")
Back to Journals » International Journal of Nanomedicine » Volume 11
Mechanistic analysis of Zein nanoparticles/PLGA triblock in situ forming implants for glimepiride
Authors Ahmed OAA , Zidan A, Khayat M
Received 3 November 2015
Accepted for publication 17 December 2015
Published 3 February 2016 Volume 2016:11 Pages 543—555
DOI https://doi.org/10.2147/IJN.S99731
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Osama Abdelhakim Aly Ahmed,1,2 Ahmed Samir Zidan,1,3 Maan Khayat4
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Saudi Arabia; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Minia University, Minia, Egypt; 3Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Zagazig University, Zagazig, Egypt; 4Department of Pharmaceutical Chemistry, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Saudi Arabia
Objectives: The study aims at applying pharmaceutical nanotechnology and D-optimal fractional factorial design to screen and optimize the high-risk variables affecting the performance of a complex drug delivery system consisting of glimepiride–Zein nanoparticles and inclusion of the optimized formula with thermoresponsive triblock copolymers in in situ gel.
Methods: Sixteen nanoparticle formulations were prepared by liquid–liquid phase separation method according to the D-optimal fractional factorial design encompassing five variables at two levels. The responses investigated were glimepiride entrapment capacity (EC), particle size and size distribution, zeta potential, and in vitro drug release from the prepared nanoparticles. Furthermore, the feasibility of embedding the optimized Zein-based glimepiride nanoparticles within thermoresponsive triblock copolymers poly(lactide-co-glycolide)-block-poly(ethylene glycol)-block-poly(lactide-co-glycolide) in in situ gel was evaluated for controlling glimepiride release rate.
Results: Through the systematic optimization phase, improvement of glimepiride EC of 33.6%, nanoparticle size of 120.9 nm with a skewness value of 0.2, zeta potential of 11.1 mV, and sustained release features of 3.3% and 17.3% drug released after 2 and 24 hours, respectively, were obtained. These desirability functions were obtained at Zein and glimepiride loadings of 50 and 75 mg, respectively, utilizing didodecyldimethylammonium bromide as a stabilizer at 0.1% and 90% ethanol as a common solvent. Moreover, incorporating this optimized formulation in triblock copolymers-based in situ gel demonstrated pseudoplastic behavior with reduction of drug release rate as the concentration of polymer increased.
Conclusion: This approach to control the release of glimepiride using Zein nanoparticles/triblock copolymers-based in situ gel forming intramuscular implants could be useful for improving diabetes treatment effectiveness.
Keywords: glimepiride, Zein, nanoparticles, quality by design, in situ implants
Introduction
Both types of diabetes mellitus can be a lead cause for blindness, kidney failure, neuropathic diseases, foot ulceration, gas gangrene in lower limbs, foot amputations, and cardiovascular insufficiency such as ischemia, stroke, etc.1 Strict control of blood glucose would be the most effective approach to prevent and reduce the occurrence and progression of these manifestations of diabetic microangiopathy. Glimepiride provides better chance of glycemic control by stimulating insulin release from β islets of Langerhans of the pancreas and by boosting the sensitivity of peripheral receptors to endogenous insulin.2 It also facilitates glucose transport from the blood into the peripheral tissues for energy consumption.3 Glimepiride is practically insoluble in water, with a maximum solubility not exceeding 0.00027 mg/mL, and hence is considered as a Biopharmaceutics Classification System Class II drug.4 Following its oral administration, low and irregular bioavailability was shown due to its low aqueous solubility. The molecular weight of glimepiride is 490.616 g/mol, with an octanol/water partition coefficient of 3.5. It has a short half-life of ~5 hours due to the extensive hepatic oxidative metabolism to its major metabolite, cyclohexylhydroxymethyl derivative (M1).5 This intrinsic criterion following oral administration makes it a good candidate for extended release via intramuscular or subcutaneous implantation. In an attempt to overcome these drawbacks, Bhulli and Sharma6 proposed an ethosomal long-acting transdermal formulation for glimepiride with 42%–78% entrapment efficiency, optimal nanometric size range, low polydispersity index, and high permeability and flux through rat skin. However, the ethosomal components loosened the skin through a dissociation of hydrogen bonding network of ceramides within lipid bilayers of the stratum corneum. Other transdermal formulations with high surfactant concentrations were also proposed in the literature to facilitate the percutaneous permeation of glimepiride.7,8 Nevertheless, the higher surface activity induced by the surfactants would contribute to undesirable dermal sensitivity, irritation, and toxicity. For example, long-term surfactant exposure to the skin might cause protein denaturation and swelling of stratum corneum, solubilization of fluid lipids and abstraction of calcium ions to reduce corneocyte adhesion, disorganization of skin lipids, and maturation of keratinocytes and Langerhans cells.9
To decrease the frequency of glimepiride administration, several studies have been proposed to demonstrate the potential application of biodegradable polymers as sustained-release carriers. Polylactic acid and its copolymers with polyglycolic acid and poly(D,L-lactide-co-glycolide) (PLGA) have been widely utilized as excipients for controlled release of injectable drugs. These polymers are formulated as solids, gels, or liquid forms. PLGA-based solid biodegradable microparticles have been proposed as depot-forming delivery systems of small-molecule active ingredients, peptides, and proteins.10 Because of PLGA-based solid biodegradable microparticles coarse size, they require not only a complex aseptic manufacturing process but also a carrier suspension as well for sterilization of these injectable microparticles. On the other hand, using these formulations in a gel or liquid form would require not only fewer manufacturing steps but terminal filter sterilization as well.11 An in situ gel-forming formulation usually consists of a dispersion medium to dissolve and/or disperse the polymeric fraction and/or the drug. This system exists in a liquid form at temperatures lower than the body temperature but is changed to gel form when injected intramuscularly or subcutaneously to form a depot for a controlled delivery of the incorporated drug. The thermoresponsive triblock copolymers (PLGA–polyethylene glycol [PEG]–PLGA copolymers) of PLGA (A-block) and PEG (B-block) are the most attractive in situ gel-forming agents due to their biodegradable and safety profile.12 Even though the entrapment of hydrophilic drugs as well as tailoring the polymer blocks chain length have been investigated to control drug release, formulation combinations of thermoresponsive triblock copolymers (PLGA–PEG–PLGA copolymers) have not been thoroughly investigated. In this study, an investigation of the embedment of Zein-based glimepiride nanoparticles within PLGA–PEG–PLGA copolymers was proposed. Zein is a corn prolamin composed of a group of amino acids with a large proportion of proline, leucine, glutamine, and alanine. Zein is a hydrophobic material and only soluble in ethanol at concentrations over 70%.13 Zein is capable of self-association in water to encapsulate both hydrophilic and hydrophobic species.14 Hence, it was employed in this study along with D-optimal fractional factorial design to screen and optimize the high-risk variables affecting the performance of glimepiride-loaded Zein-based nanoparticles. The responses investigated were glimepiride entrapment capacity (EC), particle size and size distribution, zeta potential, and in vitro drug release from the prepared nanoparticles. Furthermore, the feasibility of embedding the optimized Zein-based glimepiride nanoparticles within PLGA–PEG–PLGA copolymers was evaluated for controlling glimepiride release rate.
Materials and methods
Materials
Glimepiride was kindly supplied by Spimaco Addwaeih, Riyadh, Saudi Arabia. Zein, PLGA (50:50, molecular weight of 38,000–54,000 g/mol), ethanol, methanol, dichloromethane (DCM), sodium lauryl sulfate (SLS), didodecyldimethylammonium bromide (DDAB), stannous 2-ethylhexanoate, PEG 1500, and N-methyl-2-pyrrolidone (NMP) were purchased from Sigma Aldrich Corporation, St Louis, MO, USA. All other chemicals were of analytical grade and were used as received.
