Back to Journals » International Journal of Nanomedicine » Volume 13
Matrix metalloproteinase 2/9-triggered-release micelles for inhaled drug delivery to treat lung cancer: preparation and in vitro/in vivo studies
Authors Wang X, Chen Q, Zhang X, Ren X, Zhang X, Meng L, Liang H, Sha X, Fang X
Received 26 February 2018
Accepted for publication 30 April 2018
Published 13 August 2018 Volume 2018:13 Pages 4641—4659
DOI https://doi.org/10.2147/IJN.S166584
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Xiaofei Wang,1,2 Qinyue Chen,1 Xiaoyan Zhang,1 Xiaoqing Ren,1 Xiulei Zhang,1 Lin Meng,1 Huihui Liang,1 Xianyi Sha,1 Xiaoling Fang1
1Key Laboratory of Smart Drug Delivery, Ministry of Education of China, School of Pharmacy, Fudan University, Shanghai, People’s Republic of China; 2Shanghai Omni Pharmaceuticall Co., Ltd., Shanghai, People’s Republic of China
Background: Improvement in drug accumulation in the lungs through inhalation administration and high expression of MMP2 and MMP9 in lung tumors have both been widely reported.
Methods: MMP2/9-triggered-release micelles were constructed and in vitro and in vivo studies of inhalation administration against lung tumor carried out. Pluronic P123 (P123) was modified with GPLGIAGQ-NH2 (GQ8) peptide to obtain P123-GQ8 (PG). MMP2/9-triggered-release micelles were constructed using PG and succinylated gelatin (SG) and loading paclitaxel (Ptx). To study biodistribution of micelles, DiR encapsulated in micelles was dosed to rats via intravenous injection or inhalation before ex vivo imaging for detecting DiR quantity in lungs. And B16F10 lung cancer-bearing nude mice were chosen as animal models to evaluate in vivo efficacy of MMP2/9-triggered-release micelles.
Results: Ptx-release efficiency from PG-SG-Ptx micelles was MMP2/9-concentration-dependent. For A549 cells, PG-SG-Ptx cytotoxicity was significantly greater (P<0.001) compared to P123-Ptx. Aerosol inhalation was chosen as the method of administration. In biodistribution experiment, DiR quantity in lungs was 5.8%±0.4% of that in major organs, while the ratio was 38.8%±0.5% for inhalation. For B16F10 lung cancer-bearing nude mice, the efficacy of inhalation of PG-SG-Ptx was significantly higher (P<0.001) than Taxol inhalation and injected PG-SG-Ptx. Inhaled PG-SG-Ptx also significantly inhibited the expression of Pgp in lung cancer.
Conclusion: Inhalation of MMP2/9-triggered-release micelles increased tumor sensitivity to chemotherapeutics and reduced the toxicity of chemotherapy to healthy lung cells, which has great potential in lung cancer therapy.
Keywords: matrix metalloproteinases, inhalation, Pluronic, paclitaxel, P-glycoprotein
Introduction
As the second-most prevalent cancer worldwide,1 lung cancer is the leading cause of cancer death in the USA and People’s Republic of China.2,3 The World Health Organization divides lung cancer into two major categories, based on biological characteristics, therapeutic measures, and prognosis: non-small-cell lung cancer (NSCLC) and SCLC, with NSCLC accounting for >80% of all lung cancer cases.3 In 2017, in an estimation on 222,500 cases of lung and bronchial cancer, 116,990 males and 105,510 females were found diagnosed, and 155,870 deaths, including 84,590 men and 71,280 women, were estimated to have occurred because of the disease in the USA.2 In 2015, an estimated 733,300 new cases of lung cancer were diagnosed and 610,200 deaths estimated to occur because of the disease in People’s Republic of China.3 Compared with the 5-year survival rate of lung cancer 15 years ago,4 which was 5%–10%, it has been improved, but is still limited at 17.7%.3 Although surgery is the best treatment for lung cancer, especially for NSCLC, unfortunately only a small proportion (10%–20%) of patients are suitable for curative resection, while most patients must rely on nonsurgical and adjuvant therapies.4 Lung cancer patients after surgery or patients with unresectable tumors are often treated with chemotherapy to improve quality of life or control disease development.5 Intravenous injection or oral administration of cytotoxic drugs, including paclitaxel (Ptx), docetaxel, doxorubicin, and camptothecin, are the main approaches of chemotherapy. Generally, these therapies have poor selectivity and systemic toxicity, and low drug concentration in the tumor sites may lead to the development of multidrug resistance.6 In order to attain an ideal concentration of drug to kill the tumor, high-dosage drug administration is necessary. However, high doses may cause serious adverse reactions, leading to chemotherapy intolerance in patients.7
Inhalation is a direct delivery of drug to the lung via an oral or nasal route to achieve therapeutic efficacy, which is the most effective way to treat respiratory diseases, such as asthma and chronic obstructive pulmonary disease. Inhaled delivery of chemotherapeutic drugs has attracted growing attention in the past few decades, and inhaled doxorubicin has completed Phase II clinical study.8–10 Compared with oral and intravenous administration, inhalation can change drug distribution and improve the accumulation of a larger proportion of drug in the lung.11 Inhalation administration reduces drug exposure to the whole system and thus reduces systemic toxicity. Most adverse effects associated with inhaled chemotherapy have been reported to be localized and dose- and time-dependent. High concentrations of an anticancer drug in healthy lung cells may result in undesirable local toxicity, though toxicity data for inhaled chemotherapy impairing healthy lung cells remain to be studied.1 Inhaled chemotherapy toxicity to healthy lung cells remains uncertain, which makes the safety of inhaled chemotherapy in constant dispute. Therefore, promoting drug uptake in cancer cells and reducing accumulation in healthy cells are effective ways to ensure the safety and efficacy of inhalation chemotherapy.1 Moreover, how to determine the dosage of inhaled drug referring to the dosage of injection or oral administration is another key problem in research and clinical practice.
It is believed that the extracellular expression of such proteases as MMPs, which are specific markers of malignant tumors, is responsible for the hydrolysis of the extracellular matrix and basement membrane, both of which are related to processes involved in angiogenesis and tumor development.12,13 MMPs are highly conserved proteolytic enzymes that degrade the extracellular matrix. Much tumor tissue expresses one or more subtypes of MMPs, which play an important role in the complex process of tumor invasion and metastasis. Studies have shown that tumors with high MMP expression have high malignancy, poor sensitivity to treatment, and poor prognosis.14,15 Overexpression of MMP9 in lung tissue is a risk factor for advanced T category, tumor stage, and poor outcome for NSCLC.16 Therefore, it is possible to achieve the goal of reducing toxicity to healthy lung cells, the oral cavity, and upper respiratory tract and enhancing therapeutic efficacy by inhaled delivery of an MMP2/9-triggered-release drug-delivery system.
With a general formula of HO(C2H4O)a(C3H6O)b(C2H4O)aOH, Pluronic has a three-block structure of polyoxyethylene (PEO)–polyoxypropylene–PEO. All Pluronic formulations are chemically similar in composition and different only in the relative amounts of PEO and polyoxypropylene. More recently, Pluronic has been used in drug-delivery systems.17 Pluronic P123 can effectively reduce the microviscosity of the cell membrane and decrease the ATP pool available for Pgp.18,19 Consequently, P123 can inhibit the activity of Pgp and thus reverse multidrug resistance in tumors. Compared with other types of Pluronic, P123 also has higher drug-loading (DL) capability.17
Succinylated gelatin (SG), also known as modified fluid gelatin, substitutes intravascular volume deficits caused by losses of blood or plasma. Its isoelectric point is at pH 4.5, with negative charge in vivo. Like gelatin, SG can also be degraded by MMP2/9. It has a structural fragment similar to the peptide that can be degraded by MMP2/9 and can interact with structurally similar peptides to form a more stable structure.20 Therefore, SG will lose the ability enhancing structural stability in the presence of MMP2/9.
In this study, we modified Pluronic P123 (P123) with GPLGIAGQ-NH2 (GQ8) peptide, which can be degraded by MMP9,21 to obtain P123-GQ8 (PG). Construction of MMP2/9-triggered-release micelles was achieved by using PG and SG, and then Ptx was loaded into the micelles to form the complex of PG-SG-Ptx. The synthesis, preparation, and characterization of PG-SG-Ptx micelles are presented in this study. To establish in vivo evaluation, many aspects were studied, including method of inhalation administration and effects of different nebulizers, different flow rates on droplet-size distribution (DSD) after nebulization, and nebulization on micelle stability. The relationship between inhalation dosage and intravenous administration in vivo was investigated in male Sprague Dawley (SD) rats by studying the distribution of the fluorescent probes in the main organs. In vivo antitumor effects in B16F10 and A549 lung tumor-bearing nude mice were examined. Pgp expression in lung tissue after treatment was also evaluated.
Methods
Materials and animals
The triblock copolymers P123 and polyoxyl 40 castor oil (Cremophor EL) were procured from BASF (Ludwigshafen, Germany), GQ8 from GL Biochem Ltd (Shanghai, People’s Republic of China), Ptx from Yew Pharmaceutical Co., Ltd (Wuxi, People’s Republic of China), Ptx injection (Taxol) from Bristol-Myers Squibb (New York, NY, USA), collagenase type IV and BSA from Sigma-Aldrich (St Louis, MO, USA). SG (Gelofusine) from B Braun Medical (Melsungen, Germany), DiR, BCA protein-quantification kit, and Western blot detection kit from Beyotime Biotechnology Co., Ltd (Haimen, People’s Republic of China); RPMI 1640 medium, DMEM, FBS, and penicillin–streptomycin solution (5 kU/mL) from Thermo Fisher Scientific (Waltham, MA, USA), GM6001 from MedChemExpress (Monmouth Junction, NJ, USA), H&E from Boster Biological Technology Co., Ltd (Pleasanton, CA, USA), and nitrophenyl chloroformate (NPC), anhydrous dichloromethane (DCM), anhydrous dimethylformamide (DMF), anhydrous triethylamine (TEA), and polysorbate 80 from Aladdin (Shanghai, People’s Republic of China). All other chemicals used were analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, People’s Republic of China).
Human umbilical vein endothelial cells (HUVECs), murine melanoma cells (B16F10), human non-small cell lung adenocarcinoma cells (A549) and human breast adenocarcinoma cells (MCF7) were purchased from the Cell Culture Center of Shanghai Institutes of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). Human multidrug-resistant breast adenocarcinoma cells (MCF7/ADR) was purchased from KeyGen Biotech Co., Ltd. (Nanjing, People’s Republic of China). HUVECs, B16F10, and MCF7 were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin under 95% humidity and 5% CO2 at 37°C. A549 was cultured in RPMI 1640, and other conditions were same as HUVECs. Except for containing 1 μg/mL doxorubicin in medium, MCF7/ADR was cultured the same as A549 cells. All experiments were performed using cells in the logarithmic phase of growth.
