutrophils from ARDS patients revealed that MAPK pathways have a key role in ARDS pathogenesis. The p38 MAPK pathway is one of the main canonical pathways of neutrophil gene signatures in ARDS.333,334
Surfactant
Respiratory distress syndrome in preterm infants is the most common surfactant-deficient disorder. Although in neonates it can be treated with exogenous surfactant administration, in adults with ARDS, this approach has not proven successful.335,336 The lungs of preterm neonates differ developmentally from adults. Exogenous surfactant replacement therapy in adult patients with ARDS is still a work in progress. Until now, all clinical trials examining surfactant replacement therapy in adult patients has not resulted in improvement in the patients’ oxygenation or mortality. In future large clinical trials, factors such as dosing, delivery route, timing, and novel pharmaceutical surfactants should be examined.60,337–339 In a post hoc analysis of five studies with recombinant surfactant therapy in ARDS patients, the subgroup of patients with severe ARDS due to pneumonia or aspiration showed improved oxygenation and reduced mortality.340
Ion and Water Transport
Exacerbated pulmonary edema formation has been associated with adverse outcomes in ARDS patients.341 A SNP present in the AQP5 promoter in patients with ARDS has been associated with reduced inflammatory responses and improved survival.342 ARDS patients with the highest alveolar fluid clearance rates had a significantly lower mortality risk and required a shorter period of mechanical ventilation support.343 In a Phase II clinical trial, inhalation of an enhancer of epithelial sodium channels in mechanically ventilated ARDS patients, reduced the extravascular lung water index of ARDS patients with a Sequential Organ Failure Assessment (SOFA) score ≥11.344
Sepsis-Induced ARDS
Sepsis is a life-threatening and heterogeneous syndrome. Important cellular pathways are activated, including both pro- and anti-inflammatory, coagulant, metabolic, and hormonal, and when the phenomena are overwhelming, they could lead to organ dysfunction and failure. Septic shock is a subtype of sepsis defined by dysregulated circulatory and cellular functions and an increased mortality risk.85,345,346 Sepsis is a major risk factor for ARDS development and is associated with worse clinical outcomes.347,348 Currently, there is no specific treatment approved for sepsis. Therefore, prognostic biomarkers and molecular and metabolomic signatures could aid the early diagnosis of sepsis, as well as the identification of patients at risk for ARDS development.85,349,350
Oxidative Stress
Ten SNPs have been associated with the likelihood of ARDS establishment in septic patients, seven of which are located in histone marks or in transcriptional sites, and one in the NRF2 promoter region.351
NETs
As mentioned above, NETs have an important role in sepsis. Sepsis-induced ARDS patients had higher NET levels than healthy controls and septic patients, and furthermore NET formation was associated with ARDS severity and adverse outcome. Moreover, lower circulating levels of DNase I has been linked to the development of sepsis-induced ARDS.105,352
MAPK Pathways
Analysis of the datasets of blood polymorphonuclear neutrophils (PMNs) from patients with ARDS and from sepsis patients, showed a total of 220 differential expressed genes that overlapped between the two groups. Among the key identified genes and pathways were the MAPK signaling pathways.353
Surfactant
Several studies and clinical trials have investigated the effect of surfactant replacement as a therapy in sepsis-induced ARDS. However, like in ARDS caused by other stimuli, the results were disappointing. In all studies, surfactant administration was well tolerated, yet a significant clinical benefit was not reported.354–356
Ion and Water Transport
Patients with septic shock and ARDS had lower rates of alveolar fluid clearance compared to patients with septic shock without ARDS.357 In a study examining AQP1 expression in septic patients admitted to the ICU, some of whom developed ARDS, AQP1 levels were induced in leukocytes of patients with ICU-acquired sepsis, and the highest expression was exhibited in septic shock.358 Additionally, one SNP and one epigenetic modification present in the promoter of AQP5 have been associated with sepsis mortality.359,360
Ischemia/Reperfusion
As mentioned above, the disruption of blood circulation or ventilation results in an oxygen shortage that leads to tissue hypoxia and cell death. Restoration of blood and oxygen supply may worsen the injured lungs by introducing inflammatory and oxidative stress mediators to the former ischemic tissue. These mediators can also travel through the circulation to distant organs and cause systemic inflammation and multiple organ failure. Non-cardiogenic pulmonary edema, oxygenation impairment, and radiographic findings similar to ARDS characterize lung ischemia/reperfusion injury.361,362 A clinically significant type of I/R is lung transplantation and allograft rejection. Primary lung graft dysfunction is a complication after lung transplantation that increases the patient’s risk of morbidity and mortality. Like other lung injuries with an acute onset, it develops in the first few days after lung transplantation and is characterized by edema formation, hypoxemia, and pathological radiographic patterns.363
NETs
BALF and plasma from patients who underwent lung transplantation revealed that in the first 24-hrs after transplantation, patients who developed more severe forms of primary graft dysfunction had higher levels of NETs in their BALF compared to those who did not experience primary graft dysfunction. The extracellular DNA levels in the plasma remained unaltered before and after transplantation.140
Surfactant
Lung transplantation disturbed surfactant function shortly after transplantation; this dysfunction persisted in a long-term course in the lungs of the transplant recipients.364 Contrary to ARDS and sepsis, surfactant replacement therapy in patients who underwent lung transplantation improved the impaired surfactant function, the clinical parameters post reperfusion, including oxygenation, ventilation period, boosted post-transplantation recovery, and prognosis.365–368
Ion and Water Transport
A study investigated the role of the alveolar epithelium in reperfusion edema following orthotopic lung transplantation. It was revealed that the cause of the pulmonary edema induced post-transplantation was attributed to increased alveolar–capillary barrier permeability. However, in the majority of the included patients, alveolar epithelial fluid transport was preserved and did not improve recovery. The extent of the edema was correlated with the graft’s ischemic preservation period.369
Smoke Inhalation
Fire accidents, terrorist attacks, and military operations are the main causes of smoke inhalation-induced lung injuries. Inhaled products from the incomplete combustion of carbon, hot vapors, toxins, dust, and airborne microparticles result in airway and pulmonary injury, and the worst scenario could lead to ARDS development. The time of exposure, the inhaled mixture consistency, and the host’s parameters determine damage severity.160–162 Smoke inhalation injuries remain a complex clinical problem. The mechanisms are not fully understood and the improvement in mortality rates is attributed rather to advances in critical care medicine practices rather than smoke targeted treatments.370
To our knowledge, no studies exist on clinical acute lung inflammation and the mechanisms we have selected to present. Hence, we will briefly discuss reviews that have dealt with the selected mechanisms in chronic illness caused by tobacco smoke.
Oxidative Stress
The role of NRF2 and NADPH oxidases in human studies of inflammation and chronic lung illnesses caused by tobacco smoke have been extensively reviewed.371,372
MAPK Pathways
The review by Tamimi et al examined the clinical implications of tobacco smoke exposure on chronic lung diseases and the activation of MAPK signaling pathways, as well as possible therapeutic implications.373
Surfactant
Pulmonary surfactant levels are negatively affected by tobacco smoke. The cited review provides a thorough understanding of the impact of surfactant dysfunction in chronic lung diseases.374
Acid Aspiration
Aspiration-induced lung injury is a clinically relevant complication among critically ill patients in the ICU and can cause ARDS. The diagnosis of aspiration-induced lung injuries in patients still remains elusive, as does a standard therapeutic scheme.7,186,188 Acid aspiration lung injury is characterized by two distinct yet difficult to distinguish clinical entities; aspiration pneumonitis and aspiration bacterial pneumonia. The difference lays in the presence of pathogens in the aspirated gastric contents. Critically ill patients have a great susceptibility to aspiration. Parameters such as the patient’s positioning and nasogastric intubation increase the risk of aspiration and aspiration pneumonia.188,375,376 Relatively little research has been carried out in the lung injury mechanisms discussed in the present review in adult patients with aspiration-induced lung injury.
NETs
One study investigated the relationship of NET formation and acid aspiration-induced ARDS; the data showed that NET levels in gastric acid aspiration-induced ARDS patients are associated with disease severity.192
Radiation
As mentioned above, there is an early response and a late response following irradiation, called acute pneumonitis and fibrosis, respectively. Patients who received ionizing treatment may not experience symptoms of pneumonitis at first, however they later develop fibrosis.203,204 Radiation-induced lung injury is caused by two distinct mechanisms; the first is induced by the cytokines released at the site of radiation, causing radiation-induced pneumonitis. The second is known as sporadic radiation pneumonitis, and is the result of innate immunity activation as a response to localized lung exposure to radiation.377 One of the first studies that linked radiation-induced lung injury and ARDS was about two case reports. These patients briefly after radiation exposure suffered from respiratory failure, hypoxemia, and diffuse bilateral infiltrates.378 In another study, a neural network model consisting of 235 patients with lung cancer was used in order to predict the possibility of lung radiation-induced pneumonitis development.379 New radiotherapy techniques and engaging advances in radiotherapy could improve the impact of radiation on the lungs, and minimize the radiation-induced pulmonary damage.380
Transfusion-Related Acute Lung Injury (TRALI)
The majority of TRALI incidences occur during surgeries and in the ICU. Patients who develop TRALI, apart from pulmonary edema, display ARDS symptoms accompanied by hypoxemia and hemodynamic abnormalities.221 Up to date, the underlying mechanisms accountable for TRALI are not clear, however neutrophil activation seems to be the driving force. Neutrophils’ interaction with the lung endothelium causes an increase in capillary leakage, neutrophil infiltration into small pulmonary vessels and capillary vasculature, and the formation of interstitial and intra-alveolar edema. Several hypotheses have been proposed in order to explain the pathophysiological events leading to TRALI, in which at least six different pathways of antibody-mediated TRALI are involved.381 Both recipient’s/ patient’s and donor’s factors may be involved in the pathogenesis of TRALI, including the patient’s underlying condition and genetic susceptibility, leukocyte antibodies, cytokines, lipids, and factors that promote pulmonary endothelial cell permeability.382
Oxidative Stress
NADPH oxidase activity was shown to be elevated in the serum of TRALI patients.383
NETs
Increased NET formation and biomarkers were present in the plasma of patients who developed TRALI.224,231
Surfactant
A case report of a neonate who was diagnosed with TRALI following exchange transfusion for hyperbilirubinemia showed that after surfactant replacement, the patient’s condition improved indicating a possible therapeutic intervention for TRALI patients.384
Influenza
The first stage of influenza A infection is the insult of the epithelium of the airway and the alveoli where the virus replicates; at the same time the first immune response mechanisms are activated, triggering a cytokine storm. Following this, the adaptive immune defences are stimulated in order to promote viral clearance. The robust activation of the immune responses causes damage to the alveolar endothelial and epithelial cells. Post-mortem lung examination of deceased patients has revealed diffused alveolar damage. Finally, when immunity against the infecting virus strain develops, restoration and regeneration of the damaged lung tissue occurs. During this time, the risk of a secondary bacterial infection is increased.385
Oxidative Stress
The expression of NRF2 and HO-1, and the production of ROS were elevated in human transdifferentiated alveolar epithelial cells infected with influenza A virus. Overexpression of NRF2 reduced viral replication and oxidative stress, indicating that NRF2 has a protective role by inducing the expression of antioxidant genes.386
NETs
Hospitalized patients with severe H7N9 and H1N1 influenza infections exhibited increased plasma levels of NET formation and NET markers on admission day, which positively correlated with infection severity and poor prognosis.387 Increased NET markers, cell free DNA and histone-bound DNA were present in the BALF and serum of patients with severe H1N1 infection. The high plasma NET levels could discriminate between severe and mild infections, while even higher NET levels could be used as prognosis markers.388
Surfactant
In influenza A infected patients, genetic variants of the SP-A2 gene were associated with the need for mechanical ventilation, ARDS progression, and infection severity.389 A SNP in the SP-B gene was associated with the severity of influenza A infection in a Chinese population.390 Moreover, a gene expression profile analysis of the lungs of deceased H5N1 influenza-infected patients revealed that pulmonary SP-D was downregulated; this was also confirmed by quantitative RT-PCR. SP-D levels were considerably lower in influenza-infected lungs than in ARDS lungs, suggesting a possible role in pathophysiology.391 However, in a recently published study, serum levels of SP-D were found to be elevated in severe pandemic influenza patients and furthermore, SP-D could be used as a biomarker to distinguish severe influenza A infection from COVID-19.392
Streptococcus (S.) pneumoniae
Different pneumococcal serotypes are responsible for different clinical manifestations and disease severity. For example, infection with some serotypes leads to invasive disease, and with others to lobar pneumonia; some serotypes are more prevalent in specific age groups.283 S. pneumoniae vaccine development and the implementation of children vaccination strategies, has altered the serotypes that cause invasive pneumococcal disease among adults. These new invasive serotypes are mainly those not included in the vaccines, and are associated with disease severity and an increased risk of sepsis progression.393 Pneumonia is one of the leading causes of hospitalization and mortality worldwide. Since this review does not focus on pathogens and their related mechanisms, we chose to explore S. pneumoniae, which is among the pathogens that can infect the lower respiratory tract and cause pneumonia. Two of the most common categories of S. pneumoniae-induced disease are hospital-acquired pneumonia (HAP) and community-acquired pneumonia (CAP).394 In HAP, the infection in the lower tract occurs within 2-days post-admission, while in CAP, infection of the lungs is acquired outside of a hospital setting.394,395 Critically ill patients are at a very high risk of developing HAP, which increases hospitalization length, mortality, and sepsis development risk.393,395 It is worth mentioning that in the ICU, HAP is more frequently caused by hospital-acquired gram-negative bacilli and Staphylococcus aureus (including methicillin-resistant strains). S. pneumoniae is the main pathogen responsible for CAP. Another clinically relevant phenomenon is secondary bacterial infection following influenza A infection. Pneumococci take advantage of the dysregulated lung physiology and the changes in host immune responses in order to invade and cause a bacterial super-infection.396
Oxidative Stress
Gene expression analysis of septic patients secondary to CAP revealed differential expression of NADPH oxidase genes. Distinct gene expression patterns were observed in survivors and deceased patients.397
NETs
Patients with CAP exhibited increased admission serum levels of cell-free nucleosomes markers of NETosis. This pronounced NET formation was associated with prolonged hospitalization and adverse patients’ outcome.331
Surfactant
Genetic variability, SNPs and several haplotypes, in the genes of surfactant proteins have been shown to modulate the susceptibility and outcome of CAP patients.398 Serum levels of SP-D have been proposed as a biomarker of CAP severity in pediatric patients. Elevated SP-D levels were detected in critically ill patients and were correlated with severity scores.399
Coronavirus Infectious Disease 2019-Induced ARDS
SARS-CoV-2 and SARS-CoV have greater structural and pathogenicity similarities than MERS-CoV. The symptoms include fever, cough, fatigue, and myalgia. In patients with more severe symptomatology, worsening of dyspnea and hypoxemia can lead to ARDS development.305 In the early phases of SARS-CoV infection, diffuse alveolar damage is observed, which in later phases can be accompanied by acute fibrinous and organizing pneumonia (AFOP). The histological changes induced by MERS-CoV consist of diffuse alveolar damage, hyaline membranes, pulmonary edema formation, and interstitial pneumonia.400 Apart from the involvement of the respiratory and immune systems in the SARS-CoV-2 infection, other systems, such as the cardiovascular and central nervous systems, are also affected. Prominent characteristics of coronavirus disease 2019 (COVID-19) are the inflammatory response and endotheliopathy.401–403 Severe COVID-19 infection is usually accompanied by an increase in cytokine levels, which many have identified as a distinct SARS-CoV-2 symptom. However, several studies have compared the cytokine levels in severe COVID-19 and non-COVID-19 inflammatory syndromes, such as ARDS and sepsis and have found their levels comparable.404,405 Histopathologic findings in the lungs of SARS-CoV-2 patients are similar to those described above for SARS-CoV and MERS-CoV. Diffuse alveolar damage is the predominant histopathologic finding, followed by hyaline membrane formation, and, in a later phase, edema and fibroblast proliferation. The elderly and patients with comorbidities are at risk of developing more severe disease.406–408
The novel disease, COVID-19, is caused by SARS-CoV-2. While COVID-19 usually manifests with mild symptoms and signs, about 10 to 20% of patients rapidly progress to ARDS, and multiple organ dysfunction, requiring treatment in the ICU.
