Back to Journals » International Journal of Nanomedicine » Volume 11
Matrix metalloproteinases-2/9-sensitive peptide-conjugated polymer micelles for site-specific release of drugs and enhancing tumor accumulation: preparation and in vitro and in vivo evaluation
Authors Zhang X, Wang X, Zhong W, Ren X, Sha X, Fang X
Received 22 November 2015
Accepted for publication 31 January 2016
Published 22 April 2016 Volume 2016:11 Pages 1643—1661
DOI https://doi.org/10.2147/IJN.S101030
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Lei Yang
Xiaoyan Zhang, Xiaofei Wang, Weitong Zhong, Xiaoqing Ren, Xianyi Sha, Xiaoling Fang
Key Laboratory of Smart Drug Delivery, Ministry of Education and People’s Liberation Army of China, School of Pharmacy, Fudan University, Shanghai, People’s Republic of China
Abstract: Since elevated expression of matrix metalloproteinase (MMP)-2 and MMP-9 is commonly observed in several malignant tumors, MMPs have been widely reported as key factors in the design of drug delivery systems. Several strategies have been proposed to develop MMPs-responsive nanoparticles to deliver chemotherapeutics to malignant solid tumors. A stimuli-responsive drug delivery system, which could be cleaved by MMPs, was proposed in this study. By inserting an MMP-2/9 cleavable oligopeptide GPVGLIGK-NH2 (GK8) as spacer between α-tocopherol succinate (α-TOS) and methoxy-polyethylene glycol molecular weight (MW 2000 Da) activated by N-hydroxysuccinimide (mPEG2K-NHS), mPEG2K-GK8-α-TOS (TGK) was synthesized as the primary ingredient for MMP-2/9-sensitive micelles composed of D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) and TGK (n:n =40:60, TGK micelles). mPEG2K-α-TOS (T2K) was similarly synthesized as nonsensitive control. The TGK micelles showed better stability than nonsensitive micelles composed of TPGS and T2K (n:n =40:60, T2K micelles) owing to the inserted peptide. Fluorescence resonance energy transfer results indicated that TGK micelles could be successfully cleaved by MMP-2/9. Effective drug release was demonstrated in the presence of collagenase type IV, a mixture of MMP-2 and MMP-9. Compared with nonsensitive micelles, docetaxel (DTX)-loaded TGK micelles showed a fold higher cellular uptake in HT1080 cells. While the half-maximal inhibitory concentration (IC50) of TGK and T2K micelles were similar (P>0.05) in MCF-7 cells (MMP-2/9 underexpression), the IC50 values of the aforementioned micelles were 0.064±0.006 and 0.122±0.009 µg/mL, respectively, in HT1080 cells (MMP-2/9 overexpression). The MMP-2/9-sensitive micelles also demonstrated desired tumor targeting and accumulation ability in vivo. The results of in vivo antitumor effect evaluation indicate that TGK micelles are potent against solid tumors while maintaining minimum systemic toxicity compared with T2K micelles and DTX.
Keywords: matrix metalloproteinases, stimuli responsive, nanocarriers, TPGS, FRET
Introduction
Over past decades, the application of nanomaterial and related devices in the field of chemotherapy have been proven to be effective in minimizing side effects, thereby improving therapeutic efficacy and achieving targeted drug delivery.1,2 Ideal nanocarriers should maintain chemical and physical stabilities in circulation and release the encapsulated drugs at tumor site.3 Nanocarriers responsive to internal or external stimuli are of great interest since they allow more efficient delivery of drugs to pathological regions.4 Based on several significant differences between the microenvironment of tumor sites and normal tissues including acidic pH, altered redox potential, and overexpressed proteins, various stimuli-responsive nanocarriers have been studied.5 Tumor-associated enzyme-triggered drug release is one of the most selective and powerful strategies for achieving effective drug delivery to tumor site.6 Matrix metalloproteinases (MMPs), which degrade extracellular matrix, are secreted by cancer cells and play an important role in tumor progression.7,8 MMP-2 and MMP-9, commonly associated with tumor invasion and metastasis, have been considered as biomarkers in many types and stages of cancer.9–12 Since activated MMP-2 and MMP-9 were observed extracellularly in several malignant tumors such as breast, colon, and brain cancers,13 they have been widely reported as targets for drug delivery. Several strategies have been proposed to develop MMP-2/9-responsive nanoparticles for drug delivery to solid tumors.14 However, based on the reported data, further improvement is required in terms of site-specific release and higher drug accumulation in tumors.15
D-α-Tocopheryl polyethylene glycol 1000 succinate (TPGS, Figure S1) and its analogs were chosen for the development of nanocarriers in this study for several reasons. First, TPGS is an amphiphilic polymer containing hydrophobic vitamin E succinate and hydrophilic poly(ethylene oxide) blocks that can self-assemble into micelles in aqueous environment.16 The bulky hydrophobic cores formed by vitamin E succinate ensure a relatively high encapsulation rate in TPGS micelles.16 Second, TPGS also acts as an anticancer agent inducing apoptosis and producing a synergistic effect with other anticancer agents.17 D-α-Tocopherol succinate (α-TOS) and its analogs can induce reactive oxygen species generation, subsequently inducing apoptosis in cancer cells while being nontoxic toward normal cells.18,19 TPGS has also been approved by the US Food and Drug Administarion as a safe pharmaceutical adjuvant used in drug formulation.20 Besides, it was reported that TPGS could be efficient in overcoming multidrug resistance.21 However, in spite of the suggested advantages, critical micelles concentration (CMC) value of TPGS is as high as 0.02%, resulting in poor stability of single TPGS micelles.22 Additionally, the polyethyleneglycol (PEG) chain of TPGS is too short to extend the circulation of micelles.23 Thus, TPGS has always been incorporated with other amphiphilic polymers to form mixed micelles for drug delivery.16 It has been reported that PEG-derived vitamin E with longer PEG chain possesses much lower CMC value than TPGS.24 Mixing these conjugates with TPGS to form micelles might contribute to overcoming the disadvantages of TPGS micelles.25 Various studies have illustrated the importance of surface modification with PEG chains with a molecular weight (MW) of over 2,000 Da for the extended circulation of nanoparticles.26 Combined with the impact of PEG chain on CMC value and drug loading efficiency (DL), PEG with MW of 2,000 Da was chosen to synthesize MMP-2/9-sensitive conjugate.
Docetaxel (DTX) is one of the most potent antineoplastic agents in chemotherapy owing to its ability to inhibit the microtubule depolymerization of free tubulins.27 To overcome the solubility concerns and reduce the adverse effects of DTX, various drug delivery systems have been proposed. However, drug retention, drug release control, and permeation in tissue still remain the major challenges.28
In this study, we proposed that MMPs could be utilized to trigger drug release from polymer micelles. GPVGLIGK-NH2 (GK8) has been conjugated with poloxamer to yield MMP-2/9-sensitive multi block copolymers successfully.7 By inserting GK8 peptide sequence between α-TOS and methoxy-polyethylene glycol (MW 2,000 Da) activated by N-hydroxysuccinimide (mPEG2K-NHS) as linker, mPEG2K-GK8-α-TOS (TGK) was synthesized. mPEG2K-α-TOS (T2K) was synthesized similarly as control. MMP-2/9-sensitive micelles were prepared by TPGS and TGK at the ratio of 40:60 (n:n, TGK micelles), while nonsensitive micelles composed of TPGS and T2K (T2K micelles) were used as control. DTX was encapsulated in the hydrophobic core physically. Upon administration, the micelles accumulated in tumor site via enhanced permeability and retention effect followed by exposure to MMP-2/9 in the extracellular matrix, thereby inducing collapse of micelles and site-specific release of the loaded drug, resulting in an increased local drug concentration.29 Compared with traditional polymeric micelles, TGK micelles have a low risk of drug leakage in circulation as well as enhanced anti-tumor efficacy.
The synthesis, preparation, and characterization of TGK micelles are presented in this study. The in vitro cellular uptake and cytotoxicity against human fibrosarcoma cell line HT1080 (MMP-2/9 overexpression) and human breast adenocarcinoma cell line MCF-7 (MMP-2/9 underexpression) were investigated. Furthermore, biodistribution study using near-infrared fluorescence imaging and in vivo antitumor effect in HT1080 tumor-bearing nude mice model was examined in the study.
Materials and methods
Materials and animals
GPVGLIGK-NH2 was synthesized by Shanghai Jier Biochem Co., Ltd. (Shanghai, People’s Republic of China). DTX was purchased from Meilun Biological Technology Co., Ltd. (Dalian, Liaoning Province, People’s Republic of China). mPEG2K-NHS (MW 2,000 Da) and mPEG2K-NH2 (MW 2000 Da) were purchased from Seebio Biochem Co., Ltd. (Shanghai, People’s Republic of China). Anhydrous dimethylformamide (DMF), triethylamine (TEA), α-TOS, collagenase type IV, coumarin-6, bovine serum albumin (BSA), and 3-(4,5-dimethylthiazol-2-y1)-2,5-di-phenyltetrazolium bromide (MTT) were obtained from Sigma-Aldrich (St Louis, MO, USA). O-(6-Chlorobenzotriazol-1-yl)-N,N,N′,N′,-tetramethyluronium tetrafluoroborate (TCTU) was purchased from Adamas Reagent Co., Ltd (Shanghai, People’s Republic of China). 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), LysoTracker Red, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine (DiR), and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) were purchased from Beyotime Biotechnology (Shanghai, People’s Republic of China). TPGS was provided by BASF Corporation (Ludwigshafen, Germany). Dulbecco’s Modified Eagle’s Medium (DMEM), penicillin–streptomycin, phosphate buffered saline (PBS), and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, CA, USA). GM 6001 was purchased from Medchemexpress (Monmouth Junction, NJ, USA). All other reagents and chemicals were of analytical grade.
Human fibrosarcoma cell line HT1080 and human breast adenocarcinoma cell line MCF-7 were purchased from the Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Science, Chinese Academy of Sciences, Shanghai, People’s Republic of China. The cells were cultured in DMEM medium supplemented with 10% FBS, 100 U/mL of penicillin, and 100 U/mL of streptomycin under 95% humidity and 5% CO2 at 37°C. All experiments were performed using cells in the logarithmic phase of growth.
Male BALB/c nude mice (20–22 g) were supplied by Laboratory Animal Center, Fudan University, Shanghai, People’s Republic of China. Animals were kept under specific pathogen free conditions. Ethical approval was obtained from the Ethics Committee of Fudan University for the experiments in the study involving human cell line and animals.