Formulation of glimepiride-loaded Zein nanoparticles
Sixteen experimental runs of glimepiride-loaded Zein-based nanoparticle formulations were prepared by liquid–liquid phase separation method as described by Hashem et al15 with slight modification (Table 1). The specified amounts of glimepiride and Zein were dissolved in 3 mL DCM and 9 mL 90% v/v ethanol in water, respectively. The two solutions were homogenized using a probe sonicator (VCX 750; 750 watts; Sonics and Materials Inc., Newtown, CT, USA) for 5 minutes at a controlled temperature of 10°C to prevent protein denaturation. The obtained emulsion was then added dropwise to 20 mL phosphate-buffered saline (pH 7.2) containing a specified concentration and type of stabilizer, as listed in Table 1, while stirring at 2,000 rpm at room temperature for 3 hours, followed by evaporation overnight under reduced pressure using rotary evaporator at room temperature till complete ethanol evaporation. The nanodispersion was centrifuged for 60 minutes at 20,000 rpm to harvest the prepared nanoparticles. The formed residue was then freeze-dried for 72 hours (alpha 1–2 LD plus freeze dryer with a condenser temperature of −55°C; Martin Christ Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany) using mannitol as a cryoprotectant.
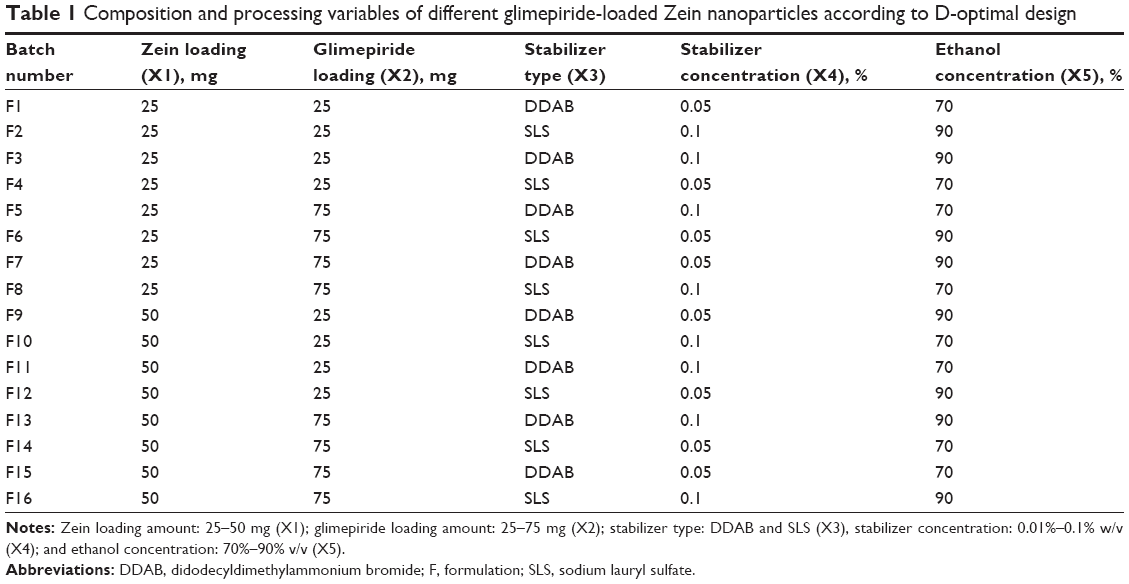 | Table 1 Composition and processing variables of different glimepiride-loaded Zein nanoparticles according to D-optimal design |
Experimental design
The D-optimal experimental design was constructed using the response surface methodology procedure in the Statistical Analysis System (JMP statistical discovery, version 11.1.1; SAS Institute Inc., Cary, NC, USA). The single and interactive effects of five different factors on glimepiride EC, particle size and size distribution, zeta potential, and in vitro drug release from the prepared nanoparticles were investigated. The tested factors were Zein loading amount (X1; 25–50 mg), glimepiride loading amount (X2; 25–75 mg), stabilizer type (X3; DDAB and SLS), stabilizer concentration (X4; 0.01%–0.1% w/v), and ethanol concentration (X5; 70%–90% v/v). The D-optimal design generated 16 nanoparticle trials consisting of various combinations of five factors in random order (Table 1). The selection of levels for independent variables was based on the preliminary risk assessment study as well as a review of the relevant literature. Optimization process was employed using a generalized desirability function to maximize glimepiride EC, zeta potential while minimizing particle size, skewness of size distribution, and drug release percentages. The optimized formulation was then incorporated into the PLGA–PEG–PLGA thermoresponsive triblock copolymers for in situ gel formation.
Formulation of glimepiride-loaded PLGA–PEG–PLGA in situ gel
PLGA–PEG–PLGA triblock copolymers were prepared by ring-opening pathway as described by Zentner et al16 with slight modification. PEG 1500 was added to a chemical reactor and heated for 2 hours at 150°C under vacuum. The PLGA was then added while heating at 150°C under vacuum for 30 minutes. Stannous 2-ethylhexanoate as catalyst was then added while heating at 160°C for 11 hours under vacuum. The precipitated copolymers were then dissolved in cold water (4°C) to remove any water-soluble impurities and then heated again to 80°C to allow solidification. This purification step was repeated three times and the purified triblock copolymer was then kept dried at 37°C for further experimentation.
The optimized Zein-based glimepiride nanoparticles were then embedded within PLGA–PEG–PLGA copolymers using the following procedure. The dried copolymer was dissolved in NMP to prepare three polymeric solutions of 10%, 20%, and 30% w/v. In particular, the sealed vial containing a known amount of copolymer and 3 mL solvent was placed in a shaker water bath overnight at 37°C till complete dissolution. The optimized freeze-dried Zein-based glimepiride nanoparticles were then added to the polymeric solutions and mixed by homogenization at 8,000 rpm for 2 minutes. All formulations were prepared with glimepiride loading equivalent to 50 mg and were readily injectable through 21-gauge needle.
Characterization of glimepiride-loaded Zein nanoparticles
EC was determined directly by destroying the nanoparticles, followed by aqueous extraction of entrapped drug per unit mass of each nanoparticle formulation. In brief, 20 mg of the freeze-dried nanoparticles were dissolved in 10 mL of 90% v/v ethanol in water at 45°C. Probe sonication was used as needed to break down any aggregates and lumps that may be formed until a clear solution was obtained. After appropriate dilution with the mobile phase, glimepiride content in this solution was assessed utilizing a developed and validated in-house high-performance liquid chromatography analytical method. A high-performance liquid chromatography instrument (Agilent Technologies, Santa Clara, CA, USA) with HP 1200 UV detector set at 238 nm wavelength was used. The chromatographic separation was carried out by injecting 10 μL sample into RP-18 Luna2 (250×4.6 mm, 5 μm packing) column (Phenomenex Inc., Torrance, CA, USA). Acetonitrile–phosphate buffer 0.01 M (pH 3.5; 45:55, v/v) with a flow rate of 1.0 mL/minute was used as the mobile phase. The detected mass of glimepiride (Cm) loaded within unit mass of Zein matrix (Ct) was used to express the EC according to the following equation:
|
|
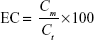
Particle size and electrical properties of the prepared nanoparticles were determined by dynamic light scattering using a Zetatrac analyzer (Microtrac Inc., Montgomerville, PA, USA) after appropriate sample dilution with distilled water. Release of glimepiride from Zein nanoparticles was studied utilizing automated Franz diffusion cell apparatus (MicroettePlus; Hanson Research, Chatsworth, CA, USA) with 1.76 cm2 diffusion area and 7 mL receptor chamber volume. Freeze-dried nanoparticles were reconstituted with phosphate-buffered saline (pH 7.2) and the obtained dispersion was added to the donor chamber. A synthetic LoProdyne LP nylon membrane (0.2 μm pore size, 51–56 psi water bubble point, and 139.7–177.8 μm thickness; Pall Corporation, Port Washington, NY, USA) was mounted in between the donor and receptor chambers. The receptor medium was phosphate-buffered saline (pH 7.2) maintained at 37°C±0.5°C, with a stirring rate of 400 rpm. Aliquots were withdrawn at 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 hours by the autosampler and analyzed for glimepiride diffused using the previously mentioned in-house developed and validated analytical chromatographic method. Three replicates of each experiment were performed. The in vitro release data were then evaluated for the mechanism of drug release by using a nonlinear computer program (Scientist®, version 3; MicroMath Scientific Software, Saint Louis, MO, USA). The cumulative least square release data (>10% and up to 80%) over time was fit to different mathematical release models such as power law, zero order, first order, and Higuchi’s diffusion. The higher value of coefficient of determination (r2) would indicate a superiority of the release profile and mechanism fitting to the model.
