Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
Mechanisms Underlying Hepatocellular Carcinoma Progression in Patients with Type 2 Diabetes
Authors Shi T ![]() , Kobara H, Oura K, Masaki T
, Kobara H, Oura K, Masaki T
Received 2 November 2020
Accepted for publication 25 January 2021
Published 11 February 2021 Volume 2021:8 Pages 45—55
DOI https://doi.org/10.2147/JHC.S274933
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Tingting Shi, Hideki Kobara, Kyoko Oura, Tsutomu Masaki
Department of Gastroenterology and Neurology, Faculty of Medicine, Kagawa University, Kida, Kagawa, 761-0793, Japan
Correspondence: Tsutomu Masaki
Department of Gastroenterology and Neurology, Kagawa University, 1750-1 Ikenobe, Miki, Kida, Kagawa, 761-0793, Japan
Tel +81-87-891-2156
Fax +81-87-891-2158
Email [email protected]
Abstract: Hepatocellular carcinoma (HCC) ranks third in cancer-related deaths from solid tumors worldwide. The incidence of type 2 diabetes mellitus (T2DM) has increased worldwide in conjunction with the expansion of the Western lifestyle. Furthermore, patients with T2DM have been documented to have an increased risk of HCC, as well as bile tract cancer. Growing evidence shows that T2DM is a strong additive metabolic risk factor for HCC, but how diabetes affects the incidence of HCC requires additional investigation. In this review, we discuss the underlying mechanisms of HCC in patients with T2DM. Topics covered include abnormal glucose and lipid metabolism, hyperinsulinemia, and insulin resistance; the effect of activated platelets; hub gene expression associated with HCC; inflammation and signaling pathways; miRNAs; altered gut microbiota and immunomodulation. The evidence suggests that reducing obesity, diabetes, and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis through efficient measures of prevention may lead to decreased rates of T2DM-related HCC.
Keywords: abnormal metabolism, activated platelets, hub gene, miRNAs, signaling, inflammation
Introduction
Hepatocellular carcinoma (HCC) ranks third in cancer-related deaths from solid tumors worldwide, causing nearly one million deaths every year.1 The incidence and mortality of HCC are increasing in North America and in several European regions.2 Hepatitis B virus (HBV) and hepatitis C virus (HCV) infections remain the main burden factors for HCC; however, patients with type 2 diabetes mellitus (T2DM) have been shown to have an increased risk of HCC,3 as well as bile tract cancer.4 There is an increasing amount of evidence showing that T2DM is a strong additive metabolic risk factor for HCC.5,6 The global prevalence of diabetes mellitus (DM) in adults has risen by 3.8% from 1980 to 2014, with 90–95% of cases being T2DM.7 Meanwhile, a meta-analysis and systematic review demonstrated that concurrent T2DM is strongly associated with an increased HCC risk among patients with chronic HBV8 or HCV9 infection. Recently, T2DM has also been associated with a significant increased risk of non-cirrhotic HCC with a hazard ration (HR) of 3.05 and 95% confidence interval (CI) of 1.41–6.62.10 HCC represents an ever-increasing challenge, in part due to T2DM being a risk factor for HCC and contributing to the rise in HCC incidence. However, the means by which diabetes affects the incidence of HCC still needs to be further investigated. In this review, we summarize what is currently known regarding the mechanisms underlying the progression of HCC in patients with T2DM (Figure 1).
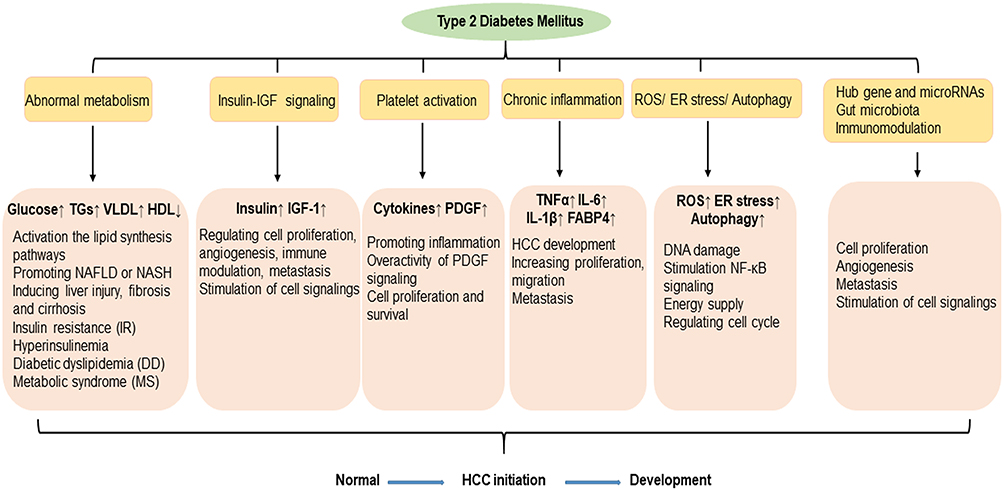 |
Figure 1 Potential mechanisms linking type 2 diabetes to hepatocellular carcinoma. |
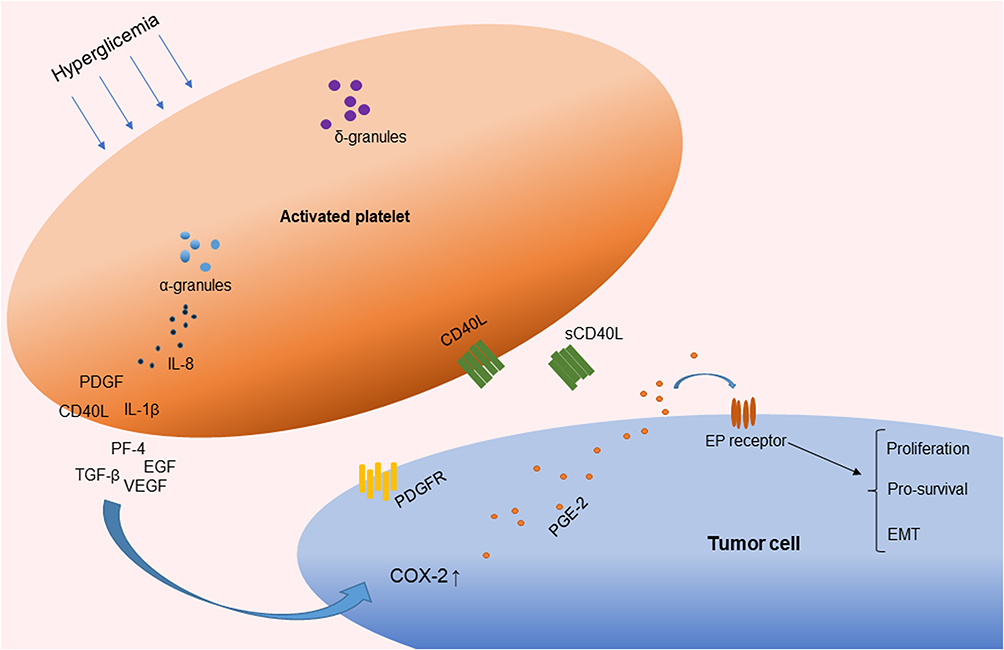 |
Figure 2 Activated platelets involved in chronic inflammation by the release of cytokines. Abbreviations: PDGF, platelet-derived growth factor; TGF-β, transforming growth factor β; EGF, endothelial growth factor; VEGF, vascular endothelial growth factor; IL-1β, interleukin-1β; sCD40L, soluble CD40 ligand; IL-8, interleukin-8; PF4, platelet factor 4; COX, cyclooxygenase; PGE2, prostaglandin E2; EMT, epithelial–mesenchymal transition. |
Epidemiology