Synthesis, purification, and characterization of mPEG2K-GK8-α-TOS conjugate
The synthesis of mPEG2K-GK8-α-TOS involves two steps as shown in Figure 1. mPEG2K-NHS (MW 2,000 Da) and C-terminus-protected peptide GK8 (n:n =1:1.2) were dissolved in 5 mL of DMF and then 20 μL TEA was added. The synthesis was carried out under nitrogen protection at 40°C overnight. The unreacted peptide was removed by dialysis (molecular weight cut off [MWCO] 1,000 Da) against deionized water. The purified mPEG2K-GK8 conjugate was lyophilized and characterized by 1H-nuclear magnetic resonance spectroscopy (1H NMR) in CDCl3 at 400 MHz (Mercury Plus; Varian, Palo Alto, CA, USA). Then, mPEG2K-GK8 conjugate and α-TOS (n:n =1:1.2) were dissolved in anhydrous DMF in the presence of 5% TCTU (catalytic agent) and 20 μL TEA. Reaction was carried out under nitrogen protection at room temperature overnight. Diethyl ether (0°C) was added to the reaction system to precipitate the product and remove any unreacted α-TOS.23 The precipitate was redissolved in deionized water before being dialyzed (MWCO 2,000 Da) against deionized water for 48 hours to remove any TCTU. The product was lyophilized before characterization by 1H NMR.
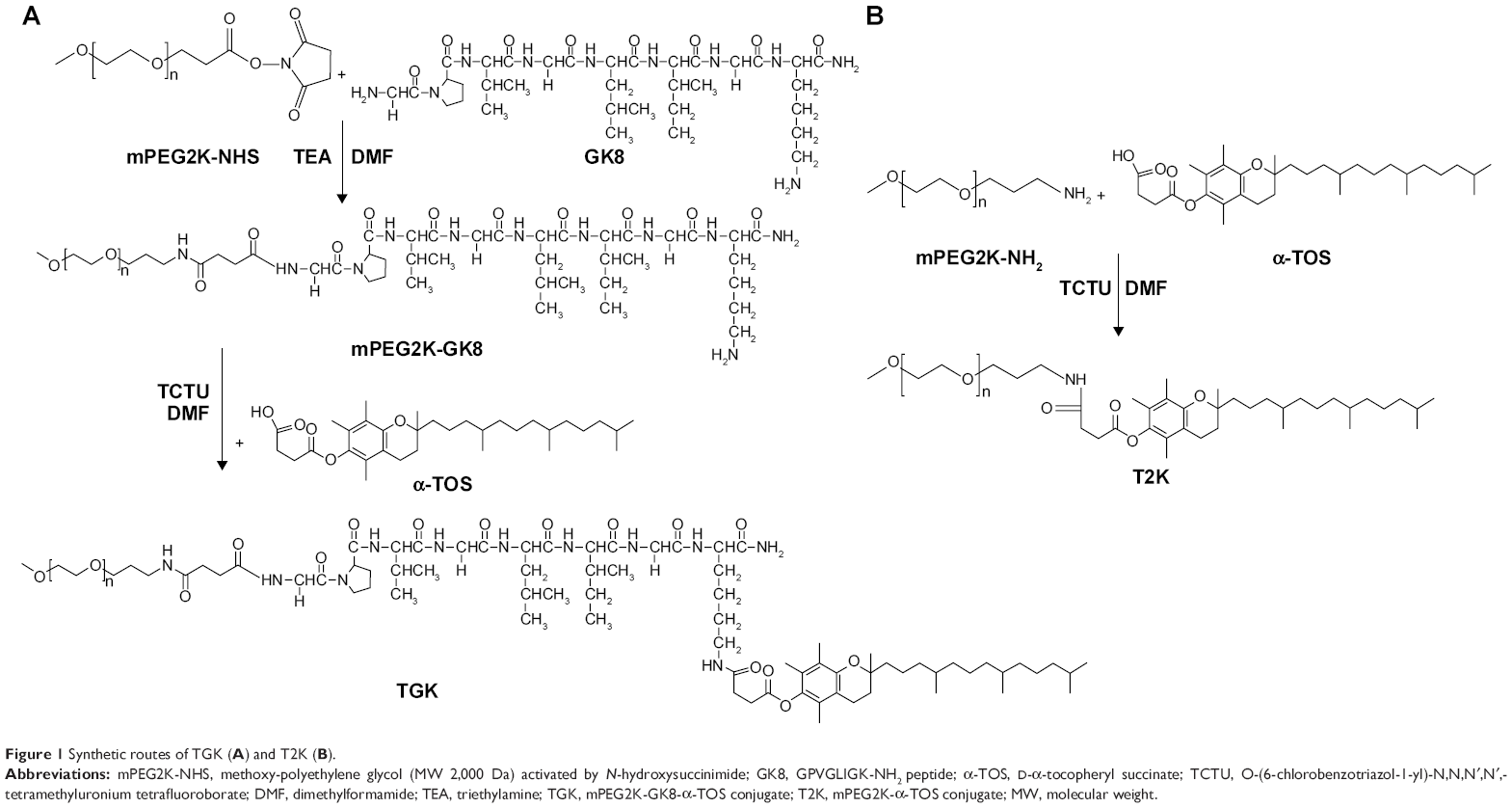 | Figure 1 Synthetic routes of TGK (A) and T2K (B). |
The T2K conjugate was synthesized as control following a similar protocol wherein mPEG2K-NH2 (MW 2,000 Da) and α-TOS were mixed at a molar ratio of 1:1.2, respectively, in 5 mL DMF with 5% TCTU and 20 μL TEA. Similarly, the reaction system was treated with 0°C diethyl ether, dialyzed against deionized water and lyophilized before characterization by 1H NMR.
Preparation of DTX-loaded micelles
Thin-film hydration method was chosen to prepare micelles. Briefly, 20 mg of TPGS, 65 mg of TGK (n:n =40:60), and 3 mg of DTX were dissolved in 5 mL of chloroform in a round-bottomed flask to prepare DTX-loaded TGK micelles. The organic solvent was removed by rotary evaporation at 40°C in a water bath to form a thin film. DTX-loaded micelles were formed by suspending the thin film in 5 mL deionized water for 30 minutes after vacuum drying overnight. The unloaded DTX was removed by filtration through 0.22 μm sterile filter. The DTX-loaded T2K micelles were prepared following a similar procedure wherein 20 mg of TPGS, 50 mg of T2K (n:n =40:60), and 3 mg of DTX were mixed in 5 mL of chloroform. The micelles solution was lyophilized for further use. Coumarin-6-loaded T2K micelles and TGK micelles were prepared following the same procedure.
Characterization of DTX-loaded MMP-2/9-sensitive micelles
CMC value determination
The CMC values of blank T2K and TGK micelles were measured by ultraviolet-visible spectroscopy method, using iodine (I2) as a hydrophobic probe.25 The I2/KI standard solution was prepared by dissolving 1 g of I2 and 2 g of KI in 100 mL of deionized water. Samples of blank T2K and TGK micelles with concentration ranging from 0.0001% to 0.1% were prepared before 5 μL of I2 standard solution was added to 1 mL of the sample solution. After incubating for 24 hours in the dark at 37°C, the absorbance of samples at 366 nm was measured by a microplate reader (Synergy 2; Bio Tek, Winooski, VT, USA). CMC value was calculated by corresponding polymer concentration at which a sharp increase in absorbance was observed. The experiment was performed in triplicate.
Particle size and distribution
Particle size, distribution, and zeta-potential of DTX-loaded micelles were determined by dynamic light scattering at 25°C by using Malvern Zetasizer (Nano ZS90; Malvern Instruments, Worcestershire, UK).
Determination of drug encapsulation efficiency and drug loading efficiency
The DTX encapsulated in the micelles was measured by reverse-phase high-performance liquid chromatography (RP-HPLC) assay with 10-Avp HPLC system (Shimadzu Corporation, Kyoto, Japan). A reverse-phase column (Gemini 5u C18 110A, 4.6×150 mm, 5 μm; Phenomenex, Torrance, CA, USA) was used. The mobile phase consisted of 0.043 mol/L ammonium acetate solution and acetonitrile (v:v =55:45), and was delivered at a rate of 1.0 mL/min. The injection volume was 20 μL, and absorbance at 232 nm was measured by SPD-10AVP UV-vis-detector (Shimadzu Corporation, Kyoto, Japan). The encapsulation efficiency (EE) and DL of the micelles were calculated by the following equations, respectively:
|
|
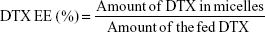
|
|
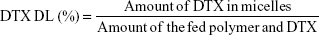
Stability of the MMP-2/9-sensitive micelles
The TGK micelles were incubated for 48 hours in the dark at 25°C. The micelles solution (500 μL) was collected at predetermined intervals. After being filtered through a 0.22 μm filter membrane, the amount of DTX remaining in the filtrate was analyzed by RP-HPLC. Both experiments were performed in triplicate. MMP-2/9-nonsensitive T2K micelles were used as control. The percentage of DTX remaining in the micelles was calculated by the following equation:
|
|
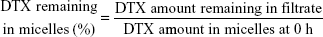
Cleavage study of MMP-2/9-sensitive micelles
To confirm whether TGK micelles can be cleaved by MMP-2/9, fluorescence resonance energy transfer (FRET) analysis was conducted. A pair of FRET hydrophobic dyes, DiI and DiO, were incorporated into the mixed micelles with DiO as the donor and DiI as the acceptor. Simultaneous encapsulation of DiI and DiO will result in a strong signal of DiI to be observed due to the close proximity of DiI and DiO in the micelles’ core. After micelles’ decomposition, FRET will be no longer possible due to DiI and DiO couldn’t retain closely anymore. The DiO-/DiI-coloaded micelles containing 0.2% DiO and 0.2% DiI were prepared following the same procedure as DTX-loaded micelles. DiO-/DiI-coloaded micelles (900 μL) and 100 μL of MMP-2/9 solution (1 mg/mL) were mixed and incubated at 37°C for 2 hours. The fluorescence emission spectrum from 500 to 650 nm and intensity at 510 nm (IG) and 565 nm (IR) with excitation wavelength of 484 nm were recorded using a fluorescence spectrophotometer (Cary Eclipse; Agilent, Santa Clara, CA, USA).
In vitro drug release profile
The in vitro drug release profile of DTX-loaded T2K and TGK micelles was analyzed by dialysis method. Briefly, 900 μL of DTX-loaded micelle suspensions and 100 μL of MMP-2/9 solution (1 mg/mL), MMP-2/9 solution (100 μg/mL) or BSA (1 mg/mL) were premixed and loaded into dialysis bags (MWCO 2,000 Da), respectively. The micelle suspensions were dialyzed against 25 mL PBS (pH 7.4) containing 0.1% Tween 80 in order to maintain sink condition.30 The experiment was carried out in triplicate at 37°C with shaking at 100 rpm, for 48 hours. Release medium (500 μL) was collected and replaced with the same amount of PBS containing 0.1% Tween 80, at appropriate intervals. The samples were analyzed by RP-HPLC as described previously.