Oxidative Stress
Soluble levels of NOX2-derived peptides have been shown to be elevated in COVID-19 patients and NOX2 activation has been associated with disease severity.409 It has also been suggested that dysregulated NOX-dependent signaling pathways prior to SARS-CoV-2 infection, are associated with COVID-19 disease severity and outcome.410 Several published reviews have investigated the role of the NRF2 signaling pathway in COVID-19. The suggested therapeutic strategies based on NRF2 activators include restoring oxygenation balance and suppressing inflammation-induced responses.411–414 In lung biopsies of deceased COVID-19 patients, NRF2 expression was suppressed.415
NETs
Several studies have highlighted the important role of NET formation in patients with ARDS secondary to COVID-19.416–418 High levels of NETs were present in the lower respiratory tract and peripheral blood of critically ill COVID-19 patients.419–421 Uncontrolled NET formation exacerbated alveolar and endothelial lung damage, triggered hyperinflammation and immunothrombosis, and adverse outcomes in critically ill COVID-19 patients.422–425 Elevated levels of cell-free DNA and specific markers for NET remnants, myeloperoxidase-DNA, citrullinated histone H3, and high anti-NET activity in the IgG and IgM fractions were found in the serum of hospitalized COVID-19 patients. These findings correlated with several clinical and severity parameters.426,427 Novel therapeutic strategies for severe COVID-19 targeting NETs have been proposed, yet further studies and clinical trials need to be carried out.425,428–431
MAPK Pathways
In COVID-19, SARS-CoV-2 induces p38 activity in order to support its replication. p38 mediates uncontrolled pro-inflammatory cytokine production, pulmonary edema formation and, in the worst scenario, could lead to ARDS development and death.432–435 Several p38 inhibitors have been examined as pharmacological therapies in ARDS, and might also provide therapeutic benefit in COVID-19 patients.433,436
Surfactant
A study showed that SP-D was significantly elevated in SARS-type pneumonia, impelling the authors to propose that monitoring systemic SP-D may be useful in monitoring the alveolar integrity in SARS-type pneumonia.437 Transcriptome analysis of lung biopsies from COVID-19 patients revealed that genes implicated in lung surfactant metabolism are dysregulated. In drug enrichment analysis conducted by the same researchers, it was suggested that surfactant replacement is a possible therapeutic strategy in COVID-19 patients.438 Several studies have investigated the effects of surfactant therapy in patients with COVID-19-induced ARDS. The results were encouraging,439–441 contrary to the effect of surfactant replacement therapy in ARDS patients. This difference might be attributed to the fact that COVID-19-induced ARDS is pathophysiologically different from ARDS, and has more common elements with neonatal respiratory distress syndrome. Therefore, novel treatments leveraging surfactant beneficial effects should be investigated.442
Ion and Water Transport
It appears that SARS-CoV-2 negatively regulates Na, K-ATPase, disturbing the alveolar epithelial barrier, and promoting alveolar fluid collection. Inhibitors of Na, K-ATPase might be a beneficial therapeutic target for COVID-19-induced ARDS patients.443 The possibility of using AQPs as pharmacological treatment in COVID-19 is discussed in a recently published article. The authors propose that therapies based on AQP function in fluid transport and edema clearance could be used in order to eliminate the inflammation-induced symptomatology in COVID-19.444
Table 2 provides the major findings of the studies on the lung injury mechanisms discussed in the clinical studies presented.
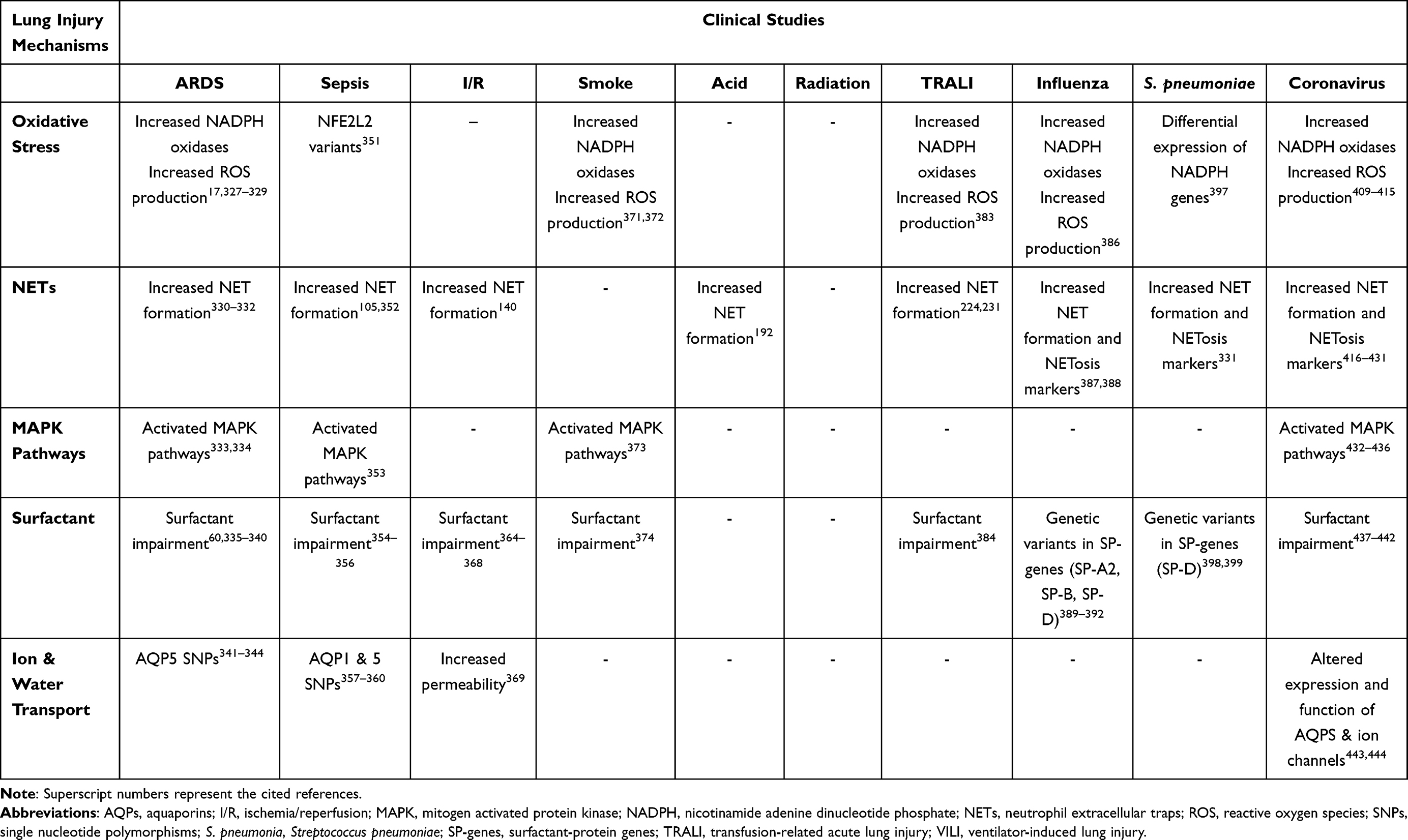 |
Table 2 Major Aspects of the Lung Injury Mechanisms in Clinical Studies |
Emerging Techniques in Lung Injury
The most common treatment strategies for ARDS are mainly pressure- and volume-limited ventilation, higher PEEP, and prone positioning for the most severe cases of ARDS, with no effective pharmacological therapy. Thus, there is an imperative need to identify new biomarkers, including genetic factors, which may help stratify ICU patients at risk, predict their prognosis, and, possibly, serve as more specific therapeutic targets. With the progression of molecular biology and bioinformatics, omics research methods have been applied to ARDS research, including genomics, transcriptomics, proteomics, and metabolomics. Figure 2 summarizes these techniques.
 |
Figure 2 Emerging techniques in lung injury. The application of emerging high throughput techniques that analyze DNA (genomics) and its modifications (epigenomics), RNA (transcriptomics), proteins (proteomics), metabolites (metabolomics), microbiomes (metagenomics), and systems (multiomics) could provide important information for the stratification of critically ill patients, in terms of risk evaluation, prognosis prediction, and guidance for novel therapeutic approaches. |
Genomics – Measuring DNA Variation
Apart from the molecular pathophysiology of lung inflammation, genomic approaches applicable to lung inflammation and injury are still on the rise. The focus of most genetic studies to date has been the identification of inherited gene risk variants involved in the immune response, vascular permeability and metabolism, coagulation, oxidative stress response, and cell development.445 The following genomic approaches have been applied to experimental lung injury models and ARDS.
Candidate-Gene Association Studies
These studies aim to investigate the association between genetic variants of genes of interest and the disease. The main disadvantages of candidate-gene association studies are the lack of reproducibility and the difficulty in interpreting their findings.
Genome-Wide Association Studies (GWAS)
GWAS explore the association between genetic polymorphisms across the genome and the disease. GWAS have been proven useful in the identification of numerous new disease genes. However, the application of GWAS to ARDS and lung injury has been limited.446–449
Whole-Exome Sequencing (WES) and Whole-Genome Sequencing
WES identifies the association between genetic variants across the exome and the disease, whereas whole-genome sequencing, identifies the association between genetic variants across the genome and the disease. Whole-exome and whole-genome sequencing studies are becoming key approaches to elucidate the genetic variants involved in ARDS.450,451 A recent bioinformatics analysis identified 201 ARDS candidate genes involved mainly in inflammatory pathways, innate immunity, and endothelial vascular signaling pathways.452
Over 80 selected genes have been associated with ARDS risk or outcome since 2000. The genes were identified through a candidate gene approach,347,453 whole-genome genotyping,447 and whole-exome sequencing.451 The most commonly detected genetic variants for ARDS risk involve polymorphisms of genes encoding pro- and anti-inflammatory cytokines, vascular injury markers, innate immunity pathway members, and markers of respiratory epithelial injury.454
The review by Giannini and Meyer discusses the latest advances in lung injury/ARDS genomics.455
Transcriptomics –Measuring RNA Expression
Transcriptomics is used to assess alterations in gene expression and biological pathways in disease states, focusing on particular targets or using array or sequencing-based approaches. Most studies to date correspond to animal models, however there are few transcriptomic analyses in ARDS patients.347,456,457
Transcriptomes were first characterized using microarrays, while RNA sequencing was introduced following the arrival of next-generation sequencing (NGS). Circular RNAs (circRNAs) and microRNAs (miRNAs), types of non-coding RNA (ncRNA) molecules, regulate gene expression at both transcriptional and post-transcriptional levels. Hence, the ncRNAs could serve as therapeutic targets, biomarkers, or provide insight to the better understanding of ARDS pathogenesis. Transcriptome sequencing has revealed differential expression of micro and circular RNAs in ALI/ARDS.458,459
Single Cell Sequencing
Single-cell RNA sequencing (scRNA-seq) is applied to individual cells with optimized NGS technologies, thus finding cellular differences in more detail, and providing a better understanding of the function of an individual cell in the context of its microenvironment. scRNA-seq facilitates the assessment of complex cellular dynamics. The previously used bulk RNA sequencing provided limited insights into disease. In contrast, scRNA-seq can examine diseases including ARDS at a higher resolution.460–462
Transcriptome-Wide Association Studies (TWAS)
TWAS investigate the associations between genetically regulated gene expression and diseases. Grigoryev et al were the first to apply an expression-based genome-wide association study (eGWAS) in ARDS, by utilizing more than 120 publicly available microarray samples of ARDS, and also discovered 14 new candidate genes associated with ARDS. The authors proposed that the study of the new candidate genes could help identify mechanisms triggered by ARDS that are evolutionarily conserved.463
Epigenomics – Measuring DNA Alterations
Epigenomics explores the processes in which gene activity is changed without alteration of its DNA sequence. In the context of lung injury/ARDS, mainly DNA methylation has been studied. Over 40,000 DNA methylation alterations, and 29 different mRNAs have been found between ARDS and healthy controls. Thirty (30) DNA methylation sites were related to the imbalance of inflammation, immunity, endothelial function, epithelial function and/or coagulation.464 The DNA methylation sites may be potentially used to improve the therapeutic management of ARDS. It is still unknown whether epigenetic modifications can influence disease susceptibility or outcome.
Proteomics – Measuring Protein Expression
Proteomics captures all the proteins expressed in an organism, including isoforms and post-translational modifications. The identification of differentially expressed proteins can help us comprehend the protein changes that occur during disease, find out the key targets, and study the corresponding genes and metabolites, hence aiding in exploring the pathogenesis, early diagnosis and treatment of the disease. Proteomics is becoming an essential tool in the study of ARDS, since it has the potential to identify key pathways and novel drug targets. A recent review summarized the advances in the application of proteomics to ARDS.465
Metabolomics – Measuring Metabolite Levels
Metabolomics measures the amount of metabolites present in biological samples. Metabolomics in the context of lung injury has revealed various metabolite mediators associated with lung injury and repair.270,466,467
Metagenomics – Measuring Microbial Composition
Metagenomics is an emerging and essential research and diagnostic tool for infectious diseases. This –omics technology is used to assess the collective microbial composition of samples from genomic data; metagenomic next-generation sequencing (mNGS) can rapidly detect pathogens for infectious diseases in different samples at once.468,469 mNGS technology has been used in ARDS patients with pneumonia for the rapid identification of pathogens.470 It has been proposed that mNGS can determine the type of microbial infection in patients with ARDS caused by severe pneumonia; this could potentially improve the diagnostic accuracy and prognosis.471
Multiomics
Omics data from genomics, transcriptomics, DNA methylation data, and proteomics were used to identify early and intermediate biomarkers for ARDS mortality. Available “omics” data identified a set of 9 genes that could differentiate ARDS survivors and non-survivors.472
Conclusions
In this review, we described a number of selected lung injury mechanisms in experimental and clinical lung injury/acute lung inflammation. We selected the major lung injury models that can lead to ARDS, including ventilation-induced lung injury (VILI), sepsis, ischemia/reperfusion, smoke, acid aspiration, radiation, transfusion-related acute lung injury (TRALI), influenza and S. pneumoniae infection, and then described emerging pathological mechanisms that contribute to acute lung inflammation, including oxidative stress, neutrophil extracellular traps, mitogen-activated protein kinase (MAPK) pathways, surfactant, and water and ion channels. We also explored whether these mechanisms are clinically relevant. In addition, we examined the abovementioned mechanisms in the pandemics caused by coronaviruses at both experimental and clinical levels.
The lack of successful pharmacological treatments in ARDS has emphasized the need for biomarkers predicting ARDS mortality and for novel therapeutics to reduce ARDS mortality. ARDS is a complex syndrome that will definitely benefit from the application of high throughput technologies. Future studies employing high throughput approaches to assay DNA, RNA, proteins, metabolites, microbiomes, and systems will help identify biomarkers to improve the prognosis of ARDS patients, and to find more effective treatments, and diagnostic methods. There is no doubt that the use of the emerging –omics technologies in future studies will help in the better stratification of ICU patients.
Acknowledgment
The authors would like to thank Ms. Dimitra Patsatzi for her significant assistance in adapting Figure 1.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Barré-Sinoussi FMX. Animal models are essential to biological research: issues and perspectives. Future Sci OA. 2015;1(4):FSO63. doi:10.4155/fso.15.63
2. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020;383(23):2255–2273. doi:10.1056/NEJMra2026131
3. Delclaux C, Azoulay E. Inflammatory response to infectious pulmonary injury. Eur Respir J. 2003;22(42 suppl):10s. doi:10.1183/09031936.03.00420203
4. Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. 2020;11:1722. doi:10.3389/fimmu.2020.01722
5. Moldoveanu B, Otmishi P, Jani P, et al. Inflammatory mechanisms in the lung. J Inflamm Res. 2009;2:1–11.
6. Goodman RB, Pugin J, Lee JS, Matthay MA. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14(6):523–535. doi:10.1016/S1359-6101(03)00059-5
7. Matute-Bello GFC, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L379–399. doi:10.1152/ajplung.00010.2008
8. Villar JBJ, Zhang H, Slutsky AS. Ventilator-induced lung injury and sepsis: two sides of the same coin? Minerva Anestesiol. 2011;77(6):647–653.
9. Rocco PRM, Marini JJ. What have we learned from animal models of ventilator-induced lung injury? Intensive care Medicine. Dec. 2020;46(12):2377–2380.