Epidemiological data from Kagawa University (our department) indicated 802 patients with HCC that were newly diagnosed from 2003 to 2017, among which 246 were early-stage, 286 were intermediate-stage, and 270 were advanced-stage. The proportion of male patients with advanced-stage disease was 76.7%. Meanwhile, non-B, non-C HCC patients comprised 41.1% in the advanced-stage group compared with 11.8% in the early stage group. The incidence of non-B, non-C HCC is increasing in Japan, instead of HCV HCC. A nationwide survey in Japan also revealed that HCC of non-viral etiologies rapidly increased between 1991 and 2010, as well as the proportion of patients with DM, which increased from 46.1% to 51.6%.11 In addition, all-cause mortality is significantly higher in T2DM patients compared to that in the general population of Japan, with HCC conferring the highest mortality risk in T2DM patients [standardized mortality ratio (SMR) 3.57, 95% CI 2.41–5.10].12 Data from the Swedish National Diabetes Register show that the risk for severe liver disease is higher in patients with T2DM than that in the general population (HR, 2.28; 95% CI, 2.21–2.36).13 A single-center retrospective analysis of clinical features of adult patients from a large transplant center who underwent liver transplantation for steatohepatitis due to non-alcoholic steatohepatitis (NASH) and alcoholic causes indicated that patients with HCC are more likely to have T2DM compared to those without HCC (56.3% vs 37.1%; P = 0.004).14 Moreover, a greater adherence to a T2DM prevention diet is associated with a lower risk of developing HCC among US men and women.15
Abnormal Glucose and Lipid Metabolism, Hyperinsulinemia, and Insulin Resistance
Abnormal glucose and lipid metabolism are characteristics of DM. As the condition progresses, chronic hyperglycemia and an inadequate response to circulatory insulin by peripheral tissues lead to insulin resistance (IR). IR and subsequent compensatory hyperinsulinemia result in an increased production of insulin-like growth factor 1 (IGF-1), which further promotes hepatic cellular proliferation and inhibits cellular apoptosis within the liver. Furthermore, IR and hyperinsulinemia have been shown to have a key role in the pathogenesis of non-alcoholic fatty liver disease (NAFLD; renamed as metabolic dysfunction associated fatty liver disease, MAFLD)-related HCC.16 Hyperinsulinemia also plays a key role in the pathophysiology of HCC by activating insulin receptor substrate-1 (IRS-1) to regulate multiple cytokine pathways.17 The evidence indicates that IR is involved in the progression of hepatic steatosis and hepatic fibrosis by the presence of increased circulating levels of free fatty acids, which promote NAFLD and NASH. Additionally, NAFLD exacerbates hepatic/peripheral IR and induces cell death and inflammation.17,18 Furthermore, the prevalence of diabetic dyslipidemia (DD) among patients with T2DM is approximately 72–85%. In T2DM, hyperinsulinemia, frequently insulin resistance, and β cell failure are related to DD.19 The main characteristics of DD are increased triglyceride (TG) levels, reduced high-density lipoprotein cholesterol (HDL-C), normal or slightly increased low-density lipoprotein cholesterol (LDL-C), increased very-low-density lipoprotein subfraction (VLDL1), small-dense LDL-C particles, susceptibility to oxidation, and glycation of apolipoproteins.20 Metabolic syndrome (MS), characterized by abdominal obesity, insulin resistance, hypertension, and hyperlipidemia, is also related to T2DM.21 In a recent study, the rate of severe morbidity was higher in an MS-related HCC group compared to that of an HBV-HCC group.22 In addition, approximately 90% of individuals with diabetes or obesity in a comparative study had significant intrahepatic fat accumulation.23 Another study showed that the prevalence of NAFLD is much higher in T2DM patients compared to that in T1DM patients.24 In longitudinal NAFLD cohorts with paired liver biopsies, the presence of diabetes is associated with increased fibrosis progression, while improved glycemic control correlates with fibrosis regression.10 A study based on a population-based database indicated NAFLD is becoming a major cause of HCC and that NAFLD is associated with shorter survival time, more advanced tumor stage, and a lower possibility of receiving a liver transplant.25 Furthermore, another meta-analysis showed that the strongest independent predictor of a diagnosis of HCC or cirrhosis is the baseline diagnosis of diabetes.26 A large amount of evidence supports the concept that abnormal lipid metabolism increases the prevalence of NAFLD and NASH and furthers HCC development.25,27,28 To summarize the entire process, abnormal glucose and lipid metabolism, hyperinsulinemia, and insulin resistance cause intrahepatic fat accumulation, which promotes the development of HCC.
