Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Mechanical Forces and Mechanotransduction in COPD: Pathogenesis, Clinical Phenotypes, and Therapeutic Implications
Authors Li J ![]() , Qin E, Zhang C
, Qin E, Zhang C ![]() , Yu Y, Liu L, Sun J, Pu G, Tang J
, Yu Y, Liu L, Sun J, Pu G, Tang J
Received 10 January 2026
Accepted for publication 2 April 2026
Published 13 April 2026 Volume 2026:21 595107
DOI https://doi.org/10.2147/COPD.S595107
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Jingli Li,1,2 E Qin,1 Chunyi Zhang,1 Yuefang Yu,1 Lingjing Liu,3 Jian Sun,1 Guimei Pu,1 Jixian Tang1
1Department of Pulmonary and Critical Care Medicine, Shaoxing People’s Hospital, Shaoxing, Zhejiang, 312000, People’s Republic of China; 2Suzhou Medical College, Soochow University, Suzhou, Jiangsu, 215000, People’s Republic of China; 3Department of Pulmonary and Critical Care Medicine, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, 325000, People’s Republic of China
Correspondence: Jixian Tang, Department of Pulmonary and Critical Care Medicine, Shaoxing People’s Hospital, Shaoxing, Zhejiang, 312000, People’s Republic of China, Email [email protected]
Abstract: Chronic obstructive pulmonary disease (COPD) remains a leading cause of global morbidity and mortality. Despite advances in therapy, its complex pathogenesis involves mechanisms beyond the traditional paradigms of inflammation and protease–antiprotease imbalance. Emerging evidence indicates that COPD is also shaped by important mechanobiological processes, in which altered airway mechanics, parenchymal destruction, and respiratory muscle dysfunction create a pathological physical environment. In this narrative review, we synthesize current knowledge on how abnormal mechanical forces are sensed by key mechanosensors—including integrins, Piezo channels, and YAP/TAZ—and transduced into biochemical signals that drive chronic inflammation, fibrosis, and defective repair. We further discuss how these mechanotransduction feedback loops perpetuate structural injury and may help explain the clinical heterogeneity observed across airflow obstruction, emphysema, and exacerbation-prone phenotypes. Furthermore, we discuss therapeutic strategies, positioning pulmonary rehabilitation, lung volume reduction, and ventilation as interventions that restore mechanical homeostasis. Finally, we highlight the emerging possibility of targeting mechanosensitive pathways (e.g. ROCK and YAP/TAZ inhibitors) and utilizing mechanobiology-informed regenerative medicine. By integrating biomechanics with clinical management, this review provides a conceptual framework that may inform future efforts to move beyond symptomatic palliation toward more mechanism-based and potentially disease-modifying strategies in COPD.
Keywords: chronic obstructive pulmonary disease, COPD, mechanotransduction, extracellular matrix remodeling, YAP/TAZ, mechanical homeostasis
Introduction
Chronic obstructive pulmonary disease (COPD) is a major global health challenge characterized by persistent airflow limitation, chronic airway inflammation, and progressive structural changes in the lungs.1 According to the Global Burden of Disease (GBD) study, COPD remains one of the leading causes of morbidity and mortality worldwide, imposing a substantial socioeconomic burden.2 Despite advances in pharmacological and non-pharmacological therapies, disease progression continues in many patients, highlighting the need for deeper mechanistic insights beyond the traditional paradigms of inflammation, oxidative stress, and protease–antiprotease imbalance.3,4
In recent years, increasing attention has been directed toward the role of mechanical forces in lung physiology and pathology.5,6 The respiratory system is inherently exposed to dynamic mechanical stimuli, including cyclic stretching of alveoli during ventilation, shear stress generated by airflow, and compressive forces from chest wall and respiratory muscles.7 In physiological homeostasis, these mechanical cues are essential for maintaining tissue homeostasis, regulating epithelial integrity, and supporting immune defense.8,9 However, in COPD, profound alterations in lung structure and compliance significantly remodel the mechanical environment, which in turn may amplify inflammation,10 accelerate tissue destruction, and contribute to disease progression.11 The concept of mechanotransduction—the process by which cells sense and convert mechanical signals into biochemical responses—provides a useful framework for understanding these interactions.12,13 Airway epithelial cells, fibroblasts, smooth muscle cells, and immune cells all express mechanosensitive structures such as integrins, Piezo1/2 ion channels, and cytoskeletal complexes.5 Activation of these mechanosensors triggers downstream signaling cascades, including mitogen-activated protein kinase (MAPK), Rho-associated coiled-coil containing protein kinase (Rho/ROCK), and Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) pathways,14 thereby influencing cellular functions such as proliferation, apoptosis, fibrosis, and immune activation. Given that COPD involves repeated cycles of tissue injury and repair under abnormal mechanical stress, mechanotransduction likely plays a pivotal role in shaping the disease phenotype.
Several pathological features of COPD exemplify the importance of mechanical forces. Small airway narrowing and loss of alveolar attachments result in dynamic hyperinflation, exposing the parenchyma to excessive tensile stress.15 Alterations in extracellular matrix (ECM) composition, such as elastin degradation and aberrant collagen deposition, change tissue stiffness and disturb the balance between elastic recoil and airway collapse.16 At the same time, increased respiratory muscle load and reduced chest wall compliance impose abnormal strain on the diaphragm and accessory muscles.17,18 Together, these factors establish a self-perpetuating feedback loop of impaired mechanics that perpetuates both structural damage and functional decline. Importantly, the influence of mechanical forces in COPD is not limited to structural remodeling but also extends to clinical outcomes and therapeutic strategies. For instance, ventilatory support can both alleviate respiratory muscle fatigue and, if improperly adjusted, exacerbate ventilator-induced lung injury (VILI) by delivering injurious mechanical stress.19,20 Similarly, pulmonary rehabilitation and respiratory muscle training may partially restore mechanical efficiency, highlighting the translational relevance of this research field.21,22 Moreover, emerging studies suggest that targeting mechano-sensitive pathways could offer novel therapeutic avenues, particularly in modulating fibrosis and airway remodeling.6,23
Despite these insights, comprehensive understanding of the interplay between mechanical forces and COPD pathophysiology remains limited. Existing studies are often fragmented, focusing on isolated cell types, signaling pathways, or clinical observations.24,25 A systematic overview of how mechanical cues shape COPD onset, progression, and treatment response is therefore timely and necessary. This review aims to integrate current evidence from cellular, molecular, and clinical perspectives, elucidating the role of mechanical forces in COPD pathogenesis and progression. To provide a focused narrative synthesis, we considered evidence from experimental, translational, and clinical studies addressing the mechanical environment of COPD, mechanotransduction pathways, clinical phenotypes, and therapeutic implications. Throughout, we distinguish between mechanisms supported by substantial experimental or clinical evidence and emerging concepts that remain more speculative. Furthermore, we will explore potential therapeutic implications, ranging from respiratory rehabilitation to pharmacological interventions targeting mechanotransduction pathways, and highlight future directions for research in this emerging field.
Literature Search and Study Selection
This article was prepared as a narrative review. To provide a focused overview of the field, we performed a structured literature search in PubMed and Web of Science using combinations of the terms “COPD,” “chronic obstructive pulmonary disease,” “mechanical stress,” “mechanotransduction,” “airway mechanics,” “emphysema,” “extracellular matrix,” “Piezo,” “YAP/TAZ,” “Rho/ROCK,” and “lung remodeling.” We considered English-language literature published from database inception to January 2015.
Studies were prioritized if they provided mechanistic insight, translational relevance, or direct clinical evidence related to the role of mechanical forces and mechanotransduction in COPD pathogenesis, phenotypic heterogeneity, or therapeutic strategies. We also included selected seminal studies to provide conceptual and historical context. Conference abstracts and purely epidemiological studies were not emphasized, as the primary aim of this review was to synthesize mechanobiological rather than descriptive or population-level evidence. As this was a narrative review, study selection was guided by thematic relevance and conceptual contribution rather than by formal systematic review criteria.
Alterations of the Mechanical Environment in COPD
The lung is constantly exposed to a spectrum of mechanical stimuli, and the ability of its tissues to maintain homeostasis relies on a finely tuned balance between structural integrity and adaptive mechanotransduction. In COPD, chronic inflammation, ECM remodeling, and progressive airway and parenchymal destruction profoundly disrupt this balance, resulting in pathological changes in mechanics across multiple anatomical compartments.26 These mechanical derangements not only exacerbate airflow limitation and impair gas exchange but also feed back to cellular and molecular processes, reinforcing disease progression. This section summarizes the major alterations in the mechanical environment of COPD at the levels of the small airways, alveoli, ECM, and respiratory muscles.
Small Airway Dysfunction and Alveolar Stress Concentration
Irreversible narrowing of small conducting airways (<2 mm in diameter) is a defining feature of COPD and substantially increases airflow resistance.27 During expiration, the loss of alveolar attachments reduces radial traction that normally stabilizes airway patency.28 As a consequence, airway walls are subjected to abnormal compressive and shear stresses, which promote dynamic airway collapse, particularly during forced expiration and exercise.29 Computational fluid dynamics studies further demonstrate markedly increased turbulence and shear stress in narrowed COPD airways, potentially contributing to epithelial injury and mucus hypersecretion.
At the alveolar level, destruction of alveolar walls reduces elastic recoil and increases lung compliance, favoring air trapping and hyperinflation.30 The remaining alveolar units become exposed to excessive cyclic stretch during tidal breathing.11 Repeated overdistension has been shown to induce epithelial apoptosis, disrupt tight junctions, and stimulate the release of pro-inflammatory mediators such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α).7 In parallel, abnormal tensile forces accelerate protease activity and ECM degradation, reinforcing emphysematous remodeling.10
A hallmark of COPD mechanics is the heterogeneity of regional behavior.31 Severely emphysematous regions exhibit abnormally high compliance and reduced resistance, whereas adjacent preserved areas are stiffer and subject to compensatory overinflation.30 This heterogeneity produces uneven stress distribution and the phenomenon of “stress focusing,” in which mechanical forces concentrate at the interfaces between diseased and relatively normal tissue.32 Such focal stress concentration is thought to drive progressive alveolar rupture and bullae formation, providing a mechanical explanation for the patchy progression of emphysema.33 Collectively, these observations support a spatially heterogeneous strain field in COPD lungs, within which stress-focusing interfaces operate as biomechanical “hotspots” for progressive failure.
Extracellular Matrix Remodeling as a Driver of Tissue Stiffening
Chronic exposure to cigarette smoke and inflammatory mediators promotes an imbalance between proteases (eg., neutrophil elastase, matrix metalloproteinases) and antiproteases.34 The resulting degradation of elastin fibers diminishes parenchymal recoil, altering the stress–strain relationship of the lung. Reduced elastic recoil decreases the tethering forces that keep small airways open, further predisposing them to collapse.
Paradoxically, while some lung regions lose architectural integrity, others undergo fibrosis-like remodeling characterized by excessive collagen deposition.35 This results in increased local stiffness and disrupted viscoelastic properties of the ECM. In vitro studies have shown that fibroblasts cultured on stiff substrates adopt an activated myofibroblast phenotype, producing additional ECM components and secreting profibrotic mediators such as transforming growth factor-β (TGF-β).36 This establishes a self-reinforcing cycle of stiffening and remodeling that contributes to airway wall thickening and progressive fibrosis.
ECM remodeling also alters the transmission of mechanical cues through integrins and focal adhesion complexes, thereby modifying downstream mechanotransduction.37 As a consequence, epithelial cells, fibroblasts, and smooth muscle cells are exposed to aberrant mechanical inputs that regulate their proliferation, survival, and inflammatory responses. Stiffened ECM may additionally impair mechanosensitive ion channels such as Piezo1, altering calcium influx and downstream signaling.38 Importantly, cytoskeletal tension and substrate rigidity directly gate αvβ6-mediated activation of latent TGF-β, meaning that as the matrix stiffens, the mechanical threshold required for TGF-β activation is lowered. This creates a positive feedback loop in which increased rigidity enhances TGF-β signaling, driving further ECM remodeling and fibrotic progression.39
Respiratory Muscle Overload and Impaired Chest Wall Mechanics
In COPD, hyperinflation flattens the diaphragm, reducing its curvature and mechanical efficiency.40 This geometric disadvantage increases the inspiratory load and forces the diaphragm to contract against elevated lung volumes. Chronic overloading leads to structural adaptations, including fiber-type transformation and mitochondrial dysfunction, which compromise contractile capacity.41 Over time, the diaphragm becomes fatigued and less responsive to neural drive, contributing to dyspnea and exercise intolerance.42
As the diaphragm weakens, accessory muscles of respiration, such as the sternocleidomastoids and scalene muscles, are increasingly recruited.43 Persistent use of these muscles imposes abnormal mechanical strain on the chest wall, leading to structural remodeling such as barrel-chest configuration.44 Thoracic cage stiffening further reduces chest wall compliance, exacerbating the mechanical disadvantage of the respiratory muscles.