10. Wilson MRPB, Takata M. Ventilation with “clinically relevant” high tidal volumes does not promote stretch-induced injury in the lungs of healthy mice. Crit Care Med. 2012;40(10):2850–2857. doi:10.1097/CCM.0b013e31825b91ef
11. Caironi P, Langer T, Carlesso E, Protti A, Gattinoni L. Time to generate ventilator-induced lung injury among mammals with healthy lungs: a unifying hypothesis. Intensive Care Med. 2011;37(12):1913–1920. doi:10.1007/s00134-011-2388-9
12. Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis. 1974;110(5):556–565. doi:10.1164/arrd.1974.110.5.556
13. Laffey JG, Kavanagh BP. Fifty years of research in ARDS. Insight into acute respiratory distress syndrome. from models to patients. Am J Respir Crit Care Med. 2017;196(1):18–28. doi:10.1164/rccm.201612-2415CI
14. Tejero J, Shiva S, Gladwin MT. Sources of vascular nitric oxide and reactive oxygen species and their regulation. Physiol Rev. 2019;99(1):311–379. doi:10.1152/physrev.00036.2017
15. Lenaz G. Mitochondria and reactive oxygen species. Which Role in Physiology and Pathology? Adv Exp Med Biol. 2012;942:93–136. doi:10.1007/978-94-007-2869-1_5
16. Sies H. Oxidative stress: from basic research to clinical application. Am J Med. 1991;91:31S–38S. doi:10.1016/0002-9343(91)90281-2
17. Chow CW, Herrera Abreu MT, Suzuki T, Downey GP. Oxidative stress and acute lung injury. Am J Respir Cell Mol Biol. 2003;29(4):427–431. doi:10.1165/rcmb.F278
18. Ottolenghi S, Sabbatini G, Brizzolari A, Samaja M, Chiumello D. Hyperoxia and oxidative stress in anesthesia and critical care medicine. Minerva Anestesiol. 2020;86(1):64–75. doi:10.23736/S0375-9393.19.13906-5
19. Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem. 2015;30(1):11–26. doi:10.1007/s12291-014-0446-0
20. Garcia-Gimenez J-L, Garces C, Roma-Mateo C, Pallardo FV. Oxidative stress-mediated alterations in histone post-translational modifications. Free Radic Biol Med. 2021;170:6–18. doi:10.1016/j.freeradbiomed.2021.02.027
21. Bedard KKK. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi:10.1152/physrev.00044.2005
22. Brandes RP, Schroder K. Differential vascular functions of Nox family NADPH oxidases. Curr Opin Lipidol. 2008;19(5):513–518. doi:10.1097/MOL.0b013e32830c91e3
23. Itoh K, Chiba T, Takahashi S, et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem Biophys Res Commun. 1997;236(2):313–322. doi:10.1006/bbrc.1997.6943
24. Itoh KWN, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi:10.1101/gad.13.1.76
25. Mizumura K, Maruoka S, Shimizu T, Gon Y. Role of Nrf2 in the pathogenesis of respiratory diseases. Respir Investig. 2020;58(1):28–35. doi:10.1016/j.resinv.2019.10.003
26. Chapman KESS, Zhuang D, Hassid A, Desai LP, Waters CM. Cyclic mechanical strain increases reactive oxygen species production in pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L834–841. doi:10.1152/ajplung.00069.2005
27. Zeng Q, Ye L, Ling M, et al. TLR4/TRAF6/NOX2 signaling pathway is involved in ventilation-induced lung injury via endoplasmic reticulum stress in murine model. Int Immunopharmacol. 2021;96:107774. doi:10.1016/j.intimp.2021.107774
28. Fisher AB, Dodia C, Chatterjee S, Peptide A. Inhibitor of Peroxiredoxin 6 Phospholipase A2 Activity Significantly Protects against Lung Injury in a Mouse Model of Ventilator Induced Lung Injury (VILI). Antioxidants. 2021;10(6):3466. doi:10.3390/antiox10060925
29. Papaiahgari S, Yerrapureddy A, Reddy SR, et al. Genetic and pharmacologic evidence links oxidative stress to ventilator-induced lung injury in mice. Am J Respir Crit Care Med. 2007;176(12):1222–1235. doi:10.1164/rccm.200701-060OC
30. Ruan H, Li W, Wang J, et al. Propofol alleviates ventilator-induced lung injury through regulating the Nrf2/NLRP3 signaling pathway. Exp Mol Pathol. 2020;114:104427. doi:10.1016/j.yexmp.2020.104427
31. Shan Y, Akram A, Amatullah H, et al. ATF3 protects pulmonary resident cells from acute and ventilator-induced lung injury by preventing Nrf2 degradation. Antioxid Redox Signal. 2015;22(8):651–668. doi:10.1089/ars.2014.5987
32. Sun Z, Wang F, Yang Y, et al. Resolvin D1 attenuates ventilator-induced lung injury by reducing HMGB1 release in a HO-1-dependent pathway. Int Immunopharmacol. 2019;75:105825. doi:10.1016/j.intimp.2019.105825
33. Tao S, Rojo de la Vega M, Quijada H, et al. Bixin protects mice against ventilation-induced lung injury in an NRF2-dependent manner. Sci Rep. 2016;6:18760. doi:10.1038/srep18760
34. Veskemaa LGJ, Pickerodt PA, Taher M, Boemke W, González-López A, Francis RCE. Tert-butylhydroquinone augments Nrf2-dependent resilience against oxidative stress and improves survival of ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2021;320(1):L17–L28. doi:10.1152/ajplung.00131.2020
35. Xu J, Li HB, Chen L, et al. BML-111 accelerates the resolution of inflammation by modulating the Nrf2/HO-1 and NF-kappaB pathways in rats with ventilator-induced lung injury. Int Immunopharmacol. 2019;69:289–298. doi:10.1016/j.intimp.2019.02.005
36. Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79(2):319–326. doi:10.1172/JCI112815
37. Brinkmann VGC, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi:10.1126/science.1092385
38. Porto BN, Stein RT. Neutrophil Extracellular Traps in Pulmonary Diseases: too Much of a Good Thing? Front Immunol. 2016;7:311. doi:10.3389/fimmu.2016.00311
39. Saffarzadeh M, Juenemann C, Queisser MA, et al. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: a Predominant Role of Histones. PLoS One. 2012;7(2):e32366. doi:10.1371/journal.pone.0032366
40. Abrams ST, Zhang N, Manson J, et al. Circulating Histones Are Mediators of Trauma-associated Lung Injury. Am J Respir Crit Care Med. 2013;187(2):160–169. doi:10.1164/rccm.201206-1037OC
41. Dreyfuss DSG. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med. 1998;157(1):294–323. doi:10.1164/ajrccm.157.1.9604014
42. Imanaka HSM, Shimaoka M, Matsuura N, Nishimura M, Ohta H, Kiyono H. Ventilator-induced lung injury is associated with neutrophil infiltration, macrophage activation, and TGF-beta 1 mRNA upregulation in rat lungs. Anesth Analg. 2001;92(2):428–436. doi:10.1097/00000539-200102000-00029
43. Belperio JA, Keane MP, Burdick MD, et al. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest. 2002;110(11):1703–1716. doi:10.1172/JCI0215849
44. Rossaint JHJ, Herter JM, Van Aken H, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap–mediated sterile inflammation. Blood. 2014;123(16):2573–2584. doi:10.1182/blood-2013-07-516484
45. Li H, Pan P, Su X, et al. Neutrophil Extracellular Traps Are Pathogenic in Ventilator-Induced Lung Injury and Partially Dependent on TLR4. Biomed Res Int. 2017;2017:8272504. doi:10.1155/2017/8272504
46. Yildiz CPN, Otulakowski G, Khan MA, et al. Mechanical ventilation induces neutrophil extracellular trap formation. Anesthesiology. 2015;122(4):864–875. doi:10.1097/ALN.0000000000000605
47. Nailwal NP, Doshi GM. Role of intracellular signaling pathways and their inhibitors in the treatment of inflammation. Inflammopharmacology. 2021;29(3):617–640. doi:10.1007/s10787-021-00813-y
48. Widmann CGS, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79(1):143–180. doi:10.1152/physrev.1999.79.1.143
49. Zhang WLH. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18. doi:10.1038/sj.cr.7290105
50. Ip YTDR. Signal transduction by the c-Jun N-terminal kinase (JNK)-from inflammation to development. Curr Opin Cell Biol. 1998;10(2):205–219. doi:10.1016/S0955-0674(98)80143-9
51. Iwaki M, Ito S, Morioka M, et al. Mechanical stretch enhances IL-8 production in pulmonary microvascular endothelial cells. Biochem Biophys Res Commun. 2009;389(3):531–536. doi:10.1016/j.bbrc.2009.09.020
52. Oudin SPJ. Role of MAP kinase activation in interleukin-8 production by human BEAS-2B bronchial epithelial cells submitted to cyclic stretch. Am J Respir Cell Mol Biol. 2002;27(1):107–114. doi:10.1165/ajrcmb.27.1.4766
53. Dolinay T, Wu W, Kaminski N, et al. Mitogen-activated protein kinases regulate susceptibility to ventilator-induced lung injury. PLoS One. 2008;3(2):e1601. doi:10.1371/journal.pone.0001601
54. Li LF, Yu L, Quinn DA. Ventilation-induced neutrophil infiltration depends on c-Jun N-terminal kinase. Am J Respir Crit Care Med. 2004;169(4):518–524. doi:10.1164/rccm.200305-660OC
55. Uhlig U, Haitsma JJ, Goldmann T, Poelma DL, Lachmann B, Uhlig S. Ventilation-induced activation of the mitogen-activated protein kinase pathway. Eur Respir J. 2002;20(4):946–956. doi:10.1183/09031936.02.01612001
56. Woods SJWA, O’Dea KP, Halford P, Takata M, Wilson MR. Kinetic profiling of in vivo lung cellular inflammatory responses to mechanical ventilation. Am J Physiol Lung Cell Mol Physiol. 2015;308(9):L912–921. doi:10.1152/ajplung.00048.2015
57. Wright JRCJ. Metabolism and turnover of lung surfactant. Am Rev Respir Dis. 1987;136(2):426–444. doi:10.1164/ajrccm/136.2.426
58. Han S, Mallampalli RK. The Role of Surfactant in Lung Disease and Host Defense against Pulmonary Infections. Ann Am Thorac Soc. 2015;12(5):765–774. doi:10.1513/AnnalsATS.201411-507FR
59. Wright J. Immunomodulatory functions of surfactant. Physiol Rev. 1997;77(4):931–962. doi:10.1152/physrev.1997.77.4.931
60. Raghavendran K, Willson D, Notter RH. Surfactant therapy for acute lung injury and acute respiratory distress syndrome. Crit Care Clin. 2011;27(3):525–559. doi:10.1016/j.ccc.2011.04.005
61. Parker JCHL, Peevy KJ. Mechanisms of ventilator-induced lung injury. Crit Care Med. 1993;21(1):131–143. doi:10.1097/00003246-199301000-00024
62. Verbrugge SJ, Bohm SH, Gommers D, Zimmerman LJ, Lachmann B. Surfactant impairment after mechanical ventilation with large alveolar surface area changes and effects of positive end-expiratory pressure. Br J Anaesth. 1998;80(3):360–364. doi:10.1093/bja/80.3.360
63. Taskar V, John J, Robertson B, Jonson B. Surfactant dysfunction makes lungs vulnerable to repetitive collapse and reexpansion. Am J Respir Crit Care Med. 1997;1:313–320. doi:10.1164/ajrccm.155.1.9001330
64. Vazquez de Anda GF, Gommers D, Verbrugge SJ, Haitsma J, Lachmann B. Treatment of ventilation-induced lung injury with exogenous surfactant. Intensive Care Med. 2001;27(3):559–565. doi:10.1007/s001340000838
65. Haitsma JJ, Uhlig S, Lachmann U, Verbrugge SJ, Poelma DL, Lachmann B. Exogenous surfactant reduces ventilator-induced decompartmentalization of tumor necrosis factor alpha in absence of positive end-expiratory pressure. Intensive Care Med. 2002;28(8):1131–1137. doi:10.1007/s00134-002-1377-4
66. Krause MFHT. Timing of surfactant administration determines its physiologic response in a rabbit model of airway lavage. Biol Neonate. 2000;77(3):196–202. doi:10.1159/000014216
67. Vassiliou AG, Kotanidou A, Dimopoulou I, Orfanos SE. Endothelial Damage in Acute Respiratory Distress Syndrome. Int J Mol Sci. 2020;21(22). doi:10.3390/ijms21228793
68. Bartoszewski RMS, Collawn JF. Ion channels of the lung and their role in disease pathogenesis. Am J Physiol Lung Cell Mol Physiol. 2017;313(5):L859–L872. doi:10.1152/ajplung.00285.2017
69. Zea Borok ASV. Lung Edema Clearance: 20 Years of Progress Invited Review: role of aquaporin water channels in fluid transport in lung and airways. J Appl Physiol. 2002;93(6):2199–2206. doi:10.1152/japplphysiol.01171.2001
70. Folkesson HG, Matthay MA, Hasegawa H, Kheradmand F, Verkman AS. Transcellular water transport in lung alveolar epithelium through mercury-sensitive water channels. Proc Natl Acad Sci U S A. 1994;91(11):4970–4974. doi:10.1073/pnas.91.11.4970
71. Nielsen S, King LS, Christensen BM, Agre P. Aquaporins in complex tissues. II. Subcellular distribution in respiratory and glandular tissues of rat. Am J Physiol. 1997;273(5):C1549–1561. doi:10.1152/ajpcell.1997.273.5.C1549
72. Vassiliou AG, Manitsopoulos N, Kardara M, Maniatis NA, Orfanos SE, Kotanidou A. Differential Expression of Aquaporins in Experimental Models of Acute Lung Injury. vivo. 2017;31(5):885–894.
73. Wittekindt OH, Dietl P. Aquaporins in the lung. Pflugers Archiv. 2019;471(4):519–532. doi:10.1007/s00424-018-2232-y
74. Verkman AS. More than just water channels: unexpected cellular roles of aquaporins. J Cell Sci. 2005;118(15):3225–3232. doi:10.1242/jcs.02519
75. Vadász IRS, Sznajder JI. Alveolar epithelium and Na,K-ATPase in acute lung injury. Intensive Care Med. 2007;33(7):1243–1251. doi:10.1007/s00134-007-0661-8
76. Morty REEO, Seeger W. Alveolar fluid clearance in acute lung injury: what have we learned from animal models and clinical studies? Intensive Care Med. 2007;33(7):1229–1240. doi:10.1007/s00134-007-0662-7
77. Hales C, Du H, Volokhov A, Mourfarrej R, Quinn DA. Aquaporin channels may modulate ventilator-induced lung injury. Respir Physiol Neurobiol. 2001;124(2):159–166. doi:10.1016/S0034-5687(00)00193-6
78. Fabregat G, Garcia-de-la-asuncion J, Sarria B, et al. Increased expression of AQP 1 and AQP 5 in rat lungs ventilated with low tidal volume is time dependent. PLoS One. 2014;9(12):e114247. doi:10.1371/journal.pone.0114247
79. Fabregat G, Garcia-de-la-asuncion J, Sarria B, et al. Expression of aquaporins 1 and 5 in a model of ventilator-induced lung injury and its relation to tidal volume. Exp Physiol. 2016;101(11):1418–1431. doi:10.1113/EP085729
80. Jin L-D, Wang L-R, Wu L-Q, et al. Effects of COX-2 inhibitor on ventilator-induced lung injury in rats. Int Immunopharmacol. 2013;16(2):288–295. doi:10.1016/j.intimp.2013.03.031
81. Liu Y, Wang Y, Song X, Dong L, Wang W, Wu H. P38 mitogen-activated protein kinase inhibition attenuates mechanical stress induced lung injury via up-regulating AQP5 expression in rats. Biotechnol Biotechnol Equip. 2019;33(1):472–480. doi:10.1080/13102818.2019.1590159
82. Adir Y, Factor P, Dumasius V, Ridge KM, Sznajder JI. Na,K-ATPase gene transfer increases liquid clearance during ventilation-induced lung injury. Am J Respir Crit Care Med. 2003;168(12):1445–1448. doi:10.1164/rccm.200207-702OC
83. Liang ZD, Yin XR, Cai DS, Zhou H, Pei L. Autologous transplantation of adipose-derived stromal cells ameliorates ventilator-induced lung injury in rats. J Transl Med. 2013;11:179. doi:10.1186/1479-5876-11-179
84. Chamorro-Marín V, García-Delgado M, Touma-Fernández A, Aguilar-Alonso E, Fernández-Mondejar E. Intratracheal dopamine attenuates pulmonary edema and improves survival after ventilator-induced lung injury in rats. Crit Care. 2008;12(2):R39. doi:10.1186/cc6829
85. Vassiliou AG, Orfanos SE, Kotanidou A. Clinical Assays in Sepsis: prognosis, Diagnosis, Outcomes, and the Genetic Basis of Sepsis. Sepsis. 2017;1:54.
86. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. doi:10.1038/nri3399
87. Guillon A, Preau S, Aboab J, et al. Preclinical septic shock research: why we need an animal ICU. Ann Intensive Care. 2019;9(1):66. doi:10.1186/s13613-019-0543-6
88. Raetz CR, Ulevitch RJ, Wright SD, Sibley CH, Ding A, Nathan CF. Gram-negative endotoxin: an extraordinary lipid with profound effects on eukaryotic signal transduction. FASEB J. 1991;5(12):2652–2660. doi:10.1096/fasebj.5.12.1916089
89. Remick DG, Ward PA. Evaluation of endotoxin models for the study of sepsis. Shock. 2005;Suppl 1:7–11. doi:10.1097/01.shk.0000191384.34066.85
90. Walley KRLN, Standiford TJ, Strieter RM, Kunkel SL. Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect Immun. 1996;64:4733–4738. doi:10.1128/iai.64.11.4733-4738.1996
91. Gnidec AG, Sibbald WJ, Cheung H, Metz CA. Ibuprofen reduces the progression of permeability edema in an animal model of hyperdynamic sepsis. J Appl Physiol. 1988;65(3):1024–1032. doi:10.1152/jappl.1988.65.3.1024
92. Lomas-Neira JL, Chung CS, Wesche DE, Perl M, Ayala A. In vivo gene silencing (with siRNA) of pulmonary expression of MIP-2 versus KC results in divergent effects on hemorrhage-induced, neutrophil-mediated septic acute lung injury. J Leukoc Biol. 2005;77(6):846–853. doi:10.1189/jlb.1004617
93. Chimenti L, Morales-Quinteros L, Puig F, et al. Comparison of direct and indirect models of early induced acute lung injury. Intensive Care Med Exp. 2020;8(Suppl 1):62. doi:10.1186/s40635-020-00350-y
94. Jiang J, Huang K, Xu S, Garcia JGN, Wang C, Targeting CH. NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. 2020;36:101638. doi:10.1016/j.redox.2020.101638
95. Palumbo SSY, Ahmad K, Desai AA, et al. Dysregulated Nox4 ubiquitination contributes to redox imbalance and age-related severity of acute lung injury. Am J Physiol. 2017;312(3):L297–L308. doi:10.1152/ajplung.00305.2016
96. Li D, Cong Z, Yang C, Zhu X. Inhibition of LPS-induced Nox2 activation by VAS2870 protects alveolar epithelial cells through eliminating ROS and restoring tight junctions. Biochem Biophys Res Commun. 2020;524(3):575–581. doi:10.1016/j.bbrc.2020.01.134
97. Thimmulappa RK, Lee H, Rangasamy T, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116(4):984–995. doi:10.1172/JCI25790
98. Kong X, Thimmulappa R, Craciun F, et al. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med. 2011;184(8):928–938. doi:10.1164/rccm.201102-0271OC
99. Cen M, Ouyang W, Zhang W, et al. MitoQ protects against hyperpermeability of endothelium barrier in acute lung injury via a Nrf2-dependent mechanism. Redox Biol. 2021;41:101936. doi:10.1016/j.redox.2021.101936
100. Liu Q, Ci X, Wen Z, Peng L. Diosmetin Alleviates Lipopolysaccharide-Induced Acute Lung Injury through Activating the Nrf2 Pathway and Inhibiting the NLRP3 Inflammasome. Biomol Ther (Seoul). 2018;26(2):157–166. doi:10.4062/biomolther.2016.234
101. Yu Y, Yang Y, Yang M, Wang C, Xie K, Yu Y. Hydrogen gas reduces HMGB1 release in lung tissues of septic mice in an Nrf2/HO-1-dependent pathway. Int Immunopharmacol. 2019;69:11–18. doi:10.1016/j.intimp.2019.01.022
102. Czaikoski PG, Mota JM, Nascimento DC, et al. Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PLoS One. 2016;11(2):e0148142. doi:10.1371/journal.pone.0148142
103. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. 2018;3(3):e98178. doi:10.1172/jci.insight.98178
104. Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–469. doi:10.1038/nm1565
105. Zhang H, Zhou Y, Qu M, et al. Tissue Factor-Enriched Neutrophil Extracellular Traps Promote Immunothrombosis and Disease Progression in Sepsis-Induced Lung Injury. Front Cell Infect Microbiol. 2021;11:677902. doi:10.3389/fcimb.2021.677902
106. Asaduzzaman M, Wang Y, Thorlacius H. Critical role of p38 mitogen-activated protein kinase signaling in septic lung injury. Crit Care Med. 2008;36(2):482–488. doi:10.1097/01.CCM.0B013E31816204FA
107. Fang W, Cai SX, Wang CL, et al. Modulation of mitogen-activated protein kinase attenuates sepsis-induced acute lung injury in acute respiratory distress syndrome rats. Mol Med Rep. 2017;16(6):9652–9658. doi:10.3892/mmr.2017.7811
108. Mannam P, Zhang X, Shan P, et al. Endothelial MKK3 is a critical mediator of lethal murine endotoxemia and acute lung injury. J Immunol. 2013;190(3):1264–1275. doi:10.4049/jimmunol.1202012
109. Qian FDJ, Gantner BN, Flavell RA, Dong C, Christman JW, Ye RD. Map kinase phosphatase 5 protects against sepsis-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L866–874. doi:10.1152/ajplung.00277.2011
110. Kolomaznik MNZ, Nova Z, Calkovska A. Pulmonary surfactant and bacterial lipopolysaccharide: the interaction and its functional consequences. Physiol Res. 2017;66(Suppl 2):S147–S157. doi:10.33549/physiolres.933672
111. Lewis JFVR, Possmayer F. Altered alveolar surfactant is an early marker of acute lung injury in septic adult sheep. Am J Respir Crit Care Med. 1994;150(1):123–130. doi:10.1164/ajrccm.150.1.8025737
112. Huang W, McCaig LA, Veldhuizen RA, Yao LJ, Lewis JF. Mechanisms responsible for surfactant changes in sepsis-induced lung injury. Eur Respir J. 2005;26(6):1074–1079. doi:10.1183/09031936.05.00085805
113. Nieman G. Surfactant replacement in the treatment of sepsis-induced adult respiratory distress syndrome in pigs. Crit Care Med. 1996;24(6):1025–1033. doi:10.1097/00003246-199606000-00024
114. Guo R, Li Y, Han M, Liu J, Sun Y. Emodin attenuates acute lung injury in Cecal-ligation and puncture rats. Int Immunopharmacol. 2020;85:106626. doi:10.1016/j.intimp.2020.106626
115. Hu X, Liu S, Zhu J, Ni H. Dachengqi decoction alleviates acute lung injury and inhibits inflammatory cytokines production through TLR4/NF-κB signaling pathway in vivo and in vitro. J Cell Biochem. 2019;120(6):8956–8964. doi:10.1002/jcb.27615
116. Liang W, Guo L, Liu T, Qin S. MEF2C alleviates acute lung injury in cecal ligation and puncture (CLP)-induced sepsis rats by up-regulating AQP1. Allergolog et Immunopathol. 2021;49(5):117–124. doi:10.15586/aei.v49i5.477
117. Liu LDWX, Tao BD, Wang N, Zhang J. Protective effect and mechanism of hydrogen treatment on lung epithelial barrier dysfunction in rats with sepsis. Genet Mol Res. 2016;15(1):65.