Activated Platelets
A previous study suggested that glucose itself drives platelet activation.29 In accordance with this, platelets can be activated during the postprandial phase of patients with low insulin levels or insulin-resistant diabetic patients, but not in healthy individuals.30 Platelet function is not only associated with hemostasis but is also involved in chronic inflammation and thrombotic pathological processes. Cytokines such as interleukin-1β (IL-1β), interleukin-8 (IL-8), and growth factors such as transforming growth factor β (TGF-β) and vascular endothelial growth factor (VEGF) are stored in alpha-granules, dense granules, and lysosomes and are involved in the cyclooxygenase 2 (COX-2) pathway in cancer cells.31,32 The subcellular machinery of de novo protein synthesis is involved in inflammatory pathways and includes IL-1β, plasminogen activator inhibitor-1 (PAI-1), and tissue factor (TF).32 Activated platelets also release soluble CD40 ligand (CD40L), which is structurally related to tumor necrosis factor (TNF)-α superfamily. More than 95% of the circulating CD40L is derived from platelets.33 CD40L stimulates resting platelets via CD40/CD40L interaction, which is involved in the expression of many pro-inflammatory and prothrombotic factors.34 In addition, platelet levels are an independent risk factor for advanced fibrosis in T2DM patients with NAFLD (odds ratio, 0.985; 95% CI, −0.972–0.999; P = 0.034).28 Platelet-derived growth factor (PDGF) stimulates wound healing and regulates tissue homeostasis. However, overactivity of PDGF signaling is associated with malignancies and other diseases characterized by excessive cell proliferation. In certain tumors, genetic or epigenetic alterations of genes for PDGF ligands and receptors drive tumor cell proliferation and survival.35 Results from an animal model suggest that hyperactivated platelets constitute a major source of serum amyloid A (SAA) proteins during the pretumoral stage and promote the initiation of colitis-associated cancer by enhancing myeloid cell-dependent immunosuppression.36 Moreover, another study showed that activated platelets release functional high molecular weight epidermal growth factor (HMW-EGF), which stimulates platelet epidermal growth factor receptor (EGFR) autophosphorylation, phosphatidylinositol-3 kinase (PI3k)-dependent AKT phosphorylation, and a transient increase in Ca2+ that is blocked by EGFR tyrosine kinase inhibition. There is a connection between activated platelets and tumorigenesis through the rapid and prolonged autocrine-stimulated release of HMW-EGF by tumor-associated platelets (Figure 2).37
Gene Expression
A recent bioinformatic analysis of key genes associated with DM and HCC identified nine hub genes.38 The hub genes include cerebral dopamine neurotrophic factor (CDNF), cysteine-rich with EGF-like domains 2 (CRELD2), DnaJ (Hsp40) homologue subfamily B member 11 (DNAJB11), denticleless E3 ubiquitin-protein ligase homologue (DTL), GINS complex subunit 2 (GINS2), mesencephalic astrocyte-derived neurotrophic factor (MANF), protein disulfide isomerase family A member 4 (PDIA4), protein disulfide isomerase family A member 6 (PDIA6), and valosin-containing protein (VCP). Among them, CDNF, CRELD2, DNAJB11, MANF, PDIA4, PDIA6, and VCP are associated with the endoplasmic reticulum (ER) stress response, which is recognized as a key driver in sustaining malignant cells and promoting cancer progression.39 Meanwhile, DTL is a widely accepted oncogene that regulates cell cycle progression and genome stability40 and GINS2 is critical for the initiation of DNA replication.41 Another bioinformatic analysis comparing T2DM with non-diabetes and HCC with the control groups indicated that genes ST3GAL2 and ZNF613 are highly methylated and positively correlate with the HCC stage.42 Furthermore, nuclear receptor co-activator 5 (NCOA5) is a possible susceptibility gene for both T2DM and HCC by regulating IL-6 during pathogenesis.43 In addition, genes CCNA2, CCNB1, MAD2L1, RACGAP1, NCAPG, and TTK are found to be overexpressed in HCC, while CCNA2, CCNB1, CHEK1, are significantly downregulated in metformin-treated samples, suggesting these genes may be mechanistically involved in the metformin treatment of T2DM and HCC.44 In previous reports, metformin was shown to have anti-HCC effects45–47 and is associated with improved survival among HCC patients with T2DM.48 Based on these findings, it is suggested that these hub genes may be associated with one of the underlying pathophysiological mechanisms of HCC and T2DM. Additionally, hypermethylation of the E-cadherin-1 (CDH-1) gene is related to an increased incidence of NAFLD-related HCC. Genetic variations in the patatin-like phospholipase domain-containing protein-3 (PNPLA3) and the transmembrane 6 superfamily 2 (TM6SF2) gene are found to be closely associated with increased risk of NAFLD progression, especially with the risk of hepatic fibrosis. Moreover, greater levels of recurrent mutations in the cell cycle regulator genes TP53 and CDKN2A and genes of the β-catenin/WNT signaling pathway are detected in patients with NASH.17,49–51
Molecular Mechanism of Inflammation and Signaling Pathways
HCC in T2DM patients always occurs in conjunction with the effects of inflammation and signaling pathways. There are several factors involved in this process, such as abnormal lipid metabolism inducing DD, MS, NASH, and NAFLD; activated platelets releasing cytokines involved in chronic inflammation and thrombotic pathological processes; insulin resistance; and the effect of hub-gene expression. Cell death and inflammation are key drivers of fibrosis in many forms of chronic liver disease52 and fibrosis is the main factor of HCC.
Stimulation of Insulin-Like Growth Factor-1 Signaling
Hepatocyte differentiation, proliferation, and apoptosis are affected by growth factors produced in the liver. Insulin-like growth factors 1 and 2 (IGF1 and IGF2) act in response to growth hormone (GH) and both insulin and IGF-1 are increased in T2DM patients with insulin resistance.53,54 In addition, IGF-1 enhances cancer progression by regulating cell proliferation, angiogenesis, immune modulation, and metastasis.55 IGF-1 activates PI3k/CCND1, which increases cell proliferation and may also indirectly modulate IGF-2 and IGF2R.56 Furthermore, IGF-1 promotes the phosphorylation of extracellular-signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK)57 and JNK/MAPK pathway.54 The mitogenic activity of IGF-1R is mediated through the Ras and AKT pathways58 and crosstalk occurs between IGF-1R and the erbB family of receptors, which include erbB1 (EGFR) and erbB2 (HER2/neu).59 In addition, insulin activates a cascade of intracellular signaling via binding of the insulin receptor (IR), leading to it phosphorylating itself and the insulin receptor substrates. IRS1 and IRS2 are the main mediators of insulin signaling in the liver, where they control insulin sensitivity.18 The IR has significant over-all homology to IGF-1R and 84% homology with the tyrosine kinase domains. Hybrid-IRs may increase the functional pool of receptors capable of activating the IGF system and provide further growth advantages.58 Interestingly, decreased serum levels, decreased tissue expression of IGF1, elevated production of IGF1R, and variable IGF2 expression has been noted in epidemiological and clinicopathological studies of chronic liver disease (CLD), including NAFLD, cirrhosis, and HCC.54 However, there has been no analysis of CLD with T2DM (Figure 3).