Adult male SD rats (initial weight 180–220 g) and BALB/c nude mice (initial weight 20–25 g) were supplied by the Laboratory Animal Center of Fudan University (Shanghai, People’s Republic of China). Animals were kept under specific pathogen-free conditions. All animal experiments were in accordance with animal-care regulations of Fudan University, followed the guidelines of China’s animal-welfare act, and were approved by the ethics committee of the School of Pharmacy, Fudan University.
Synthesis and characterization of PG
NPC was first used to activate P123 by forming active intermediate P123-NPC. P123 and NPC (1:2.4 mol:mol) were dissolved in 10 mL DCM and then 100 μL TEA was added. The reaction was carried out without light under nitrogen protection at 0°C overnight and P123-NPC was obtained by applying vacuum-rotary evaporation to remove the remaining DCM at 25°C. P123-NPC and GQ8 (1:2 mol/mol) were dissolved in 10 mL DMF in the presence of 20 μL TEA and reacted without light at 0°C under nitrogen protection overnight. Finally, the solution was dialyzed (molecular weight cutoff 3,500 Da) for 48 hours against deionized water to remove the unreacted reactants and then filtered through 0.22 μm filter. The product was lyophilized before characterization by 1H nuclear magnetic resonance spectroscopy (NMR) in D2O at 400 MHz (Mercury Plus; Varian, Palo Alto, CA, USA) and matrix-assisted laser desorption–ionization time-of-flight mass spectrometry (MALDI-TOF MS) (AB Sciex, Framingham, MA, USA). The critical micelle concentration (CMC) of PG was determined by ultraviolet-visible spectrometry with iodine as a hydrophobic probe. The KI/I2 standard solution was prepared by dissolving 1 g KI and 0.5 g I2 in 50 mL deionized water. Samples of PG with concentrations of 0.0005%–0.25% were prepared, and then 5 μL KI/I2 standard solution was added to 1 mL sample solution. After incubation at 37°C for 24 hours sheltered from light, the absorbance of samples was measured by a microplate reader (Synergy 2; BioTek, Winooski, VT, USA) at 366 nm. When absorbance increased dramatically, the corresponding concentration was the CMC of PG. The experiment was performed in triplicate.
Western blotting analysis
B16F10 cells in six-well culture-plates were incubated with P123 and PG of 0.005%–1% (w:v) for 24 hours. Cells were washed with PBS and lysed in radioimmunoprecipitation-assay buffer containing a protease inhibitor. Protein concentrations were detected with the BCA protein-quantification kit and equal amounts of protein separated on 10% sodium dodecyl sulfate–polyacrylamide gel, then transferred to polyvinylidene difluoride membranes. Then, membranes were blocked with 5% nonfat dry milk for about 1 hour and incubated with a primary antibody at 4°C overnight. After the membranes had been washed three times with Tris-buffered saline–Tween (TBST), they were incubated with a secondary antibody for 2 hours at room temperature. Finally, membranes were washed by TBST and an electrochemiluminescence Western blotting substrate used for detection.
Preparation and characterization of micelles
Preparation of P123-DiR micelles
Thin-film hydration was used to prepare micelles. P123 (200 mg) and 1 mg DiR were weighed and then dissolved in 20 mL chloroform in a round-bottomed flask to prepare P123-DiR micelles. The chloroform was removed by vacuum evaporation at 25°C to form the thin film. P123-DiR micelles were obtained by adding 40 mL 0.9% NaCl solution into the flask, then stirring for 30 minutes. Micelles were filtered through 0.22 μm sterile filters. The micelle solution was lyophilized for further use. The whole process was shielded from light.
Preparation of drug-loaded micelles
P123 (200 mg) and 10 mg Ptx were weighed and then dissolved in 20 mL anhydrous ethanol in a round-bottomed flask to prepare P123-Ptx micelles. The organic solvent was removed by vacuum evaporation at 50°C to form the film. P123-Ptx micelles were obtained by adding 40 mL 0.9% NaCl solution into the flask, then stirring for 30 minutes. P123 was replaced with PG, and Ptx-loaded PG micelles (PG-Ptx) were prepared according to the process of preparing P123-Ptx. The PG-SG-Ptx micelles were prepared by following a similar procedure, where 200 mg PG and 10 mg Ptx were mixed in 20 mL anhydrous ethanol, while the solution for hydration was 0.5% SG solution prepared by diluting SG with 0.9% NaCl solution. These micelles were filtered through 0.22 μm sterile filters and solutions lyophilized for further use with lyophilization parameters of freezing point −0.29°C, prefreezing temperature −80°C, primary drying temperature −40°C, chamber pressure −0.12 mPa, and freezing time 48 hours. P123, P123-SG, PG, and PG-SG placebo micelles were also prepared in a similar process.
Particle-size distribution
Particle-size distribution of Ptx-loaded micelles and placebo micelles were determined by dynamic light scattering at 25°C with a Zetasizer (Nano ZS90; Malvern Instruments, Malvern, UK).
Determination of drug-loading and drug-entrapment efficiency
The Ptx encapsulated in the micelles was analyzed by reverse-phase high-performance liquid chromatography (RP-HPLC). DL efficiency and entrapment efficiency (EE) of the micelles were calculated:
|
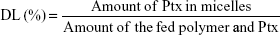
|
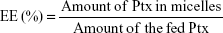
Stability of drug-loaded micelles
P123-Ptx, PG-Ptx and PG-SG-Ptx micelles were incubated for 24 hours at 37°C. Micelle solutions were collected at predetermined intervals. These fluids were filtered through a 0.22 μm filter and the Ptx in the collected fluids measured by RP-HPLC on a 10Avp HPLC system (Shimadzu, Kyoto, Japan). A reverse-phase column (Gemini 5u C18 110A, 4.6×150 mm, 5 μm; Phenomenex, Torrance, CA, USA) was used. The mobile phase consisted of acetonitrile and water (v:v 55:45) with flow rate at 1 mL/min. Injection volume was 20 μL, and absorbance at 227 nm was measured with an SPD-10AVP ultraviolet-visible detector (Shimadzu). The percentage of Ptx in the micelles was calculated:
|
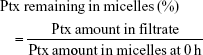
In vitro drug-release profile
In vitro drug-release profiles of PG-SG-Ptx micelles were investigated by dialysis. Briefly, 90 μL 1 mg/mL MMP2/9 solution, 90 μL of 100 μg/mL MMP2/9 solution, 90 μL 1 mg/mL BSA solution, and 90 μL PBS were added to 810 μL PG-SG-Ptx micelle suspensions, and then these mixtures were loaded into dialysis bags (molecular weight cutoff 3,500 Da). These bags were dialyzed against 30 mL PBS (pH 7.4) containing 0.5% polysorbate 80. Release was performed in triplicate at 37°C while samples were being shaken at 100 rpm for 24 hours. The medium was collected and substituted with the same volume of release medium containing polysorbate 80 at a predetermined interval. These collected fluids were filtered through 0.22 μm filters and the Ptx in the collected fluids measured by RP-HPLC, as described previously. In vitro drug-release profiles of P123-Ptx and PG-Ptx micelles were also performed.
In vitro cytotoxicity
In vitro cytotoxicity of P123, PG, PG-SG, and SG and the cytotoxicity of the drug-loading micelles P123-Ptx and PG-SG-Ptx were evaluated by MTT assay. HUVECs and A549 cells were seeded in a 96-well plate at a density of 5,000 cells per well. After HUVECs had been incubated for 24 hours, cells were incubated with P123, PG, PG-SG, and SG at concentrations of 0.01–3 mg/mL (dilute with culture medium) for 48 hours, and cells incubated with culture medium were used as control. Then, 100 μL MTT solution (0.5 mg/mL diluted with PBS) was added to each well and incubated for 4 hours. Finally, the cells were dissolved in 200 μL dimethyl sulfoxide and the absorbance of each well measured at 570 nm by a microplate reader (Synergy 2) after incubation for 10 minutes at 37°C. After A549 cells had been incubated for 24 hours, they were incubated with P123-Ptx, PG-Ptx, and PG-SG-Ptx (Ptx was diluted with culture medium to reach concentrations of 1–500 ng/mL) for 24 hours, and cells incubated with culture medium were used as control. Other procedures were the same as described earlier.
In vitro cellular uptake
To evaluate the intracellular delivery of P123, PG, and PG-SG micelles, cellular uptake of coumarin 6 (Cou6)-loaded P123 and PG-SG micelles was investigated in A549 (MMP2/9-overexpression) cells. Cou6-loaded P123 (P123-Cou6) micelles and Cou6-loaded PG-SG (PG-SG-Cou6) micelles were prepared in a similar process of drug-loaded micelle preparation and the amount of loaded Cou6 in micelles determined by RP-HPLC. A549 cells were seeded in a 24-well cell-culture plate (104/well) and inoculated for 24 hours. Then, cell monolayers were washed with PBS twice and cultured in fresh RPMI 1640 (without FBS) containing P123-Cou6 or PG-SG-Cou6 micelles (with 100 ng/mL Cou6) for 30 minutes. After incubation, cells were washed thrice with cooled PBS, followed by fixation for 15 minutes by adding 300 μL 4% paraformaldehyde solution. Then, cells were washed thrice with PBS and photographed using fluorescence microscopy. To confirm the cellular uptake mechanism, cellular uptake was investigated in the presence of GM6001 at the same time. MCF7 (MMP2/9-underexpression) and MCF7/ADR (MMP2/9-overexpression) cells that had been precultured were washed thrice and incubated with 100 nM GM6001 for 1 hour before being treated with PG-SG-Cou6.
Optimization of micelle-inhalation administration
Droplet-size distribution
Aqueous droplet-size distribution (DSD) of the nebulized aerosol was determined with a HELOS/BF inhaler laser-diffraction particle analyzer (Sympatec GmbH, Clausthal-Zellerfeld, Germany). Studies were performed at 3 or 6 cm from the mouthpiece of the nebulizer. D10, D50, D90, geometric SD (GSD), and volume mean diameter (VMD) values were recorded.
Effect of different nebulizers and compressors on DSD
Deionized water (2 mL) was added to three types of nebulizer (Pari, Germany) equipped with different compressors. Then, the DSD of nebulized water aerosol was tested.
Effect of compressor flow rate on DSD
Deionized water (2 mL) was added to one nebulizer and the compressor set to control the flow rate at 6 and 9 L/min, and DSD was measured at different flow rates.
DSD at different distances from nebulizer mouthpiece
Aqueous DSD of the nebulized aerosol was determined at distances of 3 and 6 cm from the mouthpiece of the nebulizer.
Effect of different solutes and concentration on DSD
DSD of nebulized aerosol of 0.9% NaCl solution, 5% glucose solution, SG solution, and P123 at different concentrations was measured.
DSD of Ptx injection, P123-Ptx and PG-SG-Ptx micelles, and effect of nebulization on drug-loading micelle stability
The Ptx injection was diluted fivefold with 0.9% NaCl, while P123-Ptx and PG-SG-Ptx were diluted to 0.5% polymers. Then, all solutions were nebulized and DSD of these nebulized aerosols was measured. The aerosol and micelles remaining in the nebulizer were collected using 0.9% NaCl solution (Figure 1). Fluids were filtered through 0.22 μm filters and the Ptx in them analyzed by RP-HPLC. Ptx in the unnebulized micelles with the same concentration was used as the control.