118. Hasan B, Li F, Siyit A, et al. Expression of aquaporins in the lungs of mice with acute injury caused by LPS treatment. Respir Physiol Neurobiol. 2014;200:40–45. doi:10.1016/j.resp.2014.05.008
119. Jiao G, Li E, Yu R. Decreased expression of AQP1 and AQP5 in acute injured lungs in rats. Chin Med J. 2002;115(7):963–967.
120. Su X, Song Y, Jiang J, Bai C. The role of aquaporin-1 (AQP1) expression in a murine model of lipopolysaccharide-induced acute lung injury. Respir Physiol Neurobiol. 2004;142(1):1–11. doi:10.1016/j.resp.2004.05.001
121. Rump K, Brendt P, Frey UH, et al. Aquaporin 1 and 5 expression evoked by the β2 adrenoreceptor agonist terbutaline and lipopolysaccharide in mice and in the human monocytic cell line THP-1 is differentially regulated. Shock. 2013;40(5):430–436. doi:10.1097/SHK.0000000000000035
122. Guo C, Wu T, Zhu H, Gao L. Aquaporin 4 Blockade Attenuates Acute Lung Injury Through Inhibition of Th17 Cell Proliferation in Mice. Inflammation. 2019;42(4):1401–1412. doi:10.1007/s10753-019-01002-4
123. Berger G, Guetta J, Klorin G, et al. Sepsis impairs alveolar epithelial function by downregulating Na-K-ATPase pump. Am J Physiol Lung Cell Mol Physiol. 2011;301(1):L23–30. doi:10.1016/S2213-2600(20)30404-5
124. Fisher BJ, Kraskauskas D, Martin EJ, et al. Mechanisms of attenuation of abdominal sepsis induced acute lung injury by ascorbic acid. Am J Physiol Lung Cell Mol Physiol. 2012;303(1):L20–32. doi:10.1152/ajplung.00300.2011
125. Emr BM, Roy S, Kollisch-Singule M, et al. Electroporation-mediated gene delivery of Na+,K+ -ATPase, and ENaC subunits to the lung attenuates acute respiratory distress syndrome in a two-hit porcine model. Shock. 2015;43(1):16–23. doi:10.1097/SHK.0000000000000228
126. de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia-reperfusion-induced lung injury. Am J Respir Crit Care Med. 2003;167(4):490–511. doi:10.1164/rccm.200207-670SO
127. Fard N, Saffari A, Emami G, Hofer S, Kauczor HU, Mehrabi A. Acute respiratory distress syndrome induction by pulmonary ischemia-reperfusion injury in large animal models. J Surg Res. 2014;189(2):274–284. doi:10.1016/j.jss.2014.02.034
128. Koike KME, Moore FA, Read RA, Carl VS, Banerjee A. Gut ischemia/reperfusion produces lung injury independent of endotoxin. Crit Care Med. 1994;22(9):1438–1444. doi:10.1097/00003246-199409000-00014
129. Sakao YKO, Martin TR, Nakahara Y, Hadden WA. Association of IL-8 and MCP-1 with the development of reexpansion pulmonary edema in rabbits. Ann Thorac Surg. 2001;71(6):1825–1832. doi:10.1016/S0003-4975(01)02489-4
130. Chatterjee SNG, Christie JD, Fisher AB. Shear stress-related mechanosignaling with lung ischemia: lessons from basic research can inform lung transplantation. Am J Physiol Lung Cell Mol Physiol. 2014;307(9):L668–680. doi:10.1152/ajplung.00198.2014
131. Sharma AKLD, Stone ML, Zhao Y, Mehta CK, Kron IL, Laubach VE. NOX2 Activation of Natural Killer T Cells Is Blocked by the Adenosine A2A Receptor to Inhibit Lung Ischemia-Reperfusion Injury. Am J Respir Crit Care Med. 2016;193(9):988–999. doi:10.1164/rccm.201506-1253OC
132. Sharma AK, Mulloy DP, Le LT, Laubach VE. NADPH oxidase mediates synergistic effects of IL-17 and TNF-α on CXCL1 expression by epithelial cells after lung ischemia-reperfusion. Am J Physiol. 2014;306(1):L69–L79. doi:10.1152/ajplung.00205.2013
133. Cui Y, Wang Y, Li G, et al. The Nox1/Nox4 inhibitor attenuates acute lung injury induced by ischemia-reperfusion in mice. PLoS One. 2018;13(12):e0209444. doi:10.1371/journal.pone.0209444
134. Pak O, Sydykov A, Kosanovic D, et al. Lung Ischaemia-Reperfusion Injury: the Role of Reactive Oxygen Species. Adv Exp Med Biol. 2017;967:195–225. doi:10.1007/978-3-319-63245-2_12
135. Chai D, Zhang L, Xi S, Cheng Y, Jiang H, Hu R. Nrf2 Activation Induced by Sirt1 Ameliorates Acute Lung Injury After Intestinal Ischemia/Reperfusion Through NOX4-Mediated Gene Regulation. Cell Physiol Biochem. 2018;46(2):781–792. doi:10.1159/000488736
136. Dong HQZ, Chai D, Peng J, Xia Y, Hu R, Jiang H. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging. 2020;12(13):12943–12959. doi:10.18632/aging.103378
137. Fan J, Lv H, Li J, et al. Roles of Nrf2/HO-1 and HIF-1alpha/VEGF in lung tissue injury and repair following cerebral ischemia/reperfusion injury. J Cell Physiol. 2019;234(6):7695–7707. doi:10.1002/jcp.27767
138. Meng Q-T, Cao C, Wu Y, et al. Ischemic post-conditioning attenuates acute lung injury induced by intestinal ischemia–reperfusion in mice: role of Nrf2. Labor Investig. 2016;96(10):1087–1104. doi:10.1038/labinvest.2016.87
139. Yan J, Li J, Zhang L, et al. Nrf2 protects against acute lung injury and inflammation by modulating TLR4 and Akt signaling. Free Radic Biol Med. 2018;121:78–85. doi:10.1016/j.freeradbiomed.2018.04.557
140. Sayah DM, Mallavia B, Liu F, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med. 2015;191(4):455–463. doi:10.1164/rccm.201406-1086OC
141. Scozzi D, Wang X, Liao F, et al. Neutrophil extracellular trap fragments stimulate innate immune responses that prevent lung transplant tolerance. Am J Transplant. 2019;19(4):1011–1023. doi:10.1111/ajt.15163
142. Wolf PS, Merry HE, Farivar AS, McCourtie AS, Mulligan MS. Stress-activated protein kinase inhibition to ameliorate lung ischemia reperfusion injury. J Thorac Cardiovasc Surg. 2008;135(3):656–665. doi:10.1016/j.jtcvs.2007.11.026
143. Wang T, Liu C, Pan LH, et al. Inhibition of p38 MAPK Mitigates Lung Ischemia Reperfusion Injury by Reducing Blood-Air Barrier Hyperpermeability. Front Pharmacol. 2020;11:569251. doi:10.3389/fphar.2020.569251
144. Xiong -L-L, Tan Y, Ma H-Y, et al. Administration of SB239063, a potent p38 MAPK inhibitor, alleviates acute lung injury induced by intestinal ischemia reperfusion in rats associated with AQP4 downregulation. Int Immunopharmacol. 2016;38:54–60. doi:10.1016/j.intimp.2016.03.036
145. Zheng D-Y, Zhou M, Jin J, et al. Inhibition of P38 MAPK Downregulates the Expression of IL-1βto Protect Lung from Acute Injury in Intestinal Ischemia Reperfusion Rats. Mediators Inflamm. 2016;2016:1–8.
146. Tan J, Liu D, Lv X, et al. MAPK mediates inflammatory response and cell death in rat pulmonary microvascular endothelial cells in an ischemia-reperfusion model of lung transplantation. J Heart Lung Transplant. 2013;32(8):823–831. doi:10.1016/j.healun.2013.05.005
147. Wang J, Tan J, Liu Y, Song L, Li D, Cui X. Amelioration of lung ischemia reperfusion injury by JNK and p38 small interfering RNAs in rat pulmonary microvascular endothelial cells in an ischemia-reperfusion injury lung transplantation model. Mol Med Rep. 2018;17(1):1228–1234. doi:10.3892/mmr.2017.7985
148. Ochs M, Nenadic I, Fehrenbach A, et al. Ultrastructural alterations in intraalveolar surfactant subtypes after experimental ischemia and reperfusion. Am J Respir Crit Care Med. 1999;160(2):718–724. doi:10.1164/ajrccm.160.2.9809060
149. Novick RJ, MacDonald J, Veldhuizen RA, et al. Evaluation of surfactant treatment strategies after prolonged graft storage in lung transplantation. Am J Respir Crit Care Med. 1996;154(1):98–104. doi:10.1164/ajrccm.154.1.8680706
150. van der Kaaij NP, Haitsma JJ, Kluin J, et al. Surfactant pretreatment ameliorates ischemia-reperfusion injury of the lung. Eur J Cardio. 2005;27(5):774–782. doi:10.1016/j.ejcts.2004.12.034
151. Dreyer N, Muhlfeld C, Fehrenbach A, et al. Exogenous surfactant application in a rat lung ischemia reperfusion injury model: effects on edema formation and alveolar type II cells. Respir Res. 2008;9:5. doi:10.1186/1465-9921-9-5
152. Mühlfeld C, Becker L, Bussinger C, et al. Exogenous surfactant in ischemia/reperfusion: effects on endogenous surfactant pools. J Heart Lung Transplant. 2010;29(3):327–334. doi:10.1016/j.healun.2009.07.019
153. Mühlfeld C, Schaefer IM, Becker L, et al. Pre-ischaemic exogenous surfactant reduces pulmonary injury in rat ischaemia/reperfusion. Eur Respir J. 2009;33(3):625–633. doi:10.1183/09031936.00024108
154. van Putte BP, Cobelens PM, van der Kaaij N, et al. Exogenous surfactant attenuation of ischemia-reperfusion injury in the lung through alteration of inflammatory and apoptotic factors. J Thorac Cardiovasc Surg. 2009;137(4):824–828. doi:10.1016/j.jtcvs.2008.08.046
155. Ge H, Zhu H, Xu N, et al. Increased Lung Ischemia-Reperfusion Injury in Aquaporin 1-Null Mice Is Mediated via Decreased Hypoxia-Inducible Factor 2alpha Stability. Am J Respir Cell Mol Biol. 2016;54(6):882–891. doi:10.1165/rcmb.2014-0363OC
156. Qi YC, Chen W, Li XL, Wang YW, Xie XH. H2S Protecting against Lung Injury following Limb Ischemia-reperfusion by Alleviating Inflammation and Water Transport Abnormality in Rats. Biomed Environ Sci. 2014;27(6):410–418. doi:10.3967/bes2014.070
157. Calikoglu M, Tamer L, Sucu N, et al. The effects of caffeic acid phenethyl ester on tissue damage in lung after hindlimb ischemia-reperfusion. Pharmacol Res. 2003;48(4):397–403. doi:10.1016/S1043-6618(03)00156-7
158. Koksel O, Ozdulger A, Aytacoglu B, et al. The influence of iloprost on acute lung injury induced by hind limb ischemia-reperfusion in rats. Pulm Pharmacol Ther. 2005;18(4):235–241. doi:10.1016/j.pupt.2004.12.005
159. Lan CC, Peng CK, Tang SE, Huang KL, Wu CP. Carbonic anhydrase inhibitor attenuates ischemia-reperfusion induced acute lung injury. PLoS One. 2017;12(6):e0179822. doi:10.1371/journal.pone.0179822
160. David PDD, Lu J, Moochhala S. Animal models of smoke inhalation induced injuries. Front Biosci. 2009;1(14):4618–4630.
161. Guo B, Bai Y, Ma Y, et al. Preclinical and clinical studies of smoke-inhalation-induced acute lung injury: update on both pathogenesis and innovative therapy. Ther Adv Respir Dis. 2019;13:1753466619847901. doi:10.1177/1753466619847901
162. Rabinowitz PMSM. Acute inhalation injury. Clin Chest Med. 2002;23(4):707–715. doi:10.1016/S0272-5231(02)00025-4
163. de Carvalho FO, Felipe FA, de Melo Costa ACS, et al. Inflammatory Mediators and Oxidative Stress in Animals Subjected to Smoke Inhalation: a Systematic Review. Lung. 2016;194(4):487–499. doi:10.1007/s00408-016-9879-y
164. Hikichi M, Mizumura K, Maruoka S, Gon Y. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J Thorac Dis. 2019;11(Suppl 17):S2129–S2140. doi:10.21037/jtd.2019.10.43
165. Bhalla DK, Hirata F, Rishi AK, Gairola CG. Cigarette smoke, inflammation, and lung injury: a mechanistic perspective. J Toxicol Environ Health B Crit Rev. 2009;12(1):45–64. doi:10.1080/10937400802545094
166. Lu Q, Gottlieb E, Rounds S. Effects of cigarette smoke on pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2018;314(5):L743–L756. doi:10.1152/ajplung.00373.2017
167. Lee TS, Liu YJ, Tang GJ, Yien HW, Wu YL, Kou YR. Wood smoke extract promotes both apoptosis and proliferation in rat alveolar epithelial type II cells: the role of oxidative stress and heme oxygenase-1. Crit Care Med. 2008;36(9):2597–2606. doi:10.1097/CCM.0b013e318184979c
168. Perng DW, Chang TM, Wang JY, et al. Inflammatory role of AMP-activated protein kinase signaling in an experimental model of toxic smoke inhalation injury. Crit Care Med. 2013;41(1):120–132. doi:10.1097/CCM.0b013e318265f653
169. Liu PLCY, Chen YH, Lin SJ, Kou YR. Wood smoke extract induces oxidative stress-mediated caspase-independent apoptosis in human lung endothelial cells: role of AIF and EndoG. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L739–749. doi:10.1152/ajplung.00099.2005
170. Müller T, Hengstermann A. Nrf2: friend and foe in preventing cigarette smoking-dependent lung disease. Chem Res Toxicol. 2012;25(9):1805–1824. doi:10.1021/tx300145n
171. Boutten A, Goven D, Artaud-Macari E, Boczkowski J, Bonay M. NRF2 targeting: a promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol Med. 2011;17(7):363–371. doi:10.1016/j.molmed.2011.02.006
172. Kim M, Han CH, Lee MY. NADPH oxidase and the cardiovascular toxicity associated with smoking. Toxicol Res. 2014;30(3):149–157. doi:10.5487/TR.2014.30.3.149
173. Zou Y, Chen X, He B, et al. Neutrophil extracellular traps induced by cigarette smoke contribute to airway inflammation in mice. Exp Cell Res. 2020;389(1):111888. doi:10.1016/j.yexcr.2020.111888
174. Qiu SL, Zhang H, Tang QY, et al. Neutrophil extracellular traps induced by cigarette smoke activate plasmacytoid dendritic cells. Thorax. 2017;72(12):1084–1093. doi:10.1136/thoraxjnl-2016-209887
175. Choi WISO, Kwon KY, Quinn DA, Hales CA. JNK activation is responsible for mucus overproduction in smoke inhalation injury. Respir Res. 2010;11(1):172. doi:10.1186/1465-9921-11-172
176. Syrkina OLQD, Jung W, Ouyang B, Hales CA. Inhibition of JNK activation prolongs survival after smoke inhalation from fires. Am J Physiol Lung Cell Mol Physiol. 2007;292(4):L984–991. doi:10.1152/ajplung.00248.2006
177. Banerjee A, Koziol-White C, Panettieri R. p38 MAPK inhibitors, IKK2 inhibitors, and TNFα inhibitors in COPD. Curr Opin Pharmacol. 2012;12(3):287–292. doi:10.1016/j.coph.2012.01.016
178. Mercer BA, D’Armiento JM. Emerging role of MAP kinase pathways as therapeutic targets in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):137–150. doi:10.2147/copd.2006.1.2.137
179. Crotty Alexander LE, Shin S, Hwang JH. Inflammatory Diseases of the Lung Induced by Conventional Cigarette Smoke: a Review. Chest. 2015;148(5):1307–1322. doi:10.1378/chest.15-0409
180. Oulton MRJD, MacDonald JM, Faulkner GT, Scott JE. Effects of smoke inhalation on alveolar surfactant subtypes in mice. Am J Pathol. 1994;145(4):941–950.