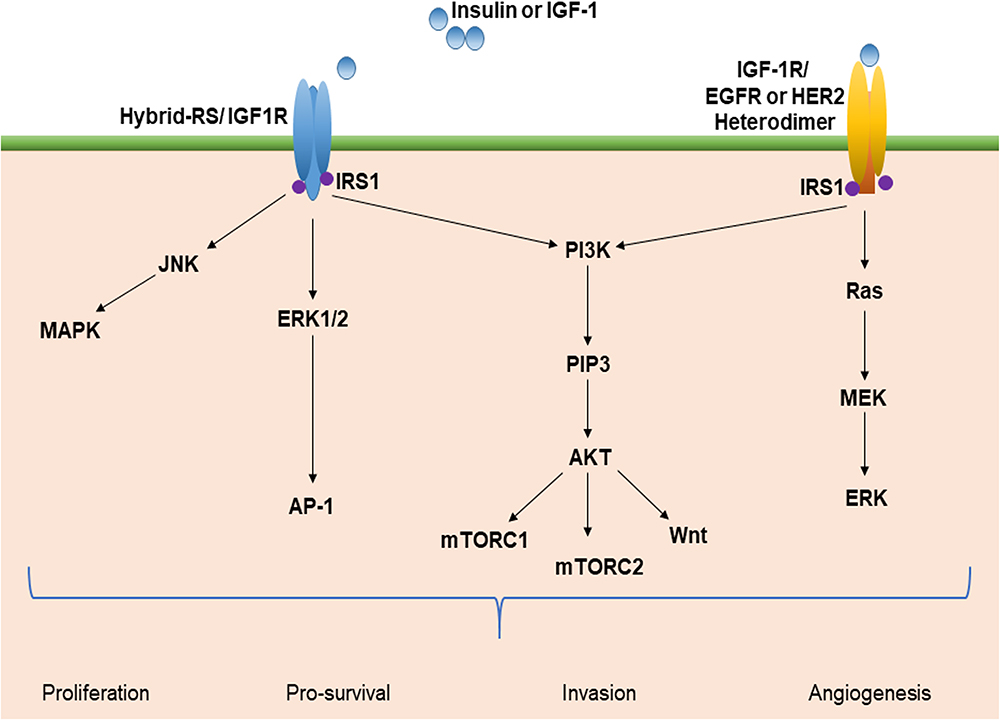 |
Figure 3 Insulin-like factor-1 (IGF-1) signaling and the multiple pathway axis. Insulin resistance and hyperglycemia stimulate IGF-1 signaling and activate multiple pathways. This leads to the activation of oncogenic signaling pathways, such as PI3K/Akt, JNK/MAPK, Wnt, mTOR, and Ras/ERK. These pathways promote cellular proliferation, increased invasion and angiogenesis, and decreased apoptosis that may foster the development of HCC. Abbreviations: IRS-1, insulin receptor substrate-1; mTOR, mammalian target of rapamycin; PI3k, phosphatidylinositol-3 kinase; JNK, c-jun-N-terminal kinase; ERK, extracellular-signal-regulated kinase; MAPK, mitogen-activated protein kinase; PIP3, phosphoinositide 3-kinases; Akt, protein kinase B; EGFR, epidermal growth factor receptor; AP-1, activator protein 1. |
Metabolic Stress, Endoplasmic Reticulum (ER) Stress, and Oxidative Stress Response Pathways
The ER stress response constitutes a cellular process triggered by a variety of conditions that disturb the folding of proteins in the ER. In cases where ER stress-induced damage cannot be reversed, cellular functions deteriorate, often leading to cell death.60 ER stress can also activate the JNK pathway, which promotes the nuclear factor kappa-B (NF-κB) signaling cascade, and increase mRNA and protein levels of pro-inflammatory cytokines, such as TNF-α and IL-6.61 Additionally, JNK acts synergistically with NF-κB and JAK/STAT to promote cell survival.62 Hyperglycemia can then increase oxidative stress,63 which can inhibit insulin signaling through the activation of an inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ) and JNK. Oxidative stress has been shown to compromise the two major mechanisms of DM, insulin secretion and insulin action.64 One-carbon metabolism plays an important role in nucleotide synthesis, methylation, reductive metabolism, and the Warburg effect.65 It has also been found in the inflammation process and carcinogenesis. For instance, in macrophages, serine is required for optimal lipopolysaccharide (LPS) induction of IL-1β mRNA expression, but not inflammasome activation.66 In addition, B vitamins involved in one-carbon metabolism have been implicated in the development of inflammation- and angiogenesis-related chronic diseases, such as colorectal cancer (Figure 4).67
Reactive Oxygen Species (ROS)
Accumulated ROS are found in DM and are important mediators for the activation of pro-inflammatory signaling pathways. Obesity and hyperglycemia-induced ROS production may favor the induction of M1-like pro-inflammatory macrophages during diabetes onset and progression. Moreover, ROS induce MAPK, STAT1, STAT6, and NFκB signaling (Figure 4), interfere with macrophage differentiation via epigenetic reprogramming,68 induce cellular cytotoxicity and DNA damage genes, including c-JNK, p-53, c-FOS, c-RAS, c-Myc,69 and induce autophagy.70 Meanwhile, clinical data71 and in vitro studies69 indicate high levels of ROS production and DNA damage, along with upregulation of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, NF-κB, and COX-2 expression, and downregulation of IL-10, with an increased risk of tumorigenesis.
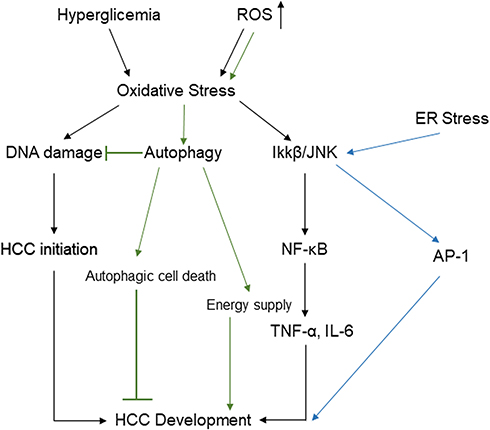 |
Figure 4 Potential pathways linking hyperglycemia and accumulated reactive oxygen species to HCC through oxidative stress, ER stress, and autophagy. Abbreviations: IL-6, interleukin-6; NF-κB, nuclear factor kappa B; TNF-a, tumor necrosis factor-a; IKK-β, inhibitor of nuclear factor kappa-B kinase subunit beta; AP-1, activator protein 1; ROS, reactive oxygen species; HCC, hepatocellular carcinoma; ER, endoplasmic reticulum. |
Pro-Inflammatory Cytokines
Inflammatory cytokines are associated with HCC progression, including well-known cytokines such as TNF-α, the IL-6 family, and IL-1β. TNF is a major inflammatory cytokine associated with NF-κB signaling. Meanwhile, IL-6 levels are well correlated with the tumor stage with IL-6 exerting many of its functions via activation of STAT3, a transcription factor found to be important for HCC development.72 Undetectable IL-17A is associated with extrahepatic metastasis. Among the cytokines, IL-6 remains an independent predictive factor for the survival of patients with HCC.73
Hypoxia and inflammation are two critical factors that shape the HCC microenvironment. Epithelial–mesenchymal transition (EMT) of HCC cells is enhanced by hypoxia-inducible factor-1α (HIF-1α)/IL-1β signaling and IL-1β promotes HCC metastasis in mouse models and is predictive of poor prognosis in HCC patients.74 A number of studies have shown that plasma concentrations of fatty acid-binding protein 4 (FABP4) are increased in obesity and T2DM, and linked with insulin resistance and dyslipidemia.75 The involvement of FABP4 in the pathogenesis of obesity and insulin resistance seems to be mediated via inhibition of FABP4-dependent peroxisome proliferator-activated receptor γ (PPARγ).75 Other data show that exogenous FABP4 significantly increases the proliferation and migration of human HCC cells. FABP4 can be synthesized and secreted by both hepatocytes and HCC cells and FABP4 may play a role in regulating tumor progression in the underlying setting of obesity.76
MicroRNAs (miRNAs)