For patients requiring ventilatory support, these respiratory muscle alterations have important consequences. High levels of intrinsic positive end-expiratory pressure (PEEPi) due to dynamic hyperinflation increase the threshold load that must be overcome at the onset of inspiration.45 If ventilator settings are not carefully adjusted, the resulting excessive transpulmonary pressures can worsen alveolar overdistension and predispose to ventilator-induced lung injury.46 Thus, understanding the altered respiratory muscle and chest wall mechanics in COPD is crucial for optimizing ventilatory strategies.
Self-Perpetuating Cycles of Mechanical Injury
The alterations described above interact to create self-perpetuating vicious cycles. Loss of alveolar integrity increases hyperinflation, which in turn flattens the diaphragm and impairs ventilation, leading to further air trapping. ECM stiffening enhances abnormal mechanical signaling, which stimulates additional inflammation and fibrosis. Mechanical heterogeneity drives stress concentration at tissue borders, causing progressive alveolar rupture and structural failure. Collectively, these changes illustrate how COPD progression is reinforced by a complex interplay of mechanical forces, making biomechanics a critical but often underappreciated dimension of disease pathophysiology. The major alterations in the mechanical environment of COPD across airway, alveolar, extracellular matrix, and respiratory muscle compartments are summarized in Figure 1.
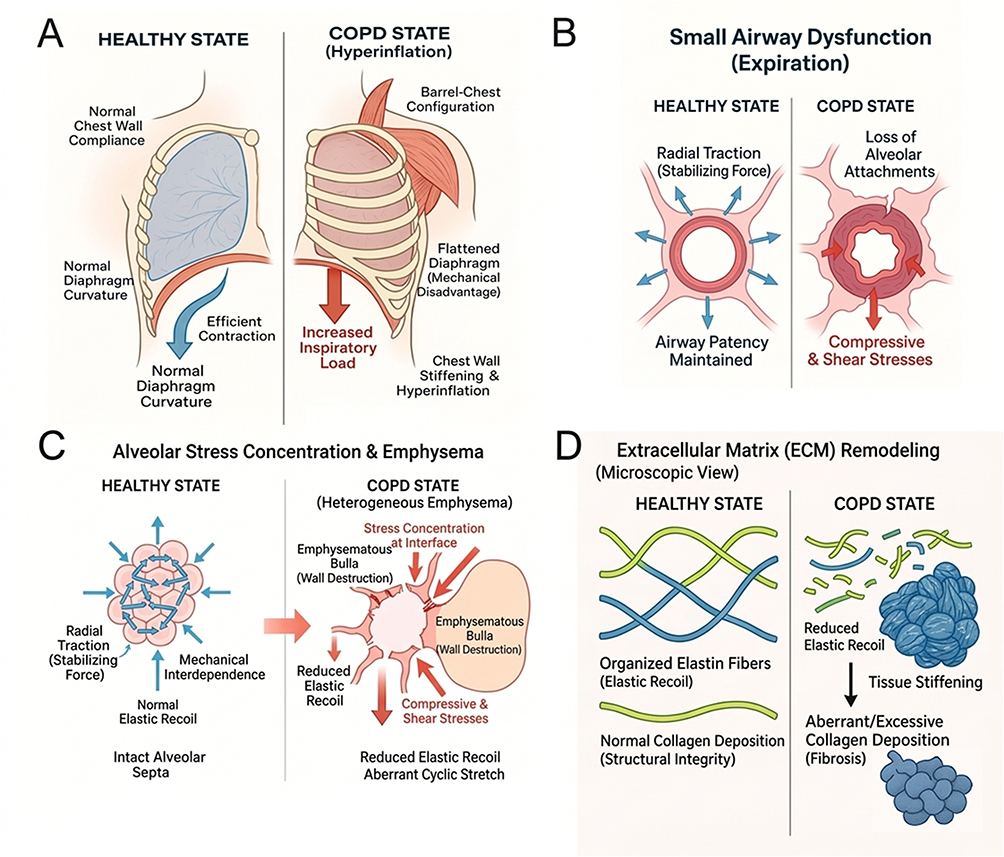 |
Figure 1 Alterations of the mechanical environment in COPD across multiple anatomical scales. (A) Respiratory muscle overload and chest wall mechanics. In the healthy state (left), the diaphragm maintains a normal curvature for efficient contraction. In COPD (right), air trapping leads to dynamic hyperinflation, resulting in a barrel-chest configuration and diaphragm flattening. This mechanical disadvantage increases the inspiratory load and recruits accessory muscles, while chest wall stiffening further impairs compliance. (B) Small airway dysfunction. Healthy airways (left) are stabilized by radial traction from alveolar attachments. In COPD (right), the loss of these attachments reduces radial traction, rendering the airway wall susceptible to dynamic collapse, particularly during expiration, and exposing it to abnormal compressive and shear stresses. (C) Alveolar stress concentration and emphysema. Healthy alveoli (left) benefit from mechanical interdependence, distributing stress evenly. In heterogeneous emphysema (right), the destruction of alveolar walls creates bullae. Mechanical forces concentrate at the interface between the bulla and preserved tissue (“stress concentration”), driving progressive rupture and aberrant cyclic stretch. (D) Extracellular matrix (ECM) remodeling. The healthy ECM (left) features organized elastin and collagen networks that provide recoil and structural integrity. In COPD (right), an imbalance of proteases leads to elastin degradation (reduced recoil) and aberrant collagen deposition (fibrosis/stiffening), disrupting the mechanical homeostasis of the lung parenchyma. |
Mechanotransduction and Cellular Responses in COPD
Mechanical forces influence virtually every aspect of lung biology, and their pathological alterations in COPD exert profound effects at the cellular and molecular levels. The process by which cells detect and convert mechanical stimuli into biochemical signals—mechanotransduction—provides a mechanistic framework linking tissue mechanics to inflammation, remodeling, and impaired repair.47 This section summarizes the key mechanosensors, signaling pathways, and cellular responses relevant to COPD pathogenesis.
Mechanosensors Orchestrating Cellular Responses
A wide array of mechanosensors enables lung cells to detect and respond to physical forces, and their dysregulation plays a critical role in COPD. Among these, integrins represent the most extensively characterized. These transmembrane receptors connect the ECM to the cytoskeleton and mediate bidirectional signaling in response to mechanical cues.48 In COPD, ECM remodeling alters ligand availability and increases matrix stiffness, leading to aberrant integrin activation.49 Engagement of β1 and β3 integrins promotes focal adhesion assembly and recruitment of focal adhesion kinase (FAK), which subsequently triggers downstream cascades such as MAPK and phosphoinositide 3-kinase (PI3K).50 In fibroblasts, enhanced integrin signaling fosters a profibrotic phenotype with excessive ECM production, whereas in epithelial cells it influences wound repair and barrier integrity, contributing to abnormal remodeling.51
Mechanosensitive ion channels constitute another important class of force sensors.52 Piezo1 and Piezo2, recently identified in airway epithelial and immune cells, respond to membrane tension by mediating Ca2⁺ influx.53 The rise in intracellular calcium activates calcineurin–nuclear factor of activated T cells (NFAT) signaling, promotes cytokine release, and under chronic stimulation may drive cell injury and apoptosis.38 In COPD, hyperinflation and repetitive cyclic stretch are likely to activate Piezo-dependent pathways, although their precise contribution to long-term tissue remodeling is still under investigation.5
YAP/TAZ function as nuclear effectors of mechanical stress.54 These proteins shuttle between the cytoplasm and nucleus in response to cytoskeletal tension and ECM stiffness.55 When activated, nuclear YAP/TAZ drive transcriptional programs regulating proliferation, survival, and ECM synthesis.56 In the context of COPD, where tissue stiffening and cytoskeletal rearrangements are pronounced, YAP/TAZ activity is expected to increase, thereby promoting aberrant epithelial repair and fibroblast activation and reinforcing pathogenic remodeling.
Cytoskeletal and junctional structures also contribute directly to mechanosensing. Actin filaments, microtubules, and intermediate filaments not only provide structural support but also transduce mechanical inputs by modulating cytoskeletal tension and signaling cascades.57 Cyclic stretch induces actin stress fiber formation and activates Rho GTPases, processes that regulate cell contractility and mechanotransduction.58 In airway epithelial cells, tension-sensitive junctional complexes such as E-cadherin and claudins are disrupted under abnormal mechanical stress, leading to barrier dysfunction and increased susceptibility to environmental insults.59 COPD patients commonly exhibit reduced epithelial integrity, and altered cytoskeletal mechanotransduction is increasingly recognized as a potential underlying mechanism.
Beyond canonical integrins and ion channels, focal-adhesion adaptor proteins can act as amplifier hubs for mechanically induced epithelial programs. Recent human airway data identify Hic-5 (TGFB1I1) as a mechano-responsive scaffold whose expression is rapidly induced by epithelial compression and by TGF-β1; single-cell RNA-seq further localizes this response to basal cells during bronchoconstriction. Mechanistically, Hic-5 links force sensing to cytoskeletal tension and transcriptional outputs, positioning it as an epithelial “gain control” for mechanical stress. While characterized in asthma models, the same epithelial compression and TGF-β activation occur in COPD small airways, arguing that Hic-5–centric scaffolding may generalize to COPD mechanobiology.60
Together, these mechanosensors—integrins, mechanosensitive ion channels, YAP/TAZ, and cytoskeletal elements—form a tightly interconnected system that converts abnormal mechanical cues in COPD into maladaptive cellular responses.
Signaling Cascades Linking Mechanics to Pathology
Activation of mechanosensors initiates a variety of signaling cascades, many of which are chronically dysregulated in COPD. One of the most prominent is the MAPK pathway. Mechanical stretch and shear stress activate ERK, JNK, and p38, driving transcription of pro-inflammatory mediators and sustaining airway inflammation.61 In parallel, integrin–FAK engagement and Piezo1-mediated calcium influx converge on nuclear factor-κB (NF-κB) activation, amplifying the expression of cytokines such as IL-1β, IL-6, and TNF-α, as well as chemokines including C-X-C motif chemokine ligand 8 (CXCL8/IL-8).62 This pro-inflammatory response is counterbalanced by mechanosensitive regulatory circuits. For example, a mechanosensitive microRNA, miR-146a, provides a negative-feedback brake on pressure-induced NF-κB activity. Oscillatory pressure robustly upregulates miR-146a in human small airway epithelial cells, and gain- and loss-of-function experiments demonstrate that miR-146a attenuates IL-6/IL-8 secretion by targeting interleukin-1 receptor-associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6) within the TLR–NF-κB axis. Conversely, inhibition of miR-146a amplifies the inflammatory output. The magnitude-dependence of these effects (20 vs. 5 cmH2O) further supports a dose–response relationship between mechanical stress and inflammatory signaling.63 Persistent activation of these pathways underlies the neutrophilic inflammation that is a hallmark of COPD. These mechanotransduction pathways and their convergence points are schematically summarized in Figure 2.