181. Oulton MMH, Scott JE, Janigan DT, Hajela R. Effects of smoke inhalation on surfactant phospholipids and phospholipase A2 activity in the mouse lung. Am J Pathol. 1991;138(1):195–202.
182. Sun YQX, Wu G. The effects of porcine pulmonary surfactant on smoke inhalation injury. J Surg Res. 2015;198(1):200–207. doi:10.1016/j.jss.2015.05.019
183. Scott JE. The pulmonary surfactant: impact of tobacco smoke and related compounds on surfactant and lung development. Tob Induc Dis. 2004;2(1):3–25. doi:10.1186/1617-9625-2-1-3
184. Sorensen GL. Surfactant Protein D in Respiratory and Non-Respiratory Diseases. Front Med. 2018;5:18. doi:10.3389/fmed.2018.00018
185. Chang J, Chen Z, Zhao R, Nie HG, Ji HL. Ion transport mechanisms for smoke inhalation-injured airway epithelial barrier. Cell Biol Toxicol. 2020;36(6):571–589. doi:10.1007/s10565-020-09545-1
186. Raghavendran K, Nemzek J, Napolitano LM, Knight PR. Aspiration-induced lung injury. Crit Care Med. 2011;39(4):818–826. doi:10.1097/CCM.0b013e31820a856b
187. Effros RMJE, Schapira RM, Biller J. Response of the lungs to aspiration. Am J Med. 2000;108(Suppl4a):15S–19S. doi:10.1016/S0002-9343(99)00290-9
188. Marik P. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665–671. doi:10.1056/NEJM200103013440908
189. Davidson BA, Vethanayagam RR, Grimm MJ, et al. NADPH oxidase and Nrf2 regulate gastric aspiration-induced inflammation and acute lung injury. J Immunol. 2013;190(4):1714–1724. doi:10.4049/jimmunol.1202410
190. Segal BHDB, Hutson AD, Russo TA, et al. Acid aspiration-induced lung inflammation and injury are exacerbated in NADPH oxidase-deficient mice. Am J Physiol. 2007;292(3):L760–768. doi:10.1152/ajplung.00281.2006
191. Puri G, Naura AS. Critical role of mitochondrial oxidative stress in acid aspiration induced ALI in mice. Toxicol Mech Methods. 2020;30(4):266–274. doi:10.1080/15376516.2019.1710888
192. Li HZX, Tan H, Hu Y, et al. Neutrophil extracellular traps contribute to the pathogenesis of acid-aspiration-induced ALI/ARDS. Oncotarget. 2017;9(2):1772–1784. doi:10.18632/oncotarget.22744
193. Chen Q, Huang Y, Yang Y, Qiu H. Acid‑induced cell injury and death in lung epithelial cells is associated with the activation of mitogen‑activated protein kinases. Mol Med Rep. 2013;8(2):565–570. doi:10.3892/mmr.2013.1537
194. Wang M, Cao X, Luan C, Li Z. Hydrogen Sulfide Attenuates Hydrogen Peroxide-Induced Injury in Human Lung Epithelial A549 Cells. Int J Mol Sci. 2019;20:16.
195. Davidson BA, Knight PR, Wang Z, et al. Surfactant alterations in acute inflammatory lung injury from aspiration of acid and gastric particulates. Am J Physiol Lung Cell Mol Physiol. 2005;288(4):L699–708. doi:10.1152/ajplung.00229.2004
196. Brackenbury AM, McCaig LA. Evaluation of exogenous surfactant in HCL-induced lung injury. Am J Respir Crit Care Med. 2001;163(5):1135–1142. doi:10.1164/ajrccm.163.5.2004049
197. Khalife-Hocquemiller T, Sage E, Dorfmuller P, et al. Exogenous surfactant attenuates lung injury from gastric-acid aspiration during ex vivo reconditioning in pigs. Transplantation. 2014;97(4):413–418. doi:10.1097/01.TP.0000441320.10787.c5
198. Nakajima D, Liu M, Ohsumi A, et al. Lung Lavage and Surfactant Replacement During Ex Vivo Lung Perfusion for Treatment of Gastric Acid Aspiration-Induced Donor Lung Injury. J Heart Lung Transplant. 2017;36(5):577–585. doi:10.1016/j.healun.2016.11.010
199. Chen CL, Li TP, Zhu LH. [Effect of MAPK signal transduction pathway inhibitor U0126 on aquaporin 4 expression in alveolar type II cells in rats with oleic acid-induced acute lung injury]. Nan Fang Yi Ke Da Xue Xue Bao. 2009;29(8):1525–1528. Chinese.
200. Song Y, Fukuda N, Bai C, Ma T, Matthay MA, Verkman AS. Role of aquaporins in alveolar fluid clearance in neonatal and adult lung, and in oedema formation following acute lung injury: studies in transgenic aquaporin null mice. J Physiol. 2000;525 Pt 3(Pt3):771–779. doi:10.1111/j.1469-7793.2000.00771.x
201. Gonçalves-de-albuquerque CF, Burth P, Silva AR, et al. Oleic acid inhibits lung Na/K-ATPase in mice and induces injury with lipid body formation in leukocytes and eicosanoid production. J Inflamm. 2013;10(1):34. doi:10.1186/1476-9255-10-34
202. Chen HI, Hsieh NK, Kao SJ, Su CF. Protective effects of propofol on acute lung injury induced by oleic acid in conscious rats. Crit Care Med. 2008;36(4):1214–1221. doi:10.1097/CCM.0b013e31816a0607
203. Giuranno L, Ient J, De Ruysscher D, Vooijs MA. Radiation-Induced Lung Injury (RILI). Front Oncol. 2019;9:877. doi:10.3389/fonc.2019.00877
204. Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol. 2008;49(4–6):119–133. doi:10.1016/j.vph.2008.06.009
205. Beach TA, Groves AM, Williams JP, Finkelstein JN. Modeling radiation-induced lung injury: lessons learned from whole thorax irradiation. Int J Radiat Biol. 2020;96(1):129–144. doi:10.1080/09553002.2018.1532619
206. Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327(1–2):48–60. doi:10.1016/j.canlet.2011.12.012
207. Spitz DRAE, Li JJ, Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23(3–4):311–322. doi:10.1023/B:CANC.0000031769.14728.bc
208. Park S, Ahn JY, Lim MJ, et al. Sustained expression of NADPH oxidase 4 by p38 MAPK-Akt signaling potentiates radiation-induced differentiation of lung fibroblasts. J Mol Med. 2010;88(8):807–816. doi:10.1007/s00109-010-0622-5
209. Sakai Y, Yamamori T, Yoshikawa Y, et al. NADPH oxidase 4 mediates ROS production in radiation-induced senescent cells and promotes migration of inflammatory cells. Free Radic Res. 2018;52(1):92–102. doi:10.1080/10715762.2017.1416112
210. Zhang Y, Zhang X, Rabbani ZN, Jackson IL, Vujaskovic Z. Oxidative stress mediates radiation lung injury by inducing apoptosis. Int J Radiat Oncol Biol Phys. 2012;83(2):740–748. doi:10.1016/j.ijrobp.2011.08.005
211. Hong ZY, Li S, Liu X, et al. Blocking C-Raf alleviated high-dose small-volume radiation-induced epithelial mesenchymal transition in mice lung. Sci Rep. 2020;10(1):11158. doi:10.1038/s41598-020-68175-z
212. Liang X, Gu J, Yu D, et al. Low-Dose Radiation Induces Cell Proliferation in Human Embryonic Lung Fibroblasts but not in Lung Cancer Cells. Dose-Response. 2016;14(1):155932581562217. doi:10.1177/1559325815622174
213. Jung JW, Hwang SY, Hwang JS, Oh ES, Park S, Han IO. Ionising radiation induces changes associated with epithelial-mesenchymal transdifferentiation and increased cell motility of A549 lung epithelial cells. Eur J Cancer. 2007;43(7):1214–1224. doi:10.1016/j.ejca.2007.01.034
214. Malaviya R, Gow AJ, Francis M, Abramova EV, Laskin JD, Laskin DL. Radiation-induced lung injury and inflammation in mice: role of inducible nitric oxide synthase and surfactant protein D. Toxicol Sci. 2015;144(1):27–38. doi:10.1093/toxsci/kfu255
215. Rubin P, Siemann DW, Shapiro DL, Finkelstein JN, Penney DP. Surfactant release as an early measure of radiation pneumonitis. Int J Radiat Oncol Biol Phys. 1983;9(11):1669–1673. doi:10.1016/0360-3016(83)90420-0
216. Christofidou-Solomidou M, Pietrofesa RA, Arguiri E, Koumenis C, Segal R. Radiation Mitigating Properties of Intranasally Administered KL4 Surfactant in a Murine Model of Radiation-Induced Lung Damage. Radiat Res. 2017;188(5):491–504. doi:10.1667/RR14686.1
217. Sun CY, Zhao YX, Zhong W, et al. The expression of aquaporins 1 and 5 in rat lung after thoracic irradiation. J Radiat Res. 2014;55(4):683–689. doi:10.1093/jrr/rru008
218. Li Y, Lu H, Lv X, et al. Blockade of Aquaporin 4 Inhibits Irradiation-Induced Pulmonary Inflammation and Modulates Macrophage Polarization in Mice. Inflammation. 2018;41(6):2196–2205. doi:10.1007/s10753-018-0862-z
219. Verheye-Dua FA, Böhm L. Influence of ouabain on cell inactivation by irradiation. Strahlentherapie und Onkologie. 1996;172(3):156–161.
220. Looney MR, Gropper MA, Matthay MA. Transfusion-related acute lung injury: a review. Chest. 2004;126(1):249–258. doi:10.1378/chest.126.1.249
221. Popovsky MA, Moore SB. Diagnostic and pathogenetic considerations in transfusion-related acute lung injury. Transfusion. 1985;25(6):573–577. doi:10.1046/j.1537-2995.1985.25686071434.x
222. Fung YL, Tung JP. How different animal models help us understand TRALI. ISBT Sci Series. 2018;13(3):197–205. doi:10.1111/voxs.12423
223. Silliman CC, Ambruso DR, Boshkov LK. Transfusion-related acute lung injury. Blood. 2005;105(6):2266–2273. doi:10.1182/blood-2004-07-2929
224. Thomas GM, Carbo C, Curtis BR, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood. 2012;119(26):6335–6343. doi:10.1182/blood-2012-01-405183
225. Lögdberg LE, Vikulina T, Zimring JC, Hillyer CD. Animal Models of Transfusion-Related Acute Lung Injury. Transfus Med Rev. 2009;23(1):13–24. doi:10.1016/j.tmrv.2008.09.002
226. Looney MR, Matthay MA. Animal models of transfusion-related acute lung injury. Crit Care Med. 2006;34(5):548. doi:10.1097/01.CCM.0000214287.58444.2D
227. Silliman CC, Thurman GW, Ambruso DR. Stored blood components contain agents that prime the neutrophil NADPH oxidase through the platelet-activating-factor receptor. Vox Sang. 1992;63(2):133–136. doi:10.1111/j.1423-0410.1992.tb02500.x
228. Bayat B, Tjahjono Y, Sydykov A, et al. Anti–Human Neutrophil Antigen-3a Induced Transfusion-Related Acute Lung Injury in Mice by Direct Disturbance of Lung Endothelial Cells. Arterioscler Thromb Vasc Biol. 2013;33(11):2538–2548. doi:10.1161/ATVBAHA.113.301206
229. McQuinn ER, Smith SA, Viall AK, Wang C, LeVine DN. Neutrophil extracellular traps in stored canine red blood cell units. J Vet Intern Med. 2020;34(5):1894–1902. doi:10.1111/jvim.15876
230. Rebetz J, Semple JW, Kapur R. The Pathogenic Involvement of Neutrophils in Acute Respiratory Distress Syndrome and Transfusion-Related Acute Lung Injury. Transfus Med Hemother. 2018;45(5):290–298. doi:10.1159/000492950
231. Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122(7):2661–2671. doi:10.1172/JCI61303
232. Curtis BR, McFarland JG. Mechanisms of transfusion-related acute lung injury (TRALI): anti-leukocyte antibodies. Crit Care Med. 2006;34(5):548. doi:10.1097/01.CCM.0000214293.72918.D8
233. Álvarez P, Carrasco R, Romero-Dapueto C, Castillo RL. Transfusion-Related Acute Lung Injured (TRALI): current Concepts. Open Respir Med J. 2015;9:92–96. doi:10.2174/1874306401509010092
234. Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their Fcγ receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest. 2006;116(6):1615–1623. doi:10.1172/JCI27238
235. Krammer F, Smith GJD, Fouchier RAM, et al. Influenza. Nat Rev Dis Primers. 2018;4(1):3. doi:10.1038/s41572-018-0002-y
236. Barnard DL. Animal models for the study of influenza pathogenesis and therapy. Antiviral Res. 2009;82(2):A110–A122. doi:10.1016/j.antiviral.2008.12.014
237. van Vught LA, Klein klouwenberg PMC, Spitoni C, et al. Incidence, Risk Factors, and Attributable Mortality of Secondary Infections in the Intensive Care Unit After Admission for Sepsis. JAMA. 2016;315(14):1469–1479. doi:10.1001/jama.2016.2691
238. Bouvier NM, Lowen AC. Animal Models for Influenza Virus Pathogenesis and Transmission. Viruses. 2010;2(8):798. doi:10.3390/v20801530
239. Yageta Y, Ishii Y, Morishima Y, et al. Role of Nrf2 in Host Defense against Influenza Virus in Cigarette Smoke-Exposed Mice. J Virol. 2011;85(10):4679–4690. doi:10.1128/JVI.02456-10
240. Simon PF, McCorrister S, Hu P, et al. Highly Pathogenic H5N1 and Novel H7N9 Influenza A Viruses Induce More Profound Proteomic Host Responses than Seasonal and Pandemic H1N1 Strains. J Proteome Res. 2015;14(11):4511–4523. doi:10.1021/acs.jproteome.5b00196
241. Shoji M, Arakaki Y, Esumi T, et al. Bakuchiol Is a Phenolic Isoprenoid with Novel Enantiomer-selective Anti-influenza A Virus Activity Involving Nrf2 Activation *. J Biol Chem. 2015;290(46):28001–28017. doi:10.1074/jbc.M115.669465
242. Ma -L-L, Wang H-Q, Wu P, et al. Rupestonic acid derivative YZH-106 suppresses influenza virus replication by activation of heme oxygenase-1-mediated interferon response. Free Radical Biol Med. 2016;96:347–361. doi:10.1016/j.freeradbiomed.2016.04.021
243. Dai J, Gu L, Su Y, et al. Inhibition of curcumin on influenza A virus infection and influenzal pneumonia via oxidative stress, TLR2/4, p38/JNK MAPK and NF-κB pathways. Int Immunopharmacol. 2018;54:177–187. doi:10.1016/j.intimp.2017.11.009
244. Dai J-P, Wang Q-W, Su Y, et al. Emodin Inhibition of Influenza A Virus Replication and Influenza Viral Pneumonia via the Nrf2, TLR4, p38/JNK and NF-kappaB Pathways. Molecules. 2017;22:10. doi:10.3390/molecules22101754
245. Guo Y, Tu YH, Wu X, et al. ResolvinD1 Protects the Airway Barrier Against Injury Induced by Influenza A Virus Through the Nrf2 Pathway. Front Cell Infect Microbiol. 2020;10:616475. doi:10.3389/fcimb.2020.616475
246. Snelgrove RJ, Edwards L, Rae AJ, Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur J Immunol. 2006;36(6):1364–1373. doi:10.1002/eji.200635977
247. Vlahos R, Stambas J, Bozinovski S, Broughton BR, Drummond GR, Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 2011;7(2):e1001271. doi:10.1371/journal.ppat.1001271
248. Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–249. doi:10.1016/j.cell.2008.02.043
249. Amatore D, Sgarbanti R, Aquilano K, et al. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell Microbiol. 2015;17(1):131–145. doi:10.1111/cmi.12343
250. Selemidis S, Seow HJ, Broughton BR, et al. Nox1 oxidase suppresses influenza a virus-induced lung inflammation and oxidative stress. PLoS One. 2013;8(4):e60792. doi:10.1371/journal.pone.0060792
251. Narasaraju T, Yang E, Samy RP, et al. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am J Pathol. 2011;179(1):199–210. doi:10.1016/j.ajpath.2011.03.013
252. Ashar HK, Mueller NC, Rudd JM, et al. The Role of Extracellular Histones in Influenza Virus Pathogenesis. Am J Pathol. 2018;188(1):135–148. doi:10.1016/j.ajpath.2017.09.014
253. Garcia CC, Weston-Davies W, Russo RC, et al. Complement C5 activation during influenza A infection in mice contributes to neutrophil recruitment and lung injury. PLoS One. 2013;8(5):e64443. doi:10.1371/journal.pone.0064443
254. Chan LLY, Nicholls JM, Peiris JSM, Lau YL, Chan MCW, Chan RWY. Host DNA released by NETosis in neutrophils exposed to seasonal H1N1 and highly pathogenic H5N1 influenza viruses. Respir Res. 2020;21(1):160. doi:10.1186/s12931-020-01425-w
255. Yu J, Sun X, Goie JY, Zhang Y. Regulation of Host Immune Responses against Influenza A Virus Infection by Mitogen-Activated Protein Kinases (MAPKs). Microorganisms. 2020;8:7. doi:10.3390/microorganisms8071067