In vitro cellular uptake
To evaluate the intracellular delivery of T2K and TGK micelles, cellular uptake of DTX-loaded T2K and TGK micelles was investigated in MCF-7 (MMP-2/9 underexpression) and HT1080 (MMP-2/9 overexpression) cells. Briefly, MCF-7 or HT1080 cells (5×105/well) were seeded in a six-well cell culture plate for 24 hours. Then, the cells were washed thrice and cultured in fresh DMEM (without FBS) containing 20 μg/mL DTX-loaded T2K or TGK micelles for 30 minutes. After incubation, the cells were washed thrice with cold PBS and harvested by scrapers. The cells were dissolved in 200 μL acetonitrile before being subjected to probe-type ultrasonic treatment (400W, JY92-II; Scientz Biotechnology Co., Ltd., Ningbo, Zhejiang Province, People’s Republic of China) for DTX extraction. The extract was centrifuged at 10,000 rpm for 5 minutes before the supernatant was analyzed by RP-HPLC. Protein content was determined using BCA protein assay kit. DTX accumulation was normalized with total protein content. In order to study the cellular uptake mechanism, the cellular uptake was evaluated in the presence of GM6001 and MMP-2/9 at the same time. The cells were washed thrice and preincubated with 100 μM GM6001 or 100 μg/mL MMP-2/9 for 1 hour before being treated with DTX-loaded T2K or TGK micelles. Both experiments were performed in triplicate.
In vitro cytotoxicity
HT1080 and MCF-7 cells were seeded in a 96-well plate at a density of 5,000 cells per well in 200 μL DMEM. After 24 hours of incubation, the medium was removed and the cells were incubated for 48 hours in DMEM containing DTX, DTX-loaded T2K and TGK micelles (concentrations ranging from 0.25 to 5 μg), or blank mixed micelles of various concentrations. Cells treated with culture medium were used as control. The cytotoxicity was assessed using the MTT method. Briefly, 200 μL of MTT solution (0.5 mg/mL, diluted with FBS-free DMEM) was added to each well. The plates were incubated for 4 hours at 37°C. The supernatants were discarded carefully and 200 μL of dimethyl sulfoxide was added to each well to dissolve the formazan crystal. Similarly treated wells without cells were used as blank. The absorbance at 570 nm of each well was measured by a microplate reader (Synergy 2; Bio Tek) after incubation for 15 minutes at 37°C. The cell viability was calculated by the following equation:
|
|
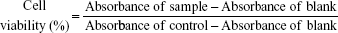
Subcellular localization of T2K and TGK micelles
HT1080 and MCF-7 cells were seeded in confocal dishes and incubated in FBS-free DMEM at 37°C for 2 hours before the experiment was conducted. Cells were incubated with coumarin-6-loaded T2K or TGK micelles (coumarin-6 concentration: 500 ng/mL) for 30 minutes and fixed with 4% paraformaldehyde solution for 15 minutes. The fixed cells were stained with 50 nM LysoTracker Red (15 minutes) and 10 μM Hoechst 33342 (15 minutes) to visualize endosome/lysosome and nuclei. Then, the cells were washed with cold PBS three times and examined by confocal laser scanning microscopy (LSM710; Carl Zeiss, Jena, Germany).
In vivo imaging studies for tumor targeting ability of MMP-2/9-sensitive micelles
The in vivo biodistribution and tumor targeting ability of T2K and TGK micelles were investigated using DiR, a near-infrared fluorescent dye. To construct HT1080 tumor-bearing nude mice, 1×105 HT1080 cells were subcutaneously inoculated into the upper right armpit. When the tumor volume reached around 100 mm3, the mice were randomly divided into three groups (three mice per group). They were then intravenously administrated DiR-loaded T2K or TGK micelles at a dosage of 1 mg/kg DiR. Mice injected with saline were used as control of autofluorescence. In vivo fluorescent imaging was performed at 2, 4, 8, 10, 12, and 24 hours of postinjection, and the excitation wavelength and emission wavelength were fixed at 780 and 800 nm, respectively, by using the IVIS Spectrum Pre-clinical In Vivo Imaging System (PerkinElmer Inc., Waltham, MA, USA). To confirm the distribution of DiR probe in different organs, the mice were euthanized at 24 hours of postinjection. Organs were collected and washed with PBS twice followed by fluorescent imaging. The data were analyzed by Living Image® Version 4.3.1 Software (Caliper Life Science, Hopkinton, MA, USA).
In vivo antitumor effect evaluation
Antitumor efficacy was evaluated on subcutaneous tumor-bearing nude mice constructed as described earlier. Tumor-bearing mice were randomized to four groups (seven mice per group) when the tumor size was approximately 100 mm3. Saline, DTX in Tween 80/dehydrated alcohol (50:50, v/v), and DTX-loaded TGK and T2K micelles were injected via tail vein every 3 days at a dosage of 10 mg/kg, three times. The tumors’ sizes were monitored every 2 days for 2 weeks. At the end of the experiment, all the treated and control nude mice were executed before the tumors were harvested, weighed, and photographed. Volume of tumors was calculated by the following formula: (volume = L × W2/2), where L is the longest and W is the shortest in tumor diameters (mm). To monitor the potential toxicity, the body weights of all groups were recorded every 2 days.
Data analysis
Data are presented as mean ± standard deviation (SD), and the difference between groups was analyzed by Student’s t-test. P<0.05 was considered statistically significant.
Results and discussion
Synthesis, purification, and characterization of mPEG2K-GK8-α-TOS conjugate
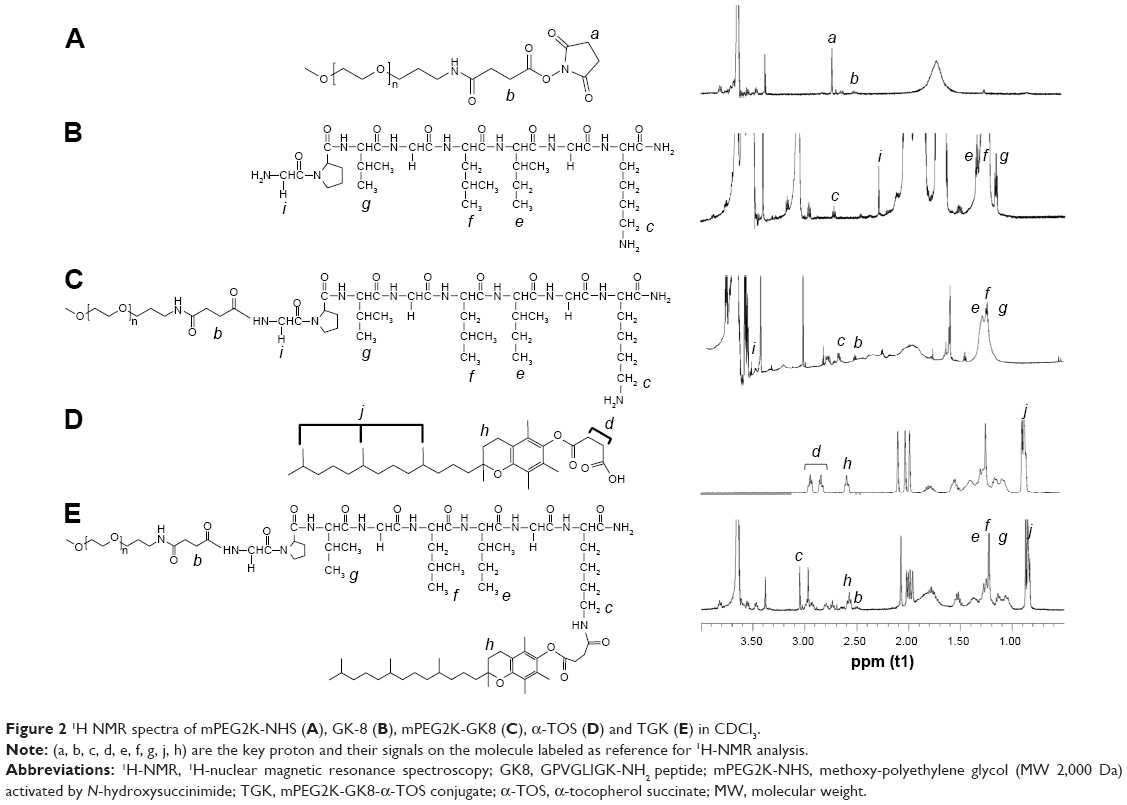
Several peptide sequences, such as GPLGIAGQ,5 GPLGV,10 GPLGVRG,32 and PVGLIG33 have been reported for the development of MMP-2/9-sensitive conjugate. MMP-2/9 susceptibility of PVGLIG has been identified by using a combinational library.34 It is reported that short peptides with a certain sequence can be cleaved by MMP-2/9 regardless of their secondary or tertiary structures.1 In order to allow conjugation at the terminal residue while maintaining MMP-2/9 susceptibility, two amino acids (glycine and lysine) were introduced at the end of PVGLIG peptide to provide amine functional groups.7 In this study, two kinds of conjugates, lysine conjugates and N terminal conjugates, are considered as the potential products of this conjugation. To control the reaction, the aqueous environment, where NHS conjugations usually take place, was replaced with proton-capturing DMF and TEA as solvents and a slightly higher temperature (40°C instead of 37°C) was chosen to enhance the nucleophilicity of the N-terminus amine groups. Considering that the N-terminus amine group possesses an advantage owing to the existence of the α-carbonyl, it is deemed to be the primary conjugation site in terms of reaction kinetics under this condition.35 T2K and TGK syntheses were confirmed by 1H NMR as shown in Figure S2 and Figure 2. The conjugation of GK8 (Figure 2B) on the N-terminal was confirmed by the absence of NHS proton peak a (s, 2H) at 2.7 ppm, the shift of proton peak i (d, 2H) from 2.2 to 3.5 ppm, and the appearance of proton peaks c (s, 2H) at 2.8 ppm, e (m, 3H), f (m, 3H), and g (m, 3H) from 1 to 1.5 ppm. The final product was determined by the appearance of proton peaks h (t, 2H) at 2.6 ppm and d (t, 2H) at 2.9 ppm of α-TOS. The shift of c and d peaks to the lower field (c [s, 2H] at 3.07ppm, d [t, 2H] at 3.01 ppm) as shown in Figure 2E proved that the α-TOS was conjugated to the lysine amine group. In terms of T2K (Figure S2C), the conjugation of mPEG2K-NH2 and α-TOS was confirmed by the chemical shift of proton c (m, 2H) changed from 2.8 to 3.09 and the appearance of proton a (t, 2H) and b (t, 2H). RP-HPLC method was used to confirm the final products as shown in Figure S3. The change of retention time was consistent with the polarity. Chromatography also indicated high purity of the final products.