Characterization of glimepiride-loaded PLGA–PEG–PLGA in situ gel
In vitro drug release studies from the prepared glimepiride-loaded PLGA–PEG–PLGA in situ gel were evaluated using a modified dialysis. For gel formation, 1 mL sample, equivalent to 15 mg glimepiride, of the homogenized triblock polymeric dispersion of glimepiride-loaded Zein nanoparticles was injected into a dialysis tube containing 10 mL of phosphate buffer solution pH 7.2. The dialysis tube (Spectra/Por 1; Spectrum Laboratories Inc., Rancho Dominguez, CA, USA) employed was a hydrophilic cellulose membrane with symmetric porosity and molecular weight cut-off of 6,000–8,000 Da. The dialysis tubes were hermetically sealed and inserted into the vessels of USP II dissolution apparatus. Phosphate buffer solution (450 mL, pH 7.2) was used as receptor medium at 37°C, with a rotational speed maintained at 100 rpm. Aliquots, 3 mL each, were withdrawn from the external medium at time intervals and replaced with the same volume of fresh medium. The samples were assayed for glimepiride content using the previously mentioned in-house developed and validated analytical chromatographic method. All the experiments were performed in triplicate, and the mean cumulative drug release ± SD were calculated.
The appearance, color, and homogeneity of the prepared triblock polymeric solutions were observed by visual observation. The viscosities were also evaluated using Brookfield DV-III Rheometer (Brookfield Engineering Laboratories Inc., Middleboro, MA, USA). Viscosity and rheological parameters were evaluated at varying shear rates with 20-second equilibration to allow for full recovery from the shear applied. The viscosity and rheological assessment was done at controlled temperature of 37°C to reflect the physiological condition. Syringeability was also assessed by determining the force needed to push the prepared triblock polymeric solutions through a 21-gauge needle using a Texture Analyzer (TA.XT Plus; Stable Micro Systems Ltd., Godalming, Surrey, UK) in the compression mode. The upper probe was set to move downward at constant speed of 1.0 mm/s with a constant down force of 0.3 N. The force displacement profiles of a 10 mm distance were then recorded and the corresponding area under the curve was used to assess the work of discharge. The rate of water diffusion into the triblock polymeric solutions for in situ gel formation was assessed using the following procedure. A transparent borosilicate glass cylinder (15 mm internal diameter) was used. Cellulose ester membrane (100 kD; Spectrum Laboratories Inc., Houston, TX, USA) was fit to one end and 1 mL sample was then added from the other side. The membrane side was immersed into 150 mL phosphate buffer solution pH 7.2 to the depth of 20 mm. The distance of water front diffused through the membrane to form a gel was determined at various time points of 0, 6, and 24 hours. The rate of water diffusion at each time point was then calculated by dividing the distance in mm by the time lapse in minutes.
Results and discussion
This investigation aimed at optimizing glimepiride-loaded Zein nanoparticles to be embedded in an in situ forming PLGA–PEG–PLGA triblock implant. The predefined critical quality attributes of the glimepiride-loaded Zein nanoparticles were glimepiride EC, particle size, and glimepiride release rate. Maximizing the EC of Zein matrix to glimepiride would allow for a reduction of manufacturing costs, dosage volume, and easiness of syringeability for in situ gel formation. Therefore, the first objective of this study was to screen and optimize the nanoparticle formulation factors using a D-optimal design for their effects to maximize EC while minimizing the produced size, size distribution, and drug release rate. In achieving this goal, minimum constraint was applied for EC to be more than 20% w/w glimepiride in Zein matrix, maximum constraint for size to be less than 100 nm, and to minimize the drug release rate. The second goal was to embed the optimized nanoparticle formulation within PLGA–PEG–PLGA triblock for in situ gel formation with acceptable gel characteristics. This would allow for an extended dosing interval as well as better patient compliance.