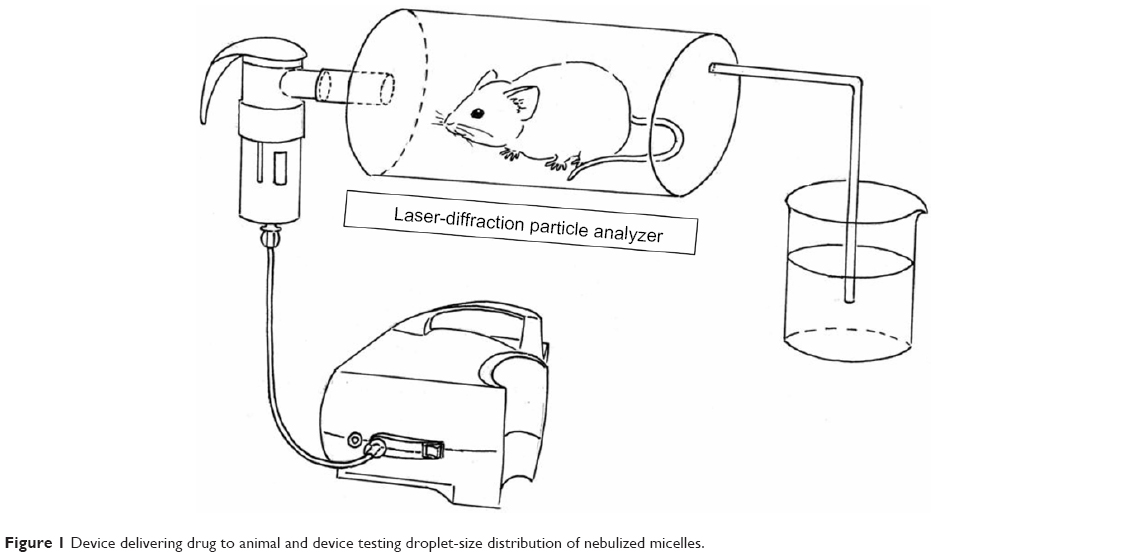 | Figure 1 Device delivering drug to animal and device testing droplet-size distribution of nebulized micelles. |
Histopathological analysis of inhalation toxicity
SD rats were grouped and intravenously administered 1% P123 aqueous solution, while aerosolized saline and aerosolized 1% P123 aqueous solution were given to rats by nebulization. Rats were placed in a special device that was connected to the nebulizer (Figure 1) and exposed to aerosol for 30 minutes. After 1 hour, rats were euthanized, and lung sections eviscerated, washed, and stained with H&E for observation of changes in lung tissue.
Biodistribution of intravenous injection and inhalation micelles in rats
SD rats were randomly grouped into two groups, where one group was intravenously administered 0.8 mL P123-DiR micelles containing 20 μg DiR and the other group was inhalation administered 4 mL P123-DiR micelles containing 100 μg DiR. After administration for 1, 6, and 12 hours, rats were killed and viscera (heart, liver, spleen, lung, and kidney) isolated and imaged with an IVIS Spectrum preclinical in vivo imaging system (PerkinElmer, Waltham, MA, USA) with an excitation wavelength of 748 nm and emission wavelength of 780 nm. Rats injected with saline were used as controls for autofluorescence.
In vivo antitumor-effect evaluation
To construct the B16F10 pulmonary metastatic model, male BALB/c nude mice were injected intravenously in the tail vein with 0.1 mL cell suspension containing 5×106 B16F10 cells. A pilot study (2017) showed that lung tumors developed after 10 days of tumor-cell injection, and MMP2/9 expression in the lung was analyzed by Western blotting. Mice were randomly divided into five groups (six per group): group 1 for saline inhalation, group 2 for Ptx inhalation (10 mg/kg), group 3 for P123-Ptx inhalation (10 mg/kg), group 4 for PG-SG-Ptx injection (10 mg/kg) and group 5 for PG-SG-Ptx inhalation (10 mg/kg). For inhalation administration, micelles were nebulized for 30 minutes using the LC Sprint junior nebulizer at a 9 L/min flow rate and the dosage of drug inhaled was equal to the amount of dosage added to the nebulizer. Formulations were administered on the 10th day (designated day 0 of administration) after the injection of cells into mice. All mice were administered formulations on days 0, 3, and 6. Body weight of mice was recorded every 2 days to monitor potential toxicity. These mice were killed on day 14 and viscera (heart, liver, spleen, lung, and kidney) immediately harvested. Lungs were weighed and stained with H&E for histopathological analysis or frozen at −80°C for Western blotting analysis. Hearts, livers, spleens, and kidneys were stained with H&E for histopathological analysis. Tumor-inhibition rates were calculated:17
|
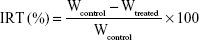
where IRT is inhibition ratio, Wtreated mean lung weight of treatment group, and Wcontrol mean lung weight of control group. The A549 pulmonary metastatic model was also constructed in male BALB/c nude mice, which were treated with inhalation of Ptx, P123-Ptx, and PG-SG-Ptx.
Statistical analysis
Data are shown as means ± SD. Statistical analysis was conducted with one-way ANOVA for three or more groups and Student’s t-test for two groups. P<0.05 was considered statistically significant.
Results and discussion
Synthesis and characterization of PG
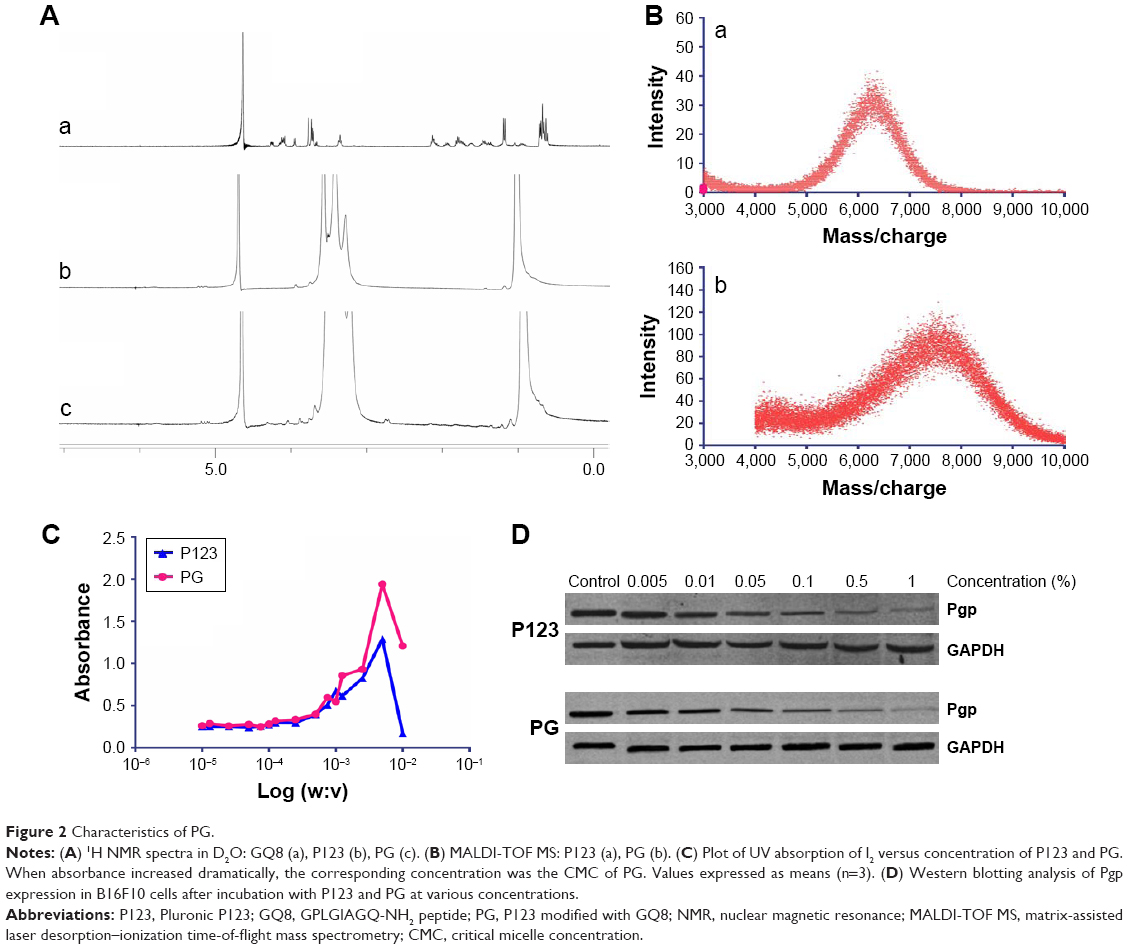
The peptide sequences GPLGV, PVGLIG, GPLGVRG, and GPLGIAGQ were used to construct MMP-triggered drug-delivery systems. P123 was modified to assemble the system. Since only the terminal hydroxyl of P123 was modified, inhibition of Pgp expression would not be affected. A novel thermosensitive multiblock copolymer comprising MMP2-degradable peptide GPVGILGK has been synthesized from a Pluronic polymer.22 In this study, the two-terminal hydroxyl of P123 was modified using peptide GPLGIAGQ-NH2, ie, the C-terminal conjugates were blocked by NH2 to avoid cross-linking, while the terminal hydroxyl of P123 was activated by NPC and then reacted with the N-terminal of GQ8 peptide. PG syntheses were confirmed a success by 1H NMR (Figure 2A). Since the polymer terminal was modified by small molecules, the proton of the modifier was not in the same order of magnitude as that of the polymer, and hence it was insufficient to characterize it with NMR. Since MALDI-TOF MS determines molecular mass distribution of polymer with high sensitivity,23 the molecular mass distributions of P123 and PG were determined (Figure 2B), with peak molecular mass-distribution difference between P123 and PG about 1,400, which was in accord with the theoretical molecular weight of NH2-QGAIGLPG-P123-GPLGIAGQ-NH2 (PG). CMC values were determined by ultraviolet-visible spectrophotometry. The results indicated that the CMC of PG was similar to P123, which was ~0.01% (Figure 2C). PG absorbance was higher than that of P123 when the concentration was higher than the CMC. Absorbance was highest in the 0.5% concentration. Surprisingly, when concentration was up to 1%, absorbance decreased, which may be have due to the fact that micelles were changed to gel and the internal structure was more hydrophilic.
 | Figure 2 Characteristics of PG. |
Western blotting analysis
B16F10 cells are highly invasive and metastatic with high Pgp expression. As shown in Figure 2D, both P123 and PG inhibited the expression of Pgp in B16F10 cells, and the inhibition was concentration-dependent. The ability of PG to inhibit the expression of Pgp was no lower than that of P123. It has been reported that the hydrophilic lypophilic balance (HLB) value of Pluronic is closely related to its ability to inhibit the expression of Pgp. Therefore, PG was likely to have a similar HLB value to P123,24 which was probably because the hydrophilicity and lipophilicity of GQ8 were balanced.