 | Figure 2 1H NMR spectra of mPEG2K-NHS (A), GK-8 (B), mPEG2K-GK8 (C), α-TOS (D) and TGK (E) in CDCl3. |
Preparation and characterization of MMP-2/9-sensitive micelles
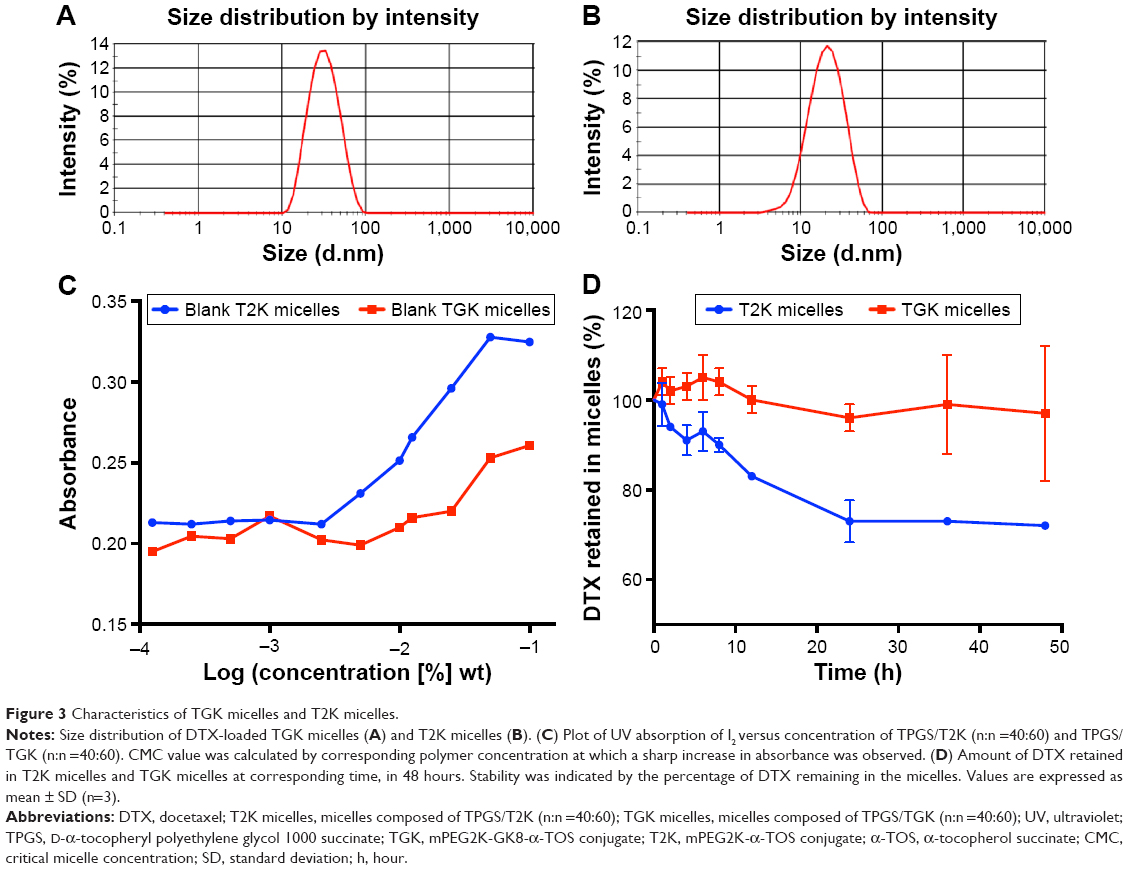
Based on the successful synthesis of TGK, micelles were prepared by thin-film hydration method as previously reported.31 The dynamic light scattering results indicated that Z-average diameter of DTX-loaded T2K micelles was 15.35±1.79 nm and the ζ-potential was −4.51±0.39 mV. The particle size of TGK micelles was slightly larger (23.65±2.17 nm), which could be attributed to the inserted GK8 peptide (Figure 3A). It is universally recognized that small-sized micelles (10–100 nm) are highly effective in passive tumor targeting through enhanced permeability and retention effect.24 Taking advantage of the suitable particle size of TGK micelles, favorable biodistribution can be predicted. The DL of T2K micelles was 3.83%±0.17%, which was higher than that of TGK micelles (1.89%±0.10%, Table 1). Since GK8 peptide is hydrophilic, the relatively lower DL could be caused by the difference in hydrophilicity between T2K and TGK conjugate. The CMC value was measured by ultraviolet-visible spectroscopy method. The results indicated that the CMC values of TPGS/T2K (n:n =40:60) and TPGS/TGK (n:n =40:60) were approximately 0.0025% and 0.005%, respectively (Figure 3C). A lower CMC value was conducive to the stability of micelles at lower concentrations. The lower CMC value suggests that T2K and TGK micelles could provide greater resistance against dilution and better stability than the single TPGS micelles.23
 | Figure 3 Characteristics of TGK micelles and T2K micelles. |
 | Table 1 Physicochemical characterization of DTX-loaded micelles |
Stability of the MMP-2/9-sensitive micelles
Following the preparation of micelles, their stability should be determined before any other characterization takes place. Figure 3D shows the stability of TGK and T2K micelles at room temperature within 48 hours. Almost 100% DTX remained in TGK micelles while only 70% remained in T2K micelles at 48 hours. The better stability of TGK micelles can be attributed to the inserted peptide sequence in TGK. Shi et al reported that arginine had an excellent stabilizing effect on DTX-loaded mPEG-poly (D,L-lactic acid) micelles.36 The stabilizing mechanism was closely related to the electrostatic interaction between amino acids and DTX as well as the hydrogen bond formation between them.36 Thus, the increased stability rendered TGK micelles more practical in future application than T2K micelles.
Cleavage study of MMP-2/9-sensitive micelles
To confirm whether TGK micelles can be cleaved by MMP-2/9, FRET analysis was conducted. A hydrophobic FRET pair, DiO as donor and DiI as acceptor, were loaded in the TGK and T2K micelle cores physically.37 When DiO/DiI-coloaded micelles were excited at 484 nm, the emission energy of DiO would be transferred to DiI, resulting in quenching of DiO’s signal and appearance of a strong DiI signal.19 The result indicated that after being incubated with 100 μg/mL MMP-2/9 for 2 hours, the ratio of IR/(IG+IR) in TGK micelles decreased significantly (P<0.01), which was not observed with the T2K micelles (Figure 4). Once the peptide was cleaved, DiI and DiO would be released from the hydrophobic core of TGK micelles, resulting in the disappearance of FRET. For T2K micelles, owing to the close proximity of DiI and DiO, IR/(IG+IR) was unaffected (P>0.05) after incubation with MMP-2/9. The change in IR/(IG+IR) before and after incubation with MMP-2/9 showed that TGK micelles were cleaved successfully.
 | Figure 4 FRET analysis of DiO/DiI-loaded T2K micelles and TGK micelles. |
In vitro drug release profile
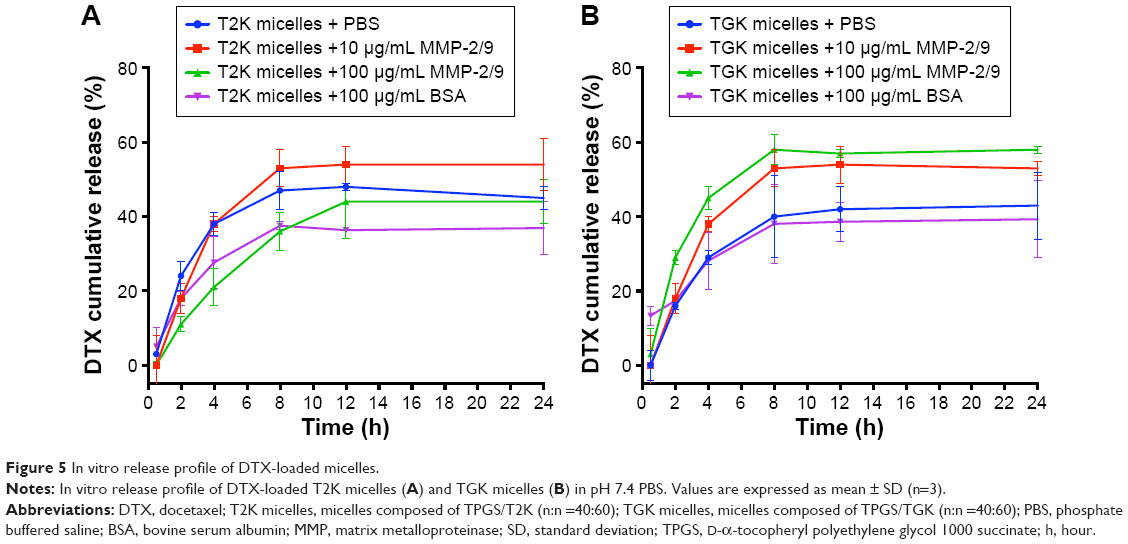
The in vitro release profiles of DTX-loaded MMP-2/9-sensitive micelles were investigated in the presence of 10 and 100 μg/mL of MMP-2/9 as well as in MMP-2/9-free medium as control. As shown in Figure 5B, the initial cumulative release of DTX from the micelles occurred rapidly within the first 8 hours, but gradually decreased, which may be attributed to the formation of aggregates between DTX and α-TOS after being cleaved by MMPs. In the absence of MMP-2/9, 42.65%±9.32% and 51.19%±2.12% of DTX were cumulatively released from TGK and T2K micelles within 24 hours, respectively. The drug release from TGK micelles increased in an MMP-2/9-concentration-dependent manner, while the release of DTX from T2K micelles was unrelated to the concentration of MMP-2/9 (Figure 5A). Interestingly, when the concentration of MMP-2/9 reached 100 μg/mL, the cumulative drug release from T2K micelles slightly decreased from 2 to 8 hours. It was believed that the high concentration of protein could form corona on the surface of nanoparticles resulting in steric stabilization.38,39 The decreasing cumulative drug release may be attributed to the stabilizing property of high concentration of MMP-2/9. In order to verify the impact of protein on drug release, drug release was conducted in the presence of 100 μg/mL BSA. It was found that DTX cumulative release of both TGK and T2K micelles slightly decreased in the presence of 100 μg/mL BSA suggesting that proteins impact on the drug release profile to some extent. It was deduced that, in MMP-2/9-free medium, TGK micelles maintained micelle structure and encapsulated DTX in their hydrophobic core, while TGK was cleaved in the presence of MMP-2/9, resulting in decomposition followed by the release of DTX. In addition, in the absence of MMP-2/9, TGK micelles presented a lower cumulative release than T2K micelles from 0 to 8 hours, which was beneficial for decreasing leakage in circulation. The MMP-2/9-dependent release profile indicated that the TGK micelles were able to rapidly release the loaded drug in the presence of MMP-2/9.
 | Figure 5 In vitro release profile of DTX-loaded micelles. |
In vitro cellular uptake
When TGK micelles are degraded by MMP-2/9, the cellular uptake profile is expected to be enhanced because the rapid passive diffusion across the cell membrane is much more effective than endocytosis pathways in terms of highly hydrophobic drugs.40 To evaluate the intracellular delivery of T2K and TGK micelles, cellular uptake of DTX-loaded T2K and TGK micelles was investigated in MCF-7 (MMP-2/9 underexpression) and HT1080 (MMP-2/9 overexpression) cells. It was clear that after incubating with free DTX and DTX-loaded micelles for 30 minutes, TGK micelles demonstrated much higher cellular uptake than T2K micelles (P<0.01, Figure 6B) in HT1080 cells, while that was similar in MCF-7 cells, which could be attributed to the differential cleavage of TGK and T2K micelles by MMP-2/9. In order to further verify MMP-2/9 degradation-induced higher uptake, the cellular uptake was also evaluated in the presence of GM6001 (a broad-spectrum MMPs inhibitor) and MMP-2/9. Interestingly, the cellular uptake of TGK micelles was significantly enhanced in the presence of 100 μg/mL of MMP-2/9 in MCF-7 cells. However, similar results could not be observed for T2K micelles neither in MCF-7 cells nor in HT1080 cells. The cellular uptake of TGK micelles declined to a level similar to T2K micelles upon the addition of 100 nM of GM6001 in HT1080 cells. These results demonstrated that TGK micelles could be cleaved by extracellular MMP-2/9 to release DTX from their hydrophobic cores, which further enhanced cellular uptake. Additional uptake assays involving NaN3 as ATP depletion agent enhanced the previously mentioned conclusion by proving that the uptake level of TGK micelles was comparable with that of DTX with the endocytosis process inhibited in HT1080 cells (Figure S4).