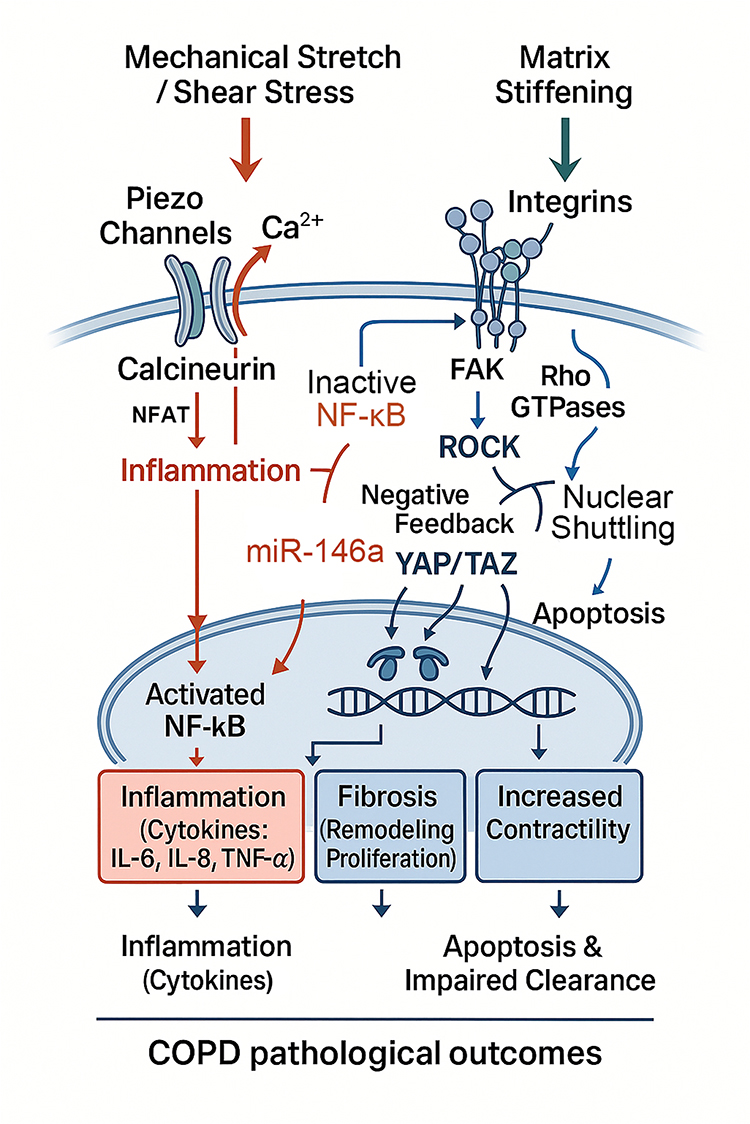 |
Figure 2 Mechanotransduction signaling cascades connecting physical forces to pathological outcomes in COPD. External mechanical stimuli activate distinct intracellular pathways. (Left) Mechanical stretch and shear stress gate Piezo channels, leading to Ca2⁺ influx. This calcium signal activates the Calcineurin/NFAT axis and NF-κB, driving the transcription of pro-inflammatory cytokines (IL-6, IL-8, TNF-α). A mechanosensitive negative feedback loop involving miR-146a serves as a brake on this inflammatory signaling. (Right) Matrix stiffening is sensed by Integrins, which recruit Focal Adhesion Kinase (FAK) and activate Rho GTPases. These pathways stimulate ROCK (enhancing contractility) and promote the nuclear shuttling of YAP/TAZ. (Bottom) Inside the nucleus, the convergence of activated NF-κB and YAP/TAZ transcriptional programs promotes chronic inflammation, fibrosis (proliferation/remodeling), and apoptosis, ultimately fueling COPD pathogenesis. |
Rho family GTPases and their downstream effector, Rho-associated protein kinase (ROCK), represent another major axis of mechanical signaling.64 By regulating actin cytoskeleton dynamics, Rho/ROCK enhances cellular contractility, motility, and stress fiber formation. In fibroblasts, this signaling promotes myofibroblast differentiation and excessive ECM deposition,65 whereas in airway smooth muscle it contributes to increased tone and hyperresponsiveness.66 Elevated ROCK activity has been observed in COPD tissues, linking abnormal mechanical stress to persistent structural remodeling.
Calcium-dependent pathways further contribute to mechanotransduction. Sustained Ca2⁺ influx through Piezo channels activates calcineurin and calmodulin-dependent kinases, which in turn stimulate transcription factors such as NFAT.38 These signals regulate apoptosis, metabolic adaptation, and inflammatory gene expression. In COPD, abnormal calcium signaling is implicated in epithelial cell death and impaired mucociliary clearance, processes that exacerbate vulnerability to environmental insults.67 Moreover, Piezo1-driven Ca2⁺ signals can interface with the Hippo pathway to modulate YAP/TAZ activity, providing a mechanistic bridge from membrane tension sensing to transcriptional programs governing proliferation and repair under mechanical load.68
Finally, the YAP/TAZ–TEAD (TEA domain transcription factor) transcriptional axis acts as a key effector of mechanical inputs. Developmental evidence highlights this effector role: epithelial loss of YAP compromises mechanical force generation during branching morphogenesis, with marked reductions in phosphorylated myosin light chain (pMLC) and failure of tissue recoil after laser ablation. Genomic and ChIP analyses further identify BCAM, S1PR2, and NUAK2 as direct YAP–TEAD targets that converge on RhoA–ROCK signaling or MLC phosphatase to sustain pMLC, delineating a YAP→RhoGEF/RhoA–ROCK→pMLC axis for contractility. Taken together, YAP functions not only as a mechanosensitive transcriptional switch but also as an active governor of actomyosin output, a logic likely relevant to chronic epithelial remodeling under mechanical load in COPD.69 Nuclear localization of YAP/TAZ under conditions of matrix stiffening and cytoskeletal tension drives the expression of genes involved in proliferation (eg., cyclin D1), survival (eg., Bcl-2), and matrix remodeling (eg., connective tissue growth factor).70 Dysregulated YAP/TAZ activity in COPD may help explain the paradoxical coexistence of fibrosis and emphysema, reflecting defective repair responses coupled with maladaptive remodeling.
Collectively, these signaling cascades demonstrate how mechanical stress is transduced into persistent inflammatory and fibrotic responses, embedding abnormal mechanics into the molecular circuitry of COPD. Rather than acting as isolated pathways, these signaling axes form an interconnected mechanotransduction network in which calcium influx, cytoskeletal tension, inflammatory amplification, and transcriptional reprogramming converge on common outputs, including barrier failure, fibroblast activation, and maladaptive repair. At the same time, not all findings are fully concordant across experimental systems. The direction and magnitude of mechanotransduction responses may vary according to cell type, anatomical compartment, disease stage, and the intensity or duration of mechanical loading, suggesting that some pathways may exert adaptive effects under physiological conditions but become maladaptive when chronically or excessively activated.
Cellular Remodeling Under Abnormal Mechanical Stress
The downstream consequences of abnormal mechanotransduction are evident across structural, mesenchymal, and immune cell populations within the lung. Airway epithelial cells, which form the first barrier to airflow and hyperinflation, are highly vulnerable to cyclic stretch.71 Excessive mechanical loading induces apoptosis, disrupts tight junctions, and reduces mucociliary clearance. These injured cells release alarmins such as IL-33 and High Mobility Group Box 1 (HMGB1), which further activate innate immune responses. In addition, abnormal mechanotransduction interferes with epithelial regeneration, driving squamous metaplasia and long-term architectural distortion of the airways.72
Fibroblasts are highly sensitive to matrix stiffness. In stiffened COPD lungs, fibroblasts acquire an activated myofibroblast phenotype characterized by α-smooth muscle actin expression and increased collagen production.73 Mechanotransduction through integrins and YAP/TAZ sustains this profibrotic program. Persistent fibroblast activation contributes to airway wall thickening and peribronchial fibrosis, reinforcing airflow limitation.74
Airway smooth muscle cells also respond strongly to abnormal mechanics. Exposure to cyclic strain enhances their proliferation and contractility, largely through Rho/ROCK and MAPK signaling. In addition, they secrete growth factors and pro-inflammatory mediators, amplifying airway remodeling and inflammation.75 The resulting increase in muscle mass narrows the airway lumen, further altering local mechanical stress and creating a reinforcing feedback loop.
Immune cells, including macrophages and neutrophils, are increasingly recognized as mechano-responsive.76 Substrate stiffness and stretch modulate macrophage polarization, often skewing them toward a pro-inflammatory M1 phenotype. Piezo1-mediated calcium influx regulates phagocytic capacity and cytokine release, suggesting that mechanical stress may directly shape innate immune responses.77 This provides a mechanistic explanation for the persistence of inflammation in COPD, independent of ongoing chemical exposures.
Endothelial cells, though less extensively studied, are also subject to abnormal forces in COPD, particularly under conditions of hyperinflation and mechanical ventilation. Excessive cyclic strain and high-tidal-volume ventilation activate Ca2⁺-entry modules—most prominently Stim1/Orai1 and the mechanosensitive channel Piezo1—driving barrier failure via VE-cadherin–centered junctional remodeling. In experimental systems, 18% cyclic stretch or injurious ventilation upregulates Stim1/Orai1 and elevates intracellular Ca2⁺, which engages PKCα and Pyk2 to activate Src and promote VE-cadherin phosphorylation, thereby increasing permeability. Complementarily, Piezo1-dependent Ca2⁺ influx signals internalization/degradation of VE-cadherin/p120-catenin, destabilizing adherens junctions under mechanical load. Junctional stability is further tuned by YAP activity; loss of endothelial YAP exaggerates VE-cadherin phosphorylation and uncoupling from catenins during mechanical ventilation, reinforcing leak. Upstream Ca2⁺ signaling converges on MLCK/ROCK to increase actomyosin tension and paracellular gaps, mechanistically linking stretch sensing to cytoskeletal contractility.78 These changes increase endothelial permeability, enhance leukocyte adhesion, and promote inflammatory infiltration, thereby contributing to vascular remodeling and pulmonary hypertension.
Taken together, these findings suggest that abnormal mechanical stress does not simply affect individual cell populations in parallel; rather, it reprograms communication across epithelial, mesenchymal, immune, and vascular compartments. In this framework, barrier disruption, matrix stiffening, smooth muscle contraction, inflammatory persistence, and endothelial leak should be viewed as mutually reinforcing features of a mechanically coupled remodeling process rather than as isolated pathological events. These cell type–specific remodeling processes under abnormal mechanical stress are schematically summarized in Figure 3.