256. Zhang R, Ai X, Duan Y, et al. Kaempferol ameliorates H9N2 swine influenza virus-induced acute lung injury by inactivation of TLR4/MyD88-mediated NF-κB and MAPK signaling pathways. Biomed Pharmacother. 2017;89:660–672. doi:10.1016/j.biopha.2017.02.081
257. Xing Z, Cardona CJ, Anunciacion J, Adams S, Dao N. Roles of the ERK MAPK in the regulation of proinflammatory and apoptotic responses in chicken macrophages infected with H9N2 avian influenza virus. J Gen Virol. 2010;91(Pt 2):343–351. doi:10.1099/vir.0.015578-0
258. Lee DC, Cheung CY, Law AH, Mok CK, Peiris M, Lau AS. p38 mitogen-activated protein kinase-dependent hyperinduction of tumor necrosis factor alpha expression in response to avian influenza virus H5N1. J Virol. 2005;79(16):10147–10154. doi:10.1128/JVI.79.16.10147-10154.2005
259. Gao W, Sun W, Qu B, et al. Distinct Regulation of Host Responses by ERK and JNK MAP Kinases in Swine Macrophages Infected with Pandemic (H1N1) 2009 Influenza Virus. PLoS One. 2012;7(1):e30328. doi:10.1371/journal.pone.0030328
260. Li Y, Xu J, Shi W, et al. Mesenchymal stromal cell treatment prevents H9N2 avian influenza virus-induced acute lung injury in mice. Stem Cell Res Ther. 2016;7(1):159. doi:10.1186/s13287-016-0395-z
261. Yu C-H, Yu W-Y, Fang J, et al. Mosla scabra flavonoids ameliorate the influenza A virus-induced lung injury and water transport abnormality via the inhibition of PRR and AQP signaling pathways in mice. J Ethnopharmacol. 2016;179:146–155. doi:10.1016/j.jep.2015.12.034
262. Geiler J, Michaelis M, Naczk P, et al. N-acetyl-l-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem Pharmacol. 2010;79(3):413–420. doi:10.1016/j.bcp.2009.08.025
263. Ding Z, Sun G, Zhu Z. Hesperidin Attenuates Influenza a virus (H1N1) Induced Lung Injury in Rats through its Anti-Inflammatory Effect. Antivir Ther. 2017;23(7):611–615. doi:10.3851/IMP3235
264. Growcott EJ, Bamba D, Galarneau JR, et al. The effect of P38 MAP kinase inhibition in a mouse model of influenza. J Med Microbiol. 2018;67(3):452–462. doi:10.1099/jmm.0.000684
265. Wang W, Yang P, Zhong Y, et al. Monoclonal antibody against CXCL-10/IP-10 ameliorates influenza A (H1N1) virus induced acute lung injury. Cell Res. 2013;23(4):577–580. doi:10.1038/cr.2013.25
266. Huang F, Zhang C, Liu Q, et al. Identification of amitriptyline HCl, flavin adenine dinucleotide, azacitidine and calcitriol as repurposing drugs for influenza A H5N1 virus-induced lung injury. PLoS Pathog. 2020;16(3):e1008341. doi:10.1371/journal.ppat.1008341
267. Li J, Jie X, Liang X, et al. Sinensetin suppresses influenza a virus-triggered inflammation through inhibition of NF-κB and MAPKs signalings. BMC Complement Med Therap. 2020;20(1):135. doi:10.1186/s12906-020-02918-3
268. Wang Q-W, Su Y, Sheng J-T, et al. Anti-influenza A virus activity of rhein through regulating oxidative stress, TLR4, Akt, MAPK, and NF-κB signal pathways. PLoS One. 2018;13(1):e0191793. doi:10.1371/journal.pone.0191793
269. Dai J-P, Wang Q-W, Su Y, et al. Oxymatrine Inhibits Influenza A Virus Replication and Inflammation via TLR4, p38 MAPK and NF-κB Pathways. Int J Mol Sci. 2018;19(4):548. doi:10.3390/ijms19040965
270. Cui L, Zheng D, Lee YH, et al. Metabolomics Investigation Reveals Metabolite Mediators Associated with Acute Lung Injury and Repair in a Murine Model of Influenza Pneumonia. Sci Rep. 2016;6:26076. doi:10.1038/srep26076
271. Woods PS, Doolittle LM, Rosas LE, Joseph LM, Calomeni EP, Davis IC. Lethal H1N1 influenza A virus infection alters the murine alveolar type II cell surfactant lipidome. Am J Physiol Lung Cell Mol Physiol. 2016;311(6):L1160–L1169. doi:10.1152/ajplung.00339.2016
272. Numata M, Mitchell JR, Tipper JL, et al. Pulmonary surfactant lipids inhibit infections with the pandemic H1N1 influenza virus in several animal models. J Biol Chem. 2020;295(6):1704–1715. doi:10.1074/jbc.RA119.012053
273. Donovan BW, Reuter JD, Cao Z, Myc A, Johnson KJ, Baker JR. Prevention of Murine Influenza a Virus Pneumonitis by Surfactant Nano-Emulsions. Antivir Chem Chemother. 2000;11(1):41–49. doi:10.1177/095632020001100104
274. Fukushi M, Yamashita M, Miyoshi-Akiyama T, Kubo S, Yamamoto K, Kudo K. Laninamivir Octanoate and Artificial Surfactant Combination Therapy Significantly Increases Survival of Mice Infected with Lethal Influenza H1N1 Virus. PLoS One. 2012;7(8):e42419. doi:10.1371/journal.pone.0042419
275. Chen X-J, Seth S, Yue G, et al. Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol. 2004;287(2):L366–L373. doi:10.1152/ajplung.00011.2004
276. Peteranderl C, Morales-Nebreda L, Selvakumar B, et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J Clin Invest. 2016;126(4):1566–1580. doi:10.1172/JCI83931
277. Chan MCW, Kuok DIT, Leung CYH, et al. Human mesenchymal stromal cells reduce influenza A H5N1-associated acute lung injury in vitro and in vivo. Proc Natl Acad Sci. 2016;113(13):3621. doi:10.1073/pnas.1601911113
278. Dockrell DH, Whyte MKB, Mitchell TJ. Pneumococcal pneumonia: mechanisms of infection and resolution. Chest. 2012;142(2):482–491. doi:10.1378/chest.12-0210
279. Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6(4):288–301. doi:10.1038/nrmicro1871
280. Brooks LRK, Mias GI. Streptococcus pneumoniae’s Virulence and Host Immunity: aging, Diagnostics, and Prevention. Front Immunol. 2018;1:9.
281. Henriques-Normark B, Tuomanen EI. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med. 2013;3(7):432. doi:10.1101/cshperspect.a010215
282. Chiavolini D, Pozzi G, Ricci S. Animal Models of Streptococcus pneumoniae Disease. Clin Microbiol Rev. 2008;21(4):666–685. doi:10.1128/CMR.00012-08
283. Borsa N, Di Pasquale M, Restrepo MI. Animal Models of Pneumococcal pneumonia. Int J Mol Sci. 2019;20:17. doi:10.3390/ijms20174220
284. Zahlten J, Kim Y-J, Doehn J-M, et al. Streptococcus pneumoniae–Induced Oxidative Stress in Lung Epithelial Cells Depends on Pneumococcal Autolysis and Is Reversible by Resveratrol. J Infect Dis. 2015;211(11):1822–1830. doi:10.1093/infdis/jiu806
285. Gomez JC, Dang H, Martin JR, Doerschuk CM. Nrf2 Modulates Host Defense during Streptococcus pneumoniae Pneumonia in Mice. J Immunol. 2016;197(7):2864–2879. doi:10.4049/jimmunol.1600043
286. Marriott HM, Jackson LE, Wilkinson TS, et al. Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am J Respir Crit Care Med. 2008;177(8):887–895. doi:10.1164/rccm.200707-990OC
287. Moorthy AN, Rai P, Jiao H, et al. Capsules of virulent pneumococcal serotypes enhance formation of neutrophil extracellular traps during in vivo pathogenesis of pneumonia. Oncotarget. 2016;7(15):19327–19340. doi:10.18632/oncotarget.8451
288. Mori Y, Yamaguchi M, Terao Y, Hamada S, Ooshima T, Kawabata S. α-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J Biol Chem. 2012;287(13):10472–10481. doi:10.1074/jbc.M111.280321
289. Narayana Moorthy A, Narasaraju T, Rai P, et al. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front Immunol. 2013;4:654. doi:10.3389/fimmu.2013.00056
290. Wartha F, Beiter K, Albiger B, et al. Capsule and d-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol. 2007;9(5):1162–1171. doi:10.1111/j.1462-5822.2006.00857.x
291. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol. 2006;16(4):401–407. doi:10.1016/j.cub.2006.01.056
292. N’Guessan PD, Schmeck B, Ayim A, et al. Streptococcus pneumoniae R6x induced p38 MAPK JNK-mediated Caspase-dependent apoptosis in human endothelial cells. Thromb Haemost. 2005;94(08):295–303. doi:10.1160/TH04-12-0822
293. Xu F, Droemann D, Rupp J, et al. Modulation of the inflammatory response to Streptococcus pneumoniae in a model of acute lung tissue infection. Am J Respir Cell Mol Biol. 2008;39(5):522–529. doi:10.1165/rcmb.2007-0328OC
294. Schmeck B, Zahlten J, Moog K, et al. Streptococcus pneumoniae-induced p38 MAPK-dependent phosphorylation of RelA at the interleukin-8 promotor. J Biol Chem. 2004;279(51):53241–53247. doi:10.1074/jbc.M313702200
295. N’Guessan PD, Hippenstiel S, Etouem MO, et al. Streptococcus pneumoniae induced p38 MAPK- and NF-κB-dependent COX-2 expression in human lung epithelium. Am J Physiol. 2006;290(6):L1131–L1138. doi:10.1152/ajplung.00383.2005
296. Szymanski KV, Toennies M, Becher A, et al. Streptococcus pneumoniae-induced regulation of cyclooxygenase-2 in human lung tissue. Eur Respir J. 2012;40(6):1458–1467. doi:10.1183/09031936.00186911
297. Boyd AR, Shivshankar P, Jiang S, Berton MT, Orihuela CJ. Age-related defects in TLR2 signaling diminish the cytokine response by alveolar macrophages during murine pneumococcal pneumonia. Exp Gerontol. 2012;47(7):507–518. doi:10.1016/j.exger.2012.04.004
298. Jounblat R, Kadioglu A, Iannelli F, Pozzi G, Eggleton P, Andrew PW. Binding and agglutination of Streptococcus pneumoniae by human surfactant protein D (SP-D) vary between strains, but SP-D fails to enhance killing by neutrophils. Infect Immun. 2004;72(2):709–716. doi:10.1128/IAI.72.2.709-716.2004
299. Jounblat R, Clark H, Eggleton P, Hawgood S, Andrew PW, Kadioglu A. The role of surfactant protein D in the colonisation of the respiratory tract and onset of bacteraemia during pneumococcal pneumonia. Respir Res. 2005;6(1):126. doi:10.1186/1465-9921-6-126
300. Tyrrell C, McKechnie SR, Beers MF, Mitchell TJ, McElroy MC. Differential alveolar epithelial injury and protein expression in pneumococcal pneumonia. Exp Lung Res. 2012;38(5):266–276. doi:10.3109/01902148.2012.683321
301. LaCanna R, Liccardo D, Zhang P, et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J Clin Invest. 2019;129(5):2107–2122. doi:10.1172/JCI125014
302. Witzenrath M, Gutbier B, Hocke AC, et al. Role of pneumolysin for the development of acute lung injury in pneumococcal pneumonia. Crit Care Med. 2006;34(7):87. doi:10.1097/01.CCM.0000220496.48295.A9
303. Ross JT, Nesseler N, Leligdowicz A, et al. The ex vivo perfused human lung is resistant to injury by high-dose S. pneumoniae bacteremia. Am J Physiol Lung Cell Mol Physiol. 2020;319(2):L218–L227. doi:10.1152/ajplung.00053.2020
304. Czikora I, Alli AA, Sridhar S, et al. Epithelial Sodium Channel-α Mediates the Protective Effect of the TNF-Derived TIP Peptide in Pneumolysin-Induced Endothelial Barrier Dysfunction. Front Immunol. 2017;8:842. doi:10.3389/fimmu.2017.00842
305. Rabaan AA, Al-Ahmed SH, Haque S, et al. SARS-CoV-2, SARS-CoV, and MERS-COV: a comparative overview. Le infezioni in medicina. Ahead Print. 2020;28(2):174–184.
306. Abdool Karim SS, de Oliveira T. New SARS-CoV-2 Variants — clinical, Public Health, and Vaccine Implications. N Engl J Med. 2021;384(19):1866–1868. doi:10.1056/NEJMc2100362
307. Gong S, Bao L. The battle against SARS and MERS coronaviruses: reservoirs and Animal Models. Anim Models Exp Med. 2018;1(2):125–133. doi:10.1002/ame2.12017
308. Pandey K, Acharya A, Mohan M, Ng CL, Reid SP, Byrareddy SN. Animal models for SARS-CoV-2 research: a comprehensive literature review. Transbound Emerg Dis. 2021;68(4):1868–1885. doi:10.1111/tbed.13907
309. Becker K, Beythien G, de Buhr N, et al. Vasculitis and Neutrophil Extracellular Traps in Lungs of Golden Syrian Hamsters With SARS-CoV-2. Front Immunol. 2021;12:640842. doi:10.3389/fimmu.2021.640842
310. Adrover JM, Carrau L, Daßler-Plenker J, et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight. 2022;7(5):53. doi:10.1172/jci.insight.157342
311. Jimenez-Guardeño JM, Nieto-Torres JL, DeDiego ML, et al. The PDZ-binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Pathog. 2014;10(8):e1004320. doi:10.1371/journal.ppat.1004320
312. Chang YJ, Liu CY, Chiang BL, Chao YC, Chen CC. Induction of IL-8 release in lung cells via activator protein-1 by recombinant baculovirus displaying severe acute respiratory syndrome-coronavirus spike proteins: identification of two functional regions. J Immunol. 2004;173(12):7602–7614. doi:10.4049/jimmunol.173.12.7602
313. Kopecky-Bromberg SA, Martinez-Sobrido L, Palese P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J Virol. 2006;80(2):785–793. doi:10.1128/JVI.80.2.785-793.2006
314. Li SW, Wang CY, Jou YJ, et al. SARS coronavirus papain-like protease induces Egr-1-dependent up-regulation of TGF-β1 via ROS/p38 MAPK/STAT3 pathway. Sci Rep. 2016;6:25754. doi:10.1038/srep25754
315. Gu T, Zhao S, Jin G, et al. Cytokine Signature Induced by SARS-CoV-2 Spike Protein in a Mouse Model. Front Immunol. 2020;11:621441. doi:10.3389/fimmu.2020.621441
316. Gralinski LE, Bankhead A, Jeng S, et al. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio. 2013;4(4):515. doi:10.1128/mBio.00271-13
317. Suresh V, Mohanty V, Avula K, et al. Quantitative proteomics of hamster lung tissues infected with SARS-CoV-2 reveal host factors having implication in the disease pathogenesis and severity. FASEB J. 2021;35(7):e21713. doi:10.1096/fj.202100431R
318. Hsieh M-H, Beirag N, Murugaiah V, et al. Human Surfactant Protein D Binds Spike Protein and Acts as an Entry Inhibitor of SARS-CoV-2 Pseudotyped Viral Particles. Front Immunol. 2021;12:641360. doi:10.3389/fimmu.2021.641360
319. Nagata N, Iwata N, Hasegawa H, et al. Mouse-passaged severe acute respiratory syndrome-associated coronavirus leads to lethal pulmonary edema and diffuse alveolar damage in adult but not young mice. Am J Pathol. 2008;172(6):1625–1637. doi:10.2353/ajpath.2008.071060
320. Yu P, Xu Y, Deng W, et al. Comparative pathology of rhesus macaque and common marmoset animal models with Middle East respiratory syndrome coronavirus. PLoS One. 2017;12(2):e0172093. doi:10.1371/journal.pone.0172093
321. Allnoch L, Beythien G, Leitzen E, et al. Vascular Inflammation Is Associated with Loss of Aquaporin 1 Expression on Endothelial Cells and Increased Fluid Leakage in SARS-CoV-2 Infected Golden Syrian Hamsters. Viruses. 2021;13(4):548. doi:10.3390/v13040639
322. Gattinoni L, Marini JJ, Collino F, et al. The future of mechanical ventilation: lessons from the present and the past. Crit Care. 2017;21(1):183. doi:10.1186/s13054-017-1750-x
323. Hubmayr RD, Kallet RH. Understanding Pulmonary Stress-Strain Relationships in Severe ARDS and Its Implications for Designing a Safer Approach to Setting the Ventilator. Respir Care. 2018;63(2):219–226. doi:10.4187/respcare.05900
324. InterNational consensus conferences in intensive care medicine: Ventilator-associated Lung Injury in ARDS. This official conference report was cosponsored by the American Thoracic Society, The European Society of Intensive Care Medicine, and The Societé de Réanimation de Langue Française, and was approved by the ATS Board of Directors 1999. Am J Respir Crit Care Med. 1999;160(6):2118–2124. doi:10.1164/ajrccm.160.6.ats16060.