Preparation and characterization of micelles
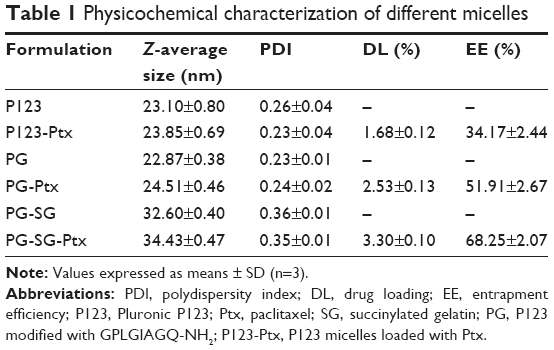
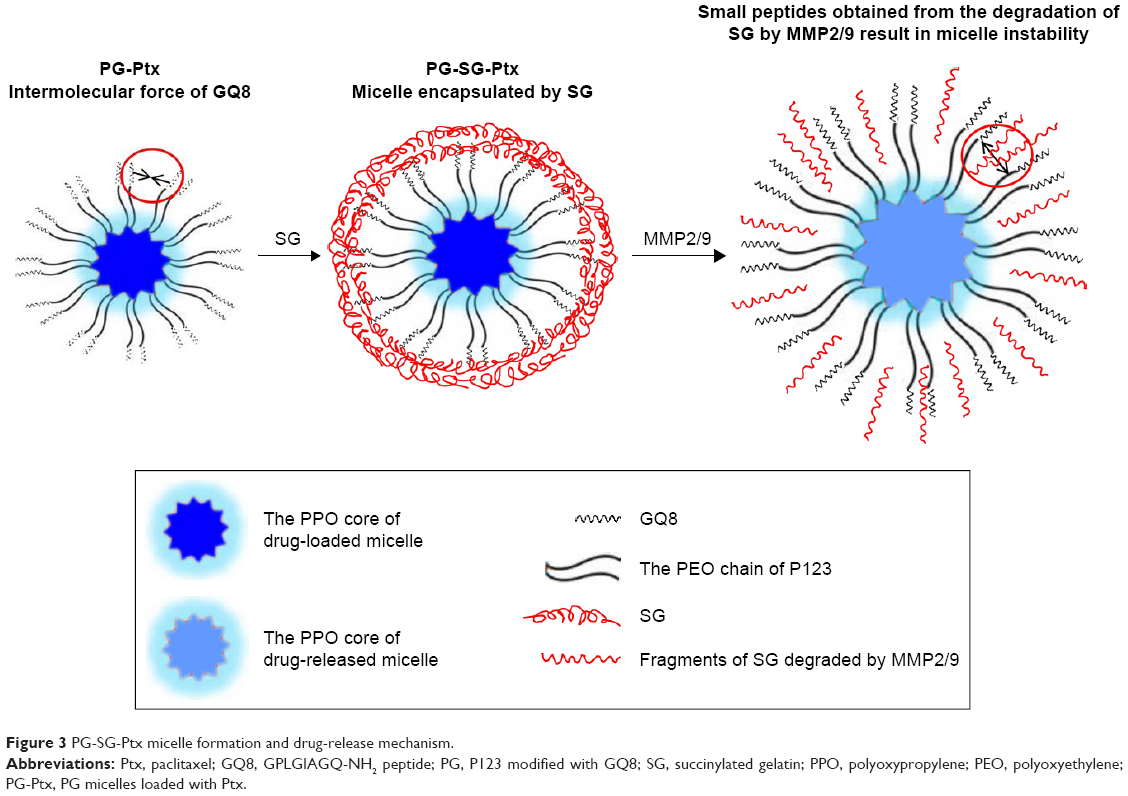
P123-Ptx and PG-SG-Ptx were prepared by thin-film hydration. The DL results (Table 1) indicated that Z-average diameter of P123-Ptx micelles was 23.85±0.69 nm, while the size of P123 micelles was 23.10±0.80 nm. The size of PG-SG-Ptx micelles was larger (34.43±0.47 nm) and similar to PG-SG (32.60±0.40 nm), which could have been due to the participation of SG in the construction of micelles, and the size of PG-SG was larger than that of PG (22.87±0.38 nm) (Figure S1). The DL of PG-SG-Ptx micelles (3.3%±0.1%) was higher than that of P123-Ptx micelles (1.68%±0.12%) and PG-Ptx micelles (2.53%±0.13%) (Table 1). The higher DL of PG-Ptx could be attributed to the interaction force between GQ8 peptides in the PG being greater than the interaction force between PEO in P123 (Figure 3). There were structural fragments in SG similar to GQ8, which enabled SG to interact with GQ8 in PG to form a more stable structure (Figure 3). Consequently, the particle size of the PG-SG micelles increased, and DL of PG-SG-Ptx was highest. The stability results of different micelles also indicated that PG-SG-Ptx was stablest (Figure 4A).
 | Table 1 Physicochemical characterization of different micelles |
 | Figure 3 PG-SG-Ptx micelle formation and drug-release mechanism. |
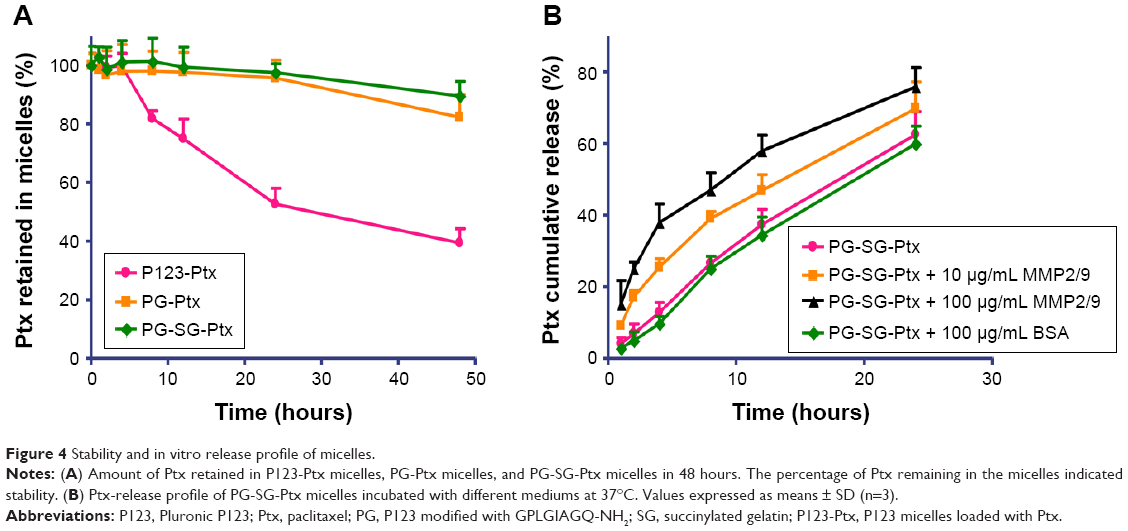 | Figure 4 Stability and in vitro release profile of micelles. |
In vitro drug-release profile
The in vitro drug-release experiment of PG-SG-Ptx was performed in the presence of different concentrations of MMP2/9, BSA, and PBS medium, which was used as control. As shown in Figure 4B, cumulative-release efficiency of Ptx in 100 μg/mL MMP2/9, 10 μg/mL MMP2/9, 100 μg/mL BSA, and PBS at 24 hours was 75.8%±1.8%, 69.7%±2.5%, 59.9%±1.6%, and 62.5%±2.1%, respectively. The release rate of Ptx in 100 μg/mL BSA was slightly slower (P>0.05) than that in PBS. Compared with the release profile in the medium without MMP2/9, Ptx release from 10 μg/mL MMP2/9 medium was faster (P<0.05), and in 100 μg/mL MMP2/9 the release of Ptx achieved its greatest speed (P<0.01). Ptx-release efficiency from PG-SG-Ptx micelles was MMP2/9-concentration-dependent. Since the studies on P123-Ptx and PG-Ptx showed that the release profile was almost MMP2/9-concentration-independent (Figure S2), this result indicates that SG was involved in the construction of the micelle structure. It is suggested that SG interacts with the peptide in PG and forms a stable protective layer in the outer layer of PG micelles by intermolecular force (Figure 3).
In vitro cytotoxicity
To evaluate the safety of materials for micelle formation, in vitro cytotoxicity assays were performed. As presented in Figure 5A, for HUVECs, P123, PG, PG-SG, and SG exhibited low cytotoxicity. IC50 values for P123, PG, and PG-SG were >3 mg/mL, and SG exhibited almost no cytotoxicity at 3 mg/mL concentration, which was consistent with effective clinical application of SG. For A549 cells, the cytotoxicity of PG-SG-Ptx (IC50 140.3±13.6 ng/mL) was significantly greater (P<0.001) compared to P123-Ptx (IC50 371.2±34.5 ng/mL) (Figure 5B). The higher cytotoxicity of PG-SG-Ptx was attributed to the rapid release of Ptx from PG-SG-Ptx, since the high expression of MMP2/9 in A549 cells25 triggered the release to occur.