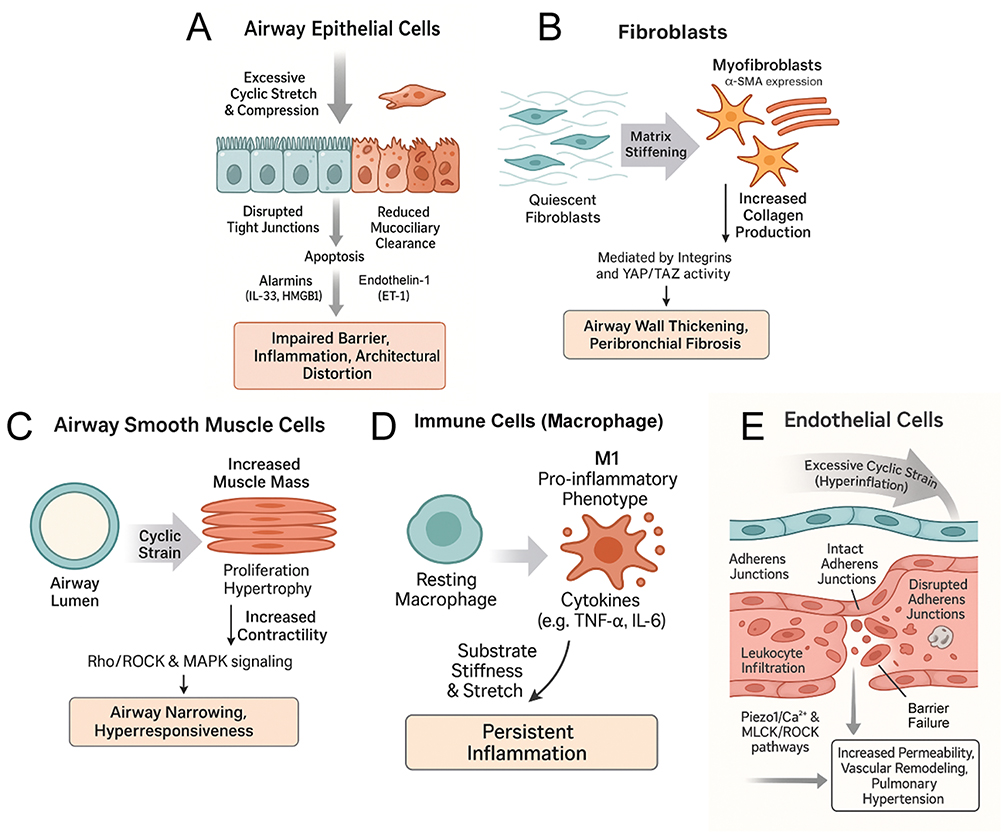 |
Figure 3 Cell type–specific remodeling and dysfunction induced by abnormal mechanical stress in COPD. (A) Airway Epithelial Cells: Excessive cyclic stretch and compression disrupt tight junctions and induce apoptosis, leading to reduced mucociliary clearance. Injured cells release alarmins (e.g., IL-33, HMGB1) and Endothelin-1 (ET-1), resulting in impaired barrier function and architectural distortion. (B) Fibroblasts: Matrix stiffening promotes the transition of quiescent fibroblasts to an activated myofibroblast phenotype (characterized by α-SMA expression). Mediated by integrins and YAP/TAZ activity, these cells produce excessive collagen, driving airway wall thickening and peribronchial fibrosis. (C) Airway Smooth Muscle Cells: Cyclic strain induces cellular proliferation and hypertrophy. Signaling via Rho/ROCK and MAPK pathways increases contractility, contributing to airway narrowing and hyperresponsiveness. (D) Immune Cells (Macrophages): Mechanical cues such as substrate stiffness and stretch drive macrophage polarization toward a pro-inflammatory M1 phenotype, perpetuating persistent inflammation through cytokine release (e.g., TNF-α, IL-6). (E) Endothelial Cells: Excessive cyclic strain (e.g., from hyperinflation) disrupts adherens junctions and causes barrier failure via Piezo1/Ca2⁺ and MLCK/ROCK pathways. This facilitates leukocyte infiltration and contributes to vascular remodeling and pulmonary hypertension. |
Feedback Loops Reinforcing COPD Progression
The effects of mechanotransduction in COPD are not linear but interconnected, forming complex feedback loops that perpetuate disease progression. Stiffened ECM activates fibroblasts, which in turn secrete additional collagen and matrix proteins, further increasing stiffness and driving a feed-forward profibrotic cascade. Hyperinflation-induced epithelial injury amplifies inflammation, which accelerates protease release, elastin degradation, and loss of compliance. At the same time, immune cells activated by stretch release proteases that exacerbate alveolar destruction, worsening emphysema and amplifying regional mechanical heterogeneity.
These intertwined processes exemplify the bidirectional relationship between mechanics and biology: structural changes alter local forces, which activate cellular signaling pathways, which then drive further structural remodeling. Such self-reinforcing loops provide a compelling explanation for the chronicity, progression, and heterogeneity of COPD, highlighting mechanotransduction as a central driver of the disease.
Mechanical Forces and Clinical Phenotypes of COPD
COPD is clinically defined by chronic airflow limitation that is not fully reversible, but its presentation is highly heterogeneous.79 Patients may predominantly exhibit chronic bronchitis, emphysema, frequent exacerbations, or systemic manifestations such as muscle wasting.80 Although inflammation and environmental exposures provide a unifying etiological framework, the diversity of clinical phenotypes is increasingly recognized as the outcome of complex interactions between structural remodeling, cellular responses, and altered mechanical forces.81 This section explores how aberrant mechanics contribute to major COPD phenotypes, including airflow obstruction, emphysema, and acute exacerbations, and how these processes shape disease heterogeneity.
Airflow Obstruction as a Consequence of Mechanical Collapse
Airflow obstruction in COPD primarily originates in the small conducting airways, where chronic inflammation, mucus hypersecretion, and wall thickening progressively narrow the lumen.82 The accompanying loss of alveolar attachments diminishes radial traction, leaving airways highly susceptible to dynamic collapse during expiration.83 This collapse is further aggravated under exertion, when increased expiratory flow rates impose higher shear and compressive stresses.84 These mechanical derangements directly manifest as reduced forced expiratory volume in one second (FEV1), increased residual volume, and impaired ventilatory efficiency. Epithelial compression not only injures the barrier but also programs a mechano-secretory state—exemplified by Hic-5–dependent endothelin-1 (ET-1) release—that could amplify small-airway tone. Although shown in asthma exacerbation models, analogous cycles of micro-collapse and compression likely occur in COPD small airways, suggesting a convergent epithelial–smooth-muscle feedback loop in chronic airflow limitation.60
Imaging and computational studies underscore that airway narrowing in COPD is patchy, producing regions of high resistance adjacent to relatively preserved segments. This heterogeneity results in maldistribution of ventilation, increased turbulence, and localized “hotspots” of shear injury.85 Such mechanical stress accelerates epithelial damage and perpetuates local inflammation, reinforcing airway remodeling. Clinically, this explains why airflow limitation is often disproportionate to the degree of visible emphysema or mucus plugging alone.
Dynamic hyperinflation represents a hallmark mechanical abnormality in COPD. During exercise, incomplete emptying of the lungs causes end-expiratory lung volume to rise, increasing PEEPi.86 The diaphragm becomes flattened and mechanically disadvantaged, requiring greater effort to initiate inspiration.87 This mechanical inefficiency contributes to exertional dyspnea, the cardinal symptom of COPD, and directly links disordered mechanics to functional impairment.88 Hyperinflation also increases mechanical loading on inspiratory muscles, promoting fatigue and reducing exercise tolerance.89 Thus, airflow limitation in COPD cannot be fully understood without recognizing its mechanical underpinnings.
Emphysema Shaped by Stress Concentration and Loss of Mechanical Interdependence
Emphysema is defined by irreversible alveolar wall destruction and abnormal airspace enlargement, and while protease–antiprotease imbalance and oxidative stress remain central to its pathogenesis, mechanical factors critically determine the sites and trajectory of tissue failure.11 At the interface between damaged and preserved regions, stress concentration magnifies tensile forces, predisposing alveolar septa to rupture.90 Over time, repetitive overdistension during tidal breathing accelerates tissue breakdown, explaining why emphysema often progresses in a patchy, heterogeneous fashion. Importantly, this progression is unlikely to be linear. As alveolar units are progressively lost, the mechanical load borne by the remaining septa increases disproportionately, so that once a critical threshold of structural failure is reached, local stress amplification may trigger accelerated rupture and rapid expansion of emphysematous regions.11,91
The concept of alveolar interdependence is particularly important in this context. In the healthy lung, alveoli are mechanically tethered to one another, allowing stresses to be evenly distributed. When alveolar walls are destroyed, this tethering is lost, and the remaining units bear disproportionate loads, making them more vulnerable to rupture. This loss of interdependence creates a mechanically unstable system in which each additional septal failure redistributes stress to adjacent regions, further lowering the threshold for subsequent damage.92 Clinically, this translates into increased compliance but diminished elastic recoil—a paradoxical mechanical profile that underlies both hyperinflation and airflow obstruction.93
At the cellular level, alveolar overdistension activates mechanosensors such as Piezo channels and YAP/TAZ, triggering epithelial and endothelial injury, cytokine release, and recruitment of inflammatory cells.5 These pathways reinforce local tissue damage and promote maladaptive fibroblast activation in adjacent areas, where fibrotic remodeling paradoxically coexists with emphysematous destruction.94 Thus, emphysema is not a passive structural outcome of chemical injury but an actively driven process of mechanical stress amplification and dysregulated mechanotransduction.
Exacerbations as Crises of Mechanical Failure
Acute exacerbations of COPD (AECOPD) represent episodes of rapid clinical deterioration, often precipitated by infection or environmental triggers, during which mechanical stresses escalate dramatically.95 Airway obstruction acutely worsens, airway resistance increases, and dynamic hyperinflation intensifies, sharply raising inspiratory threshold loads.96 Respiratory muscles are forced to operate near or beyond their physiological limits, predisposing patients to acute ventilatory failure. This explains why exacerbations are such a frequent cause of hospitalization and mortality.
In patients requiring ventilatory support, mechanical considerations become even more critical. Inappropriate ventilator settings, such as high tidal volumes or insufficient expiratory times, can exacerbate hyperinflation, elevate transpulmonary pressures, and induce VALI.97 The emerging concept of mechanical power, which integrates driving pressure, tidal volume, flow, and respiratory rate,98 provides a quantitative framework for assessing the risk of ventilator-induced damage. Tailoring ventilator strategies to minimize mechanical power has become an important principle in managing AECOPD.
The systemic consequences of exacerbation-related mechanical stress extend beyond the lung. Overloaded respiratory muscles undergo catabolic signaling and mitochondrial dysfunction, contributing to skeletal muscle wasting.99 Cyclic strain on pulmonary vasculature may exacerbate endothelial dysfunction, driving pulmonary hypertension and cardiovascular complications.100 These observations emphasize that AECOPD is not only an inflammatory or infectious event but also a crisis of mechanical failure, with wide-ranging implications for patient outcomes. Incorporating mechanical-power minimization into ventilatory protocols may therefore represent a tangible pathway to mitigate exacerbation-related injury.
Mechanics as a Determinant of Disease Heterogeneity
One of the most challenging aspects of COPD management is its heterogeneity: patients with similar environmental exposures often develop divergent clinical phenotypes.101 Some present with predominant chronic bronchitis, others with emphysema, and still others with frequent exacerbations. Mechanical forces may be a key determinant of this variability because different structural weak points within the respiratory system give rise to different dominant mechanical consequences. Individuals in whom the small airways are more compliant or less effectively tethered by the surrounding parenchyma may be more prone to expiratory narrowing and dynamic collapse, thereby favoring airflow obstruction, mucus retention, and chronic bronchitic features. By contrast, when the mechanically vulnerable compartment is the alveolar septal network, repetitive tensile loading and stress concentration may preferentially promote septal rupture, loss of elastic recoil, and emphysematous enlargement.102 Likewise, patients with limited respiratory muscle reserve or greater susceptibility to hyperinflation-induced loading may be more likely to develop exertional dyspnea, ventilatory failure, and frequent or severe exacerbations.103 Viewed in this way, COPD phenotypes may differ not only in inflammatory burden or exposure history, but also in the dominant site of mechanical vulnerability—small airways, alveolar septa, or respiratory muscles—which may help explain why patients with superficially similar risk profiles develop markedly different structural and clinical trajectories.
Advances in imaging have made it possible to visualize mechanical heterogeneity in vivo. Quantitative computed tomography (CT) provides detailed assessments of airway dimensions and parenchymal density,104 while magnetic resonance imaging (MRI) and hyperpolarized gas techniques reveal regional ventilation defects.105 More recently, methods such as strain mapping and elastography have been used to evaluate regional compliance and stress distribution.106 Correlations between these imaging-derived mechanical indices and clinical outcomes support the view that mechanics actively shape phenotypic expression.
Mechanical biomarkers also hold prognostic value. Dynamic hyperinflation measured during exercise predicts mortality and poor functional status.107 Biochemical markers of matrix destruction may provide complementary information. In particular, desmosine—an elastin-specific cross-linking amino acid released during elastin degradation—has emerged as a structural biomarker of parenchymal injury.108 Elevated desmosine levels in blood, urine, or sputum have been associated with increased elastin breakdown and may reflect the progression of emphysematous remodeling, thereby linking structural matrix failure to the mechanical deterioration of the lung.109,110 Imaging-based assessments of regional strain heterogeneity correlate with emphysema progression and may identify patients at higher risk of decline.29 Incorporating such mechanical and structural biomarkers into clinical evaluation could refine risk stratification and guide personalized interventions, bridging the gap between pathophysiology and precision medicine. Figure 4 provides an integrative schematic summarizing how abnormal mechanical cues drive disease progression in COPD through cell type–specific responses and tissue remodeling.