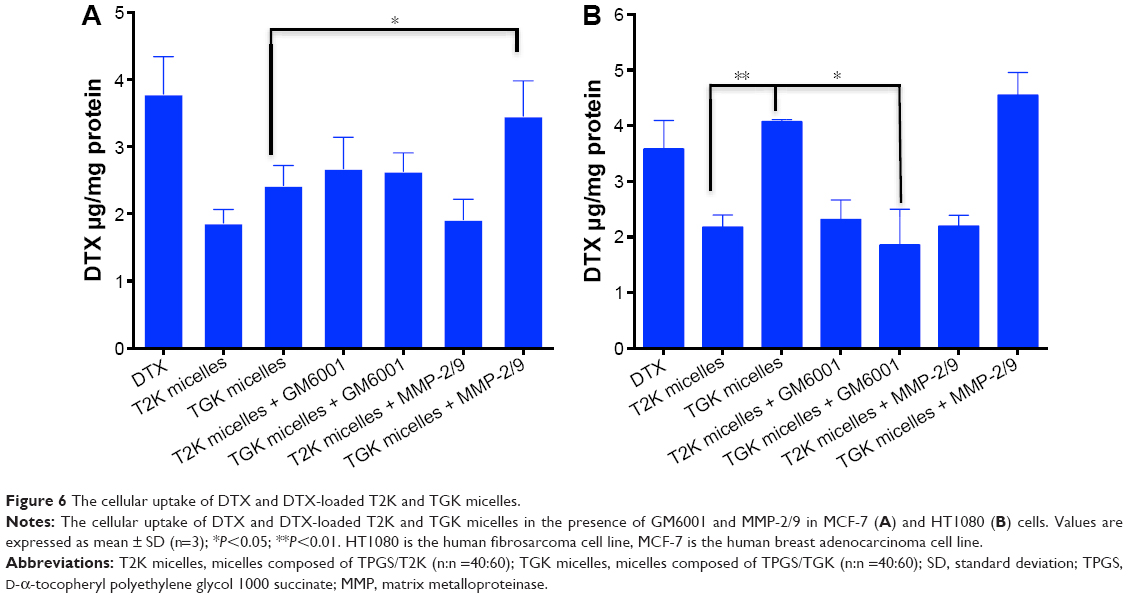 | Figure 6 The cellular uptake of DTX and DTX-loaded T2K and TGK micelles. |
In vitro cytotoxicity
To verify whether TGK micelles possess cytotoxicity advantages relative to T2K micelles, in vitro cytotoxicity assays were performed. Initially, in vitro cytotoxicity assays of blank TGK and T2K micelles were conducted with Tween 80 as the control group in MCF-7 and HT1080 cells. As shown in Figure 7, blank TGK micelles, T2K micelles, and Tween 80 displayed increasing cytotoxicity at increasing concentration. Blank TGK and T2K micelles possessed higher cytotoxicity than Tween 80 both in MCF-7 and in HT1080 cells. It has been widely reported that α-TOS and its analogs possess anticancer properties against a wide variety of human cancer cells while remaining nontoxic to normal cells (Figure S5).17 Mi et al reported the synergistic effect of T2K and DTX.23 The same conclusion could also be drawn from the cytotoxicity of DTX-loaded TGK micelles and T2K micelles (Figure 7). It was clear that DTX-loaded TGK and T2K micelles showed higher cytotoxicity than DTX both in MCF-7 and in HT1080 cells and this can be attributed to the synergistic effect. This indicates that the cytotoxicity observed in the assay could be partially attributed to the blank TGK micelles or T2K micelles. Moreover, the lower half-maximal inhibitory concentration (IC50) value of TGK micelles relative to T2K micelles in HT1080 cells can be explained by the higher uptake of TGK micelles, indicating better antiproliferative ability of TGK micelles in MMP-2/9-overexpressing tumors (Table 2). To confirm that the difference in cytotoxicity was related to MMP-2/9 cleavage, we investigated the in vitro cytotoxicity of the two micelles in MCF-7 cells (MMP-2/9 underexpression; Figure 7C). The IC50 of TGK and T2K micelles in MMP-2/9-overexpressing and underexpressing cell lines are shown in Table 2. The result revealed no difference in cytotoxicity between T2K and TGK micelles in MCF-7 cells. Combined with the similar cytotoxicity of the two micelles observed in the presence of GM6001 in HT1080 cells (Figure S6), it was convincing that higher cytotoxicity of TGK micelles was closely related to MMP-2/9 cleavage.
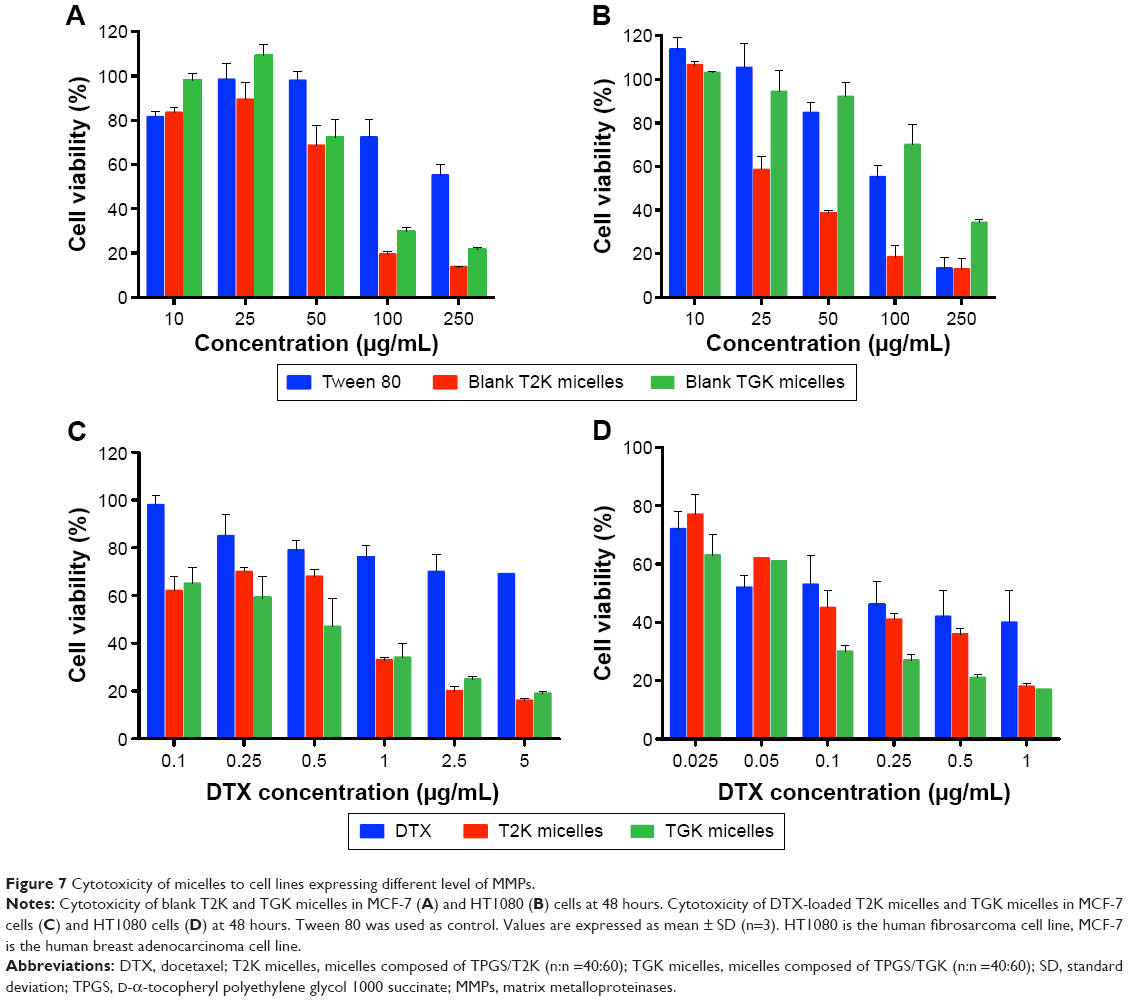 | Figure 7 Cytotoxicity of micelles to cell lines expressing different level of MMPs. |
 | Table 2 IC50 values of DTX or DTX-loaded micelles against HT1080 cells and MCF-7 cells |
Subcellular localization of MMP-2/9-sensitive micelles
In order to investigate the intracellular localization of T2K and TGK micelles after being internalized, confocal laser scanning microscopy was performed using triple labeling with fluorescent micelles (coumarin-6-loaded), acid endosome/lysosome-selective dye (LysoTracker Red), and nuclei dye (Hoechst 33342). Results shown in Figure 8 indicate that T2K micelles appear to be internalized via lysosome-mediated endocytosis, as evidenced by the colocalization of the green fluorescence of coumarin-6-loaded T2K micelles with the red fluorescence of endosome/lysosome. The suggested colocalization was lower in TGK micelles-treated HT1080 cells owing to their MMP-2/9 sensitivity. TGK micelles appeared to be disintegrated by MMP-2/9 before the loaded drug was internalized via diffusion. All formulations colocalized with the lysosome in MCF-7 cells. In order to reach their targets, anticancer drug molecules loaded in micelles must escape the relatively acidic endosome to avoid degradation.41,42 MMP-2/9-sensitive drug delivery system provides one way to circumvent this barrier.