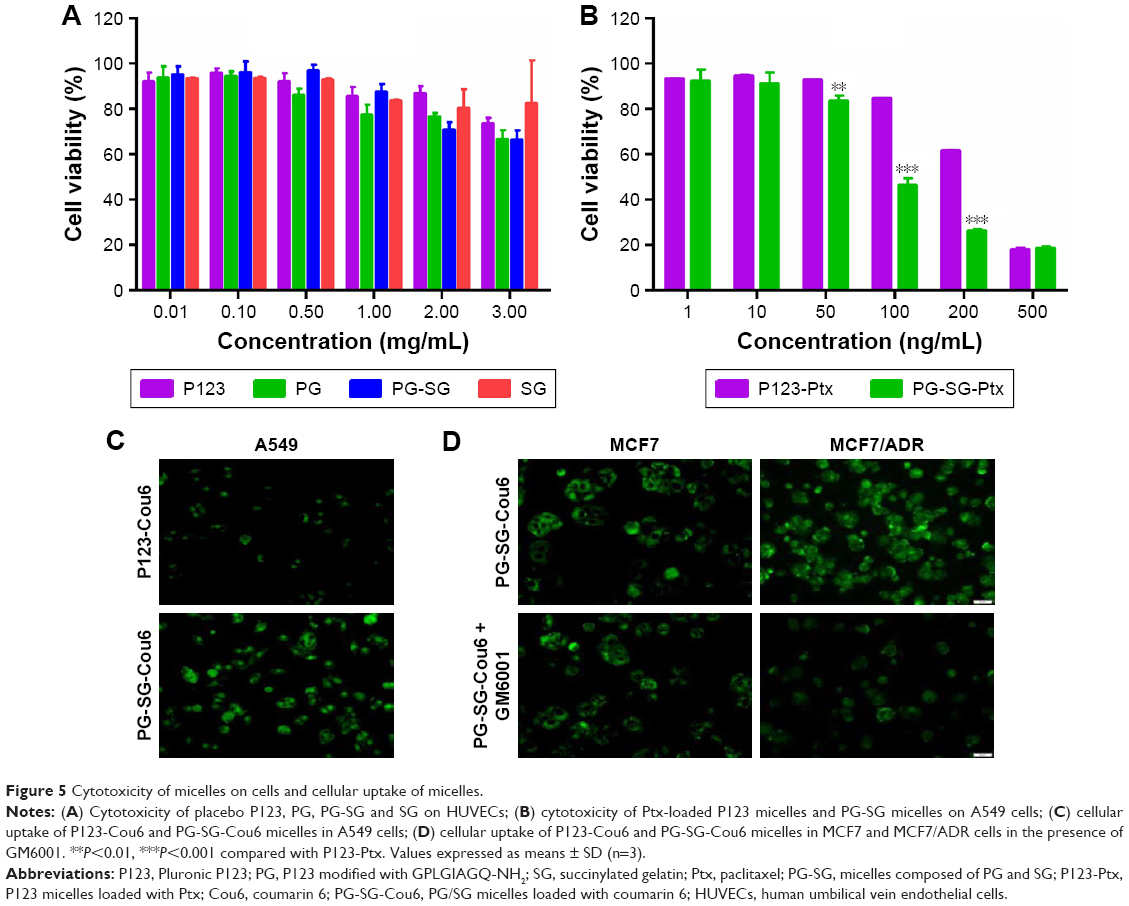 | Figure 5 Cytotoxicity of micelles on cells and cellular uptake of micelles. |
In vitro cellular uptake
To evaluate the intracellular delivery of different micelles, cellular uptake of Cou6-loaded P123 and PG-SG micelles was performed in A549 (MMP2/9-underexpression) cells. When the drug in micelles is triggered by MMP2/9 to release, cellular uptake is expected to be enhanced because of the highly hydrophobic drug, and the rapid passive diffusion across the cell membrane is much more effective than endocytosis-pathway drugs.26,27 Obviously, after incubation with Cou6-loaded micelles for 30 minutes, cellular uptake of PG-SG-Cou6 micelles was higher than that of P123-Cou6 micelles (Figure 5C) in A549 cells. When the inhibitor of MMP2/9 existed, cellular uptake in MCF7/ADR (MMP2/9-overexpression) cells decreased and cellular uptake in MCF7 (MMP2/9-underexpression) remained unchanged (Figure 5D).
Optimization of micelle inhalation
Aerodynamic particle size (APS) distribution of particles or droplets of a drug is the most important parameter to determine whether it can be inhaled into the lung and where it is deposited in the lung. For solid particles, shape and density have a great impact on the APS; therefore, it is necessary to use a cascade impactor, such as a next-generation pharmaceutical impactor, to collect and measure APS. For liquid droplets, due to their sphericity, aerodynamic size can be evaluated by laser diffraction if the density is approximate. Therefore, this study evaluated the nebulization process of nebulizers by measuring DSD through laser diffraction. As per the data in Table 2, the LC Sprint Junior and LC Sprint had better nebulization stability, and the LC Sprint Junior produced smaller droplets compared with the LC Sprint. The effect of the compressor flow rate on DSD was dependent on nebulizer design (Table 3). The variation trend was shown to be the same in the two nebulizers (Table 4), but the DSD variation trend of LC Sprint Junior nebulizer was not significant. The variation trend of DSD at different distances from the mouthpiece was independent of the nebulizer. These data (Figure S3) indicated that the effect of solute categories on the size of atomized droplets was not significant at low concentrations. There was no significant difference in droplet size after the nebulization of different micelles (Table 5). A pneumatic nebulizer would not have reduced the stability of drug-loaded micelles.
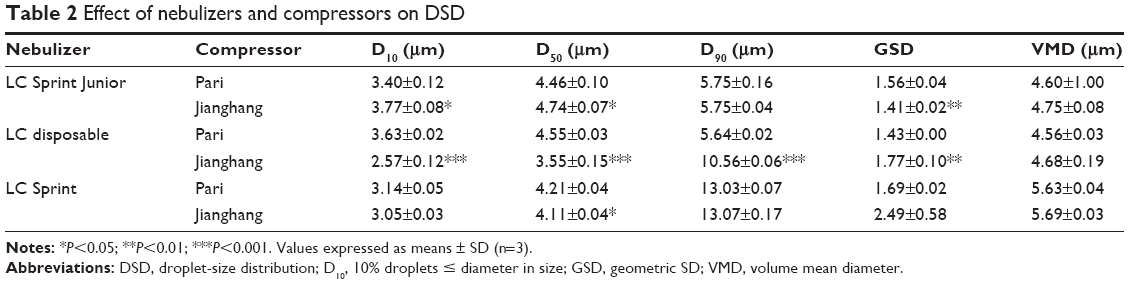 | Table 2 Effect of nebulizers and compressors on DSD |
 | Table 3 Effect of flow rate on DSD |
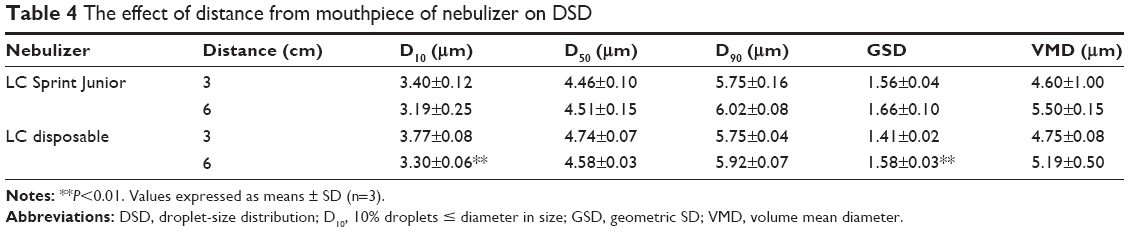 | Table 4 The effect of distance from mouthpiece of nebulizer on DSD |
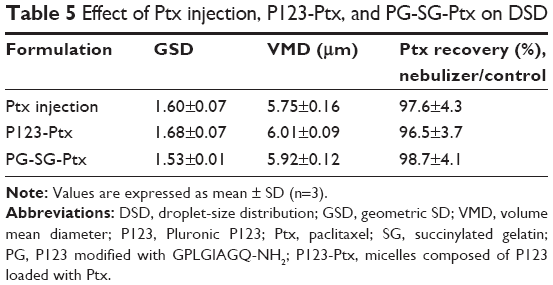 | Table 5 Effect of Ptx injection, P123-Ptx, and PG-SG-Ptx on DSD |
Histopathological analysis of inhalation toxicity
The toxicity of P123 inhalation was evaluated based on lung-inflammation responses through histopathological analysis. As shown in Figure 6, compared with parenteral administration, the lung tissue of rats inhaling saline or P123 solution displayed inflammation, but inflammatory responses between inhaled saline and P123 were indistinguishable, and no specific toxicity of P123 to the lung was observed. The tolerance of rats to humidity was poor, for the lung appeared to show mild edema after inhalation of saline for 20–30 minutes but returned to normal in a few days. However, the lung is a sensitive organ, and the toxicity of long-term pulmonary inhalation administration needs further extensive research.
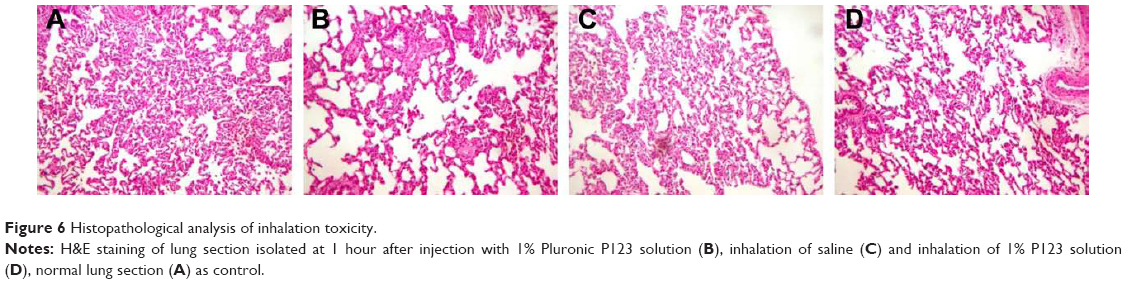 | Figure 6 Histopathological analysis of inhalation toxicity. |
Biodistribution of intravenous administration and micelle inhalation in rats
An important feature of the inhalation-delivery system is its ability to increase the local concentration of a drug in the lungs. DiR, a near-infrared fluorescent dye, was used to evaluate the in vivo biodistribution of micelle inhalation. Fluorescent imaging of the main organs (heart, liver, spleen, lung, and kidney) was performed at predetermined time intervals, as shown in Figure 7A. The vast majority of DiR was accumulated in the liver at 1 hour postinjection, followed by lung accumulation (5.8%±0.4%); 38.8%±0.5% of DiR was accumulated mainly in the lung at 1 hour after inhalation, with little in other organs (Figure 7C). As shown in Figure 7C and D, with the extension of time, P123 would slowly accumulate to the lung, inhaled drug would be redistributed slowly to other organs, and drug quantity accumulated in the lung gradually decreased. However, this process was slow: at 12 hours postinjection, fluorescence intensity in the liver was still the highest, indicating intravenous administration of the micelles mainly led to distribution in the liver, with little redistribution to other organs, though P123 has been reported to be capable of increasing drug accumulation in the lung similarly to a pulmonary surfactant.17,18
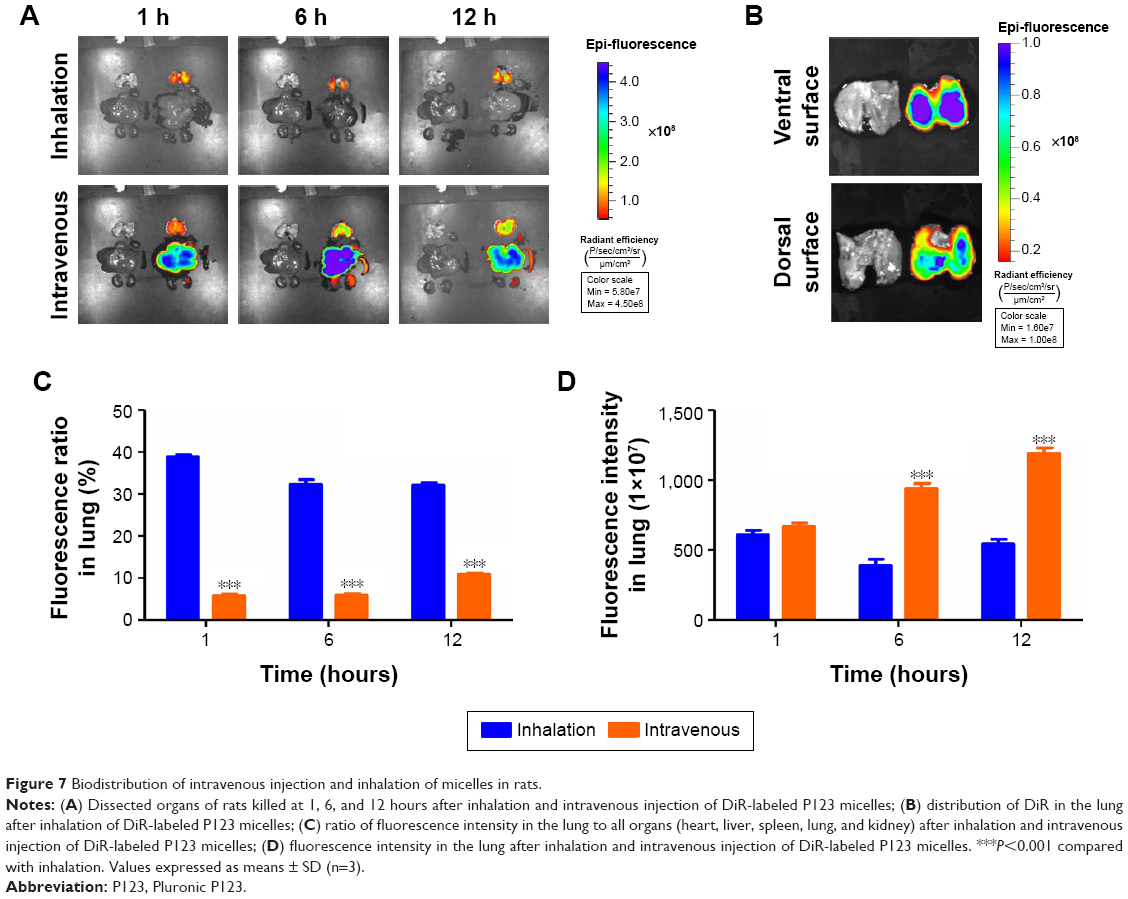 | Figure 7 Biodistribution of intravenous injection and inhalation of micelles in rats. |
Semiquantitative fluorescence-intensity results are shown in Figure 7C and D, indicating that when drug concentrations in the lung were at the same levels through both intravenous administration and inhalation, drug exposure to the whole system through intravenous injection was about sevenfold of that through inhalation. These results suggested that inhalation administration can greatly increase drug concentration in the lung while reducing the drug’s systemic toxicity, which is beneficial in curing lung disease.