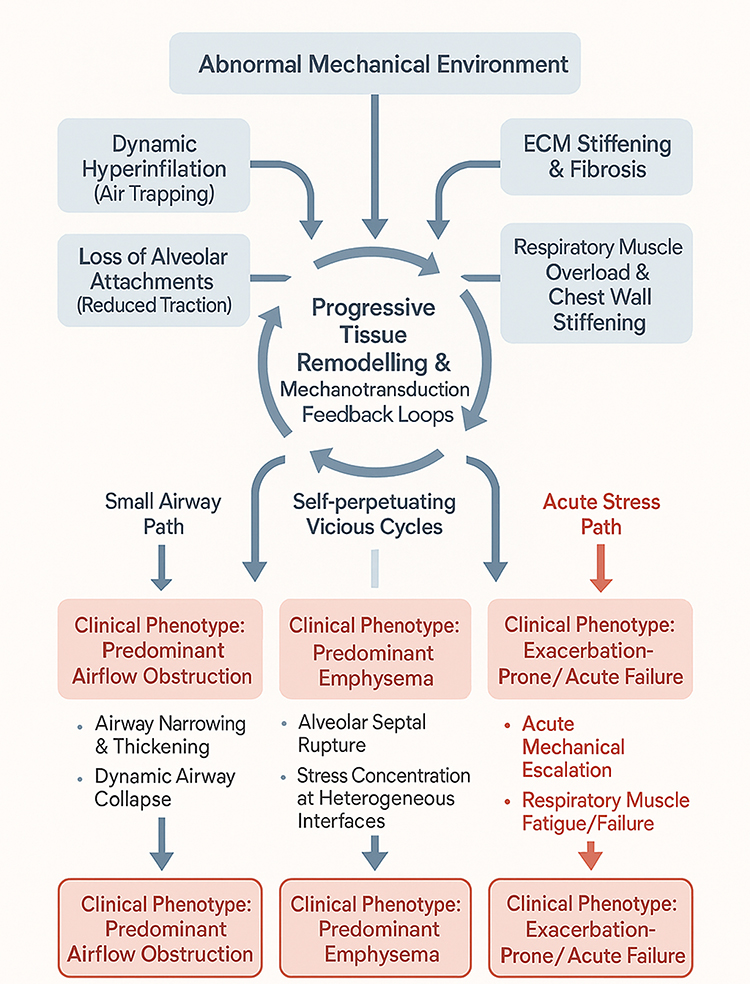 |
Figure 4 Mechanical drivers of clinical heterogeneity in COPD. The diagram illustrates how an abnormal mechanical environment fuels self-perpetuating mechanotransduction feedback loops, which subsequently diverge into distinct clinical phenotypes. (Left) Predominant Airflow Obstruction: The “Small Airway Path” is driven by airway narrowing, wall thickening, and dynamic airway collapse due to reduced radial traction. (Middle) Predominant Emphysema: Driven by “Self-perpetuating Vicious Cycles,” this phenotype involves alveolar septal rupture and stress concentration at the interfaces between diseased and preserved tissue, which accelerates parenchymal destruction. (Right) Exacerbation-Prone/Acute Failure: The “Acute Stress Path” represents a crisis of mechanical failure. It is characterized by acute mechanical escalation and respiratory muscle fatigue, often precipitated by increased inspiratory loads. |
Therapeutic Strategies Targeting Mechanical Forces in COPD
Recognition of the central role of mechanical forces in COPD pathogenesis opens new therapeutic opportunities that extend beyond conventional pharmacological suppression of inflammation or relief of bronchoconstriction.111 Instead, treatment can also aim to restore or optimize the mechanical environment, to modulate mechanotransduction pathways at the molecular level, and to harness emerging biophysical and technological innovations. This section discusses key strategies across rehabilitation, ventilation, pharmacology, technology, and integrative approaches.
Rehabilitation and Respiratory Training as Mechanical Rebalancing
Pulmonary rehabilitation remains one of the most effective interventions in COPD, not only for improving quality of life but also for directly modulating disordered mechanics.112 Inspiratory and expiratory muscle training strengthens the diaphragm and accessory muscles, counteracting the disadvantages imposed by hyperinflation.113 Randomized controlled trials consistently show that inspiratory muscle training reduces dyspnea, improves exercise tolerance, and enhances health-related quality of life measures.22 Mechanistically, these programs reduce the relative inspiratory load, improve diaphragmatic excursion, and delay fatigue, thereby rebalancing the mismatch between mechanical demand and capacity.114
Exercise training, whether endurance- or resistance-based, contributes to improved ventilatory efficiency and skeletal muscle performance.115 From a mechanical perspective, exercise training reduces dynamic hyperinflation by improving breathing pattern control, reducing respiratory rate, and enhancing tidal volume expansion.116 It also increases chest wall mobility and diaphragmatic recruitment, partially restoring normal mechanics. Simple techniques such as pursed-lip breathing or forward-leaning postures illustrate how deliberate modifications of intrathoracic pressure dynamics can reduce airway collapse and alleviate dyspnea,117 highlighting that mechanical optimization is embedded within even the most basic rehabilitation strategies.
Mechanical Ventilation and Structural Interventions
Mechanical ventilation, whether noninvasive (NIV) or invasive, plays a dual role in COPD management—unloading respiratory muscles while simultaneously risking mechanical injury if poorly adjusted. In acute exacerbations, NIV effectively reduces PEEPi, unloads fatigued inspiratory muscles, improves gas exchange, and decreases the need for intubation.118 Meta-analyses confirm reductions in mortality and hospitalization, supporting NIV as the standard of care in AECOPD.119 Chronic nocturnal NIV in patients with persistent hypercapnia has also demonstrated benefits in survival and quality of life, though its effects are heterogeneous and closely tied to mechanical adjustments.120
The concept of protective ventilation has gained traction in COPD, emphasizing minimization of injurious mechanical power by carefully titrating tidal volume, plateau pressure, and respiratory rate, while allowing sufficient expiratory time to avoid dynamic hyperinflation. Advanced monitoring techniques such as esophageal manometry and electrical impedance tomography now allow real-time assessment of lung mechanics, enabling individualized ventilator strategies.121
Beyond ventilatory support, structural interventions such as surgical or bronchoscopic lung volume reduction target mechanics at the organ level. By removing or collapsing hyperinflated and poorly ventilated regions, these approaches reduce heterogeneity, restore diaphragmatic curvature, and improve chest wall mechanics.122 Patient selection remains critical, but when applied appropriately, lung volume reduction exemplifies how direct modulation of mechanics can translate into functional gains.
Targeting Mechanotransduction with Pharmacological Agents
Pharmacological therapies targeting mechanotransduction pathways remain largely experimental but offer promising avenues. Integrins are central mediators of fibroblast activation and epithelial repair, and inhibitors of αvβ6 or αvβ1 integrins, already under investigation in fibrotic lung diseases, could attenuate maladaptive remodeling in COPD.123 Similarly, interventions aimed at modifying ECM architecture, such as inhibitors of lysyl oxidase–like enzymes that promote collagen crosslinking, may reduce pathological stiffening and restore more physiological mechanotransduction.124
Mechanosensitive ion channels, including Piezo and transient receptor potential (TRP) family members, are emerging as druggable targets. Preclinical studies suggest that modulation of these channels can blunt inflammation, reduce calcium overload, and attenuate mechanically induced injury.125 Translation of such approaches to COPD could open new therapeutic horizons.
Targeting nuclear mechanotransduction is another novel approach. Inhibition of YAP/TAZ signaling, for instance using verteporfin or small molecules that disrupt TEAD interactions, has shown potential in attenuating fibroblast activation and ECM production.73 Similarly, ROCK inhibitors, which reduce smooth muscle contractility, fibroblast activation, and vascular remodeling,126 are being tested in pulmonary hypertension and may hold promise for COPD where ROCK activity is upregulated. The major challenge remains achieving tissue specificity, as these pathways are ubiquitous across multiple organ systems. Moreover, emerging data suggest that several mechanotransduction pathways may play context-dependent roles, contributing to injury and remodeling in some settings while supporting adaptive repair or homeostasis in others. This complexity may help explain why pharmacological inhibition has shown variable results across experimental models and highlights the need for careful patient selection and compartment-specific targeting.
Technological and Biophysical Frontiers
Rapid advances in imaging and monitoring technologies are transforming how mechanical dysfunction is evaluated and managed in COPD. Quantitative CT, MRI ventilation imaging, and elastography provide regional maps of lung mechanics,127 allowing the identification of patients with specific patterns of hyperinflation or stiffness who may benefit from targeted interventions such as lung volume reduction.128 Functional imaging is also being explored to monitor therapy response and disease progression, bridging the gap between mechanistic understanding and clinical decision-making.
At the frontier of regenerative medicine, tissue engineering offers opportunities to restore lung structure with scaffolds designed to mimic the mechanical properties of healthy ECM.129 Stem-cell–based therapies are increasingly informed by mechanobiology, with culture conditions optimized to deliver mechanical cues that guide differentiation into airway or alveolar lineages.130 Though still experimental, these approaches underscore the potential of mechanobiology to shape future regenerative strategies for COPD.
Digital health technologies add another dimension. Wearable devices, home spirometry, and artificial intelligence (AI)–driven imaging analytics now enable longitudinal monitoring of mechanical parameters such as breathing pattern, chest wall motion, and hyperinflation indices.131 Integration of these data into clinical practice may allow early detection of deleterious mechanical trends and individualized adjustment of therapy.
Toward Integrative, Mechanics-Informed Management
A future vision of COPD care requires integration of mechanical interventions with established pharmacology. For example, bronchodilators reduce airway resistance and thereby improve mechanical efficiency, but their benefits are amplified when combined with pulmonary rehabilitation or respiratory muscle training. Similarly, the use of personalized ventilator strategies can be complemented by pharmacological agents targeting inflammation or fibrosis, creating a synergistic effect that neither alone could achieve.
This holistic approach positions mechanical optimization not as an alternative but as a critical complement to established COPD therapies. By uniting biomechanics, pharmacology, and rehabilitation within a single framework, management can be reframed toward re-establishing mechanical homeostasis across the respiratory system. Such integration aligns with precision medicine goals, where treatment is tailored not only to inflammatory or genetic profiles but also to individual mechanical phenotypes. Figure 5 provides an overview of therapeutic strategies targeting mechanical perturbations and their downstream pathways, highlighting opportunities for integrative and precision interventions in COPD.
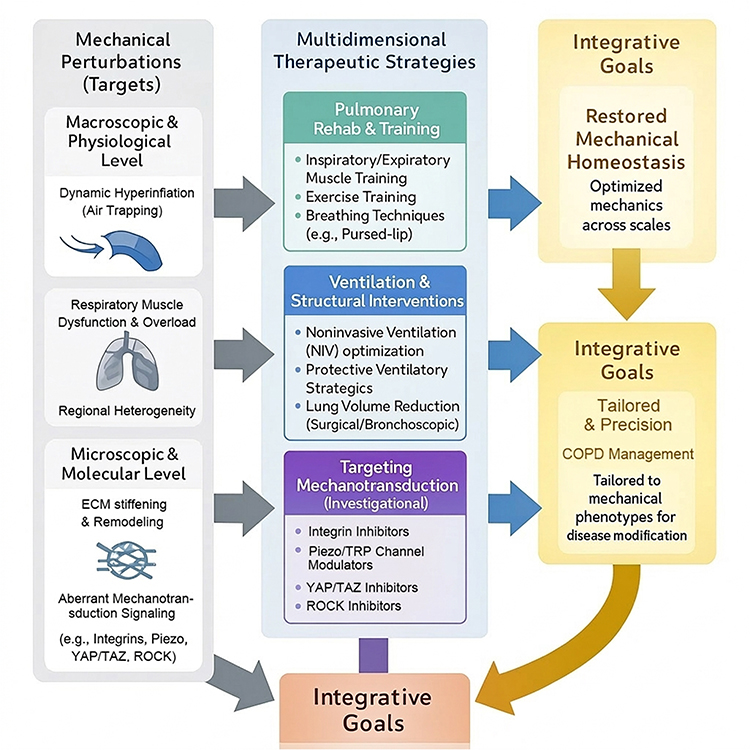 |
Figure 5 Integrative therapeutic strategies targeting mechanical perturbations in COPD. The schematic outlines a multidimensional approach to restore mechanical homeostasis. (Left) Mechanical Targets: Interventions are directed at specific abnormalities ranging from the macroscopic physiological level (e.g., dynamic hyperinflation, respiratory muscle dysfunction) to the microscopic molecular level (e.g., ECM stiffening, aberrant mechanotransduction signaling). (Middle) Multidimensional Strategies: Pulmonary Rehab & Training: Interventions such as inspiratory muscle training and pursed-lip breathing aim to strengthen respiratory muscles and optimize breathing patterns to counteract hyperinflation. Ventilation & Structural Interventions: Strategies like noninvasive ventilation (NIV) optimization and lung volume reduction target organ-level mechanics by unloading muscles and reducing regional heterogeneity. Targeting Mechanotransduction: Emerging investigational pharmacotherapies (e.g., inhibitors of integrins, Piezo channels, YAP/TAZ, and ROCK) aim to modulate the downstream cellular response to mechanical stress. (Right) Integrative Goals: The ultimate objective is to combine these modalities to restore mechanical homeostasis and achieve precision management tailored to the individual patient’s mechanical phenotype. |
Future Perspectives
The integration of mechanical biology into COPD research has revealed a new dimension of pathogenesis, yet the field remains in its early stages. Significant challenges remain in translating mechanistic discoveries into clinical practice. Future progress will require multi-scale integration, technological advances in imaging and monitoring, identification of novel therapeutic targets, and incorporation of mechanobiology into regenerative and precision medicine frameworks.