A key player in HCC and T2DM is miRNAs, which may be hidden culprits in diabetes-associated HCC. Recent studies have documented that plasma miR-17, miR-20a, miR-20b, and miR-122 are upregulated in T2DM patients with NAFLD compared to those without NAFLD.77 The miRNAs miR-34a and miR-221 are significantly upregulated and miR-16, miR-23-3p, miR-122-5p, miR-198, and miR-199a-3p are significantly downregulated in HCC-positive T2DM patients compared to liver cirrhosis-positive T2DM patients.78 Increased expression of miR-17 promotes HCC carcinogenesis through the down-regulation of sekelsky mothers against dpp3 (Smad3).79 By targeting plant homeodomain finger 2 (PHF2), miR-221 promotes HCC migration80 and acts as a promoter of the EMT process in HCC cells by targeting adiponectin receptor 1 (AdipoR1).81 Lower expression of miR-198 is related to several clinicopathological parameters in HCC patients82 and is a suppressor of HCC cell invasion by negatively regulating the HGF/c-MET pathway.83 Increased miR-199a-3p levels in HCC remarkably suppress cell proliferation and migration, induce cellular apoptosis, and downregulate the specific target gene mTOR.84 In most HCCs, miR-122 is downregulated while oncogenic SerpinB3 is upregulated, and miR-122 overexpression increases sorafenib sensitivity.85 A meta-analysis of stress-related microRNA biomarkers in T2DM revealed that 16 miRNAs are significantly related of which miR-148b, miR-223, miR-130a, miR-19a, miR-26b, and miR-27b are downregulated in both animals and humans with T2DM. In contrast, miR-146a is downregulated in humans but upregulated in animal studies, and miR-21 is upregulated in humans but downregulated in animals.86 Furthermore, miR-148b expression is decreased in HCC and is associated with vein invasion and TNM stage,87 miR-223 plays a key role in controlling steatosis-to-NASH progression by inhibiting hepatic Cxcl10 and Taz expression,88 miR-21 promotes HCC cell invasion and migration,89 and exosomal miR-21 regulates the TETs/PTENp1/PTEN pathway to promote HCC growth.90
Altered Gut Microbiota and Immunomodulation
Gut microbiota play an important role in the regulation of the immune system and host metabolism. Accumulating evidence suggests that the gut microbiome is involved in the etiology of obesity, NAFLD, insulin resistance, and T2DM.91 In healthy humans, the dominant bacterial phyla are Firmicutes (30–50%), Bacteroidetes (20–40%), and Actinobacteria (1–10%).92 Epidemiological studies have shown that the proportion of Firmicutes, including the ratio of Firmicutes to Bacteroidetes, the concentration of Faecalibacterium prausnitzii, the abundance of the class Clostridia and genus Blautia, and quantity of Lactobacillus decrease in T2DM patients, whereas the abundance of the class Betaproteobacteria increase.93–95 Changes in the gut microbiota of T2DM patients result in increased LPS levels, which causes inflammation and insulin resistance.95 Additionally, high circulating levels of LPS are observed in both animal models of carcinogen-induced hepatocarcinogenesis and HCC patients.96 Gut microbiota are known to contribute to T2DM and NAFLD/NASH, and the release of inflammatory cytokines and expression of Toll-like receptor-4 are possible mechanisms for enhanced HCC, including alterations in bile acid metabolism.97–99 Another mechanism involved in the immunopathogenesis of obesity and T2DM is alterations in the balance of T helper 17 cells/regulatory T cells (Th17/Treg). Th17 cells produce IL-17, which is implicated in the control of adipogenesis and glucose homeostasis in obesity100 and regulates insulin sensitivity.101
Anti-Diabetic Metformin as an Anti-Cancer Drug for HCC Patients with T2DM
Patients with T2DM have a significantly higher risk of developing HCC and T2DM is also involved in cancer progression. Many studies have been recently published that provide substantial insight regarding patients with T2DM. For instance, it has been reported that patients with T2DM who are using the antidiabetic drug metformin are less likely to develop various cancers.102 Metformin is a biguanide drug that through activation (phosphorylation) of AMP kinase (AMPK) suppresses gluconeogenesis and lipolysis, especially in the liver. In adipocytes, metformin reduces fatty acid synthesis and lipolysis. Insulin sensitivity increases in muscle tissue and blood glucose decreases by promoting mitochondrial oxidation, increasing fatty acid uptake, and sugar uptake.103 Metformin also affects the incidence and mortality associated with various cancers in metformin-treated patients compared to that in control patients. The corrected hazard ratios for intestinal cancer, lung cancer, and breast cancer in patients treated with metformin are 0.6, 0.7, and 0.6, respectively, indicating a reduction in the risk of developing at least these types of cancer. In addition, the corrected hazard ratios for all-cause mortality and cancer mortality were significantly reduced to 0.42 and 0.63, respectively, in patients treated with metformin.103,104 It can be easily inferred from the clinical and epidemiological data that metformin, in addition to having anti-diabetic activity, may also act to suppress cancer cell proliferation, including HCC. Therefore, metformin may prove to be an inexpensive tumor prevention agent with few side effects.
Conclusions
Epidemiological studies have documented an increase in T2DM and T2DM-related HCC over the past decades. This review aimed to review the underlying mechanisms of HCC in patients with T2DM and highlighted abnormal glucose and lipid metabolism, hyperinsulinemia, and insulin resistance; the effect of activated platelets; hub gene expression associated with HCC; inflammation and signaling pathways; miRNAs; altered gut microbiota, and immunomodulation. In addition, efforts in the prevention of obesity, diabetes, and NAFLD/NASH may lead to a decreased rate of T2DM-related HCC.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lombardi A, Grimaldi A, Zappavigna S, et al. Hepatocarcinoma: genetic and epigenetic features. Minerva Gastroenterol Dietol. 2018;64(1):14–27. doi:10.23736/S1121-421X.17.02408-4
2. Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology. 2019;156(2):477–491. doi:10.1053/j.gastro.2018.08.065
3. Gerbes A, Zoulim F, Tilg H, et al. Gut roundtable meeting paper: selected recent advances in hepatocellular carcinoma. Gut. 2018;67(2):380–388. doi:10.1136/gutjnl-2017-315068
4. Schlesinger S, Aleksandrova K, Pischon T, et al. Diabetes mellitus, insulin treatment, diabetes duration, and risk of biliary tract cancer and hepatocellular carcinoma in a European cohort. Ann Oncol. 2013;24(9):2449–2455. doi:10.1093/annonc/mdt204
5. Tseng CH. Metformin and risk of hepatocellular carcinoma in patients with type 2 diabetes. Liver Int. 2018;38(11):2018–2027. doi:10.1111/liv.13872