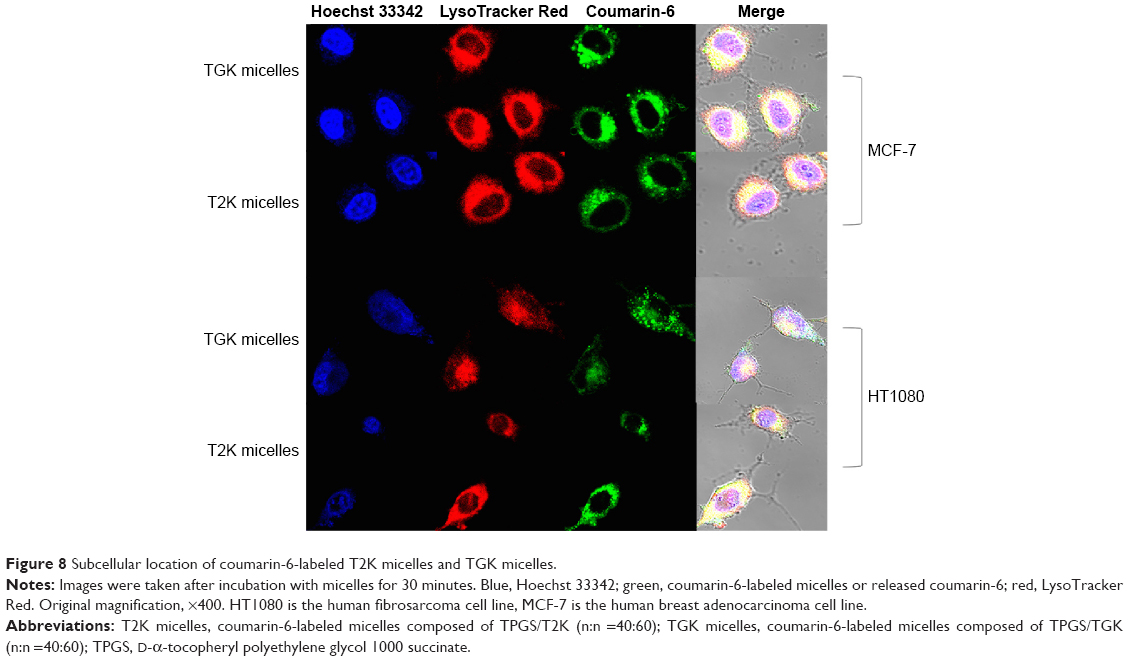 | Figure 8 Subcellular location of coumarin-6-labeled T2K micelles and TGK micelles. |
In vivo imaging studies for tumor targeting ability of MMP-2/9-sensitive micelles
An important feature of the TGK micelles delivery system is its ability to release loaded drug at tumor site; DiR, a near-infrared fluorescent dye was used to evaluate the in vivo biodistribution of TGK and T2K micelles. After intravenous injection of DiR-loaded TGK or T2K micelles via tail vein, in vivo fluorescent imaging was performed at predetermined time intervals as shown in Figure 9A. While both T2K and TGK micelles were found to be widely distributed in the whole body at 2 hours postinjection, the TGK micelles demonstrated a significantly enhanced accumulation in tumor site at 4 hours and the increased fluorescence intensity was retained even at 24 hours postinjection indicating a low clearance rate of TGK micelles in tumor site. Throughout the entire experiment, TGK micelles exhibited more and longer accumulation in tumor site compared to T2K micelles which suggested that TGK micelles possessed better tumor targeting and accumulation ability, a fact that was further confirmed by ex vivo imaging of excised organs (Figure 9B). At 24 hours postinjection, fluorescence intensity of TGK micelles in tumor tissue was a fold higher than that of the T2K micelles (Figure 9C), while the fluorescence in liver was reduced. These results demonstrated that TGK micelles have the potential to increase tumor accumulation, which might contribute to their ability to release the loaded drug/dyes in a site-specific manner.
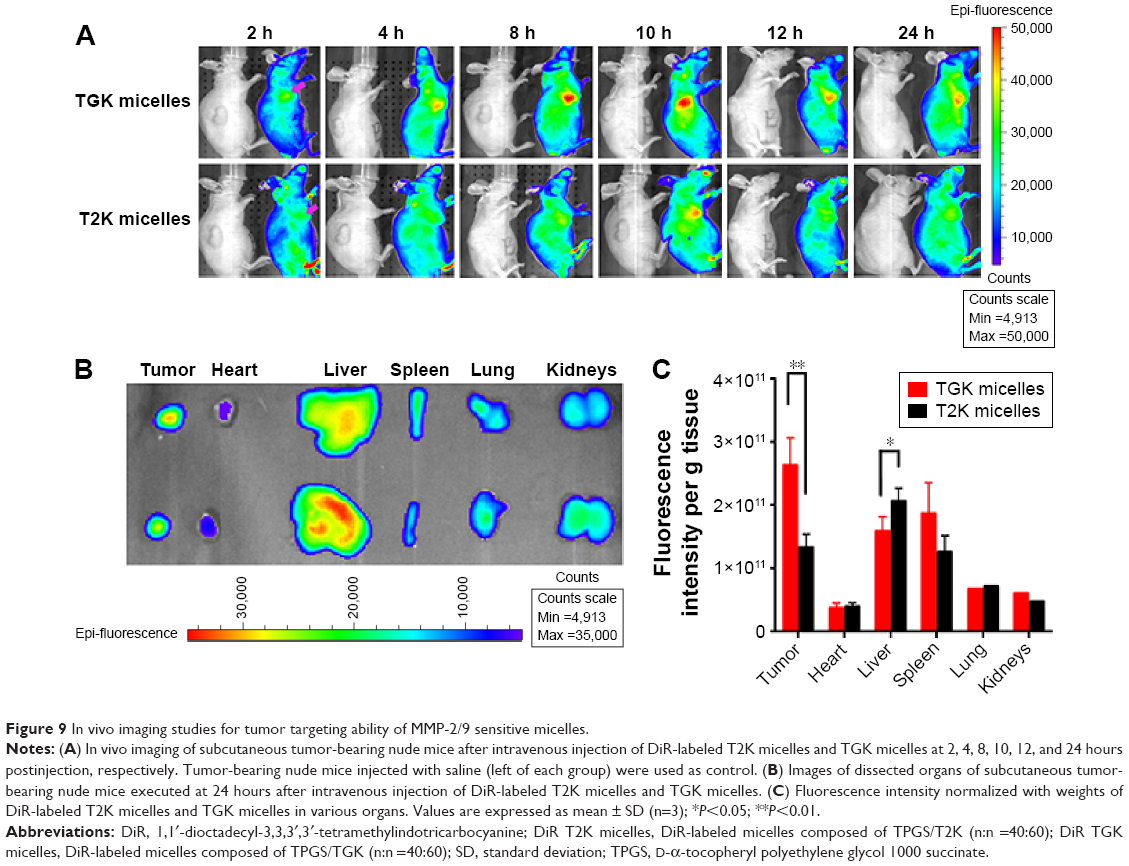 | Figure 9 In vivo imaging studies for tumor targeting ability of MMP-2/9 sensitive micelles. |
In vivo antitumor effect evaluation
To test the in vivo antitumor effect of MMP-2/9-sensitive micelles, subcutaneous tumor-bearing nude mice were administered saline, DTX in Tween 80/dehydrated alcohol (50:50, v/v), and injected with DTX loaded TGK or T2K micelles at a dosage of 10 mg/kg three times during the experiment. The interval was three days. The results indicated that compared with the baseline, continuous decrease in tumor size was observed in TGK micelles group while increases were observed in other groups simultaneously from day 0 to day 7 (Figure 10A). However, after the cessation of the treatment on day 7, the size of tumor in all groups increased. On day 14, the mice were executed humanely before the tumors were harvested and weighed. The tumor weight was 0.87±0.24 g (saline group), 0.19±0.12 g (DTX group), 0.13±0.05 g (T2K micelles group), and 0.07±0.02 g (TGK micelles group). A significant difference was observed (P<0.05) between the TGK micelles group and T2K micelles group in terms of tumor weights (Figure 10D). To monitor the potential systemic toxicity, the body weights of all groups were recorded every 2 days. It is worth noticing that two mice and one mouse died in the DTX and T2K micelles groups, respectively, during the treatment, due to systemic toxicity. Nude mice demonstrated 19.22%±13.12% and 13.93%±10.94% loss of initial weight in DTX and T2K groups, respectively, while no significant body weight changes were observed after treatment with TGK micelles (Figure 10B). All these results indicate that TGK micelles are potent against solid tumors while maintaining minimum systemic toxicity compared with T2K micelles and DTX.
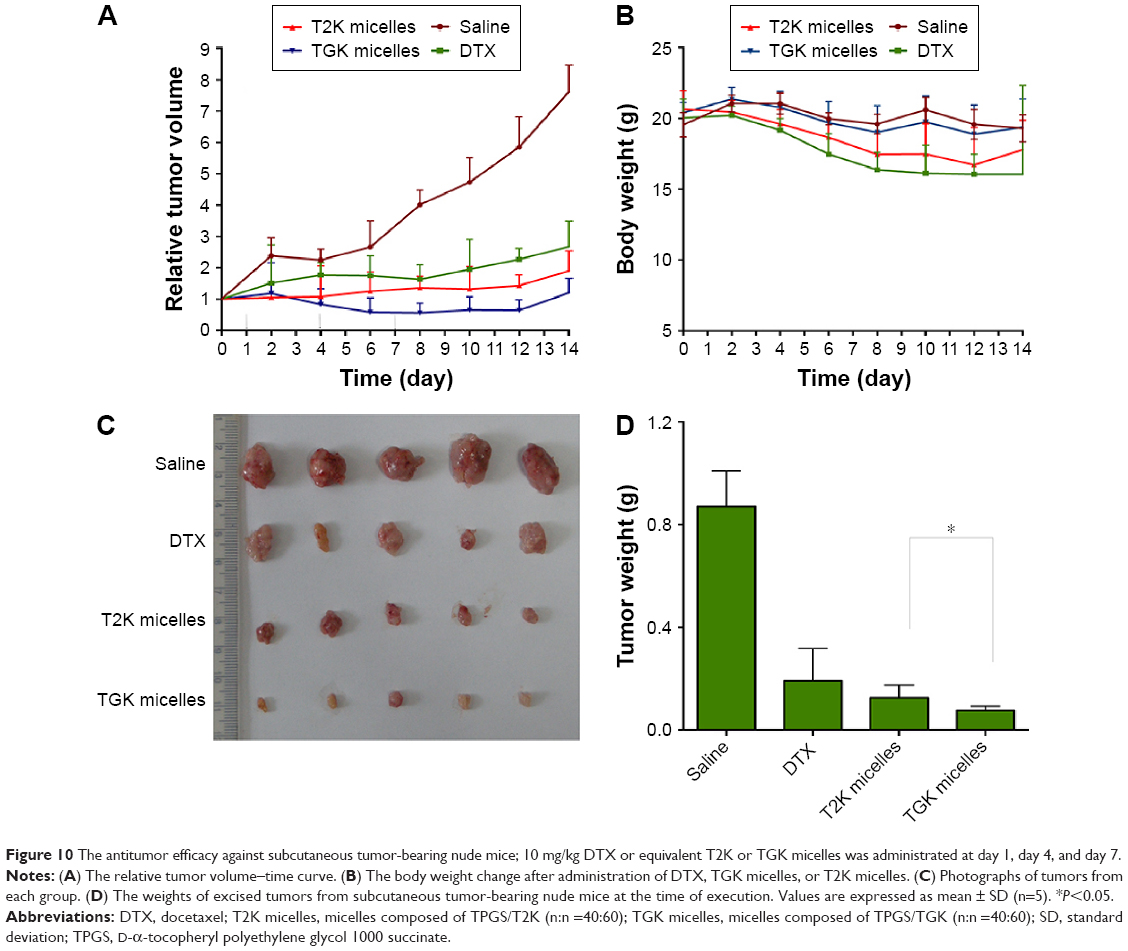 | Figure 10 The antitumor efficacy against subcutaneous tumor-bearing nude mice; 10 mg/kg DTX or equivalent T2K or TGK micelles was administrated at day 1, day 4, and day 7. |
Conclusion
In this study, MMP-2/9-sensitive mPEG2K-peptide-α-TOS conjugate TGK was successfully synthesized to develop novel stimuli-responsive micelles composed of TPGS and TGK that were carefully characterized. FRET and in vitro drug release profile indicated that TGK micelles could be disintegrated in the presence of MMP-2/9. Compared with nonsensitive micelles and DTX, TGK micelles showed higher uptake in HT1080 cells. While the IC50 of TGK and T2K micelles was similar in MCF-7 cells, the IC50 value of the aforementioned micelles were 0.064±0.006 and 0.122±0.009 μg/mL in HT1080 cells, respectively. MMP-2/9-sensitive micelles also improved tumor targeting and accumulation relative to nonsensitive micelles in vivo owing to the specific release of loaded drug at tumor site. The results of in vivo antitumor effect evaluation also indicate that TGK micelles are potent against solid tumors while maintaining minimum systemic toxicity compared with T2K micelles and DTX. While further studies regarding pharmacokinetic profile are needed to illustrate MMP-2/9-sensitive polymer micelles full potential, to the best of our knowledge, this strategy of antitumor drug delivery to MMP-2/9-overexpressing tumors is promising.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81373354).