Integrating Mechanics Across Biological Scales
COPD is fundamentally a disease of disrupted structure–function relationships,27 in which mechanics interact with inflammation, immunity, and metabolism at multiple biological scales. Future research must therefore adopt integrative models that connect molecular mechanotransduction events to tissue-level remodeling and whole-organ function. Computational modeling can simulate how microscopic changes in ECM stiffness translate into macroscopic alterations in airflow or spirometry. Similarly, systems biology approaches integrating transcriptomic, proteomic, and metabolomic data with mechanical readouts could identify new disease endotypes defined by both molecular and biomechanical signatures. Such multi-scale frameworks will not only deepen mechanistic understanding but also refine disease classification beyond traditional spirometric indices.
Next-Generation Imaging and Functional Assessment
Technological innovations are enabling in vivo quantification of mechanical heterogeneity with unprecedented resolution. CT-based strain mapping, hyperpolarized gas MRI, and ultrasound elastography allow direct visualization of regional compliance, ventilation defects, and stress distribution.132 Coupled with longitudinal cohort studies, these modalities may yield mechanical biomarkers predictive of disease progression and therapeutic response. Beyond imaging, wearable devices and digital health platforms capable of continuously monitoring breathing patterns, chest wall motion, and indices of hyperinflation could bring real-time mechanical monitoring into patients’ homes.133 Integration of these functional metrics into clinical care will be essential for early detection of deleterious changes and for guiding timely, individualized interventions.
Emerging Therapeutic Targets in Mechanotransduction
While current pharmacological therapies for COPD remain largely symptomatic, future treatments may target the mechanosensitive pathways that drive disease progression. Inhibitors of integrin signaling, Piezo ion channel modulators, YAP/TAZ antagonists, and ROCK inhibitors are already being investigated in related fibrotic or vascular diseases,134 and their translation to COPD is an active frontier. The main challenge is specificity, given that mechanotransduction pathways are ubiquitous across tissues. Inhaled nanomedicine platforms, capable of delivering targeted therapies to the lung microenvironment, may overcome this limitation by ensuring local modulation of mechanical signaling without systemic toxicity.135 Importantly, clinical trials should incorporate mechanical endpoints such as changes in hyperinflation, regional compliance, or strain heterogeneity to establish the therapeutic relevance of targeting mechanotransduction. Future studies should also directly address discordant findings across models by standardizing mechanical exposure paradigms, distinguishing cell- and compartment-specific responses, and separating acute adaptive signaling from chronic maladaptive remodeling.
Mechanobiology-Informed Regenerative Medicine
The convergence of mechanobiology with regenerative medicine represents a particularly exciting frontier. Advances in biomaterials and tissue engineering now allow the creation of scaffolds with tunable stiffness and elasticity that recapitulate healthy lung ECM.136 These scaffolds can provide an appropriate mechanical niche for stem or progenitor cells, enhancing their differentiation into functional epithelial or alveolar lineages. Likewise, organoid and lung-on-chip systems incorporating physiological mechanical forces are increasingly being used to model COPD in vitro and to test regenerative strategies. Although these approaches remain experimental, they underscore the potential of mechanical cues to guide not only disease pathogenesis but also tissue regeneration, offering hope for structural repair in advanced disease.
Toward Precision Medicine Guided by Mechanics
COPD is now recognized as a heterogeneous syndrome encompassing multiple overlapping phenotypes.137 Incorporating mechanical assessments into precision medicine strategies could greatly refine patient stratification. For example, patients with predominant small airway collapse may benefit most from stenting or lung volume reduction,138 whereas those with fibrotic stiffening may be more responsive to antifibrotic or YAP/TAZ-targeted therapies.36 Combining mechanical biomarkers with molecular and clinical data can generate multi-dimensional patient profiles, enabling individualized treatment algorithms. Artificial intelligence and machine learning are poised to play a critical role by integrating imaging, physiological, and molecular data into predictive models that guide therapy selection.139 Ultimately, precision medicine in COPD will depend not only on genetics and inflammation but equally on the mechanobiological profile of each patient.
Conclusion
Chronic obstructive pulmonary disease has long been viewed primarily through the lens of inflammation, oxidative stress, and protease–antiprotease imbalance, but it is increasingly evident that mechanical forces are also integral to its pathogenesis. COPD can also be viewed as a mechanobiological disease, in which abnormal airway and parenchymal mechanics, ECM remodeling, and respiratory muscle dysfunction interact with canonical inflammatory and proteolytic processes to perpetuate tissue injury and functional decline. These structural alterations reshape the mechanical environment, and through mechanosensors such as integrins, Piezo channels, and YAP/TAZ, they are transduced into biochemical signals that drive chronic inflammation, defective repair, and pathological remodeling. The resulting feedback loops create self-reinforcing cycles of hyperinflation, fibrosis, and emphysema, providing a mechanistic explanation for the persistence and heterogeneity of COPD.
Understanding COPD as a disorder of dysregulated mechanotransduction reframes both its clinical phenotypes and its therapeutic opportunities. Pulmonary rehabilitation, ventilatory support, and lung volume reduction can be reinterpreted as interventions that restore mechanical homeostasis. At the same time, emerging pharmacological strategies targeting integrin signaling, mechanosensitive ion channels, YAP/TAZ, and ROCK pathways highlight the possibility of modulating the molecular machinery of mechanotransduction, although most remain at an early or preclinical stage of development. Complementary advances in imaging, computational modeling, and digital health technologies now make it feasible to quantify mechanical dysfunction in vivo, providing new biomarkers for risk stratification and early intervention. The integration of these approaches with regenerative and precision medicine may, with further validation, help shift COPD management from symptomatic palliation toward more mechanism-informed and potentially disease-modifying strategies.
Moving forward, multidisciplinary collaboration will be essential. Pulmonologists, bioengineers, systems biologists, and computational scientists must work together to translate the principles of mechanobiology into clinical benefit. Such integration will not only refine our understanding of COPD heterogeneity but also guide the development of interventions tailored to each patient’s unique mechanical and molecular profile. By bridging fundamental discoveries with practical applications, the recognition of COPD as a mechanobiological disease offers a pathway to redefine its clinical management and ultimately to improve outcomes for millions of patients worldwide.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Natural Science Foundation of China (82500025), and Shaoxing People’s Hospital Cultivation Fund Project (2024PY04).
Disclosure
The authors report no other conflict of interest to disclose.
References
1. Agustí A, Celli BR, Criner GJ, et al. Global initiative for chronic obstructive lung disease 2023 report: gold executive summary. Am J Respir Crit Care Med. 2023;207(7):819–21. doi:10.1164/rccm.202301-0106PP
2. Momtazmanesh S, Moghaddam SS, Ghamari S-H. Global burden of chronic respiratory diseases and risk factors, 1990-2019: an update from the global burden of disease study 2019. EClinicalMedicine. 2023;59:101936. doi:10.1016/j.eclinm.2023.101936
3. Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012;122(8):2749–2755. doi:10.1172/jci60324
4. Barnes PJ, Burney PG, Silverman EK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1:15076. doi:10.1038/nrdp.2015.76
5. Xia T, Pan Z, Wan H, et al. Mechanisms of mechanical stimulation in the development of respiratory system diseases. Am J Physiol Lung Cell Mol Physiol. 2024;327(5):L724–l739. doi:10.1152/ajplung.00122.2024
6. Zheng M, Borkar NA, Yao Y, et al. Mechanosensitive channels in lung disease. Front Physiol. 2023;14:1302631. doi:10.3389/fphys.2023.1302631
7. Novak C, Ballinger MN, Ghadiali S. Mechanobiology of pulmonary diseases: a review of engineering tools to understand lung mechanotransduction. J Biomech Eng. 143(11). doi:10.1115/1.4051118
8. Lin C, Zheng X, Lin S, Zhang Y, Wu J, Li Y. Mechanotransduction regulates the interplays between alveolar epithelial and vascular endothelial cells in lung. Front Physiol. 2022;13:818394. doi:10.3389/fphys.2022.818394
9. Du H, Bartleson JM, Butenko S, et al. Tuning immunity through tissue mechanotransduction. Nat Rev Immunol. 2023;23(3):174–188. doi:10.1038/s41577-022-00761-w
10. Kononov S, Brewer K, Sakai H, et al. Roles of mechanical forces and collagen failure in the development of elastase-induced emphysema. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1920–1926. doi:10.1164/ajrccm.164.10.2101083
11. Suki B, Sato S, Parameswaran H, Szabari MV, Takahashi A, Bartolák-Suki E. Emphysema and mechanical stress-induced lung remodeling. Physiology. 2013;28(6):404–413. doi:10.1152/physiol.00041.2013
12. Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB j. 2006;20(7):811–827. doi:10.1096/fj.05-5424rev
13. Hsia CC, Hyde DM, Weibel ER. Lung structure and the intrinsic challenges of gas exchange. Compr Physiol. 2016;6(2):827–895. doi:10.1002/cphy.c150028
14. Cai X, Wang KC, Meng Z. Mechanoregulation of YAP and TAZ in cellular homeostasis and disease progression. Front Cell Dev Biol. 2021;9:673599. doi:10.3389/fcell.2021.673599
15. Lazarinis N, Fouka E, Linden A, Bossios A. Small airways disease in chronic obstructive pulmonary disease. Expert Rev Respir Med. 2024;18(7):539–552. doi:10.1080/17476348.2024.2380070
16. Joglekar MM, Bekker NJ, Koloko Ngassie ML, et al. The lung extracellular matrix protein landscape in severe early-onset and moderate chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2024;327(3):L304–l318. doi:10.1152/ajplung.00332.2023
17. Santana PV, Albuquerque ALP. Respiratory muscles in COPD: be aware of the diaphragm. J Bras Pneumol. 2018;44(1):1–2. doi:10.1590/s1806-37562018000010001
18. Gea J, Orozco-Levi M, Pascual-Guàrdia S, et al. Biological mechanisms involved in muscle dysfunction in COPD: an integrative damage-regeneration-remodeling framework. Cells. 2025;14(21). doi:10.3390/cells14211731