EC of glimepiride
Data in Table 2 show that glimepiride EC ranged from 15.7% (F8) to 62.6% (F10) for the different variable combinations of D-optimal design to prepare Zein nanoparticles. Using the full multiple regression equations, quantile–quantile correlation to regress the observed EC values versus the predicted one showed a linear relationship with a coefficient of multiple determination (R2) value of 0.9773 (Table 3). This step demonstrates the accuracy and robustness of the regression model to predict the EC within the investigated design space. The consequent analysis of variance demonstrated a significant prediction efficiency with Prob > F-value of 0.0003 and 0.0025 at P<0.05 and P<0.01, respectively (Table 3). After neglecting the nonsignificant factors, the following reduced prediction model was obtained to correlate the individual and interaction effects of the significant variables on glimepiride EC. Using the developed prediction model, the response surface plots, shown in Figure 1, would demonstrate the design space for predicting and subsequent monitoring of glimepiride entrapment within Zein nanoparticles. The reduced prediction equation of EC value is:
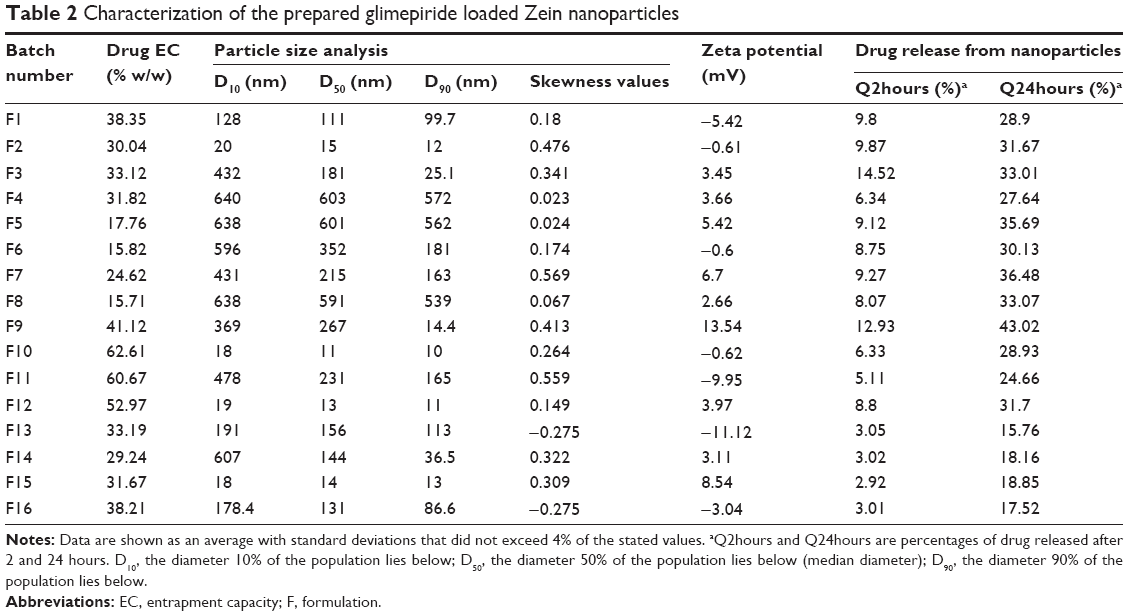 | Table 2 Characterization of the prepared glimepiride loaded Zein nanoparticles |
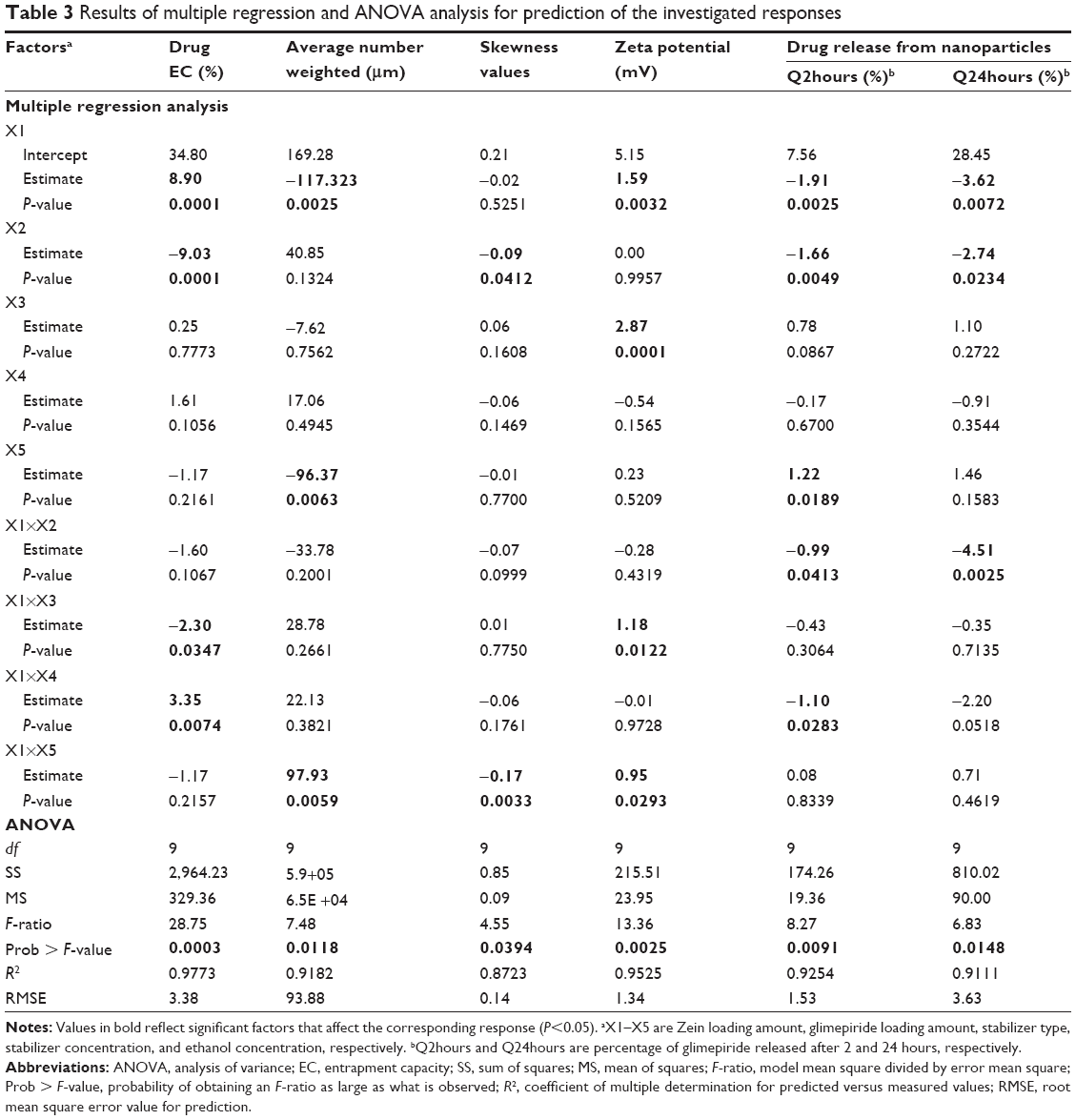 | Table 3 Results of multiple regression and ANOVA analysis for prediction of the investigated responses |
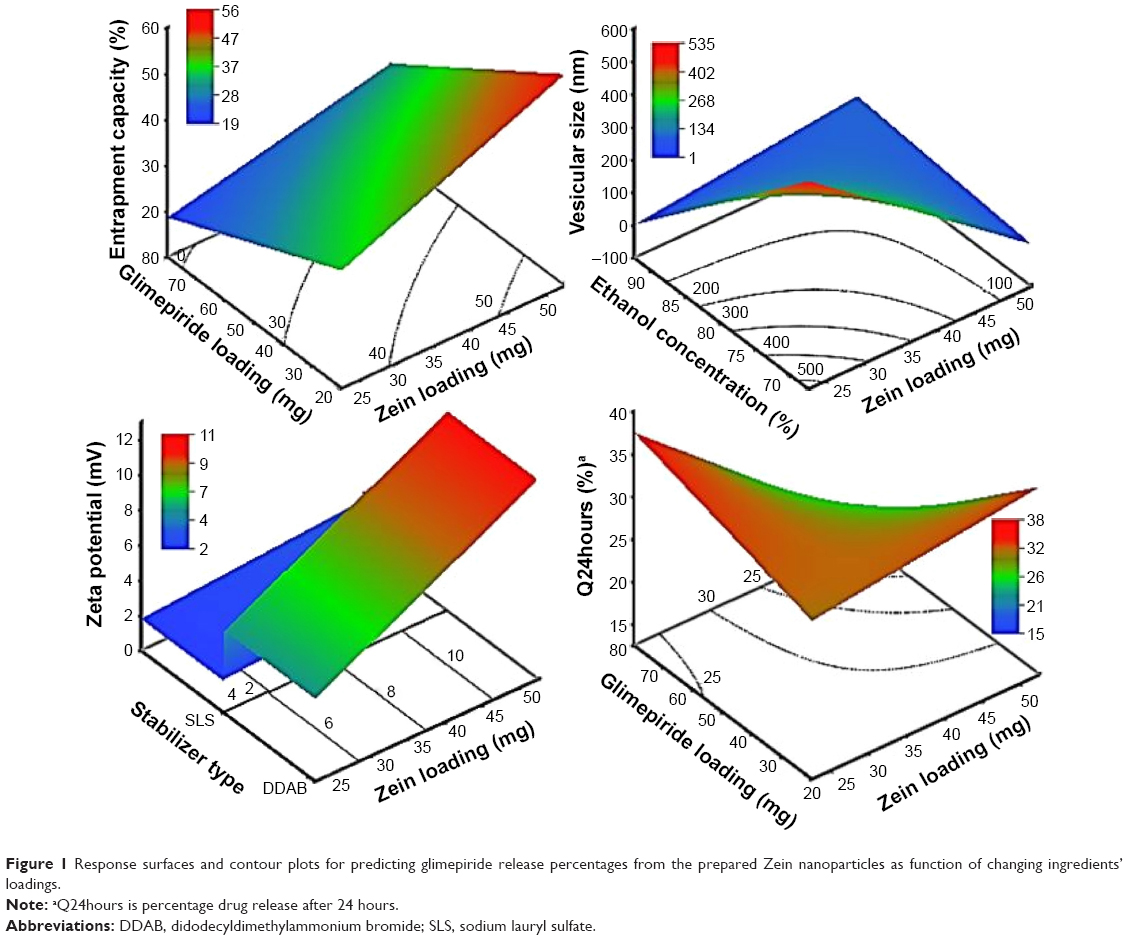 | Figure 1 Response surfaces and contour plots for predicting glimepiride release percentages from the prepared Zein nanoparticles as function of changing ingredients’ loadings. |
|
|
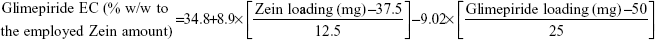
Out of five formulation and processing variables, Zein and glimepiride experimental loadings were significant for their influences on EC. Glimepiride working amounts had the most significant effect on the resultant EC with a t-ratio of −10.67, followed by Zein loading with a t-ratio of 10.52 (Figure 2 and Table 3). Positive and negative effects on EC were observed by increasing experimental Zein and drug experimental loadings (Figure 1). Maximum EC of 62.6% (F10) was obtained at 2:1 Zein to glimepiride weight ratio. In comparison with DDAB, a negative influence of SLS at low Zein loading on EC was observed for nanoparticle stabilization (Figure 1). However, the interaction profiler of Figure 3 showed that both stabilizers were comparable for their performance at higher Zein loading. Zein is composed of charged amino acids in high proportions. While coalescing at pH 7.2, protein molecules aggregated into smaller particles with decreased void spaces where the peptide chains became unfolded to expose more reactive sites for cross-linking. Therefore, the EC of glimepiride was higher than those obtained by Muthuselvi and Dhathathreyan17 for gitoxin into Zein nanoparticles. However, these EC values were in a good agreement with that obtained by Lai and Guo18 for the encapsulation of 5-fluorouracil into Zein nanoparticles. Hence, the unfolding of the protein chains at pH 7.2 accelerated the thiol–disulfide interchange reaction to enhance nanoparticle formation while inhibiting large aggregation. On the other hand, the negative effect of glimepiride of the resultant EC would be attributed to its association at the surfaces of the formed peptide aggregates. Hence, these weakly bound or adsorbed drug molecules to the relatively larger surface of nanoparticles would facilitate pore formation for leaching more drug molecules.18 The positive action of DDAB to retain glimepiride within the formed nanoparticles might be attributed to the packing of DDAB at the formed surfaces. Both the double-tailed structure of DDAB and its critical packing parameter of 0.5–1 would contribute to particle formation.19 Moreover, the high surface coverage of DDAB on the Zein nanoparticles would protect the particles and restrict the liberty of the glimepiride molecules to increase its EC.