In this study, 0.8 mL P123-DiR micelles containing 20 μg DiR were injected into rats, while 4 mL P123-DiR micelles containing 100 μg DiR were used for inhalation. The micelles placed in the nebulizer for inhalation were divided into two parts: one part was nebulized and inhaled by the rats, and the other remained in the nebulizer or was nebulized without being inhaled. Many studies have reported that pulmonary deposition of drugs accounts for 5%–15% of total drug added to nebulizers.28 The total amount of drug entering the body was calculated according to the sum of the fluorescence intensity of the major organs, and the amount that entered through injection was about 7.6-fold that through inhalation. This means that only about 2.6 μg DiR in a total of 100 μg DiR was inhaled by rats, ie, pulmonary deposition was 2.6%, which was lower than reported and might be attributed to the lung volume of rats being much smaller than that of humans. Even though the LC Sprint Junior nebulizer, specifically designed for infants, was used in our study, droplet size was still relatively large for the rats.
DiR accumulated mainly in the ventral side of the lung, while the dorsal side showed relatively less (Figure 7B), consistent with results in the literature,29 and distribution was related to the distribution of bronchi in the lung. Deposition in the lung’s left principal bronchus was also more than that at the right principal bronchus, due to the airflow disorder in the tracheal bifurcation, and the flow direction of droplets was changed, which made the droplets prone to depositing.
In vivo antitumor-effect evaluation
To evaluate the in vivo antitumor effect of PG-SG-Ptx micelles, pulmonary metastatic melanoma tumor-bearing nude mice were administered every 3 days with saline, Ptx injection, P123-Ptx and PG-SG-Ptx by inhalation, and PG-SG-Ptx intravenous injection at a dosage of 10 mg/kg. As shown in Figure 8A, compared with the saline group (0.48±0.05 g), lung weights in the Ptx-injection group (0.42±0.06 g) were not significantly decreased (P>0.05), while lung weights in the P123-Ptx- (0.22±0.04 g) and PG-SG-Ptx- (0.21±0.03 g) inhalation groups were significantly reduced (P<0.001), as well as lung weights in the PG-SG-Ptx intravenous injection group (0.37±0.05 g, P<0.01). If lung weight were used as an index to evaluate therapeutic effect on lung tumors, inhalation P123-Ptx and PG-SG-Ptx had optimal therapeutic effect. Lung sections isolated on day 14 after first treatment with the different formulations for histopathological analysis indicated that inhaled P123-Ptx and PG-SG-Ptx had the strongest antitumor ability, as shown in Figure 8C. It can be observed from the lung section in Figure 8C that mice inhaling saline had the most tumor nodules in the lung and melanin was found in most tumor tissues, while mice inhaling Ptx had the fewer nodules, but melanin in the tumor site was still obvious, but in mice inhaling P123-Ptx and PG-SG-Ptx, no large nodules or melanin were found, and no melanin in these nodules was found in mice injected with PG-SG-Ptx. Histopathological analysis results were in agreement with lung-weight results, indicating that lung weight can be used as an indicator to evaluate therapeutic effects for highly invasive tumors. Mouse body weight in the model group and treatment group decreased in 0–12 days, and the decrease in the Ptx-injection group was the most obvious. The average body-weight loss in the inhaled PG-SG-Ptx group was 5.9%, while in other groups the loss was >10% (Figure 8B). Therefore, as the body weight of mice was used as one of the markers of safety, the toxicity of inhalation Ptx was highest and inhalation PG-SG-Ptx lowest. H&E-stained sections of hearts, livers, spleens, and kidneys showed no pathological variation in multiple organs isolated after different treatments (Figure S4).
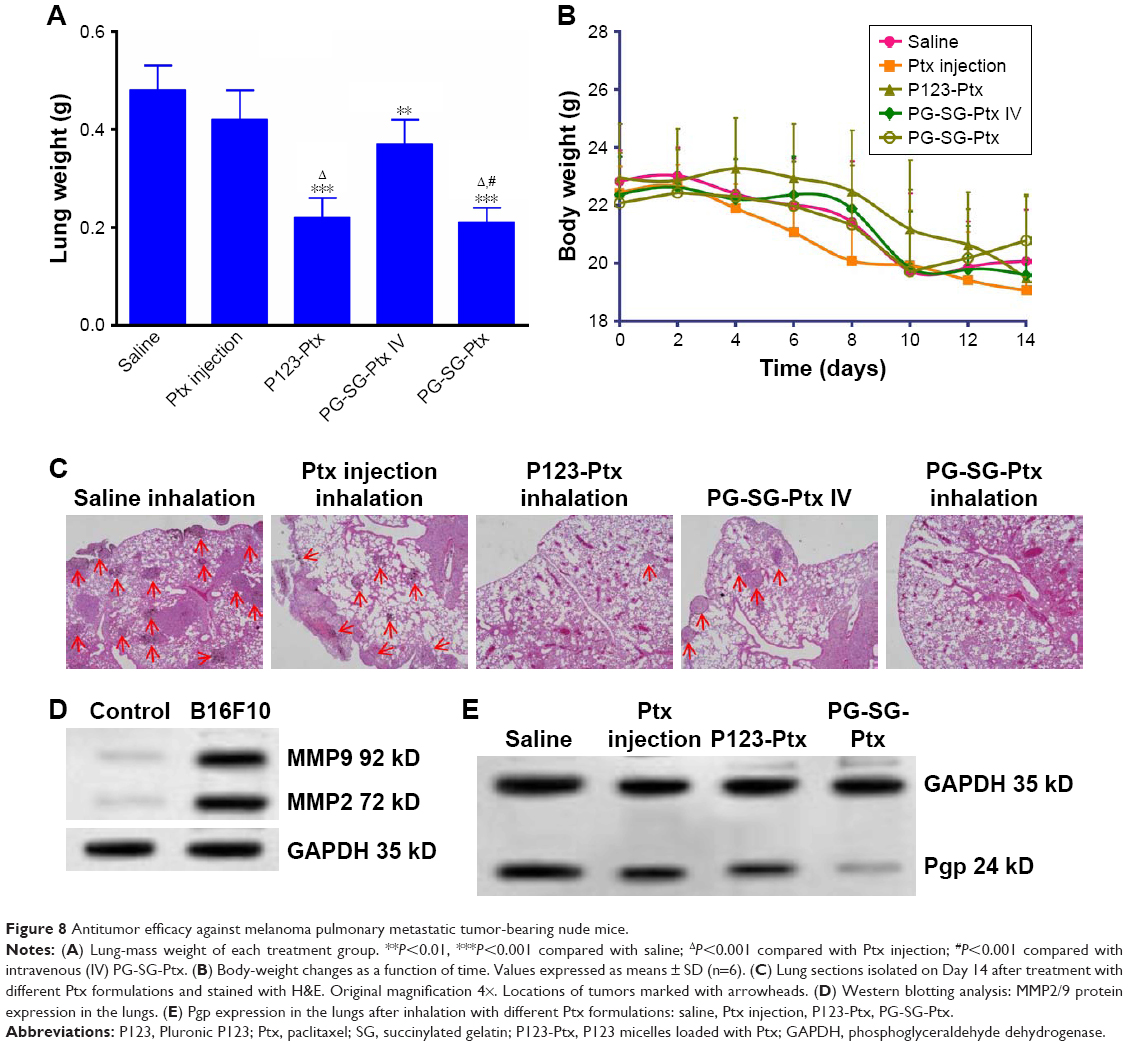 | Figure 8 Antitumor efficacy against melanoma pulmonary metastatic tumor-bearing nude mice. |
As shown in Figure 8E, while tumor-bearing nude mice inhaled saline as the control group, Pgp expression in the lungs inhibited by the inhalation Ptx, P123-Ptx, and PG-SG-Ptx were determined through Western blot. The quantity of Pgp in the treatment group was lower than that in the model group: inhaled PG-SG-Ptx significantly inhibited Pgp expression in lung cancer. Melanoma is highly malignant, invasive, and fatal. Injection of B16F10 cells through the tail vein of mice formed nodules in the lung rapidly, and then tumors spread to surrounding tissues or regionally to other organs. MMP2/9 and Pgp were highly expressed in lungs of melanoma-bearing mice (Figure 8D), which results in high invasiveness and high malignancy.30
In this study, the antitumor efficacy of Ptx injection was worst, because B16F10 was resistant to Ptx (Figure S5 and Table S1).17 Inhaled PG-SG-Ptx showed better efficacy, which was partly attributed to the inhibition of Pgp by PG increasing the sensitivity of B16F10 to Ptx. Moreover, inhaled drugs increased local concentrations in the lung, thereby improving the therapeutic effect, and the Ptx in PG-SG-Ptx was triggered to release by MMP2/9 at tumor sites. The efficacy of intravenous administration was lower than inhalation therapy, due to lower drug concentrations reaching the lung. Polyoxyl 40 castor oil, which is an important excipient in Ptx injection, might have been the main reason for the toxicity of inhaled Ptx, while MMP2/9-triggered-release micelles alleviated toxicity.