325. Force* TADT. Acute Respiratory Distress Syndrome: the Berlin Definition. JAMA. 2012;307(23):2526–2533. doi:10.1001/jama.2012.5669.
326. Bernard GRAA, Brigham KL, Carlet J, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3Pt 1):818–824. doi:10.1164/ajrccm.149.3.7509706
327. Hoidal JR, Xu P, Huecksteadt T, Sanders KA, Pfeffer K, Sturrock AB. Lung injury and oxidoreductases. Environ Health Perspect. 1998;106(suppl 5):1235–1239. doi:10.1289/ehp.98106s51235
328. Carnesecchi S, Dunand-Sauthier I, Zanetti F, et al. NOX1 is responsible for cell death through STAT3 activation in hyperoxia and is associated with the pathogenesis of acute respiratory distress syndrome. Int J Clin Exp Pathol. 2014;7(2):537–551.
329. O’Mahony DS, Glavan BJ, Holden TD, et al. Inflammation and immune-related candidate gene associations with acute lung injury susceptibility and severity: a validation study. PLoS One. 2012;7(12):e51104. doi:10.1371/journal.pone.0051104
330. Williams AECR. The mercurial nature of neutrophils: still an enigma in ARDS? Am J Physiol Lung Cell Mol Physiol. 2014;306(3):L217–230. doi:10.1152/ajplung.00311.2013
331. Ebrahimi F, Giaglis S, Hahn S, et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: secondary analysis of a randomised controlled trial. Eur Respir J. 2018;51(4):45. doi:10.1183/13993003.01389-2017
332. Park SY, Shrestha S, Youn Y-J, et al. Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation during Sepsis. Am J Respir Crit Care Med. 2017;196(5):577–589. doi:10.1164/rccm.201603-0596OC
333. Hu L, Zhao T, Sun Y, Chen Y, Bai K, Xu F. Bioinformatic identification of hub genes and key pathways in neutrophils of patients with acute respiratory distress syndrome. Medicine. 2020;99(15):e19820. doi:10.1097/MD.0000000000019820
334. Juss JK, House D, Amour A, et al. Acute Respiratory Distress Syndrome Neutrophils Have a Distinct Phenotype and Are Resistant to Phosphoinositide 3-Kinase Inhibition. Am J Respir Crit Care Med. 2016;194(8):961–973. doi:10.1164/rccm.201509-1818OC
335. Davidson WJ, Dorscheid D, Spragg R, Schulzer M, Mak E, Ayas NT. Exogenous pulmonary surfactant for the treatment of adult patients with acute respiratory distress syndrome: results of a meta-analysis. Crit Care. 2006;10(2):R41. doi:10.1186/cc4851
336. Hintz SR, Poole WK, Wright LL, et al. Changes in mortality and morbidities among infants born at less than 25 weeks during the post-surfactant era. Arch Dis Child Fetal Neonatal Ed. 2005;90(2):F128–133. doi:10.1136/adc.2003.046268
337. Meng SS, Chang W, Lu ZH, et al. Effect of surfactant administration on outcomes of adult patients in acute respiratory distress syndrome: a meta-analysis of randomized controlled trials. BMC Pulm Med. 2019;19(1):9. doi:10.1186/s12890-018-0761-y
338. Spragg RG, Lewis JF, Wurst W, et al. Treatment of acute respiratory distress syndrome with recombinant surfactant protein C surfactant. Am J Respir Crit Care Med. 2003;167(11):1562–1566. doi:10.1164/rccm.200207-782OC
339. Willson DF, Notter RH. The future of exogenous surfactant therapy. Respir Care. 2011;56(9):1369–1388. doi:10.4187/respcare.01306
340. Taut FJH, Rippin G, Schenk P, et al. A Search for Subgroups of Patients With ARDS Who May Benefit From Surfactant Replacement Therapy: a Pooled Analysis of Five Studies With Recombinant Surfactant Protein-C Surfactant (Venticute). Chest. 2008;134(4):724–732. doi:10.1378/chest.08-0362
341. Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82(3):569–600. doi:10.1152/physrev.00003.2002
342. Rahmel T, Rump K, Peters J, Adamzik M. Aquaporin 5-1364A/C Promoter Polymorphism Is Associated with Pulmonary Inflammation and Survival in Acute Respiratory Distress Syndrome. Anesthesiology. 2019;130(3):404–413. doi:10.1097/ALN.0000000000002560
343. Matthay MA. Alveolar fluid clearance in patients with ARDS: does it make a difference? Chest. 2002;122(6Suppl):340S–343S. doi:10.1378/chest.122.6_suppl.340S
344. Krenn K, Lucas R, Croizé A, et al. Inhaled AP301 for treatment of pulmonary edema in mechanically ventilated patients with acute respiratory distress syndrome: a phase IIa randomized placebo-controlled trial. Crit Care. 2017;21(1):194. doi:10.1186/s13054-017-1795-x
345. Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: international Guidelines for Management of Sepsis and Septic Shock: 2016. Crit Care Med. 2017;45(3):486–552. doi:10.1097/CCM.0000000000002255
346. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
347. Kangelaris KN, Prakash A, Liu KD, et al. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am J Physiol. 2015;308(11):L1102–L1113. doi:10.1152/ajplung.00380.2014
348. Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and Outcomes of Acute Lung Injury. N Engl J Med. 2005;353(16):1685–1693. doi:10.1056/NEJMoa050333
349. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. 2016;353:i1585. doi:10.1136/bmj.i1585
350. Leligdowicz A, Matthay MA. Heterogeneity in sepsis: new biological evidence with clinical applications. Crit Care. 2019;23(1):80. doi:10.1186/s13054-019-2372-2
351. Acosta-Herrera M, Pino-Yanes M, Blanco J, et al. Common variants of NFE2L2 gene predisposes to acute respiratory distress syndrome in patients with severe sepsis. Crit Care. 2015;19(1):256. doi:10.1186/s13054-015-0981-y
352. Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. 2018;3(3):5485.
353. Wang D, Li Y, Gu C, Liu M, Wang Y. Identification of Key Pathways and Genes of Acute Respiratory Distress Syndrome Specific Neutrophil Phenotype. Biomed Res Int. 2019;2019:9528584. doi:10.1155/2019/9528584
354. Anzueto A, Baughman RP, Guntupalli KK, et al. Aerosolized Surfactant in Adults with Sepsis-Induced Acute Respiratory Distress Syndrome. N Engl J Med. 1996;334(22):1417–1422. doi:10.1056/NEJM199605303342201
355. Walmrath D, Grimminger F, Pappert D, et al. Bronchoscopic administration of bovine natural surfactant in ARDS and septic shock: impact on gas exchange and haemodynamics. Eur Respir J. 2002;19(5):805–810. doi:10.1183/09031936.02.00243402
356. Weg JG, Balk RA, Tharratt RS, et al. Safety and Potential Efficacy of an Aerosolized Surfactant in Human Sepsis-Induced Adult Respiratory Distress Syndrome. JAMA. 1994;272(18):1433–1438. doi:10.1001/jama.1994.03520180057035
357. Zeyed YF, Bastarache JA, Matthay MA, Ware LB. The severity of shock is associated with impaired rates of net alveolar fluid clearance in clinical acute lung injury. Am J Physiol. 2012;303(6):L550–L555. doi:10.1152/ajplung.00190.2012
358. Vassiliou AG, Maniatis NA, Orfanos SE, et al. Induced expression and functional effects of aquaporin-1 in human leukocytes in sepsis. Crit Care. 2013;17(5):R199. doi:10.1186/cc12893
359. Adamzik M, Frey UH, Möhlenkamp S, et al. Aquaporin 5 Gene Promoter −1364A/C Polymorphism Associated with 30-day Survival in Severe Sepsis. Anesthesiology. 2011;114(4):912–917. doi:10.1097/ALN.0b013e31820ca911
360. Rump K, Unterberg M, Dahlke A, et al. DNA methylation of a NF-κB binding site in the aquaporin 5 promoter impacts on mortality in sepsis. Sci Rep. 2019;9(1):18511. doi:10.1038/s41598-019-55051-8
361. Li G, Zhang Y, Fan Z. Cellular Signal Transduction Pathways Involved in Acute Lung Injury Induced by Intestinal Ischemia-Reperfusion. Oxid Med Cell Longev. 2021;2021:9985701.
362. Weyker PD, Webb CA, Kiamanesh D, Flynn BC. Lung ischemia reperfusion injury: a bench-to-bedside review. Semin Cardiothorac Vasc Anesth. 2013;17(1):28–43. doi:10.1177/1089253212458329
363. Christie JD, Carby M, Bag R, et al. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction part II: definition. A consensus statement of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2005;24(10):1454–1459. doi:10.1016/j.healun.2004.11.049
364. Hohlfeld JM, Tiryaki E, Hamm H, et al. Pulmonary surfactant activity is impaired in lung transplant recipients. Am J Respir Crit Care Med. 1998;158(3):706–712. doi:10.1164/ajrccm.158.3.9708063
365. Amital A, Shitrit D, Raviv Y, et al. The Use of Surfactant in Lung Transplantation. Transplantation. 2008;86:11. doi:10.1097/TP.0b013e31818a8418
366. Kermeen FD, McNeil KD, Fraser JF, et al. Resolution of Severe Ischemia–Reperfusion Injury Post–Lung Transplantation After Administration of Endobronchial Surfactant. J Heart Lung Transplant. 2007;26(8):850–856. doi:10.1016/j.healun.2007.05.016
367. Strüber M, Fischer S, Niedermeyer J, et al. Effects of exogenous surfactant instillation in clinical lung transplantation: a prospective, randomized trial. J Thorac Cardiovasc Surg. 2007;133(6):1620–1625. doi:10.1016/j.jtcvs.2006.12.057
368. Strüber M, Hirt SW, Cremer J, Harringer W, Haverich A. Surfactant replacement in reperfusion injury after clinical lung transplantation. Intensive Care Med. 1999;25(8):862–864. doi:10.1007/s001340050967
369. Ware LB, Golden JA, Finkbeiner WE, Matthay MA. Alveolar epithelial fluid transport capacity in reperfusion lung injury after lung transplantation. Am J Respir Crit Care Med. 1999;159(3):980–988. doi:10.1164/ajrccm.159.3.9802105
370. Dries DJEFS. Inhalation injury: epidemiology, pathology, treatment strategies. J Trauma Resusc Emerg Med. 2013;21:31. doi:10.1186/1757-7241-21-31
371. Cho HY, Kleeberger SR. Nrf2 protects against airway disorders. Toxicol Appl Pharmacol. 2010;244(1):43–56. doi:10.1016/j.taap.2009.07.024
372. Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020;33:101544. doi:10.1016/j.redox.2020.101544
373. Tamimi A, Serdarevic D, Hanania NA. The effects of cigarette smoke on airway inflammation in asthma and COPD: therapeutic implications. Respir Med. 2012;106(3):319–328. doi:10.1016/j.rmed.2011.11.003
374. Zhao CZ, Fang XC, Wang D, Tang FD, Wang XD. Involvement of type II pneumocytes in the pathogenesis of chronic obstructive pulmonary disease. Respir Med. 2010;104(10):1391–1395. doi:10.1016/j.rmed.2010.06.018
375. Drakulovic MB, Torres A, Bauer TT, Nicolas JM, Nogué S, Ferrer M. Supine body position as a risk factor for nosocomial pneumonia in mechanically ventilated patients: a randomised trial. Lancet. 1999;354(9193):1851–1858. doi:10.1016/S0140-6736(98)12251-1
376. Potts RG, Zaroukian MH, Guerrero PA, Baker CD. Comparison of blue dye visualization and glucose oxidase test strip methods for detecting pulmonary aspiration of enteral feedings in intubated adults. Chest. 1993;103(1):117–121. doi:10.1378/chest.103.1.117
377. Morgan GWBS. Radiation and the lung: a reevaluation of the mechanisms mediating pulmonary injury. Int J Radiat Oncol Biol Phys. 1995;31(2):361–369. doi:10.1016/0360-3016(94)00477-3
378. Byhardt RWAR, Almagro U. The association of adult respiratory distress syndrome (ARDS) with thoracic irradiation (RT). Int J Radiat Oncol Biol Phys. 1988;15(6):1441–1446. doi:10.1016/0360-3016(88)90241-6
379. Chen S, Zhou S, Zhang J, Yin FF, Marks LB, Das SK. A neural network model to predict lung radiation-induced pneumonitis. Med Phys. 2007;34(9):3420–3427. doi:10.1118/1.2759601
380. Jarzebska N, Karetnikova ES, Markov AG, Kasper M, Rodionov RN, Spieth PM. Scarred Lung. An Update on Radiation-Induced Pulmonary Fibrosis. Front Med. 2020;7:585756. doi:10.3389/fmed.2020.585756
381. Tung J-P, Chiaretti S, Dean MM, Sultana AJ, Reade MC, Fung YL. Transfusion-related acute lung injury (TRALI): potential pathways of development, strategies for prevention and treatment, and future research directions. Blood Rev. 2022;53:100926. doi:10.1016/j.blre.2021.100926
382. Toy P, Lowell C. TRALI–definition, mechanisms, incidence and clinical relevance. Best Pract Res Clin Anaesthesiol. 2007;21(2):183–193. doi:10.1016/j.bpa.2007.01.003
383. Lenahan SE, Domen RE, Silliman CC, Kingsley CP, Romano PJ. Transfusion-Related Acute Lung Injury Secondary to Biologically Active Mediators. Arch Pathol Lab Med. 2001;125(4):523–526. doi:10.5858/2001-125-0523-TRALIS
384. Wu T-J, Teng R-J, Yau K-IT. Transfusion-related acute lung injury treated with surfactant in a neonate. Eur J Pediatr. 1996;155(7):589–591. doi:10.1007/BF01957910
385. Herold S, Becker C, Ridge KM, Budinger GR. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J. 2015;45(5):1463–1478. doi:10.1183/09031936.00186214
386. Kosmider B, Messier EM, Janssen WJ, et al. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir Res. 2012;13(1):43. doi:10.1186/1465-9921-13-43
387. Zhu L, Liu L, Zhang Y, et al. High Level of Neutrophil Extracellular Traps Correlates With Poor Prognosis of Severe Influenza A Infection. J Infect Dis. 2018;217(3):428–437. doi:10.1093/infdis/jix475
388. Zhang N, Zhu L, Zhang Y, et al. Circulating Rather Than Alveolar Extracellular Deoxyribonucleic Acid Levels Predict Outcomes in Influenza. J Infect Dis. 2020;222(7):1145–1154. doi:10.1093/infdis/jiaa241
389. Herrera-Ramos E, López-Rodríguez M, Ruíz-Hernández JJ, et al. Surfactant protein A genetic variants associate with severe respiratory insufficiency in pandemic influenza A virus infection. Crit Care. 2014;18(3):R127. doi:10.1186/cc13934
390. To KKW, Zhou J, Song Y-Q, et al. Surfactant Protein B Gene Polymorphism Is Associated With Severe Influenza. Chest. 2014;145(6):1237–1243. doi:10.1378/chest.13-1651
391. Kongchanagul A, Suptawiwat O, Boonarkart C, et al. Decreased expression of surfactant protein D mRNA in human lungs in fatal cases of H5N1 avian influenza. J Med Virol. 2011;83(8):1410–1417. doi:10.1002/jmv.22105
392. Choreño-Parra JA, Jiménez-álvarez LA, Ramírez-Martínez G, et al. Expression of Surfactant Protein D Distinguishes Severe Pandemic Influenza A(H1N1) from Coronavirus Disease 2019. J Infect Dis. 2021;224(1):21–30. doi:10.1093/infdis/jiab113
393. Burgos J, Falcó V, Borrego A, et al. Impact of the emergence of non-vaccine pneumococcal serotypes on the clinical presentation and outcome of adults with invasive pneumococcal pneumonia. Clin Microbiol Infect. 2013;19(4):385–391. doi:10.1111/j.1469-0691.2012.03895.x
394. Lanks CW, Musani AI, Hsia DW. Community-acquired Pneumonia and Hospital-acquired Pneumonia. Med Clin North Am. 2019;103(3):487–501. doi:10.1016/j.mcna.2018.12.008
395. Suaya JA, Fletcher MA, Georgalis L, et al. Identification of Streptococcus pneumoniae in hospital-acquired pneumonia in adults. J Hosp Infect. 2021;108:146–157. doi:10.1016/j.jhin.2020.09.036
396. Sender V, Hentrich K, Henriques-Normark B. Virus-Induced Changes of the Respiratory Tract Environment Promote Secondary Infections With Streptococcus pneumoniae. Front Cell Infect Microbiol. 2021;11:643326. doi:10.3389/fcimb.2021.643326
397. Nucci LA, Santos SS, Brunialti MK, et al. Expression of genes belonging to the interacting TLR cascades, NADPH-oxidase and mitochondrial oxidative phosphorylation in septic patients. PLoS One. 2017;12(2):e0172024. doi:10.1371/journal.pone.0172024
398. García-Laorden M. Influence of genetic variability at the surfactant proteins A and D in community-acquired pneumonia: a prospective, observational, genetic study. Crit Care. 2011;15(1):R57. doi:10.1186/cc10030
399. Saleh NY, Ibrahem RAL, Saleh AAH, Soliman SES, Mahmoud AAS. Surfactant protein D: a predictor for severity of community-acquired pneumonia in children. Pediatr Res. 2022;91(3):665–671. doi:10.1038/s41390-021-01492-9
400. Liu J, Zheng X, Tong Q, et al. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-nCoV. J Med Virol. 2020;92(5):491–494. doi:10.1002/jmv.25709
401. Vassiliou AG, Keskinidou C, Jahaj E, et al. ICU Admission Levels of Endothelial Biomarkers as Predictors of Mortality in Critically Ill COVID-19 Patients. Cells. 2021;10(1):87. doi:10.3390/cells10010186
402. Vassiliou AG, Zacharis A, Keskinidou C, et al. Soluble Angiotensin Converting Enzyme 2 (ACE2) Is Upregulated and Soluble Endothelial Nitric Oxide Synthase (eNOS) Is Downregulated in COVID-19-induced Acute Respiratory Distress Syndrome (ARDS). Pharmaceuticals. 2021;14(7):25. doi:10.3390/ph14070695
403. Keskinidou C, Vassiliou AG, Zacharis A, et al. Endothelial, Immunothrombotic, and Inflammatory Biomarkers in the Risk of Mortality in Critically Ill COVID-19 Patients: the Role of Dexamethasone. Diagnostics. 2021;11(7):53. doi:10.3390/diagnostics11010053
404. Leisman DE, Ronner L, Pinotti R, et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir Med. 2020;8(12):1233–1244.