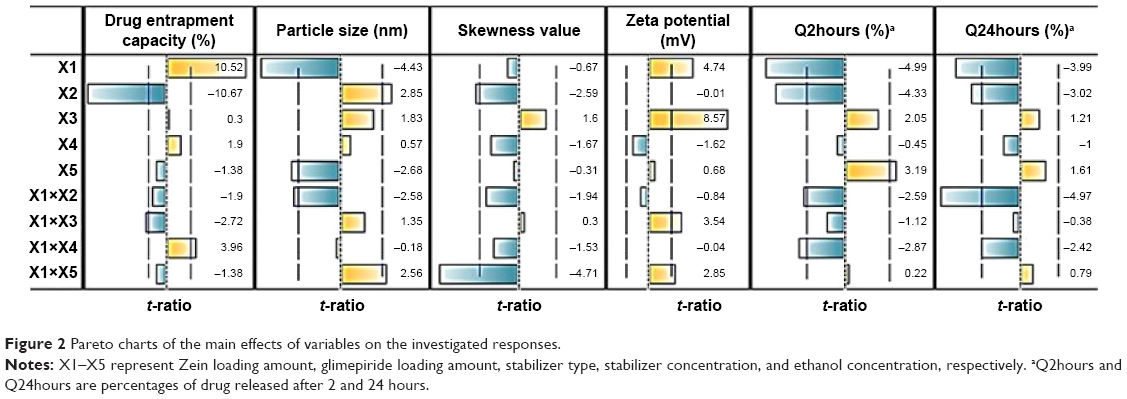 | Figure 2 Pareto charts of the main effects of variables on the investigated responses. |
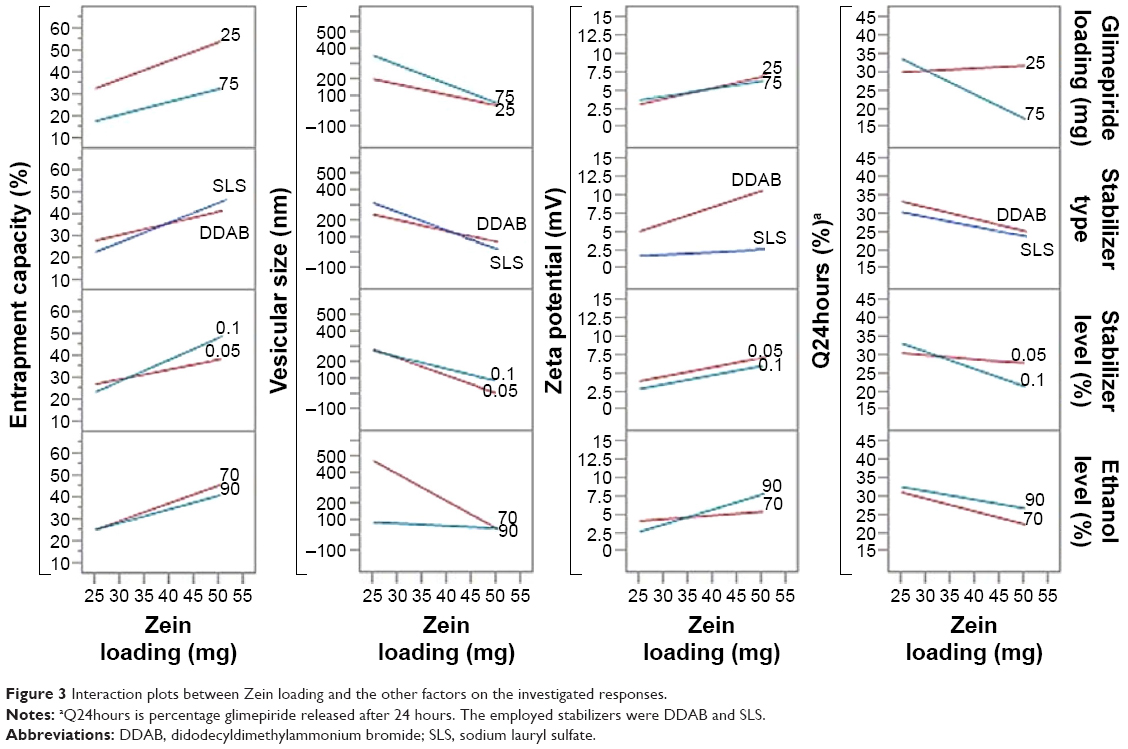 | Figure 3 Interaction plots between Zein loading and the other factors on the investigated responses. |
Nanoparticle size
Histograms of the particle size distribution of the prepared Zein nanoparticles showed a monodispersion in diameter. Table 2 lists the median nanoparticles size (D50), which ranged from 11 nm (F10) to 603 nm (F4) on changing both formulation and process variables investigated. On the other hand, skewness values for size distribution ranged from 0.023 (F4) to 0.56 (F7). Plotting the observed versus predicted values for nanoparticle sizes and skewness yielded linear relationships with R2 value of 0.9182 and 0.8723, respectively, to demonstrate that a prediction model can be constructed with an acceptable accuracy (Table 3). Analysis of variance (ANOVA) results revealed significant effects of Zein and glimepiride loadings and ethanol level on the sizes and skewness values with Prob > F-values of 0.0118 and 0.0394, respectively, P<0.05 (Table 3). The reduced prediction models to correlate individual and significant variables with the obtained sizes and corresponding skewness are shown below:
|
|
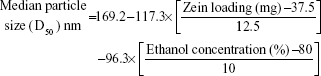
|
|

Experimental Zein loading was the most significant factor affecting the obtained size (P=0.0025), followed by ethanol level and its interaction time with Zein (Figure 2 and Table 3). Regarding the size distribution, glimepiride loading was more significant than Zein concentration with P-values of 0.0412 and 0.5251, respectively (Figure 2 and Table 3). Negative correlations were found on the resultant sizes and skewness values by increasing both Zein and ethanol loadings (Figure 1), with a lowest nanoparticle size obtained at 2:1 Zein to glimepiride weight ratio, 0.1% SLS as stabilizer, and 70% ethanol concentration (F10). A significant interaction existed between Zein loading and ethanol concentration to affect the resultant sizes (Figure 3). At low ethanol concentration, the nanoparticles sizes decreased as the amount of Zein increased, thus abolishing aggregation of Zein molecules and vice versa. This observation might be described by the unique structural configuration of hydrophobic and hydrophilic regions on the surface of Zein molecules. Studies of the orientation of Zein peptides in 70% ethanolic solution demonstrated a three-dimensional configuration with a high axial ratio.20 Hence, at low ethanol level, Zein molecules associated into an elongated prismlike shape with hydrophobic sides and hydrophilic tops and bottoms.21 This configuration would allow Zein molecules to associate in a side-by-side manner to form a large number of smaller nanoparticles rather than increasing the particle size. Despite being nonsignificant, the positive influence of glimepiride entrapment on the resultant sizes could be explained according to the law of mass action to entrap glimepiride molecules within Zein protein matrix.