The same results were obtained in in vivo evaluation of PG-SG-Ptx-treated A549 pulmonary metastatic tumor-bearing nude mice (Figure S6). The invasiveness of A549 was weaker than B16F10, and the increased lung weight in A549 pulmonary metastatic tumor-bearing nude mice was not significant compared with that of B16F10 (Figure S6A), so lung weight may not be suitable to be used as a single indicator to evaluate the efficacy of micelles treating A549 pulmonary metastatic tumors.
Conclusion
In summary, PG was successfully synthesized and characterized and MMP2/9-triggered-release micelles PG-SG-Ptx prepared using PG. In vitro drug-release data indicated the release profile of PG-SG-Ptx was MMP2/9-dependent and in vitro cytotoxicity enhanced by PG-SG-Ptx in cells with high MMP2/9 expression compared with P123-Ptx. Analysis of the influence of the nebulized aerosol DSD was carried out, and the LC Sprint Junior equipped with a 9 L/min flow-rate compressor was selected as the device for delivery of micelles to the lung. It was also found that the nebulization process did not affect the stability of drug-loaded micelles. Biodistribution analysis showed that while drug concentrations in the lung were achieved at the same levels by intravenous administration and inhalation, drug exposure to the whole system through intravenous injection was about sevenfold of that through inhalation. The relationship between inhaled and intravenous dosage was obtained by studying the distribution of DiR in vivo. About 2.6% of total drug added to nebulizers was deposited in rat lungs, which was lower than reported results of 5%–15% in human lungs. In vivo antitumor-efficacy evaluation indicated that inhaled MMP2/9-triggered-release PG-SG-Ptx micelles increased concentration in the lung, released the drug at tumor sites, and inhibited Pgp in lung cancer. Consequently, PG-SG-Ptx increased tumor sensitivity to chemotherapeutics and reduced chemotherapy toxicity to healthy lung cells. In conclusion, MMP2/9-triggered-release micelles have great potential in lung cancer therapy.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81373354).
Disclosure
The authors report no conflicts of interest in this work.
References
Mangal S, Gao W, Li T, Zhou QT. Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: challenges and opportunities. Acta Pharmacol Sin. 2017;38(6):782–797. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Ettinger D, Wood D, Aisner D, et al. Non-small cell lung cancer, version 5.2017: clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017;15(4):504–535. | ||
Spiro SG, Porter JC. Lung cancer: where are we today? Current advances in staging and nonsurgical treatment. Am J Respir Crit Care Med. 2002;166(9):1166–1196. | ||
Ahmad A, Gadgeel S, editors. Lung Cancer and Personalized Medicine: Current Knowledge and Therapies. Heidelberg: Springer; 2016. | ||
Jinturkar KA, Anish C, Kumar MK, Bagchi T, Panda AK, Misra AR. Liposomal formulations of etoposide and docetaxel for p53 mediated enhanced cytotoxicity in lung cancer cell lines. Biomaterials. 2012;33(8):2492–2507. | ||
Zarogoulidis P, Chatzaki E, Porpodis K, et al. Inhaled chemotherapy in lung cancer: future concept of nanomedicine. Int J Nanomedicine. 2012;7:1551–1572. | ||
Kuzmov A, Minko T. Nanotechnology approaches for inhalation treatment of lung diseases. J Control Release. 2015;219:500–518. | ||
Otterson GA, Villalona-Calero MA, Sharma S, et al. Phase I study of inhaled doxorubicin for patients with metastatic tumors to the lungs. Clin Cancer Res. 2007;13(4):1246–1252. | ||
Rao RD, Markovic SN, Anderson PM. Aerosol therapy for malignancy involving the lungs. Curr Cancer Drug Targets. 2003;3(4):239–250. | ||
Gagnadoux F, Hureaux J, Vecellio L, et al. Aerosolized chemotherapy. J Aerosol Med Pulm Drug Deliv. 2008;21(1):61–70. | ||
Duffy MJ, McGowan PM, Gallagher WM. Cancer invasion and metastasis: changing views. J Pathol. 2008;214(3):283–293. | ||
Chambers AF, Matrisian LM. Changing views of the role of matrix metalloproteinases in metastasis. J Natl Cancer Inst. 1997;89(17):1260–1270. | ||
Nair SA, Jagadeeshan S, Indu R, Sudhakaran PR, Pillai MR. How intact is the basement membrane? Role of MMPs. Adv Exp Med Biol. 2012;749:215–232. | ||