Disclosure
The authors report no conflicts of interest in this work.
References
Li R, Wu W, Liu Q, et al. Intelligently targeted drug delivery and enhanced antitumor effect by gelatinase-responsive nanoparticles. PLoS One. 2013;8(7):e69643. | ||
Liang D, Wang AT, Yang ZZ, Liu YJ, Qi XR. Enhance cancer cell recognition and overcome drug resistance using hyaluronic acid and alpha-tocopheryl succinate based multifunctional nanoparticles. Mol Pharm. 2015;12(6):2189–2202. | ||
Abouelmagd SA, Hyun H, Yeo Y. Extracellularly activatable nanocarriers for drug delivery to tumors. Expert Opin Drug Deliv. 2014;11(10):1601–1618. | ||
Li Y, Gao GH, Lee DS. Stimulus-sensitive polymeric nanoparticles and their applications as drug and gene carriers. Adv Healthc Mater. 2013;2(3):388–417. | ||
Zhu L, Kate P, Torchilin VP. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano. 2012;6(4):3491–3498. | ||
Andresen TL, Thompson DH, Kaasgaard T. Enzyme-triggered nanomedicine: drug release strategies in cancer therapy. Mol Membr Biol. 2010;27(7):353–363. | ||
Garripelli VK, Kim JK, Son S, Kim WJ, Repka MA, Jo S. Matrix metalloproteinase-sensitive thermogelling polymer for bioresponsive local drug delivery. Acta Biomater. 2011;7(5):1984–1992. | ||
Samuelson LE, Scherer RL, Matrisian LM, McIntyre JO, Bornhop DJ. Synthesis and in vitro efficacy of MMP9-activated NanoDendrons. Mol Pharm. 2013;10(8):3164–3174. | ||
Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278(1):16–27. | ||
Lee GY, Park K, Kim SY, Byun Y. MMPs-specific PEGylated peptide-DOX conjugate micelles that can contain free doxorubicin. Eur J Pharm Biopharm. 2007;67(3):646–654. | ||
Stankovic S, Konjevic G, Gopcevic K, Jovic V, Inic M, Jurisic V. Activity of MMP-2 and MMP-9 in sera of breast cancer patients. Pathol Res Pract. 2010;206(4):241–247. | ||
Zhu L, Torchilin VP. Stimulus-responsive nanopreparations for tumor targeting. Integr Biol. 2013;5(1):96–107. | ||
Nazli C, Demirer GS, Yar Y, Acar HY, Kizilel S. Targeted delivery of doxorubicin into tumor cells via MMP-sensitive PEG hydrogel-coated magnetic iron oxide nanoparticles (MIONPs). Colloids Surf B Biointerfaces. 2014;122:674–683. | ||
Vartak DG, Gemeinhart RA. Matrix metalloproteases: underutilized targets for drug delivery. J Drug Target. 2007;15(1):1–20. | ||
Thambi T, Park JH. Recent advances in shell-sheddable nanoparticles for cancer therapy. J Biomed Nanotechnol. 2014;10(9):1841–1862. | ||
Muthu MS, Kulkarni SA, Liu Y, Feng SS. Development of docetaxel-loaded vitamin E TPGS micelles: formulation optimization, effects on brain cancer cells and biodistribution in rats. Nanomedicine. 2012;7(3):353–364. | ||
Kanai K, Kikuchi E, Mikami S, et al. Vitamin E succinate induced apoptosis and enhanced chemosensitivity to paclitaxel in human bladder cancer cells in vitro and in vivo. Cancer Sci. 2010;101(1):216–223. | ||
Angulo-Molina A, Reyes-Leyva J, Lopez-Malo A, Hernandez J. The role of alpha tocopheryl succinate (alpha-TOS) as a potential anticancer agent. Nut Cancer. 2014;66(2):167–176. | ||
Su Z, Chen M, Xiao Y, et al. ROS-triggered and regenerating anticancer nanosystem: an effective strategy to subdue tumor’s multidrug resistance. J Control Release. 2014;196:370–383. | ||
Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Ma Y, Liu D, Wang D, et al. Combinational delivery of hydrophobic and hydrophilic anticancer drugs in single nanoemulsions to treat MDR in cancer. Mol Pharm. 2014;11(8):2623–2630. | ||
Sadoqi M, Lau-Cam CA, Wu SH. Investigation of the micellar properties of the tocopheryl polyethylene glycol succinate surfactants TPGS 400 and TPGS 1000 by steady state fluorometry. J Colloid Interface Sci. 2009;333(2):585–589. | ||
Mi Y, Liu Y, Feng SS. Formulation of docetaxel by folic acid-conjugated d-alpha-tocopheryl polyethylene glycol succinate 2000 (Vitamin E TPGS(2k)) micelles for targeted and synergistic chemotherapy. Biomaterials. 2011;32(16):4058–4066. | ||