Zeta potential
For the 16 formulations of D-optimal design, Table 2 lists the zeta potential values, which ranged from 0.61 mV (F2) to 13.54 mV (F9). The significant factors affecting the resultant zeta potentials were Zein loading and stabilizer employed, with a more predominant effect to stabilizer type (Table 3 and Figure 2). Multiple regression analysis showed positive values of both variables on the recorded zeta potentials, with a significant effect for their interaction term X1×X3. The extent of the positive charge on the formed Zein nanoparticles decreased from about +12.82 mV when using DDAB to about +2.84 mV when using SLS. This behavior was pronounced at both low and high Zein loadings. The adsorption of nonionic surfactants with its anionic impurities (such as free fatty acids) at the formed cationic Zein surfaces could reduce their charge.22 The contribution to increase the zeta potential of Zein nanoparticles dispersions would be suggestive of its stability due to the London dispersion forces. Hence, a maximized desirability function for zeta potential was applied to preserve the required electrostatic energies at nanoparticles’ surfaces. The regression equation revealed an acceptable predictability of zeta potential, with a P-value of 0.0025 and quantile–quantile correlation coefficient of 0.9525 (Table 3). The reduced prediction equation of zeta potential value is:
|
|
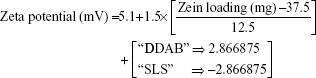
Glimepiride release
Glimepiride release profile of all 16 formulations of D-optimal design can be described as a two-step biphasic process, ie, an initial burst effect followed by subsequent slower release (Figure 4). At the initial stage, the burst release is usually ascribed to the free glimepiride or that embedded near the surface of the formed nanoparticles. The obtained release data fit to a logarithmic time-dependent release rather than Higuchi diffusion release (Figure 4). This behavior would be ascribed to the expanding transient boundary at the interface of either protein surface or DDAB-adsorbed layer and the aqueous medium.23 The following is the logarithmic equation to describe the drug release rate, where Qti is amount of cumulative drug released after time t in the aqueous medium and K is the release constant or the slope of the regressing Qti versus base-10 logarithm of time divided by 2.303.
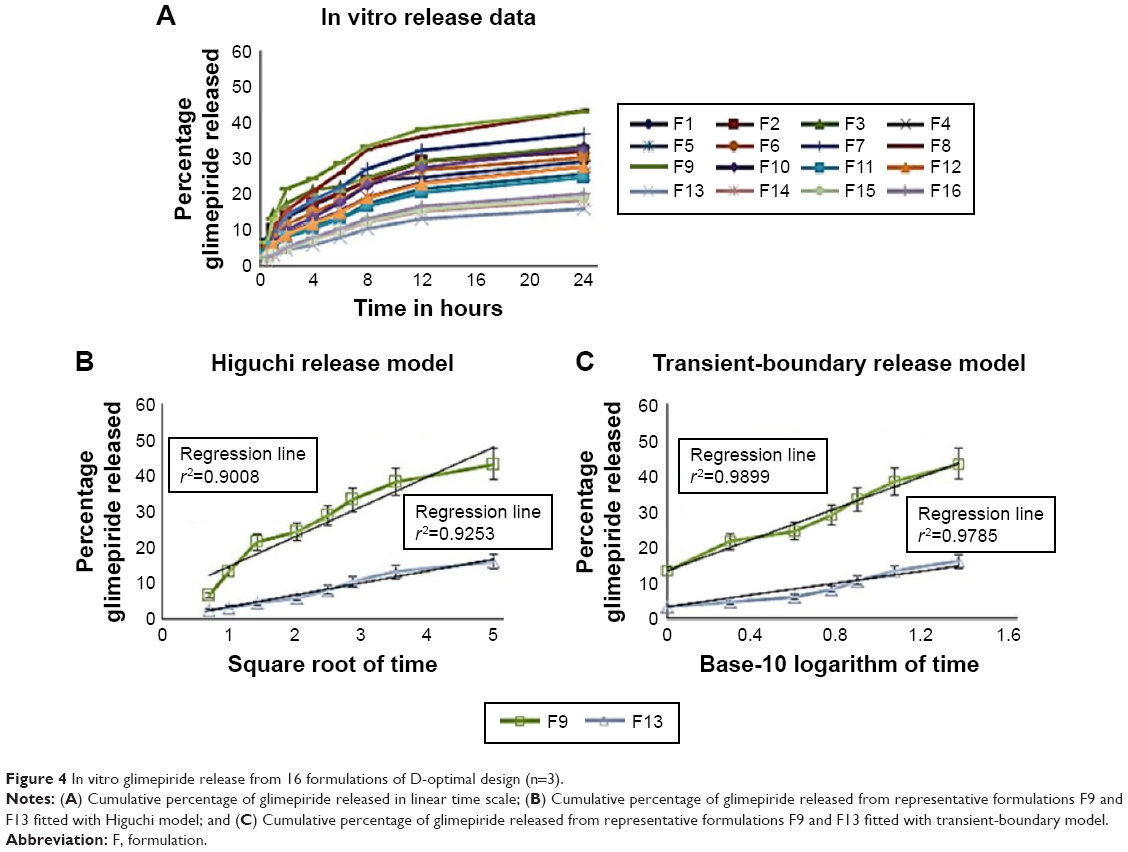 | Figure 4 In vitro glimepiride release from 16 formulations of D-optimal design (n=3). |
|
|
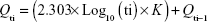
The cumulative percentages of glimepiride released after 2 hours (Q2hours) and 24 hours (Q24hours) for the 16 formulations varied from 3.02% (F16) to 14.52% (F3) and from 15.76% (F13) to 43.02% (F9), respectively (Table 2). A good correlation was established between the formulation/process parameters and glimepiride release with R2 values of 0.9254 and 0.9111 for Q2hours and Q24hours, respectively (Table 3). ANOVA results confirmed that the prediction capability was evident with a Prob > F-values of 0.0091 and 0.0148 for Q2hours and Q24hours, respectively, at P<0.05 (Table 3). More detailed effect analysis demonstrated that glimepiride release from Zein nanoparticles was predominantly affected by formulation design (Table 2 and Figure 1). Zein and glimepiride loadings significantly impacted these release parameters, with their interaction term (X1×X2) being the most important formulation parameter (P<0.05; Figures 2 and 3). Logically, higher Zein-to-glimepiride weight ratio in the nanoparticle solid state led to lower concentration of the solubilized drug (molecular state) in the transient layer, and consequently a lesser drug release. Particle size of the nanoparticle is also thought to impact the initial boundary layer thickness. A large size of the nanoparticle that is similar or greater than the thickness of the boundary may significantly increase the thickness of the initial boundary layer. Despite being insignificant for the prediction model, the employed stabilizer would likely affect glimepiride release by indirectly accelerating drug diffusion to the aqueous medium (Figure 2). The linear reduced model equations to predict both responses are given below:
|
|
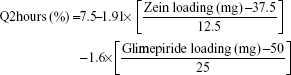
|
|
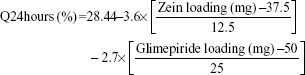
Optimization using desirability function
The optimization tool of JMP software has been selected based on a generalized desirability function that maximizes glimepiride EC and zeta potential while minimizing vesicular size and release parameters. The highest desirability was obtained at upper levels of both Zein and glimepiride loadings (50 and 75 mg, respectively), using DDAB as stabilizer at 0.1% and when the percentage of ethanol during processing was at its higher level of 90%. Under these optimization criteria, glimepiride EC of 33.6%, nanoparticle size of 120.9 nm with a skewness value of 0.2, zeta potential of 11.1 mV, Q2hours of 3.3%, and Q24hours of 17.3% were obtained. The proposed optimal conditions were then experimentally corroborated, and the results were closely correlated with the data predicted by the desirability function of the D-optimal design.