6. Tilg H, Moschen AR, Roden M. NAFLD and diabetes mellitus. Nat Rev Gastroenterol Hepatol. 2017;14(1):32–42. doi:10.1038/nrgastro.2016.147
7. Henning RJ. Type-2 diabetes mellitus and cardiovascular disease. Future Cardiol. 2018;14(6):491–509. doi:10.2217/fca-2018-0045
8. Tan Y, Wei S, Zhang W, et al. Type 2 diabetes mellitus increases the risk of hepatocellular carcinoma in subjects with chronic hepatitis B virus infection: a meta-analysis and systematic review. Cancer Manag Res. 2019;11:705–713. doi:10.2147/CMAR.S188238
9. Dyal HK, Aguilar M, Bartos G, et al. Diabetes mellitus increases risk of hepatocellular carcinoma in chronic hepatitis C virus patients: a systematic review. Dig Dis Sci. 2016;61(2):636–645. doi:10.1007/s10620-015-3983-3
10. Simon TG, King LY, Chong DQ, et al. Diabetes, metabolic comorbidities, and risk of hepatocellular carcinoma: results from two prospective cohort studies. Hepatology. 2018;67(5):1797–1806. doi:10.1002/hep.29660
11. Tateishi R, Uchino K, Fujiwara N, et al. A nationwide survey on non-B, non-C hepatocellular carcinoma in Japan: 2011–2015 update. J Gastroenterol. 2019;54(4):367–376. doi:10.1007/s00535-018-1532-5
12. Shima T, Uto H, Ueki K, et al. Hepatocellular carcinoma as a leading cause of cancer-related deaths in Japanese type 2 diabetes mellitus patients. J Gastroenterol. 2019;54(1):64–77. doi:10.1007/s00535-018-1494-7
13. Björkström K, Franzén S, Eliasson B, et al. Risk factors for severe liver disease in patients with type 2 diabetes. Clin Gastroenterol Hepatol. 2019;17(13):2769–2775.e4. doi:10.1016/j.cgh.2019.04.038
14. Zarrinpar A, Faltermeier CM, Agopian VG, et al. Metabolic factors affecting hepatocellular carcinoma in steatohepatitis. Liver Int. 2019;39(3):531–539. doi:10.1111/liv.14002
15. Luo X, Sui J, Yang W, et al. Type 2 diabetes prevention diet and hepatocellular carcinoma risk in US men and women. Am J Gastroenterol. 2019;114(12):1870–1877. doi:10.14309/ajg.0000000000000450
16. Streba LA, Vere CC, Rogoveanu I, et al. Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: an open question. World J Gastroenterol. 2015;21(14):4103–4110. doi:10.3748/wjg.v21.i14.4103
17. Mantovani A, Targher G. Type 2 diabetes mellitus and risk of hepatocellular carcinoma: spotlight on nonalcoholic fatty liver disease. Ann Transl Med. 2017;5(13):270. doi:10.21037/atm.2017.04.41.
18. Fujii H, Kawada N; Japan Study Group Of Nafld Jsg-Nafld. The role of insulin resistance and diabetes in nonalcoholic fatty liver disease. Int J Mol Sci. 2020;21(11):3863. doi:10.3390/ijms21113863
19. Vergès B. Pathophysiology of diabetic dyslipidaemia: where are we? Diabetologia. 2015;58(5):886–899. doi:10.1007/s00125-015-3525-8
20. Athyros VG, Doumas M, Imprialos KP, et al. Diabetes and lipid metabolism. Hormones (Athens). 2018;17(1):61–67. doi:10.1007/s42000-018-0014-8
21. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. 2018;20(2):12. doi:10.1007/s11906-018-0812-z.
22. Tian Y, Lyu H, He Y, et al. Comparison of hepatectomy for patients with metabolic syndrome-related HCC and HBV-related HCC. J Gastrointest Surg. 2018;22(4):615–623. doi:10.1007/s11605-017-3629-1.
23. Gastaldelli A, Cusi K, Pettiti M, et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology. 2007;133(2):496–506. doi:10.1053/j.gastro.2007.04.068
24. Cusi K, Sanyal AJ, Zhang S, et al. Non-alcoholic fatty liver disease (NAFLD) prevalence and its metabolic associations in patients with type 1 diabetes and type 2 diabetes. Diabetes Obes Metab. 2017;19(11):1630–1634. doi:10.1111/dom.12973
25. Younossi ZM, Otgonsuren M, Henry L, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology. 2015;62(6):1723–1730. doi:10.1002/hep.28123
26. Alexander M, Loomis AK, van der Lei J, et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: real-world study of 18 million patients in four European cohorts. BMC Med. 2019;17(1):95. doi:10.1186/s12916-019-1321-x
27. Liang JQ, Teoh N, Xu L, et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat Commun. 2018;9(1):4490. doi:10.1038/s41467-018-06931-6
28. Tuong TTK, Tran DK, Phu PQT, et al. Non-alcoholic fatty liver disease in patients with type 2 diabetes: evaluation of hepatic fibrosis and steatosis using fibroscan. Diagnostics (Basel). 2020;10(3):159. doi:10.3390/diagnostics10030159
29. Assert R, Scherk G, Bumbure A, et al. Regulation of protein kinase C by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia. 2001;44(2):188–195. doi:10.1007/s001250051598
30. Grandl G, Wolfrum C. Hemostasis, endothelial stress, inflammation, and the metabolic syndrome. Semin Immunopathol. 2018;40(2):215–224. doi:10.1007/s00281-017-0666-5
31. Heijnen H, van der Sluijs P. Platelet secretory behaviour: as diverse as the granules … or not? J Thromb Haemost. 2015;13(12):2141–2151. doi:10.1111/jth.13147
32. Barale C, Russo I. Influence of cardiometabolic risk factors on platelet function. Int J Mol Sci. 2020;21(2):623. doi:10.3390/ijms21020623
33. André P, Prasad KS, Denis CV, et al. CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med. 2002;8(3):247–252. doi:10.1038/nm0302-247
34. Santilli F, Simeone P, Liani R, Davì G. Platelets and diabetes mellitus. Prostaglandins Other Lipid Mediat. 2015;120:28–39. doi:10.1016/j.prostaglandins.2015.05.002
35. Heldin CH, Lennartsson J, Westermark B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J Intern Med. 2018;283(1):16–44. doi:10.1111/joim.12690
36. Servais L, Wéra O, Dibato Epoh J, et al. Platelets contribute to the initiation of colitis-associated cancer by promoting immunosuppression. J Thromb Haemost. 2018;16(4):762–777. doi:10.1111/jth.13959
37. Chen R, Jin G, Li W, et al. Epidermal growth factor (EGF) autocrine activation of human platelets promotes EGF receptor-dependent oral squamous cell carcinoma invasion, migration, and epithelial mesenchymal transition. J Immunol. 2018;201(7):2154–2164. doi:10.4049/jimmunol.1800124
38. Liu GM, Zeng HD, Zhang CY, et al. Key genes associated with diabetes mellitus and hepatocellular carcinoma. Pathol Res Pract. 2019;215(11):152510. doi:10.1016/j.prp.2019.152510
39. Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40(3):141–148. doi:10.1016/j.tibs.2015.01.002
40. Abbas T, Dutta A. CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle. 2011;10(2):241–249. doi:10.4161/cc.10.2.14530
41. Rantala JK, Edgren H, Lehtinen L, et al. Integrative functional genomics analysis of sustained polyploidy phenotypes in breast cancer cells identifies an oncogenic profile for GINS2. Neoplasia. 2010;12(11):877–888. doi:10.1593/neo.10548
42. Wei H, Wang J, Li W, et al. The underlying pathophysiology association between the Type 2-diabetic and hepatocellular carcinoma. J Cell Physiol. 2019;234(7):10835–10841. doi:10.1002/jcp.27919
43. Liu CY, Feng GS. NCOA5, a molecular link between type 2 diabetes and liver cancer. Hepatobiliary Surg Nutr. 2014;3(3):106–108. doi:10.3978/j.issn.2304-3881.2014.04.02
44. Shi Z, Xiao Z, Hu L, et al. The genetic association between type 2 diabetic and hepatocellular carcinomas. Ann Transl Med. 2020;8(6):380. doi:10.21037/atm.2020.02.13
45. Fujita K, Iwama H, Miyoshi H, et al. Diabetes mellitus and metformin in hepatocellular carcinoma. World J Gastroenterol. 2016;22(27):6100–6113. doi:10.3748/wjg.v22.i27.6100
46. Miyoshi H, Kato K, Iwama H, et al. Effect of the anti-diabetic drug metformin in hepatocellular carcinoma in vitro and in vivo. Int J Oncol. 2014;45(1):322–332. doi:10.3892/ijo.2014.2419
47. Schulte L, Scheiner B, Voigtländer T, et al. Treatment with metformin is associated with a prolonged survival in patients with hepatocellular carcinoma. Liver Int. 2019;39(4):714–726. doi:10.1111/liv.14048
48. Luo CS, Lin Y, Zhou WP, et al. Survival advantage associated with metformin usage in hepatocellular carcinoma patients with diabetes mellitus receiving radical resection: a propensity score matching analysis. Eur J Gastroenterol Hepatol. 2019;32(8):1030–1035. doi:10.1097/MEG.0000000000001610
49. Wainwright P, Scorletti E, Byrne CD. Type 2 diabetes and hepatocellular carcinoma: risk factors and pathogenesis. Curr Diab Rep. 2017;17(4):20. doi:10.1007/s11892-017-0851-x
50. Raksayot M, Chuaypen N, Khlaiphuengsin A, et al. Independent and additive effects of PNPLA3 and TM6SF2 polymorphisms on the development of non-B, non-C hepatocellular carcinoma. J Gastroenterol. 2019;54(5):427–436. doi:10.1007/s00535-018-01533-x
51. Yang J, Trépo E, Nahon P, et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int J Cancer. 2019;144(3):533–544. doi:10.1002/ijc.31910
52. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology. 2020;158(7):1913–1928. doi:10.1053/j.gastro.2019.11.311
53. NeamŢu MC, Avramescu ET, Marcu IR, et al. The correlation between insulin-like growth factor with glycemic control, glomerular filtration rate, blood pressure, hematological changes or body mass index in patients with type 2 diabetes mellitus. Rom J Morphol Embryol. 2017;58(3):857–861.
54. Adamek A, Kasprzak A. Insulin-like growth factor (IGF) system in liver diseases. Int J Mol Sci. 2018;19(5):1308. doi:10.3390/ijms19051308
55. Al-Gayyar MMH, Bagalagel A, Noor AO, et al. The therapeutic effects of nicotinamide in hepatocellular carcinoma through blocking IGF-1 and effecting the balance between Nrf2 and PKB. Biomed Pharmacother. 2019;112:108653. doi:10.1016/j.biopha.2019.108653
56. Dai C, Li N, Song G, et al. Insulin-like growth factor 1 regulates growth of endometrial carcinoma through PI3k signaling pathway in insulin-resistant type 2 diabetes. Am J Transl Res. 2016;8(8):3329–3336.
57. Teng JA, Wu SG, Chen JX, et al. The activation of ERK1/2 and JNK MAPK signaling by insulin/IGF-1 is responsible for the development of colon cancer with type 2 diabetes mellitus. PLoS One. 2016;11(2):e0149822. doi:10.1371/journal.pone.0149822
58. Weroha SJ, Haluska P. The insulin-like growth factor system in cancer. Endocrinol Metab Clin North Am. 2012;41(2):335–vi. doi:10.1016/j.ecl.2012.04.014
59. Haluska P, Carboni JM, TenEyck C, et al. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol Cancer Ther. 2008;7(9):2589–2598. doi:10.1158/1535-7163.MCT-08-0493
60. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–3470. doi:10.1016/j.bbamcr.2013.06.028
61. Hammouda MB, Ford AE, Liu Y, et al. The JNK signaling pathway in inflammatory skin disorders and cancer. Cells. 2020;9(4):857. doi:10.3390/cells9040857
62. Wu Q, Wu W, Fu B, et al. JNK signaling in cancer cell survival. Med Res Rev. 2019;39(6):2082–2104. doi:10.1002/med.21574
63. Khan RS, Bril F, Cusi K, et al. Modulation of insulin resistance in nonalcoholic fatty liver disease. Hepatology. 2019;70(2):711–724. doi:10.1002/hep.30429
64. Ighodaro OM. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed Pharmacother. 2018;108:656–662. doi:10.1016/j.biopha.2018.09.058
65. Newman AC, Maddocks ODK. One-carbon metabolism in cancer. Br J Cancer. 2017;116(12):1499–1504. doi:10.1038/bjc.2017.118
66. Rodriguez AE, Ducker GS, Billingham LK, et al. Serine metabolism supports macrophage IL-1β production. Cell Metab. 2019;29(4):1003–1011.e4. doi:10.1016/j.cmet.2019.01.014
67. Kiblawi R, Holowatyj AN, Gigic B, et al. One-carbon metabolites, B vitamins and associations with systemic inflammation and angiogenesis biomarkers among colorectal cancer patients: results from the ColoCare Study. Br J Nutr. 2020;123(10):1187–1200. doi:10.1017/S0007114520000422
68. Rendra E, Riabov V, Mossel DM, et al. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology. 2019;224(2):242–253. doi:10.1016/j.imbio.2018.11.010
69. Shah HK, Sharma T, Banerjee BD. Organochlorine pesticides induce inflammation, ROS production, and DNA damage in human epithelial ovary cells: an in vitro study. Chemosphere. 2020;246:125691. doi:10.1016/j.chemosphere.2019.125691
70. Yuan X, Wang B, Yang L, Zhang Y. The role of ROS-induced autophagy in hepatocellular carcinoma. Clin Res Hepatol Gastroenterol. 2018;42(4):306–312. doi:10.1016/j.clinre.2018.01.005
71. Tatsch E, De Carvalho JA, Hausen BS, et al. Oxidative DNA damage is associated with inflammatory response, insulin resistance and microvascular complications in type 2 diabetes. Mutat Res. 2015;782:17–22. doi:10.1016/j.mrfmmm.2015.10.003