19. Goligher EC, Dres M, Patel BK, et al. Lung- and diaphragm-protective ventilation. Am J Respir Crit Care Med. 2020;202(7):950–961. doi:10.1164/rccm.202003-0655CP
20. van den Berg MJW, Heunks L, Doorduin J, van den Berg MJW. Advances in achieving lung and diaphragm-protective ventilation. Curr Opin Crit Care. 2025;31(1):38–46. doi:10.1097/mcc.0000000000001228
21. Wouters EFM, Wouters B, Augustin IML, Houben-Wilke S, Vanfleteren L, Franssen FME. Personalised pulmonary rehabilitation in COPD. Eur Respir Rev. 27(147). doi:10.1183/16000617.0125-2017
22. Beaumont M, Forget P, Couturaud F, Reychler G. Effects of inspiratory muscle training in COPD patients: a systematic review and meta-analysis. Clin Respir J. 2018;12(7):2178–2188. doi:10.1111/crj.12905
23. Papavassiliou KA, Sofianidi AA, Spiliopoulos FG, Gogou VA, Gargalionis AN, Papavassiliou AG. YAP/TAZ signaling in the pathobiology of pulmonary fibrosis. Cells. 13(18). doi:10.3390/cells13181519
24. Stolz D, Mkorombindo T, Schumann DM, et al. Towards the elimination of chronic obstructive pulmonary disease: a lancet commission. Lancet. 2022;400(10356):921–972. doi:10.1016/s0140-6736(22)01273-9
25. Agustí A, Celli B. Natural history of COPD: gaps and opportunities. ERJ Open Res. 2017;3(4):00117–2017. doi:10.1183/23120541.00117-2017
26. Brandsma CA, Van den Berge M, Hackett TL, Brusselle G, Timens W. Recent advances in chronic obstructive pulmonary disease pathogenesis: from disease mechanisms to precision medicine. J Pathol. 2020;250(5):624–635. doi:10.1002/path.5364
27. McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–1575. doi:10.1056/NEJMoa1106955
28. Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. 2004;364(9435):709–721. doi:10.1016/s0140-6736(04)16900-6
29. Bhatt SP, Soler X, Wang X, et al. Association between functional small airway disease and fev1 decline in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;194(2):178–184. doi:10.1164/rccm.201511-2219OC
30. Gagnon P, Guenette JA, Langer D, et al. Pathogenesis of hyperinflation in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2014;9:187–201. doi:10.2147/copd.S38934
31. Castaldi PJ, San José Estépar R, Mendoza CS, et al. Distinct quantitative computed tomography emphysema patterns are associated with physiology and function in smokers. Am J Respir Crit Care Med. 2013;188(9):1083–1090. doi:10.1164/rccm.201305-0873OC
32. Moriondo A, Marcozzi C, Bianchin F, et al. Impact of mechanical ventilation and fluid load on pulmonary glycosaminoglycans. Respir Physiol Neurobiol. 2012;181(3):308–320. doi:10.1016/j.resp.2012.03.013
33. Suki B, Ito S, Stamenovic D, Lutchen KR, Ingenito EP. Biomechanics of the lung parenchyma: critical roles of collagen and mechanical forces. J Appl Physiol. 2005;98(5):1892–1899. doi:10.1152/japplphysiol.01087.2004
34. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
35. Barnes PJ. Small airway fibrosis in COPD. Int J Biochem Cell Biol. 2019;116:105598. doi:10.1016/j.biocel.2019.105598
36. Tschumperlin DJ, Ligresti G, Hilscher MB, Shah VH. Mechanosensing and fibrosis. J Clin Invest. 2018;128(1):74–84. doi:10.1172/jci93561
37. Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15(12):802–812. doi:10.1038/nrm3896
38. Solis AG, Bielecki P, Steach HR, et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature. 2019;573(7772):69–74. doi:10.1038/s41586-019-1485-8
39. Giacomini MM, Travis MA, Kudo M, Sheppard D. Epithelial cells utilize cortical actin/myosin to activate latent TGF-β through integrin α(v)β(6)-dependent physical force. Exp Cell Res. 2012;318(6):716–722. doi:10.1016/j.yexcr.2012.01.020
40. Gorman RB, McKenzie DK, Butler JE, Tolman JF, Gandevia SC. Diaphragm length and neural drive after lung volume reduction surgery. Am J Respir Crit Care Med. 2005;172(10):1259–1266. doi:10.1164/rccm.200412-1695OC
41. Levine S, Nguyen T, Taylor N, et al. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med. 2008;358(13):1327–1335. doi:10.1056/NEJMoa070447
42. O’Donnell DE, James MD, Milne KM, Neder JA. The pathophysiology of dyspnea and exercise intolerance in chronic obstructive pulmonary disease. Clin Chest Med. 2019;40(2):343–366. doi:10.1016/j.ccm.2019.02.007
43. Laghi F, Tobin MJ. Disorders of the respiratory muscles. Am J Respir Crit Care Med. 2003;168(1):10–48. doi:10.1164/rccm.2206020
44. Loring SH, Garcia-Jacques M, Malhotra A. Pulmonary characteristics in COPD and mechanisms of increased work of breathing. J Appl Physiol. 2009;107(1):309–314. doi:10.1152/japplphysiol.00008.2009
45. Haluszka J, Chartrand DA, Grassino AE, Milic-Emili J. Intrinsic PEEP and arterial PCO2 in stable patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. 1990;141(5 Pt 1):1194–1197. doi:10.1164/ajrccm/141.5_Pt_1.1194
46. Marini JJ. Dynamic hyperinflation and auto-positive end-expiratory pressure: lessons learned over 30 years. Am J Respir Crit Care Med. 2011;184(7):756–762. doi:10.1164/rccm.201102-0226PP
47. Burgess JK, Gosens R. Mechanotransduction and the extracellular matrix: key drivers of lung pathologies and drug responsiveness. Biochem Pharmacol. 2024;228:116255. doi:10.1016/j.bcp.2024.116255
48. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi:10.1016/s0092-8674(02)00971-6
49. Burgess JK, Harmsen MC. Chronic lung diseases: entangled in extracellular matrix. Eur Respir Rev. 2022;31(163). doi:10.1183/16000617.0202-2021
50. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18(9):533–548. doi:10.1038/s41568-018-0038-z
51. Koivisto L, Heino J, Häkkinen L, Larjava H. Integrins in wound healing. Adv Wound Care. 2014;3(12):762–783. doi:10.1089/wound.2013.0436
52. Martinac B. Mechanosensitive ion channels: molecules of mechanotransduction. J Cell Sci. 2004;117(Pt 12):2449–2460. doi:10.1242/jcs.01232
53. Murthy SE, Dubin AE, Patapoutian A. Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat Rev Mol Cell Biol. 2017;18(12):771–783. doi:10.1038/nrm.2017.92
54. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–183. doi:10.1038/nature10137
55. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. 2018;20(8):888–899. doi:10.1038/s41556-018-0142-z
56. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol. 2017;18(12):758–770. doi:10.1038/nrm.2017.87
57. Pegoraro AF, Janmey P, Weitz DA. Mechanical Properties of the Cytoskeleton and Cells. Cold Spring Harb Perspect Biol. 2017;9(11). doi:10.1101/cshperspect.a022038
58. Burridge K, Wittchen ES. The tension mounts: stress fibers as force-generating mechanotransducers. J Cell Biol. 2013;200(1):9–19. doi:10.1083/jcb.201210090
59. Haines J, Chua SHK, Smith J, Slinger C, Simpson AJ, Fowler SJ. Triggers of breathlessness in inducible laryngeal obstruction and asthma. Clin Exp Allergy. 2020;50(11):1230–1237. doi:10.1111/cea.13715
60. Mwase C, Deng W, Kim HJ, et al. Hic-5 transduces mechanical force that drives a vicious cycle of bronchoconstriction. Res Sq. 2025. doi:10.21203/rs.3.rs-6498980/v1
61. Correa-Meyer E, Pesce L, Guerrero C, Sznajder JI. Cyclic stretch activates ERK1/2 via G proteins and EGFR in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;282(5):L883–91. doi:10.1152/ajplung.00203.2001
62. Leng S, Zhang X, Zhao R, et al. Mechanical activation of adipose tissue macrophages mediated by Piezo1 protects against diet-induced obesity by regulating sympathetic activity. Metabolism. 2025;168:156262. doi:10.1016/j.metabol.2025.156262
63. Huang Y, Crawford M, Higuita-Castro N, Nana-Sinkam P, Ghadiali SN. miR-146a regulates mechanotransduction and pressure-induced inflammation in small airway epithelium. FASEB j. 2012;26(8):3351–3364. doi:10.1096/fj.11-199240
64. Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton. 2010;67(9):545–554. doi:10.1002/cm.20472
65. Ji H, Tang H, Lin H, et al. Rho/Rock cross-talks with transforming growth factor-β/Smad pathway participates in lung fibroblast-myofibroblast differentiation. Biomed Rep. 2014;2(6):787–792. doi:10.3892/br.2014.323
66. Connolly MJ, Aaronson PI. Key role of the RhoA/Rho kinase system in pulmonary hypertension. Pulm Pharmacol Ther. 2011;24(1):1–14. doi:10.1016/j.pupt.2010.09.001
67. Roth D, Şahin AT, Ling F, et al. Structure and function relationships of mucociliary clearance in human and rat airways. Nat Commun. 2025;16(1):2446. doi:10.1038/s41467-025-57667-z
68. Zhong M, Komarova Y, Rehman J, Malik AB. Mechanosensing Piezo channels in tissue homeostasis including their role in lungs. Pulm Circ. 2018;8(2):2045894018767393. doi:10.1177/2045894018767393
69. Lin C, Yao E, Zhang K, et al. YAP is essential for mechanical force production and epithelial cell proliferation during lung branching morphogenesis. Elife. 6. doi:10.7554/eLife.21130
70. Liu F, Lagares D, Choi KM, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308(4):L344–57. doi:10.1152/ajplung.00300.2014
71. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol. 2018;58(2):157–169. doi:10.1165/rcmb.2017-0200TR
72. Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298(6):L715–31. doi:10.1152/ajplung.00361.2009
73. Haak AJ, Kostallari E, Sicard D, et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci Transl Med. 2019;11(516). doi:10.1126/scitranslmed.aau6296
74. Hallgren O, Nihlberg K, Dahlbäck M, et al. Altered fibroblast proteoglycan production in COPD. Respir Res. 2010;11(1):55. doi:10.1186/1465-9921-11-55
75. Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? Am J Physiol Lung Cell Mol Physiol. 2013;305(12):L912–33. doi:10.1152/ajplung.00259.2013
76. Sridharan R, Cavanagh B, Cameron AR, Kelly DJ, O’Brien FJ. Material stiffness influences the polarization state, function and migration mode of macrophages. Acta Biomater. 2019;89:47–59. doi:10.1016/j.actbio.2019.02.048
77. Atcha H, Jairaman A, Holt JR, et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat Commun. 2021;12(1):3256. doi:10.1038/s41467-021-23482-5
78. Lai Y, Huang Y. Mechanisms of mechanical force induced pulmonary vascular endothelial hyperpermeability. Front Physiol. 2021;12:714064. doi:10.3389/fphys.2021.714064
79. Scichilone N, Whittamore A, White C, Nudo E, Savella M, Lombardini M. The patient journey in Chronic Obstructive Pulmonary Disease (COPD): a human factors qualitative international study to understand the needs of people living with COPD. BMC Pulm Med. 2023;23(1):506. doi:10.1186/s12890-023-02796-8
80. Vanfleteren LE, Spruit MA, Groenen M, et al. Clusters of comorbidities based on validated objective measurements and systemic inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(7):728–735. doi:10.1164/rccm.201209-1665OC
81. Wang Y, Xu J, Meng Y, Adcock IM, Yao X. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:3341–3348. doi:10.2147/copd.S176122
82. Burgel PR. The role of small airways in obstructive airway diseases. Eur Respir Rev. 2011;20(119):23–33. doi:10.1183/09059180.00010410
83. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. doi:10.1056/NEJMoa032158
84. Greulich T, Weist BJD, Koczulla AR, et al. Prevalence of comorbidities in COPD patients by disease severity in a German population. Respir Med. 2017;132:132–138. doi:10.1016/j.rmed.2017.10.007
85. Hariprasad DS, Sul B, Liu C, et al. Obstructions in the lower airways lead to altered airflow patterns in the central airway. Respir Physiol Neurobiol. 2020;272:103311. doi:10.1016/j.resp.2019.103311