Yang JM, Xu Z, Wu H, Zhu H, Wu X, Hait WN. Overexpression of extracellular matrix metalloproteinase inducer in multidrug resistant cancer cells. Mol Cancer Res. 2003;1(6):420–427. | ||
Gong L, Wu D, Zou J, et al. Prognostic impact of serum and tissue MMP-9 in non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget. 2016;7(14):18458–18468. | ||
Zhang W, Shi Y, Chen Y, Hao J, Sha X, Fang X. The potential of Pluronic polymeric micelles encapsulated with paclitaxel for the treatment of melanoma using subcutaneous and pulmonary metastatic mice models. Biomaterials. 2011;32(25):5934–5944. | ||
Zhang W, Shi Y, Chen Y, et al. Enhanced antitumor efficacy by paclitaxel-loaded Pluronic P123/F127 mixed micelles against non-small cell lung cancer based on passive tumor targeting and modulation of drug resistance. Eur J Pharm Biopharm. 2010;75(3):341–353. | ||
Batrakova EV, Li S, Alakhov VY, Miller DW, Kabanov AV. Optimal structure requirements for Pluronic block copolymers in modifying P-glycoprotein drug efflux transporter activity in bovine brain microvessel endothelial cells. J Pharmacol Exp Ther. 2003;304(2):845–854. | ||
Marsden HR, Kros A. Self-assembly of coiled coils in synthetic biology: inspiration and progress. Angew Chem Int Ed Engl. 2010;49(17):2988–3005. | ||
Zhu L, Kate P, Torchilin VP. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano. 2012;6(4):3491–3498. | ||
Garripelli VK, Kim JK, Son S, Kim WJ, Repka MA, Jo S. Matrix metalloproteinase-sensitive thermogelling polymer for bioresponsive local drug delivery. Acta Biomater. 2011;7(5):1984–1992. | ||
Na DH, Park MO, Choi SY, et al. Identification of the modifying sites of mono-PEGylated salmon calcitonins by capillary electrophoresis and MALDI-TOF mass spectrometry. J Chromatogr B Biomed Sci Appl. 2001;754(1):259–263. | ||
Sun H, Meng Q, Tang S, et al. Inhibition of breast cancer metastasis by Pluronic copolymers with moderate hydrophilic-lipophilic balance. Mol Pharm. 2015;12(9):3323–3331. | ||
Jung O, Lee J, Lee YJ, et al. Timosaponin AIII inhibits migration and invasion of A549 human non-small-cell lung cancer cells via attenuations of MMP-2 and MMP-9 by inhibitions of ERK1/2, Src/FAK and β-catenin signaling pathways. Bioorg Med Chem Lett. 2016;26(16):3963–3967. | ||
Zhang J, Liu J, Zhao Y, Wang G, Zhou F. Plasma and cellular pharmacokinetic considerations for the development and optimization of antitumor block copolymer micelles. Expert Opin Drug Deliv. 2015;12(2):263–281. | ||
Zhang XY, Wang XF, Zhong WT, Ren XQ, Sha XY, Fang XL. Matrix metalloproteinases-2/9-sensitive peptide-conjugated polymer micelles for site-specific release of drugs and enhancing tumor accumulation: preparation and in vitro and in vivo evaluation. Int J Nanomedicine. 2016;11:1643–1661. | ||
Newman S. Respiratory Drug Delivery: Essential Theory and Practice. Richmond (VA): RDD; 2010. | ||
Zhang Z, Kleinstreuer C, Kim CS, Hickey AJ. Aerosol transport and deposition in a triple bifurcation bronchial airway model with local tumors. Inhal Toxicol. 2002;(14):1111–1133. | ||
Choi EO, Cho EJ, Jeong JW, et al. Baicalein inhibits the migration and invasion of B16F10 mouse melanoma cells through inactivation of the PI3K/Akt signaling pathway. Biomol Ther (Seoul). 2017;25(2):213–221. |
Supplementary materials
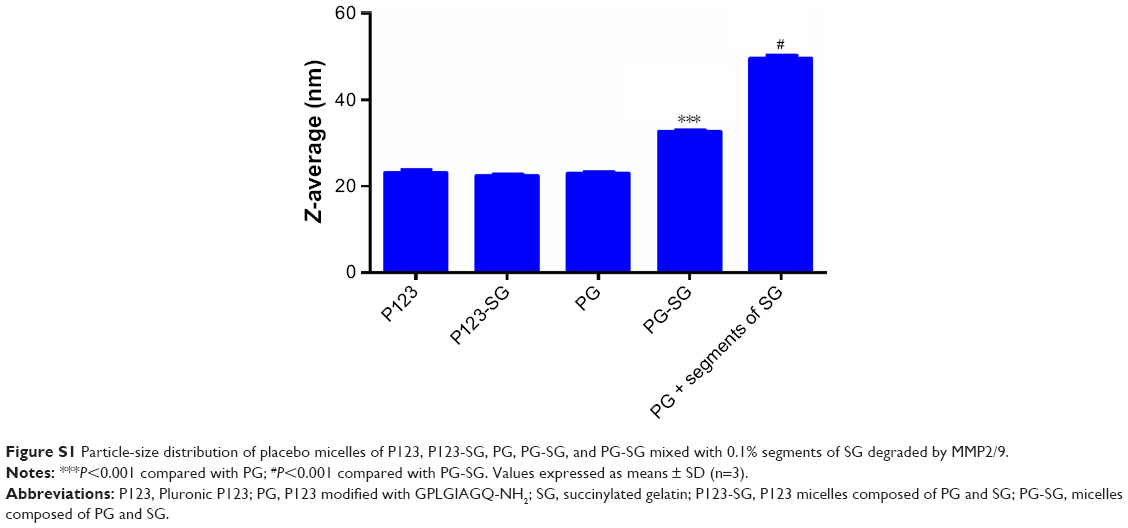 | Figure S1 Particle-size distribution of placebo micelles of P123, P123-SG, PG, PG-SG, and PG-SG mixed with 0.1% segments of SG degraded by MMP2/9. |
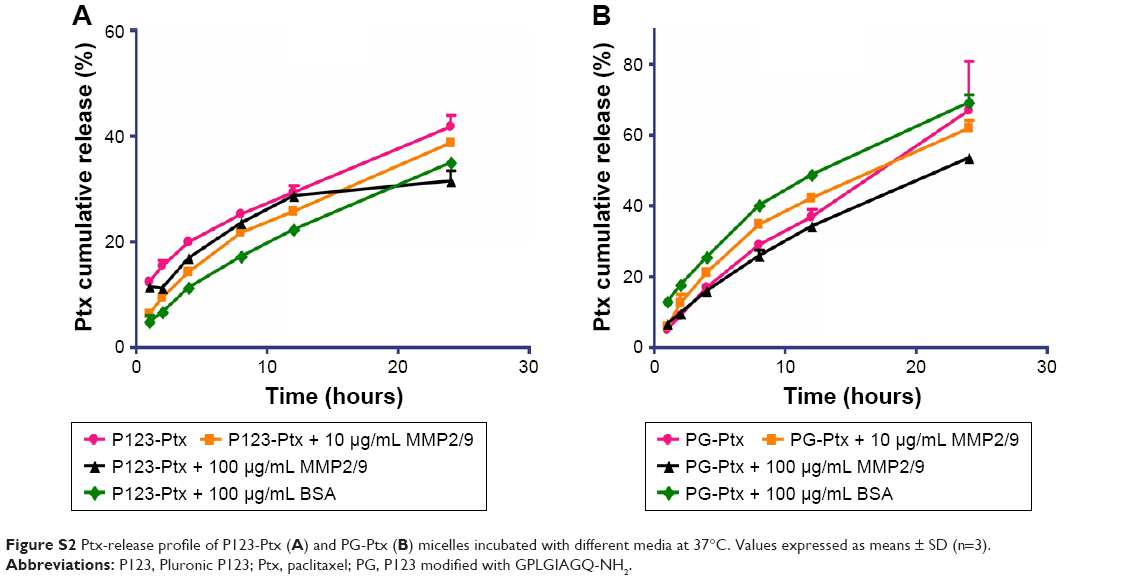 | Figure S2 Ptx-release profile of P123-Ptx (A) and PG-Ptx (B) micelles incubated with different media at 37°C. Values expressed as means ± SD (n=3). |
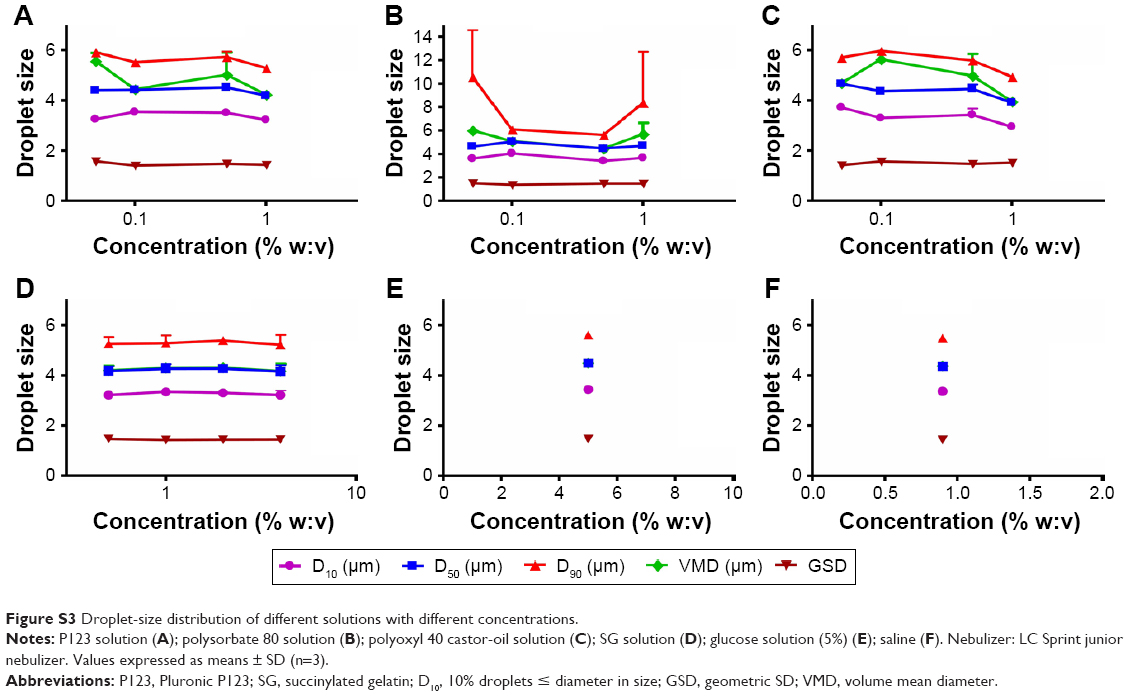 | Figure S3 Droplet-size distribution of different solutions with different concentrations. |
 | Figure S4 Histopathological analysis of heart, liver, spleen, and kidney of melanoma pulmonary metastatic tumor-bearing male nude mice. |
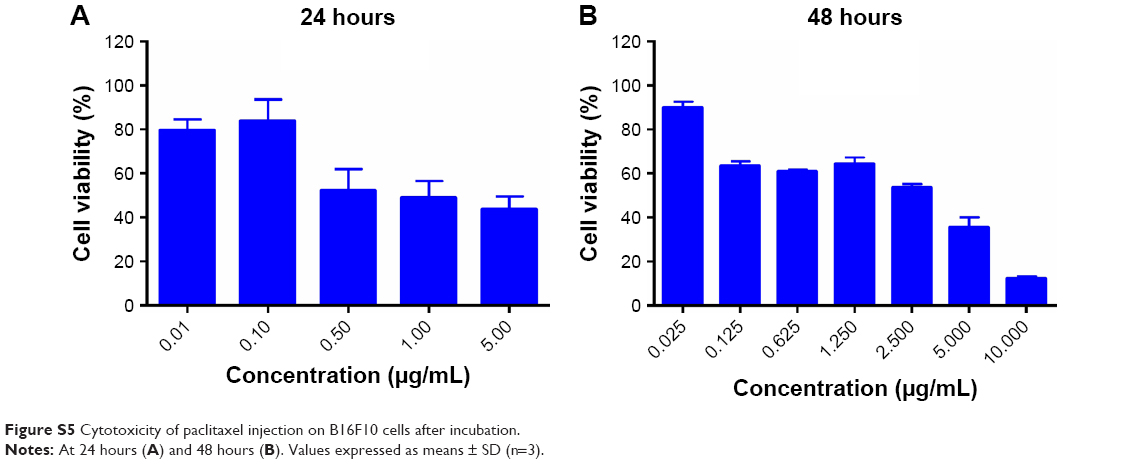 | Figure S5 Cytotoxicity of paclitaxel injection on B16F10 cells after incubation. |
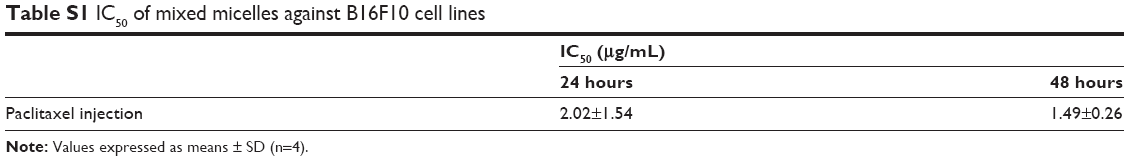 | Table S1 IC50 of mixed micelles against B16F10 cell lines |
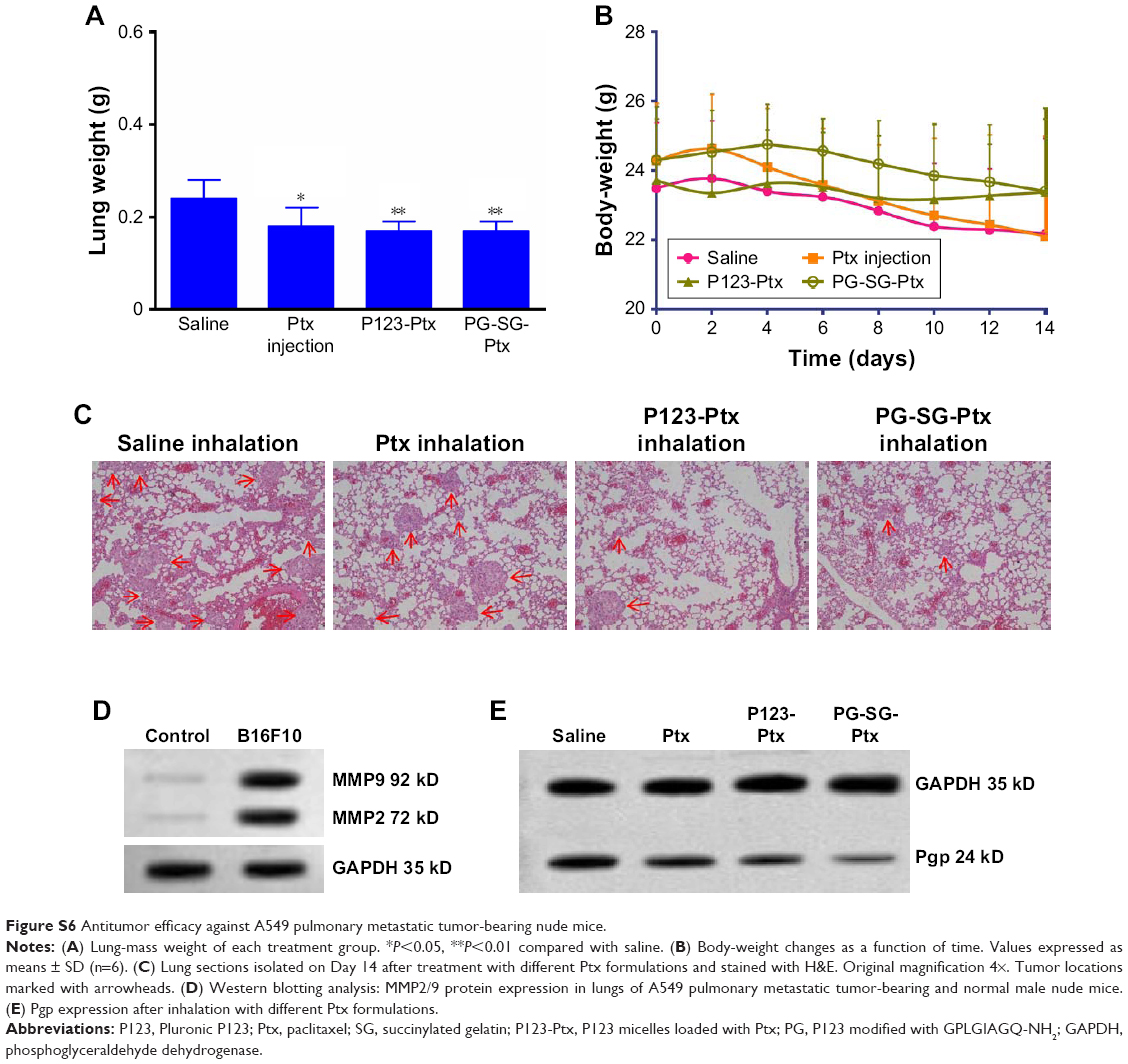 | Figure S6 Antitumor efficacy against A549 pulmonary metastatic tumor-bearing nude mice. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.