Characterization of the prepared in situ gels
Glimepiride-loaded in situ gels were prepared by incorporating the optimized Zein nanoparticles into PLGA–PEG–PLGA triblock polymer at 10%, 20%, and 30% w/v loadings. All polymeric solutions were yellowish in color, with the Zein nanoparticles suspended homogeneously with no signs of precipitations or agglomerations. The pH values of all solutions were in the range of 7.21±0.32, with no effect of nanoparticles incorporation. The viscosities of the gel formulations at 37°C are shown in Figure 5. For all samples, the apparent viscosities were higher at low shear rate than those at a high shear rate to demonstrate pseudoplastic behavior. Similar observations were observed by other researchers for ethylcellulose-based gels incorporating antimicrobial drugs.24 The significant interaction between both polymer chains and drug molecules at low temperature and the shearing rate explained this behavior. The flow behavior of the solutions was also studied by calculating the flow indices and viscosity coefficient (η) as a function of polymer concentration at 37°C (Figure 5). The flow index values of all formulations were more than 1 to confirm the non-Newtonian flow similar to that of the gel base. Regarding the viscosity coefficient (η), the amount of the polymer employed was significantly (P<0.05) increasing the resultant viscosity coefficient. Fresno et al25 explained this behavior by the deformation and change in the shape of polymer molecules and in the number of molecular entanglements observed as the shearing rate increased. Syringeability was also assessed to estimate the force needed to pass the polymeric solutions through 21-gauge needle to form hypodermic in situ gels. The obtained results showed that the expelling force significantly increased (P<0.05) as the percentage of polymer was increased. For example, 65±2.4 and 43±1.7 N/mm2 were the exerted forces of expulsion of formulations with 30% and 10% polymer concentrations, respectively. Parent et al26 explained this behavior by the dominance of the polymer–solvent interaction over the polymer–polymer interaction, therefore lowering the resistance to flow. NMP could dissolve a large amount of the polymer, thus controlling both drug diffusion and easiness of syringeability.27
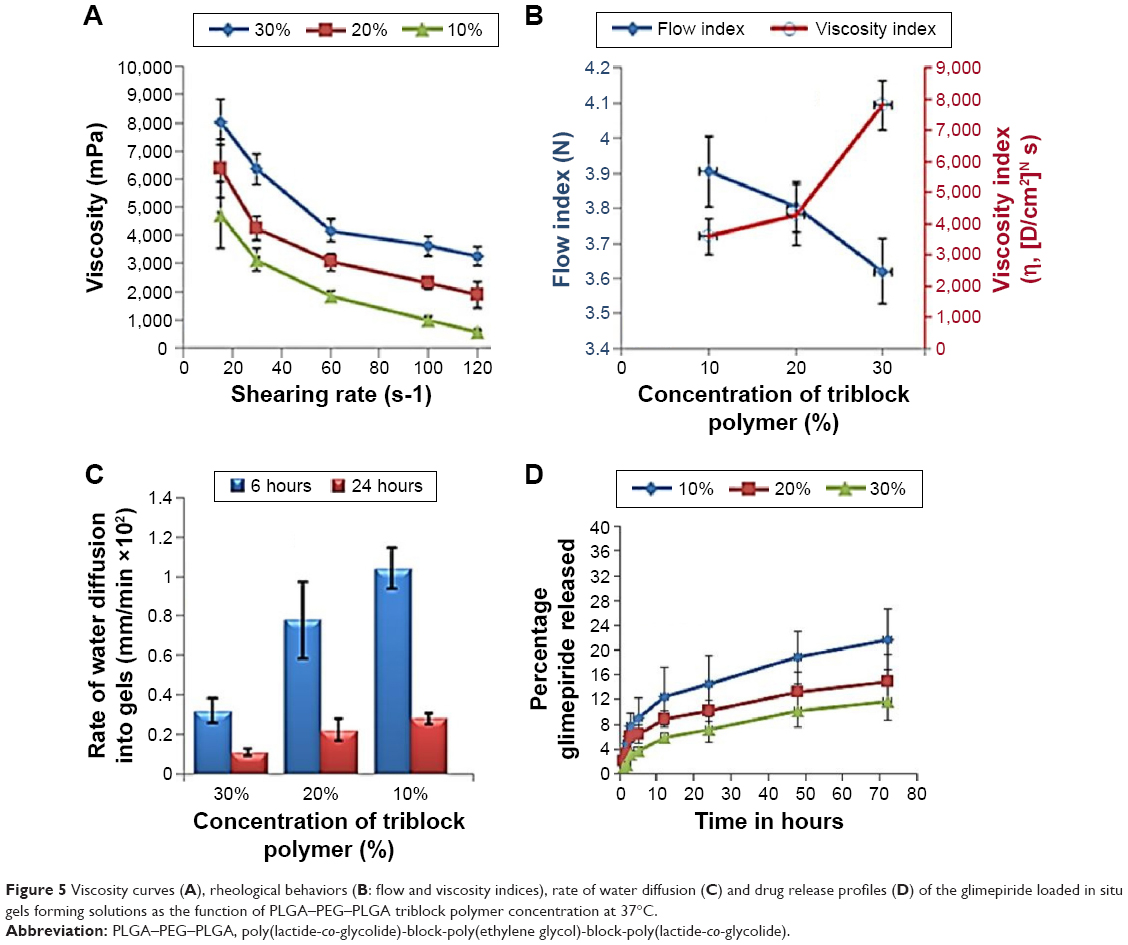 | Figure 5 Viscosity curves (A), rheological behaviors (B: flow and viscosity indices), rate of water diffusion (C) and drug release profiles (D) of the glimepiride loaded in situ gels forming solutions as the function of PLGA–PEG–PLGA triblock polymer concentration at 37°C. |
The main critical parameters of depot formation or structure change to form a gel were both temperature increase to 37°C and water diffusion rate into the Zein nanoparticles-loaded triblock polymer. The diffusion rate of the aqueous phosphate buffer pH 7.2 into the polymeric solutions decreased with increasing the triblock polymer concentration (P<0.05; Figure 5) due to the increased gel viscosity. Wang et al28 demonstrated that the fast partitioning of the employed solvent might cause fast solidification of the implant; hence, higher drug retention rate was expected. Glimepiride release from the prepared gels was conducted in phosphate buffer pH 7.2 to simulate the physiologic condition (Figure 5). In general, glimepiride release was slower from the prepared gels than from Zein nanoparticles. For example, 17.3% drug was released from the optimized nanoparticle formulation at 24 hours, whereas drug release rates in in situ gels containing 10%, 20%, and 30% w/w triblock polymer were about 14.5%, 10.2%, and 7.1% at 24 hours (Figure 5), respectively. Hence, the polymer concentration was the critical parameter to control glimepiride release. The physical entanglements between the polymer’s chains to form a dense matrix would explain this result. Liu and Venkatraman29 demonstrated that the polymer phase inversion dynamics would suppress the initial drug burst release by each increase in polymer level. The hydrophobic PLGA compartment of the triblock would slow the drug release, whereas the hydrophilic PEG would modulate the initial burst release, followed by a more rapid phase once the triblock became hydrated.30 On the other hand, a porous rubbery gel structure was formed by the NMP diffusion out of the hydrated triblock to cause burst release of glimepiride due to rapid phase inversion of PLGA.24
Conclusion
This study revealed a thorough understanding of embedding glimepiride–Zein nanoparticles into a thermoresponsive triblock copolymer to form an in situ gel. D-optimal fractional factorial design encompassing five variables at two levels was applied for the preparation of glimepiride–Zein nanoparticles. Through the systematic optimization phase, glimepiride EC of 33.6%, nanoparticle size of 120.9 nm with a skewness value of 0.2, zeta potential of 11.1 mV, and sustained release features of 3.3% and 17.3% drug released after 2 and 24 hours, respectively, were obtained. The optimized nanoparticles formulation was included in the triblock copolymers-based in situ gel that demonstrated pseudoplastic behavior. The increased concentration of triblock copolymers resulted in increase in the expelling force and reduction of drug release rate from the in situ gel formulae that could be useful for improving diabetes treatment effectiveness.