86. Vogiatzis I, Zakynthinos S. Factors limiting exercise tolerance in chronic lung diseases. Compr Physiol. 2012;2(3):1779–1817. doi:10.1002/cphy.c110015
87. Ottenheijm CA, Heunks LM, Dekhuijzen RP. Diaphragm adaptations in patients with COPD. Respir Res. 2008;9(1):12. doi:10.1186/1465-9921-9-12
88. Parshall MB, Schwartzstein RM, Adams L, et al. An official American Thoracic Society statement: update on the mechanisms, assessment, and management of dyspnea. Am J Respir Crit Care Med. 2012;185(4):435–452. doi:10.1164/rccm.201111-2042ST
89. McKenzie DK, Butler JE, Gandevia SC. Respiratory muscle function and activation in chronic obstructive pulmonary disease. J Appl Physiol. 2009;107(2):621–629. doi:10.1152/japplphysiol.00163.2009
90. Suki B, Lutchen KR, Ingenito EP. On the progressive nature of emphysema: roles of proteases, inflammation, and mechanical forces. Am J Respir Crit Care Med. 2003;168(5):516–521. doi:10.1164/rccm.200208-908PP
91. Winkler T, Suki B. Emergent structure-function relations in emphysema and asthma. Crit Rev Biomed Eng. 2011;39(4):263–280. doi:10.1615/critrevbiomedeng.v39.i4.20
92. Suki B, Stamenović D, Hubmayr R. Lung parenchymal mechanics. Compr Physiol. 2011;1(3):1317–1351. doi:10.1002/cphy.c100033
93. Ferguson GT. Why does the lung hyperinflate? Proc Am Thorac Soc. 2006;3(2):176–179. doi:10.1513/pats.200508-094DO
94. Kulkarni T, O’Reilly P, Antony VB, Gaggar A, Thannickal VJ. Matrix remodeling in pulmonary fibrosis and emphysema. Am J Respir Cell Mol Biol. 2016;54(6):751–760. doi:10.1165/rcmb.2015-0166PS
95. JAEC-C W, Miravitlles M, Hurst JR, et al. Management of COPD exacerbations: a European Respiratory Society/American Thoracic Society guideline. Eur Respir J. 2017;49(3). doi:10.1183/13993003.00791-2016
96. O’Donnell DE, Parker CM. COPD exacerbations. 3: pathophysiology. Thorax. 2006;61(4):354–361. doi:10.1136/thx.2005.041830
97. Sun S, Pan L, Wang X, Zhao J. Diaphragmatic ultrasound in guiding weaning from invasive mechanical ventilation in patients with acute exacerbation of chronic obstructive pulmonary disease. Heart Lung. 2026;77:102692. doi:10.1016/j.hrtlng.2025.102692
98. Gattinoni L, Tonetti T, Cressoni M, et al. Ventilator-related causes of lung injury: the mechanical power. Intensive Care Med. 2016;42(10):1567–1575. doi:10.1007/s00134-016-4505-2
99. Gea J, Pascual S, Casadevall C, Orozco-Levi M, Barreiro E. Muscle dysfunction in chronic obstructive pulmonary disease: update on causes and biological findings. J Thorac Dis. 2015;7(10):E418–38. doi:10.3978/j.issn.2072-1439.2015.08.04
100. MacDonald MI, Shafuddin E, King PT, Chang CL, Bardin PG, Hancox RJ. Cardiac dysfunction during exacerbations of chronic obstructive pulmonary disease. Lancet Respir Med. 2016;4(2):138–148. doi:10.1016/s2213-2600(15)00509-3
101. Han MK, Agusti A, Calverley PM, et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med. 2010;182(5):598–604. doi:10.1164/rccm.200912-1843CC
102. Nakano Y, Muro S, Sakai H, et al. Computed tomographic measurements of airway dimensions and emphysema in smokers. Correlation with lung function. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1102–1108. doi:10.1164/ajrccm.162.3.9907120
103. Vilaró J, Ramirez-Sarmiento A, Martínez-Llorens JM, et al. Global muscle dysfunction as a risk factor of readmission to hospital due to COPD exacerbations. Respir Med. 2010;104(12):1896–1902. doi:10.1016/j.rmed.2010.05.001
104. Etienne S, Hoheisel A, Agarwal P, et al. Quantitative computed tomography and predictive modelling for COPD exacerbations. Eur Respir Rev. 2025;34(178):250097. doi:10.1183/16000617.0097-2025
105. Kirby M, Pike D, McCormack DG, Lam S, Coxson HO, Parraga G. Longitudinal computed tomography and magnetic resonance imaging of COPD: thoracic imaging network of canada (tincan) study objectives. Chronic Obstr Pulm Dis. 2014;1(2):200–211. doi:10.15326/jcopdf.1.2.2014.0136
106. Amelon R, Cao K, Ding K, Christensen GE, Reinhardt JM, Raghavan ML. Three-dimensional characterization of regional lung deformation. J Biomech. 2011;44(13):2489–2495. doi:10.1016/j.jbiomech.2011.06.009
107. Casanova C, Cote C, de Torres JP, et al. Inspiratory-to-total lung capacity ratio predicts mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med.;171(6):591–597. doi:10.1164/rccm.200407-867OC.
108. Luisetti M, Ma S, Iadarola P, et al. Desmosine as a biomarker of elastin degradation in COPD: current status and future directions. Eur Respir J. 2008;32(5):1146–1157. doi:10.1183/09031936.00174807
109. Ma S, Lin YY, Turino GM. Measurements of desmosine and isodesmosine by mass spectrometry in COPD. Chest. 2007;131(5):1363–1371. doi:10.1378/chest.06-2251
110. Huang JT, Chaudhuri R, Albarbarawi O, et al. Clinical validity of plasma and urinary desmosine as biomarkers for chronic obstructive pulmonary disease. Thorax. 2012;67(6):502–508. doi:10.1136/thoraxjnl-2011-200279
111. Vogiatzis I, Rochester CL, Spruit MA, Troosters T, Clini EM. Increasing implementation and delivery of pulmonary rehabilitation: key messages from the new ATS/ERS policy statement. Eur Respir J. 2016;47(5):1336–1341. doi:10.1183/13993003.02151-2015
112. Spruit MA, Singh SJ, Garvey C, et al. An official American Thoracic Society/European Respiratory Society statement: key concepts and advances in pulmonary rehabilitation. Am J Respir Crit Care Med. 2013;188(8):e13–64. doi:10.1164/rccm.201309-1634ST
113. Bordoni B, Morabito B, Myftari V, D’Amato A, Severino P. The glymphatic system and diaphragmatic dysfunction in patients with chronic obstructive pulmonary disease and chronic heart failure: the importance of inspiratory rehabilitation training. J Cardiovasc Dev Dis. 2025;12(10). doi:10.3390/jcdd12100390
114. Petrovic M, Reiter M, Zipko H, Pohl W, Wanke T. Effects of inspiratory muscle training on dynamic hyperinflation in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2012;7:797–805. doi:10.2147/copd.S23784
115. Iepsen UW, Jørgensen KJ, Ringbaek T, Hansen H, Skrubbeltrang C, Lange P. A systematic review of resistance training versus endurance training in COPD. J Cardiopulm Rehabil Prev. 2015;35(3):163–172. doi:10.1097/hcr.0000000000000105
116. Puente-Maestu L, Stringer WW. Hyperinflation and its management in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(4):381–400. doi:10.2147/copd.2006.1.4.381
117. Nakahiro K, Murata A, Kaneko Y, Takasaki Y. Effects of bathing on lung mechanics in patients with severe COPD. Nihon Kokyuki Gakkai Zasshi. 1998;36(2):150–156.
118. Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 1995;333(13):817–822. doi:10.1056/nejm199509283331301
119. Osadnik CR, Tee VS, Carson-Chahhoud KV, Picot J, Wedzicha JA, Smith BJ. Non-invasive ventilation for the management of acute hypercapnic respiratory failure due to exacerbation of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2017;7(7):Cd004104. doi:10.1002/14651858.CD004104.pub4
120. Murphy PB, Rehal S, Arbane G, et al. Effect of home noninvasive ventilation with oxygen therapy vs oxygen therapy alone on hospital readmission or death after an acute COPD exacerbation: a randomized clinical trial. JAMA. 2017;317(21):2177–2186. doi:10.1001/jama.2017.4451
121. Mauri T, Yoshida T, Bellani G, et al. Esophageal and transpulmonary pressure in the clinical setting: meaning, usefulness and perspectives. Intensive Care Med. 2016;42(9):1360–1373. doi:10.1007/s00134-016-4400-x
122. Fessler HE, Scharf SM, Permutt S. Improvement in spirometry following lung volume reduction surgery: application of a physiologic model. Am J Respir Crit Care Med. 2002;165(1):34–40. doi:10.1164/ajrccm.165.1.2101149
123. John AE, Graves RH, Pun KT, et al. Translational pharmacology of an inhaled small molecule αvβ6 integrin inhibitor for idiopathic pulmonary fibrosis. Nat Commun. 2020;11(1):4659. doi:10.1038/s41467-020-18397-6
124. Raghu G, Brown KK, Collard HR, et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: a randomised, double-blind, controlled, Phase 2 trial. Lancet Respir Med. 2017;5(1):22–32. doi:10.1016/s2213-2600(16)30421-0
125. Zhang Y, Su SA, Li W, et al. Piezo1-mediated mechanotransduction promotes cardiac hypertrophy by impairing calcium homeostasis to activate calpain/calcineurin signaling. Hypertension. 2021;78(3):647–660. doi:10.1161/hypertensionaha.121.17177
126. Abedi F, Omidkhoda N, Arasteh O, Ghavami V, Hosseinzadeh H. The therapeutic role of rho kinase inhibitor, fasudil, on pulmonary hypertension; a systematic review and meta-analysis. Drug Res (Stuttg). 2023;73(1):5–16. doi:10.1055/a-1879-3111
127. Labaki WW, Martinez CH, Martinez FJ, et al. The Role of Chest Computed Tomography in the Evaluation and Management of the Patient with Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2017;196(11):1372–1379. doi:10.1164/rccm.201703-0451PP
128. Raja J, Menn K, Jindal G, Bokhari SAJ. Radiographic manifestations of diffuse pulmonary alveolar derecruitment. J Thorac Imaging. 2019;34(6):362–366. doi:10.1097/rti.0000000000000414
129. Wagner DE, Bonenfant NR, Parsons CS, et al. Comparative decellularization and recellularization of normal versus emphysematous human lungs. Biomaterials. 2014;35(10):3281–3297. doi:10.1016/j.biomaterials.2013.12.103
130. Kumar A, Placone JK, Engler AJ. Understanding the extracellular forces that determine cell fate and maintenance. Development. 2017;144(23):4261–4270. doi:10.1242/dev.158469
131. Castaldi PJ, Boueiz A, Yun J, et al. Machine learning characterization of COPD subtypes: insights from the copdgene study. Chest. 2020;157(5):1147–1157. doi:10.1016/j.chest.2019.11.039
132. Wielpütz M, Kauczor HU. MRI of the lung: state of the art. Diagn Interv Radiol. 2012;18(4):344–353. doi:10.4261/1305-3825.Dir.5365-11.0
133. Liu Y, Shukla D, Newman H, Zhu Y. Soft wearable sensors for monitoring symptoms of COVID-19 and other respiratory diseases: a review. Prog Biomed Eng. 4(1). doi:10.1088/2516-1091/ac2eae
134. Lynch MD, watt FM. Fibroblast heterogeneity: implications for human disease. J Clin Invest. 2018;128(1):26–35. doi:10.1172/jci93555
135. Khan MA, Tania M, Fu J. Epigenetic role of thymoquinone: impact on cellular mechanism and cancer therapeutics. Drug Discov Today. 2019;24(12):2315–2322. doi:10.1016/j.drudis.2019.09.007
136. Booth AJ, Hadley R, Cornett AM, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med. 2012;186(9):866–876. doi:10.1164/rccm.201204-0754OC
137. Agusti A, Bel E, Thomas M, et al. Treatable traits: toward precision medicine of chronic airway diseases. Eur Respir J. 2016;47(2):410–419. doi:10.1183/13993003.01359-2015
138. Klooster K, ten Hacken NH, Hartman JE, Kerstjens HA, van Rikxoort EM, Slebos DJ. Endobronchial valves for emphysema without interlobar collateral ventilation. N Engl J Med. 2015;373(24):2325–2335. doi:10.1056/NEJMoa1507807
139. Pinheira A, Casal-Guisande M, Represas-Represas C, Torres-Durán M, Comesaña-Campos A, Fernández-Villar A. Artificial intelligence applications in chronic obstructive pulmonary disease: a global scoping review of diagnostic, symptom-based, and outcome prediction approaches. Biomedicines. 2025;13(12). doi:10.3390/biomedicines13123053
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.