72. Heichler C, Scheibe K, Schmied A, et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut. 2020;69(7):1269–1282. doi:10.1136/gutjnl-2019-319200
73. Lee HL, Jang JW, Lee SW, et al. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci Rep. 2019;9(1):3260. doi:10.1038/s41598-019-40078-8
74. Zhang J, Zhang Q, Lou Y, et al. Hypoxia-inducible factor-1α/interleukin-1β signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology. 2018;67(5):1872–1889. doi:10.1002/hep.29681
75. Trojnar M, Patro-Małysza J, Kimber-Trojnar Ż, et al. Associations between fatty acid-binding protein 4¯A proinflammatory adipokine and insulin resistance, gestational and type 2 diabetes mellitus. Cells. 2019;8(3):227. doi:10.3390/cells8030227
76. Thompson KJ, Austin RG, Nazari SS, et al. Altered fatty acid-binding protein 4 (FABP4) expression and function in human and animal models of hepatocellular carcinoma. Liver Int. 2018;38(6):1074–1083. doi:10.1111/liv.13639
77. Ye D, Zhang T, Lou G, et al. Plasma miR-17, miR-20a, miR-20b and miR-122 as potential biomarkers for diagnosis of NAFLD in type 2 diabetes mellitus patients. Life Sci. 2018;208:201–207. doi:10.1016/j.lfs.2018.07.029
78. Elemeery MN, Mohamed MA, Madkour MA, et al. MicroRNA signature in patients with hepatocellular carcinoma associated with type 2 diabetes. World J Gastroenterol. 2019;25(42):6322–6341. doi:10.3748/wjg.v25.i42.6322
79. Lu Z, Li X, Xu Y, et al. microRNA-17 functions as an oncogene by downregulating Smad3 expression in hepatocellular carcinoma. Cell Death Dis. 2019;10(10):723. doi:10.1038/s41419-019-1960-z
80. Fu Y, Liu M, Li F, et al. MiR-221 promotes hepatocellular carcinoma cells migration via targeting PHF2. Biomed Res Int. 2019;2019:4371405. doi:10.1155/2019/4371405
81. Li T, Li M, Hu S, et al. MiR-221 mediates the epithelial-mesenchymal transition of hepatocellular carcinoma by targeting AdipoR1. Int J Biol Macromol. 2017;103:1054–1061. doi:10.1016/j.ijbiomac.2017.05.108
82. Huang WT, Wang HL, Yang H, et al. Lower expressed miR-198 and its potential targets in hepatocellular carcinoma: a clinicopathological and in silico study. Onco Targets Ther. 2016;9:5163–5180. doi:10.2147/OTT.S108828
83. Tan S, Li R, Ding K, et al. miR-198 inhibits migration and invasion of hepatocellular carcinoma cells by targeting the HGF/c-MET pathway. FEBS Lett. 2011;585(14):2229–2234. doi:10.1016/j.febslet.2011.05.042
84. Varshney A, Panda JJ, Singh AK, et al. Targeted delivery of microRNA-199a-3p using self-assembled dipeptide nanoparticles efficiently reduces hepatocellular carcinoma in mice. Hepatology. 2018;67(4):1392–1407. doi:10.1002/hep.29643
85. Turato C, Fornari F, Pollutri D, et al. MiR-122 targets serpinB3 and is involved in sorafenib resistance in hepatocellular carcinoma. J Clin Med. 2019;8(2):171. doi:10.3390/jcm8020171
86. Liang YZ, Li JJ, Xiao HB, et al. Identification of stress-related microRNA biomarkers in type 2 diabetes mellitus: A systematic review and meta-analysis. J Diabetes. 2020;12(9):633–644. doi:10.1111/1753-0407.12643.
87. Zhang Z, Zheng W, Hai J. MicroRNA-148b expression is decreased in hepatocellular carcinoma and associated with prognosis. Med Oncol. 2014;31(6):984. doi:10.1007/s12032-014-0984-6.
88. He Y, Hwang S, Cai Y, et al. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatology. 2019;70(4):1150–1167. doi:10.1002/hep.30645.
89. Wang J, Chu Y, Xu M, et al. miR-21 promotes cell migration and invasion of hepatocellular carcinoma by targeting KLF5. Oncol Lett. 2019;17(2):2221–2227. doi:10.3892/ol.2018.9843.
90. Cao LQ, Yang XW, Chen YB, et al. Exosomal miR-21 regulates the TETs/PTENp1/PTEN pathway to promote hepatocellular carcinoma growth. Mol Cancer. 2019;18(1):148. doi:10.1186/s12943-019-1075-2.
91. Canfora EE, Meex RCR, Venema K, Blaak EE. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev Endocrinol. 2019;15(5):261–273. doi:10.1038/s41574-019-0156-z
92. Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249. doi:10.1038/nature11552.
93. Larsen N, Vogensen FK, van den Berg FW, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085. doi:10.1371/journal.pone.0009085
94. Inoue R, Ohue-Kitano R, Tsukahara T, et al. Prediction of functional profiles of gut microbiota from 16S rRNA metagenomic data provides a more robust evaluation of gut dysbiosis occurring in Japanese type 2 diabetic patients. J Clin Biochem Nutr. 2017;61(3):217–221. doi:10.3164/jcbn.17-44
95. Li BY, Xu XY, Gan RY, et al. Targeting gut microbiota for the prevention and management of diabetes mellitus by dietary natural products. Foods. 2019;8(10):440. doi:10.3390/foods8100440
96. Wan MLY, El-Nezami H. Targeting gut microbiota in hepatocellular carcinoma: probiotics as a novel therapy. Hepatobiliary Surg Nutr. 2018;7(1):11–20. doi:10.21037/hbsn.2017.12.07.
97. Chu H, Williams B, Schnabl B. Gut microbiota, fatty liver disease, and hepatocellular carcinoma. Liver Res. 2018;2(1):43–51. doi:10.1016/j.livres.2017.11.005
98. Ponziani FR, Bhoori S, Castelli C, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology. 2019;69(1):107–120. doi:10.1002/hep.30036
99. Gupta H, Youn GS, Shin MJ, et al. Role of gut microbiota in hepatocarcinogenesis. Microorganisms. 2019;7(5):121. doi:10.3390/microorganisms7050121
100. Zúñiga LA, Shen WJ, Joyce-Shaikh B, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol. 2010;185(11):6947–6959. doi:10.4049/jimmunol.1001269.
101. Tao L, Liu H, Gong Y. Role and mechanism of the Th17/Treg cell balance in the development and progression of insulin resistance. Mol Cell Biochem. 2019;459(1–2):183–188. doi:10.1007/s11010-019-03561-4.
102. Libby G, Donnelly LA, Donnan PT, et al. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32(9):1620–1625. doi:10.2337/dc08-2175.
103. Mallik R, Chowdhury TA. Metformin in cancer. Diabetes Res Clin Pract. 2018;143:409–419. doi:10.1016/j.diabres.2018.05.023.
104. Coyle C, Cafferty FH, Vale C, et al. Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis. Ann Oncol. 2016;27(12):2184–2195. doi:10.1093/annonc/mdw410.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.