405. Vassiliou AG, Dimopoulou I, Jahaj E, et al. Selection of the Appropriate Control Group Is Essential in Evaluating the Cytokine Storm in COVID-19. In vivo. 2021;35(2):1295–1298. doi:10.21873/invivo.12381
406. Deshmukh V, Motwani R, Kumar A, Kumari C, Raza K. Histopathological observations in COVID-19: a systematic review. J Clin Pathol. 2021;74(2):76. doi:10.1136/jclinpath-2020-206995
407. Martines RB, Ritter JM, Matkovic E, et al. Pathology and Pathogenesis of SARS-CoV-2 Associated with Fatal Coronavirus Disease, United States. Emerg Infect Dis. 2020;26(9):2005–2015. doi:10.3201/eid2609.202095
408. Gibson PG, Qin L, Puah SH. COVID-19 acute respiratory distress syndrome (ARDS): clinical features and differences from typical pre-COVID-19 ARDS. Med J Aust. 2020;213(2):54–56 e51. doi:10.5694/mja2.50674
409. Violi F, Oliva A, Cangemi R, et al. Nox2 activation in Covid-19. Redox Biol. 2020;36:101655. doi:10.1016/j.redox.2020.101655
410. Damiano S, Sozio C, La Rosa G, Santillo M. NOX-Dependent Signaling Dysregulation in Severe COVID-19: clues to Effective Treatments. Front Cell Infect Microbiol. 2020;10:608435. doi:10.3389/fcimb.2020.608435
411. Cuadrado A, Pajares M, Benito C, et al. Can Activation of NRF2 Be a Strategy against COVID-19? Trends in pharmacological sciences. Sep. 2020;41(9):598–610.
412. Emanuele S, Celesia A, D’Anneo A, et al. The Good and Bad of Nrf2: an Update in Cancer and New Perspectives in COVID-19. Int J Mol Sci. 2021;22:15. doi:10.3390/ijms22157963
413. McCord JM, Hybertson BM, Cota-Gomez A, Gao B. Nrf2 activator PB125® as a carnosic acid-based therapeutic agent against respiratory viral diseases, including COVID-19. Free Radic Biol Med. 2021;175:56–64. doi:10.1016/j.freeradbiomed.2021.05.033
414. Singh E, Matada GSP, Abbas N, Dhiwar PS, Ghara A, Das A. Management of COVID-19-induced -cytokine -storm by Keap1-Nrf2 system: a review. Inflammopharmacology. 2021;29(5):1347–1355. doi:10.1007/s10787-021-00860-5
415. Olagnier D, Farahani E, Thyrsted J, et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat Commun. 2020;11(1):4938. doi:10.1038/s41467-020-18764-3
416. Arcanjo A, Logullo J, Menezes CCB, et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep. 2020;10(1):19630. doi:10.1038/s41598-020-76781-0
417. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. 2020;217(6):54. doi:10.1084/jem.20200652
418. Jing H, Chen X, Zhang S, et al. Neutrophil extracellular traps (NETs): the role of inflammation and coagulation in COVID-19. Am J Transl Res. 2021;13(8):8575–8588.
419. Masso-Silva JA, Moshensky A, Lam MTY, et al. Increased peripheral blood neutrophil activation phenotypes and NETosis in critically ill COVID-19 patients: a case series and review of the literature. Clin Infect Dis. 2021;2:532.
420. Ouwendijk WJD, Raadsen MP, van Kampen JJA, et al. High Levels of Neutrophil Extracellular Traps Persist in the Lower Respiratory Tract of Critically Ill Patients With Coronavirus Disease 2019. J Infect Dis. 2021;223(9):1512–1521. doi:10.1093/infdis/jiab050
421. Teluguakula N. Neutrophils Set Extracellular Traps to Injure Lungs in Coronavirus Disease 2019. J Infect Dis. 2021;223(9):1503–1505. doi:10.1093/infdis/jiab053
422. Middleton EA, He XY, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–1179. doi:10.1182/blood.2020007008
423. Skendros P, Mitsios A, Chrysanthopoulou A, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. 2020;130(11):6151–6157. doi:10.1172/JCI141374
424. Tomar B, Anders HJ, Desai J, Mulay SR. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells. 2020;9(6):58. doi:10.3390/cells9061383
425. Yaqinuddin A, Kashir J. Novel therapeutic targets for SARS-CoV-2-induced acute lung injury: targeting a potential IL-1β/neutrophil extracellular traps feedback loop. Med Hypotheses. 2020;143:109906. doi:10.1016/j.mehy.2020.109906
426. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5(11):87.
427. Zuo Y, Yalavarthi S, Navaz SA, et al. Autoantibodies stabilize neutrophil extracellular traps in COVID-19. JCI Insight. 2021;6(15):87.
428. Fisher J, Mohanty T, Karlsson CAQ, et al. Proteome Profiling of Recombinant DNase Therapy in Reducing NETs and Aiding Recovery in COVID-19 Patients. Mol Cell Proteom. 2021;20:100113. doi:10.1016/j.mcpro.2021.100113
429. Hazeldine J, Lord JM. Neutrophils and COVID-19: active Participants and Rational Therapeutic Targets. Front Immunol. 2021;12:680134. doi:10.3389/fimmu.2021.680134
430. Holliday ZM, Earhart AP, Alnijoumi MM, Krvavac A, Allen LH, Schrum AG. Non-Randomized Trial of Dornase Alfa for Acute Respiratory Distress Syndrome Secondary to Covid-19. Front Immunol. 2021;12:714833. doi:10.3389/fimmu.2021.714833
431. Weber AG, Chau AS, Egeblad M, Barnes BJ, Janowitz T. Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: a case series. Mol Med. 2020;26(1):91. doi:10.1186/s10020-020-00215-w
432. Goel S, Saheb Sharif-Askari F, Saheb Sharif Askari N, et al. SARS-CoV-2 Switches ‘on’ MAPK and NFκB Signaling via the Reduction of Nuclear DUSP1 and DUSP5 Expression. Frontiers in Pharmacology. 2021;12. doi:10.3389/fphar.2021.631879
433. Grimes JM, Grimes KV. p38 MAPK inhibition: a promising therapeutic approach for COVID-19. J Mol Cell Cardiol. 2020;144:63–65. doi:10.1016/j.yjmcc.2020.05.007
434. Roy RK, Sharma U, Wasson MK, Jain A, Hassan MI, Prakash H. Macrophage Activation Syndrome and COVID 19: impact of MAPK Driven Immune-Epigenetic Programming by SARS-Cov-2. Front Immunol. 2021;12:763313. doi:10.3389/fimmu.2021.763313
435. Shahgolzari M, Yavari A, Arjeini Y, et al. Immunopathology and Immunopathogenesis of COVID-19, what we know and what we should learn. Gene Reports. 2021;25:101417. doi:10.1016/j.genrep.2021.101417
436. Horie S, McNicholas B, Rezoagli E, et al. Emerging pharmacological therapies for ARDS: COVID-19 and beyond. Intensive Care Med. 2020;46(12):2265–2283. doi:10.1007/s00134-020-06141-z
437. Wu YP, Liu ZH, Wei R, et al. Elevated plasma surfactant protein D (SP-D) levels and a direct correlation with anti-severe acute respiratory syndrome coronavirus-specific IgG antibody in SARS patients. Scand J Immunol. 2009;69(6):508–515. doi:10.1111/j.1365-3083.2009.02245.x
438. Islam ABMMK, Khan M-A-A-K. Lung transcriptome of a COVID-19 patient and systems biology predictions suggest impaired surfactant production which may be druggable by surfactant therapy. Sci Rep. 2020;10(1):19395. doi:10.1038/s41598-020-76404-8
439. Avdeev SN, Trushenko NV, Chikina SY, et al. Beneficial effects of inhaled surfactant in patients with COVID-19-associated acute respiratory distress syndrome. Respir Med. 2021;185:106489. doi:10.1016/j.rmed.2021.106489
440. Heching M, Lev S, Shitenberg D, Dicker D, Kramer MR. Surfactant for the Treatment of ARDS in a Patient With COVID-19. Chest. 2021;160(1):e9–e12. doi:10.1016/j.chest.2021.01.028
441. Piva S, DiBlasi RM, Slee AE, et al. Surfactant therapy for COVID-19 related ARDS: a retrospective case–control pilot study. Respir Res. 2021;22(1):20. doi:10.1186/s12931-020-01603-w
442. Bhatt RM, Clark HW, Girardis M, Busani S. Exogenous pulmonary surfactant in COVID-19 ARDS. The similarities to neonatal RDS suggest a new scenario for an ‘old’ strategy. BMJ Open Respir Res. 2021;8(1):87. doi:10.1136/bmjresp-2020-000867
443. Kryvenko V, Vadász I. Molecular mechanisms of Na,K-ATPase dysregulation driving alveolar epithelial barrier failure in severe COVID-19. Am J Physiol Lung Cell Mol Physiol. 2021;320(6):L1186–L1193. doi:10.1152/ajplung.00056.2021
444. Mariajoseph-Antony LF, Kannan A, Panneerselvam A, Loganathan C, Anbarasu K, Prahalathan C. Could aquaporin modulators be employed as prospective drugs for COVID-19 related pulmonary comorbidity? Med Hypotheses. 2020;143:110201. doi:10.1016/j.mehy.2020.110201
445. Hernández-Beeftink T, Guillen-Guio B, Villar J, Flores C. Genomics and the Acute Respiratory Distress Syndrome: current and Future Directions. Int J Mol Sci. 2019;20:16. doi:10.3390/ijms20164004
446. Bime C, Pouladi N, Sammani S, et al. Genome-Wide Association Study in African Americans with Acute Respiratory Distress Syndrome Identifies the Selectin P Ligand Gene as a Risk Factor. Am J Respir Crit Care Med. 2018;197(11):1421–1432. doi:10.1164/rccm.201705-0961OC
447. Christie JD, Wurfel MM, Feng R, et al. Genome wide association identifies PPFIA1 as a candidate gene for acute lung injury risk following major trauma. PLoS One. 2012;7(1):e28268. doi:10.1371/journal.pone.0028268
448. Du M, Garcia JGN, Christie JD, et al. Integrative omics provide biological and clinical insights into acute respiratory distress syndrome. Intensive Care Med. 2021;47(7):761–771. doi:10.1007/s00134-021-06410-5
449. Guillen-Guio B, Lorenzo-Salazar JM, Ma SF, et al. Sepsis-associated acute respiratory distress syndrome in individuals of European ancestry: a genome-wide association study. Lancet Respir Med. 2020;8(3):258–266. doi:10.1016/S2213-2600(19)30368-6
450. Lee S, Emond MJ, Bamshad MJ, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91(2):224–237. doi:10.1016/j.ajhg.2012.06.007
451. Shortt K, Chaudhary S, Grigoryev D, et al. Identification of novel single nucleotide polymorphisms associated with acute respiratory distress syndrome by exome-seq. PLoS One. 2014;9(11):e111953. doi:10.1371/journal.pone.0111953
452. Lynn H, Sun X, Casanova N, Gonzales-Garay M, Bime C, Garcia JGN. Genomic and Genetic Approaches to Deciphering Acute Respiratory Distress Syndrome Risk and Mortality. Antioxid Redox Signal. 2019;31(14):1027–1052. doi:10.1089/ars.2018.7701
453. Kovach MA, Stringer KA, Bunting R, et al. Microarray analysis identifies IL-1 receptor type 2 as a novel candidate biomarker in patients with acute respiratory distress syndrome. Respir Res. 2015;16(1):29. doi:10.1186/s12931-015-0190-x
454. Meyer NJ. Beyond single-nucleotide polymorphisms: genetics, genomics, and other ‘omic approaches to acute respiratory distress syndrome. Clin Chest Med. 2014;35(4):673–684. doi:10.1016/j.ccm.2014.08.006
455. Giannini HM, Meyer NJ. Genetics of Acute Respiratory Distress Syndrome: pathways to Precision. Crit Care Clin. 2021;37(4):817–834. doi:10.1016/j.ccc.2021.05.006
456. Lv X, Zhang Y, Lu W, et al. Digital gene expression analysis of transcriptomes in lipopolysaccharide-induced acute respiratory distress syndrome. Clin Chim Acta. 2016;453:182–189. doi:10.1016/j.cca.2015.07.018
457. Wang M, Yan J, He X, Zhong Q, Zhan C, Li S. Candidate genes and pathogenesis investigation for sepsis-related acute respiratory distress syndrome based on gene expression profile. Biol Res. 2016;49:25. doi:10.1186/s40659-016-0085-4
458. Cao Y, Lyu YI, Tang J, MicroRNAs: LY. Novel regulatory molecules in acute lung injury/acute respiratory distress syndrome. Biomed Rep. 2016;4(5):523–527. doi:10.3892/br.2016.620
459. Guo W, Wang Z, Wang S, Liao X, Qin T. Transcriptome sequencing reveals differential expression of circRNAs in sepsis induced acute respiratory distress syndrome. Life Sci. 2021;278:119566. doi:10.1016/j.lfs.2021.119566
460. Hurskainen M, Mižíková I, Cook DP, et al. Single cell transcriptomic analysis of murine lung development on hyperoxia-induced damage. Nat Commun. 2021;12(1):1565. doi:10.1038/s41467-021-21865-2
461. Jiang Y, Rosborough BR, Chen J, et al. Single cell RNA sequencing identifies an early monocyte gene signature in acute respiratory distress syndrome. JCI Insight. 2020;5(13):87. doi:10.1172/jci.insight.135678
462. Riemondy KA, Jansing NL, Jiang P, et al. Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury. JCI Insight. 2019;5(8):287.
463. Grigoryev DN, Cheranova DI, Chaudhary S, Heruth DP, Zhang LQ, Ye SQ. Identification of new biomarkers for Acute Respiratory Distress Syndrome by expression-based genome-wide association study. BMC Pulm Med. 2015;15:95. doi:10.1186/s12890-015-0088-x
464. Zhang S, Wu Z, Xie J, Yang Y, Wang L, Qiu H. DNA methylation exploration for ARDS: a multi-omics and multi-microarray interrelated analysis. J Transl Med. 2019;17(1):345. doi:10.1186/s12967-019-2090-1
465. Wen XP, Zhang YZ, Wan QQ. Non-targeted proteomics of acute respiratory distress syndrome: clinical and research applications. Proteome Sci. 2021;19(1):5. doi:10.1186/s12953-021-00174-y
466. Gao Y, Li X, Gao J, et al. Metabolomic Analysis of Radiation-Induced Lung Injury in Rats: the Potential Radioprotective Role of Taurine. Dose Response. 2019;17(4):1559325819883479. doi:10.1177/1559325819883479
467. Stringer KA, McKay RT, Karnovsky A, Quémerais B, Lacy P. Metabolomics and Its Application to Acute Lung Diseases. Front Immunol. 2016;7:44. doi:10.3389/fimmu.2016.00044
468. Long Y, Zhang Y, Gong Y, et al. Diagnosis of Sepsis with Cell-free DNA by Next-Generation Sequencing Technology in ICU Patients. Arch Med Res. 2016;47(5):365–371. doi:10.1016/j.arcmed.2016.08.004
469. Zhang XJ, Zheng JY, Li X, Liang YJ, Zhang ZD. Usefulness of metagenomic next-generation sequencing in adenovirus 7-induced acute respiratory distress syndrome: A case report. World journal of clinical cases. 2021;9(21):6067–6072.
470. Fischer N, Rohde H, Indenbirken D, et al. Rapid metagenomic diagnostics for suspected outbreak of severe pneumonia. Emerging infectious diseases. 2014;20(6):1072-1075.
471. Zhang P, Chen Y, Li S, et al. Metagenomic next-generation sequencing for the clinical diagnosis and prognosis of acute respiratory distress syndrome caused by severe pneumonia: a retrospective study. PeerJ. 2020;8:e9623.
472. Liao SY, Casanova NG, Bime C, Camp SM, Lynn H, Garcia JGN. Identification of early and intermediate biomarkers for ARDS mortality by multi-omic approaches. Scientific reports. 2021;11(1):18874.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.