Acknowledgments
This work was supported by Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia, under grant number (166-758-D1435). The authors, therefore, gratefully acknowledge the DSR technical and financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
Kang FR, Singh J. In vitro release of insulin and biocompatibility of in situ forming gel systems. Int J Pharm. 2005;304(1–2):83–90. | ||
Li CJ, Zhang JY, Yu DM, Zhang QM. Adding glimepiride to current insulin therapy increases high-molecular weight adiponectin levels to improve glycemic control in poorly controlled type 2 diabetes. Diabetol Metab Syndr. 2014;6(1):1–7. | ||
Bahr M, Vonholtey M, Muller G, Eckel J. Direct stimulation of myocardial glucose-transport and glucose transporter-1 (Glut1) and Glut4 protein expression by the sulfonylurea glimepiride. Endocrinology. 1995; 136(6):2547–2553. | ||
Taupitz T, Dressman JB, Klein S. New formulation approaches to improve solubility and drug release from fixed dose combinations: case examples pioglitazone/glimepiride and ezetimibe/simvastatin. Eur J Pharm Biopharm. 2013;84(1):208–218. | ||
Ning X, Sun J, Han XP, et al. Strategies to improve dissolution and oral absorption of glimepiride tablets: solid dispersion versus micronization techniques. Drug Dev Ind Pharm. 2011;37(6):727–736. | ||
Bhulli N, Sharma A. Preparation of novel vesicular carrier ethosomes with glimepiride and their invistigation of permeability. Int J Ther Appl. 2012;10:1–10. | ||
Pachisia N, Agrawal SS. Formulation, development and evaluation of transdermal drug delivery system of glimepiride. Int J Pharm Pharm Sci Res. 2012;2(1):1–8. | ||
Ammar HO, Salama HA, El-Nahhas SA, Elmotasem H. Design and evaluation of chitosan films for transdermal delivery of glimepiride. Curr Drug Deliv. 2008;5(4):290–298. | ||
Som I, Bhatia K, Yasir M. Status of surfactants as penetration enhancers in transdermal drug delivery. J Pharm Bioallied Sci. 2012;4(1):2. | ||
Cleland JL, Duenas E, Daugherty A, et al. Recombinant human growth hormone poly(lactic-co-glycolic acid) (PLGA) microspheres provide a long lasting effect. J Control Release. 1997;49(2–3):193–205. | ||
Cleland JL, Daugherty A, Mrsny R. Emerging protein delivery methods. Curr Opin Biotechnol. 2001;12(2):212–219. | ||
Kim YJ, Choi S, Koh JJ, Lee M, Ko KS, Kim SW. Controlled release of insulin from injectable biodegradable triblock copolymer. Pharm Res. 2001;18(4):548–550. | ||
Elzoghby AO, Samy WM, Elgindy NA. Protein-based nanocarriers as promising drug and gene delivery systems. J Control Release. 2012; 161(1):38–49. | ||
Wu YP, Luo YG, Wang Q. Antioxidant and antimicrobial properties of essential oils encapsulated in zein nanoparticles prepared by liquid-liquid dispersion method. Lwt-Food Sci Technol. 2012;48(2):283–290. | ||
Hashem FM, Al-Sawahli MM, Nasr M, Ahmed OAA. Optimized zein nanospheres for improved oral bioavailability of atorvastatin. Int J Nanomedicine. 2015;10:4059–4069. | ||
Zentner GM, Rathi R, Shih C, et al. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. J Control Release. 2001;72(1–3):203–215. | ||
Muthuselvi L, Dhathathreyan A. Simple coacervates of zein to encapsulate Gitoxin. Colloids Surf B Biointerfaces. 2006;51(1):39–43. | ||
Lai LF, Guo HX. Preparation of new 5-fluorouracil-loaded zein nanoparticles for liver targeting. Int J Pharm. 2011;404(1–2):317–323. | ||
Zhang L, Sun X, Song Y, Jiang X, Dong S, Wang E. Didodecyldimethylammonium bromide lipid bilayer-protected gold nanoparticles: synthesis, characterization, and self-assembly. Langmuir. 2006;22(6): 2838–2843. | ||
Bugs MR, Forato LA, Bortoleto-Bugs RK, et al. Spectroscopic characterization and structural modeling of prolamin from maize and pearl millet. Eur Biophys J. 2004;33(4):335–343. | ||
Matsushima N, Danno G-i, Takezawa H, Izumi Y. Three-dimensional structure of maize α-zein proteins studied by small-angle X-ray scattering. Biochim Biophys Acta. 1997;1339(1):14–22. | ||
Hu K, McClements DJ. Fabrication of surfactant-stabilized zein nanoparticles: a pH modulated antisolvent precipitation method. Food Res Int. 2014;64:329–335. | ||
Xu X, Al-Ghabeish M, Krishnaiah YS, Rahman Z, Khan MA. Kinetics of drug release from ointments: role of transient-boundary layer. Int J Pharm. 2015;494(1):31–39. | ||
Phaechamud T, Mahadlek J. Solvent exchange-induced in situ forming gel comprising ethyl cellulose-antimicrobial drugs. Int J Pharm. 2015; 494(1):381–392. | ||
Fresno MJ, Ramirez AD, Jimenez MM. Systematic study of the flow behaviour and mechanical properties of Carbopol Ultrez 10 hydroalcoholic gels. Eur J Pharm Biopharm. 2002;54(3):329–335. | ||
Parent M, Nouvel C, Koerber M, Sapin A, Maincent P, Boudier A. PLGA in situ implants formed by phase inversion: critical physicochemical parameters to modulate drug release. J Control Release. 2013; 172(1):292–304. | ||
Chang JY, Oh YK, Kong HS, et al. Prolonged antifungal effects of clotrimazole-containing mucoadhesive thermosensitive gels on vaginitis. J Control Release. 2002;82(1):39–50. | ||
Wang L, Wang AP, Zhao XL, et al. Design of a long-term antipsychotic in situ forming implant and its release control method and mechanism. Int J Pharm. 2012;427(2):284–292. | ||
Liu H, Venkatraman SS. Cosolvent effects on the drug release and depot swelling in injectable in situ depot-forming systems. J Pharm Sci. 2012;101(5):1783–1793. | ||
Malik K, Singh I, Nagpal M, Arora S. Atrigel: A potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74–81. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.