Lu J, Huang Y, Zhao W, et al. Design and characterization of PEG-derivatized vitamin E as a nanomicellar formulation for delivery of paclitaxel. Mol Pharm. 2013;10(8):2880–2890. | ||
Saxena V, Hussain MD. Poloxamer 407/TPGS mixed micelles for delivery of gambogic acid to breast and multidrug-resistant cancer. Int J Nanomedicine. 2012;7:713–721. | ||
Owens DE 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. | ||
Chen L, Sha X, Jiang X, Chen Y, Ren Q, Fang X. Pluronic P105/F127 mixed micelles for the delivery of docetaxel against Taxol-resistant non-small cell lung cancer: optimization and in vitro, in vivo evaluation. Int J Nanomedicine. 2013;8:73–84. | ||
Yokoyama M. Polymeric micelles as drug carriers: their lights and shadows. J Drug Target. 2014;22(7):576–583. | ||
Wu XL, Kim JH, Koo H, et al. Tumor-targeting peptide conjugated pH-responsive micelles as a potential drug carrier for cancer therapy. Bioconj Chem. 2010;21(2):208–213. | ||
Yin YM, Cui FD, Mu CF, et al. Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release. 2009;140(2):86–94. | ||
Zhang W, Shi Y, Chen Y, Ye J, Sha X, Fang X. Multifunctional Pluronic P123/F127 mixed polymeric micelles loaded with paclitaxel for the treatment of multidrug resistant tumors. Biomaterials. 2011;32(11):2894–2906. | ||
Li J, Ge Z, Liu S. PEG-sheddable polyplex micelles as smart gene carriers based on MMP-cleavable peptide-linked block copolymers. Chem Commun. 2013;49(62):6974–6976. | ||
Chau Y, Padera RF, Dang NM, Langer R. Antitumor efficacy of a novel polymer-peptide-drug conjugate in human tumor xenograft models. Int J Cancer. 2006;118(6):1519–1526. | ||
Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol. 2001;19(7):661–667. | ||
Leavell MD, Novak P, Behrens CR, Schoeniger JS, Kruppa GH. Strategy for selective chemical cross-linking of tyrosine and lysine residues. J Am Soc Mass Spectrom. 2004;15(11):1604–1611. | ||
Shi J, Zhang J, Shen Y, et al. Arginine-stabilized mPEG-PDLLA (50/50) polymeric micelles of docetaxel by electrostatic mechanism for tumor-targeted delivery. Drug Deliv. 2015;22(2):168–181. | ||
Chen H, Kim S, Li L, Wang S, Park K, Cheng JX. Release of hydrophobic molecules from polymer micelles into cell membranes revealed by Forster resonance energy transfer imaging. Proc Natl Acad Sci U S A. 2008;105(18):6596–6601. | ||
Treuel L, Docter D, Maskos M, Stauber RH. Protein corona – from molecular adsorption to physiological complexity. Beilstein J Nanotechnol. 2015;6:857–873. | ||
Moore TL, Rodriguez-Lorenzo L, Hirsch V, et al. Nanoparticle colloidal stability in cell culture media and impact on cellular interactions. Chem Soc Rev. 2015;44(17):6287–6305. | ||
Zhang J, Liu J, Zhao Y, Wang G, Zhou F. Plasma and cellular pharmacokinetic considerations for the development and optimization of antitumor block copolymer micelles. Expert Opin Drug Deliv. 2015;12(2):263–281. | ||
Alexander-Bryant AA, Vanden Berg-Foels WS, Wen X. Bioengineering strategies for designing targeted cancer therapies. Adv Cancer Res. 2013;118:1–59. | ||
Sriraman SK, Aryasomayajula B, Torchilin VP. Barriers to drug delivery in solid tumors. Tissue Barriers. 2014;2:e29528. |
Supplementary materials
 | Figure S1 Chemical structure of TPGS. |
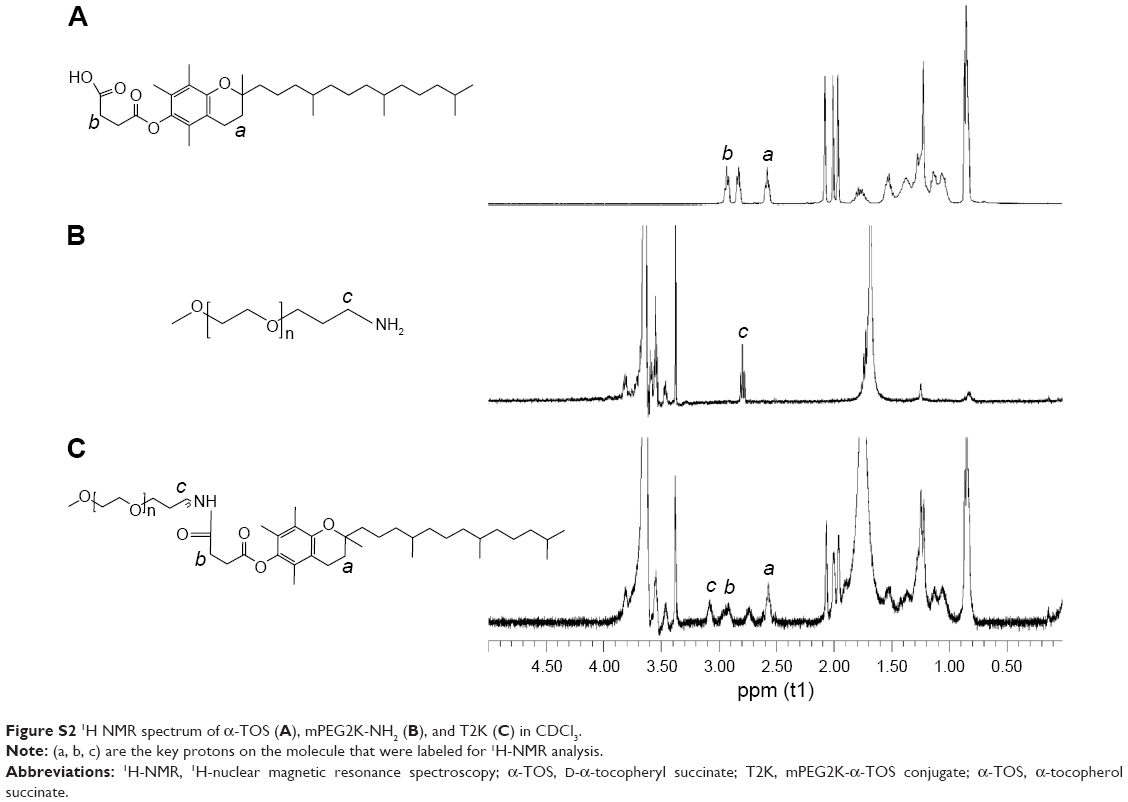 | Figure S2 1H NMR spectrum of α-TOS (A), mPEG2K-NH2 (B), and T2K (C) in CDCl3. |
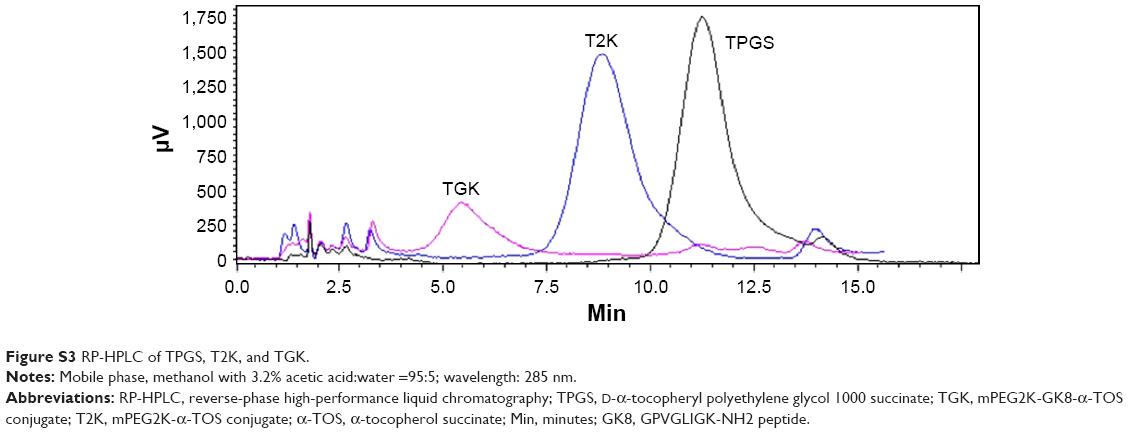 | Figure S3 RP-HPLC of TPGS, T2K, and TGK. |
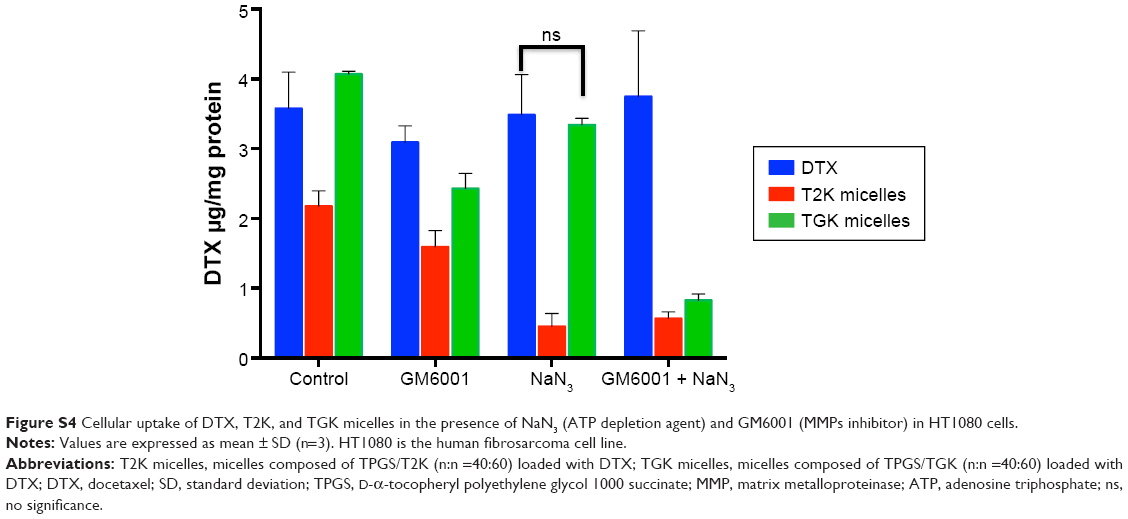 | Figure S4 Cellular uptake of DTX, T2K, and TGK micelles in the presence of NaN3 (ATP depletion agent) and GM6001 (MMPs inhibitor) in HT1080 cells. |
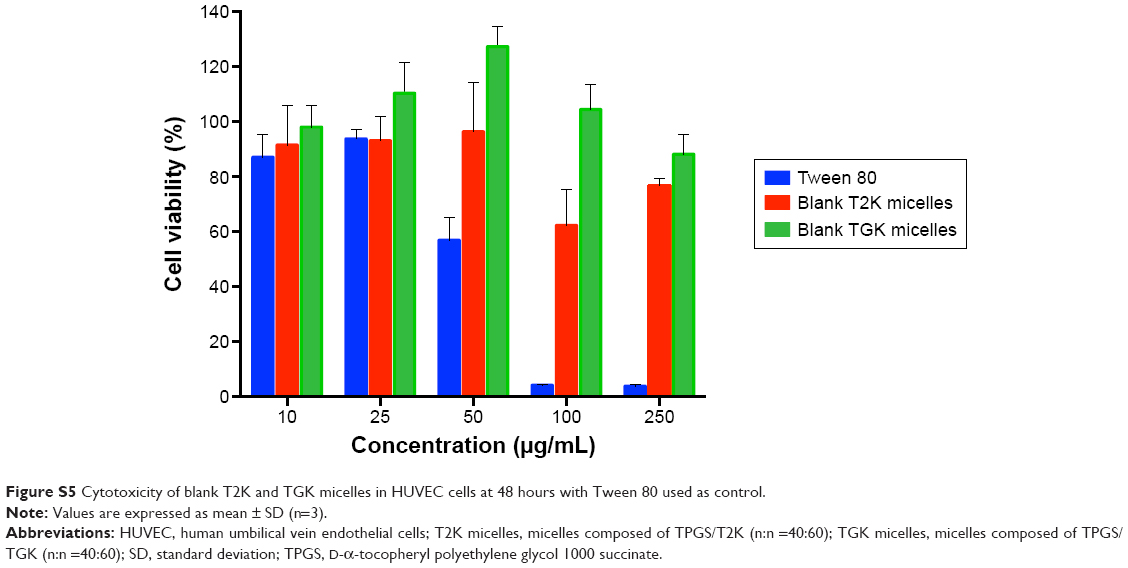 | Figure S5 Cytotoxicity of blank T2K and TGK micelles in HUVEC cells at 48 hours with Tween 80 used as control. |
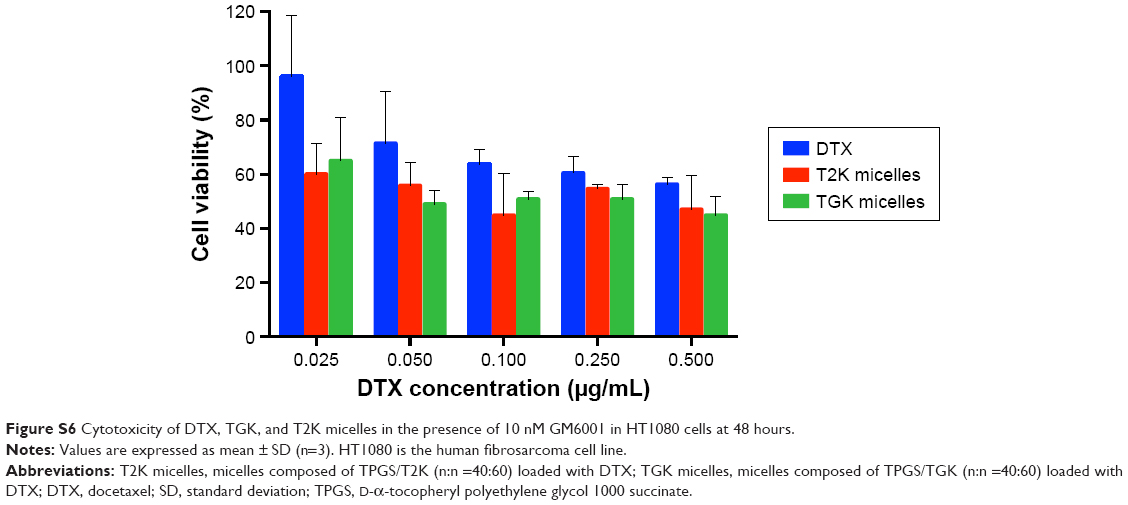 | Figure S6 Cytotoxicity of DTX, TGK, and T2K micelles in the presence of 10 nM GM6001 in HT1080 cells at 48 hours. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.