Back to Journals » Neuropsychiatric Disease and Treatment » Volume 21
Maternal Immune Activation and Neurodevelopmental Disorders: Integrating Molecular, Cellular and Systems Mechanisms
Authors Yong Q ![]() , Zhao C, Xia L, Zhu T, Xia K
, Zhao C, Xia L, Zhu T, Xia K
Received 11 April 2025
Accepted for publication 31 October 2025
Published 24 November 2025 Volume 2025:21 Pages 2575—2594
DOI https://doi.org/10.2147/NDT.S533813
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Taro Kishi
Qi Yong,1,* Chao Zhao,1,* Lu Xia,2 Tengfei Zhu,3,4 Kun Xia3
1The Seventh Affiliated Hospital Hengyang Medical School, University of South China (Hunan Provincial Veterans Administration Hospital), Changsha, Hunan, 410000, People’s Republic of China; 2Hunan Key Laboratory of Medical Genetics, MOE Key Laboratory of Pediatric Rare Diseases, School of Life Sciences, Central South University, Changsha, Hunan, 410083, People’s Republic of China; 3MOE Key Laboratory of Pediatric Rare Diseases, University of South China, Hengyang, Hunan, 421001, People’s Republic of China; 4Department of Critical Care Medicine, Shenzhen Third People’s Hospital, Shenzhen, Guangdong, 518112, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Kun Xia, MOE Key Laboratory of Pediatric Rare Diseases, University of South China, Hengyang, Hunan, 421001, People’s Republic of China, Email [email protected] Tengfei Zhu, MOE Key Laboratory of Pediatric Rare Diseases, University of South China, Hengyang, Hunan, 421001, People’s Republic of China, Email [email protected]
Abstract: Neurodevelopmental Disorders (NDDs) represent chronic cerebral dysfunctions arising from gene-environment interactions, encompassing conditions such Autism Spectrum Disorders (ASD) and attention-deficit/hyperactivity disorders (ADHD). Emerging evidence identifies Maternal immune activation (MIA) as a critical environmental risk factor for NDDs. Gestational infections, inflammatory responses, or immune dysregulation elevate maternal-fetal inflammatory mediators, which disrupt neurodevelopmental trajectories via placental-fetal signaling cascades. Preclinical models (rodents, non-human primates) demonstrate that MIA induces characteristic NDD phenotypes—including social deficits and cognitive impairments—through microglial hyperactivation, aberrant synaptic pruning, oxidative stress, and mitochondrial dysfunction. Concurrently, gut microbiota dysbiosis and Th17/Treg immune imbalance exacerbate neuroinflammatory processes. Novel therapeutic strategies targeting inflammatory pathways microglial modulation, or microbial homeostasis restoration show translational promise. Future investigations must unravel MIA’s molecular underpinnings and multifactorial interactions to enable early-risk stratification and precision interventions for NDDs.
Keywords: MIA, NDD, Microglia
Introduction
NDDs are chronic brain dysfunction syndromes arising from multifactorial genetic or acquired etiologies that impair pluripotent cerebral domains, including cognition, motor function, social adaptability, and behavioral regulation. Representative NDD subtypes encompass ASD, ADHD, intellectual disability (ID), and schizophrenia (SCZ). Large-scale twin studies estimate heritability at 0.6–0.9 for ASD and 0.76 for ADHD,1–3 with significant genetic overlap observed between ADHD and ASD. Genetic contributions account for approximately 50% of idiopathic ID cases, escalating to ≥66% in moderate-to-severe presentations. However, NDD pathogenesis reflects dynamic interplay between polygenic susceptibility and environmental exposures, with non-genetic factors demonstrating substantial etiological weight. This is evidenced by the discordance rates in ASD and SCZ twin cohorts, underscoring environmental triggers’ critical role in neurodevelopmental trajectory disruption.4
Neurodevelopmental disorders have diverse etiology and complex pathogenesis. Genetic and environmental factors are involved in the occurrence of neurodevelopmental disorders, and individual genetic susceptibility can be caused by gene mutation, chromosome abnormality and copy number variation. Hundreds of genes have been identified to be associated with neurodevelopmental disorders, including FMR1,5 UBE3A,6 CACNA1C,7 SHANK3,8 etc. Abnormal brain structure and development in NDDs patients are also considered to be one of the causes, such as abnormal development of frontal lobe and cingulate cortex in ASD patients.9 The abnormal development of the frontal lobe10,11 and the asymmetry of the head end of the bilateral caudate nucleus12 in ADHD patients are also important mechanisms for the pathogenesis of NDDs. The increased levels of inflammatory factors in the body can affect the normal growth and function of brain nerve cells and cause neurodevelopmental abnormalities. Studies have shown that there is neuroinflammation in the brain tissue of ASD patients, and the excessive activation of microglia plays a role in releasing pro-inflammatory factors, leading to nerve cell damage.13
MIA has been increasingly recognized as a critical risk factor for ASD and other neurodevelopmental disorders. MIA arises from gestational immune hyperactivation triggered by maternal infections, autoinflammatory conditions, or autoimmune diseases, encompassing but not limited to infection-induced inflammatory dysregulation.14 Notably, MIA can be initiated by both infectious stimuli (eg, viral/bacterial pathogens) and sterile triggers, including maternal asthma, obesity, and autoimmune disorders. Gestational immune perturbations exert enduring consequences, with MIA inducing persistent remodeling of developing neural circuits and altering fetal brain developmental trajectories.15 When superimposed on genetic and environmental vulnerabilities, this immune dysregulation amplifies offspring risks for NDDs,16,17 including schizophrenia, bipolar disorder, ADHD, cerebral palsy, developmental delay, cognitive dysfunction, anxiety/depression, and ASD.18 High-resolution ex vivo MRI and electron microscopy analyses in murine models reveal that late-gestation MIA exposure induces volumetric expansion in embryonic brain regions—specifically the striatum/caudoputamen, hippocampus, lateral septum, cingulate cortex, and cerebellum.19 These neuroanatomical anomalies mirror structural abnormalities identified in neuroimaging studies of ASD and schizophrenia patient.20 Furthermore, third-trimester MIA exacerbates neuroinflammation and compromises placental efficiency in prenatal rat models, as evidenced by elevated proinflammatory cytokines and impaired nutrient transport mechanisms.21
Although existing research has confirmed that MIA is a key environmental risk factor for NDDs, several limitations remain. Firstly, the diverse triggers of MIA and the lack of a standardized definition make it difficult to directly compare and integrate results across studies. Secondly, the specific molecular and cellular mechanisms through which MIA affects neurodevelopment—particularly the causal relationships and synergistic effects between inflammatory cytokine transmission across the placenta, microglial dysfunction, and gut microbiota dysbiosis—have not been fully elucidated. Additionally, how interactions between genetic background and MIA exposure contribute to the phenotypic heterogeneity of NDDs requires further in-depth investigation.
This review aims to systematically analyze the multi-level mechanisms by which MIA contributes to NDDs, with a focus on key aspects such as the disruption of neural progenitor cell differentiation by inflammatory cytokine cascades at the maternal-fetal interface (eg, the IL-17A/IL-6/TNF-α axis), microglia-mediated synaptic pruning deficits and oxidative stress damage, as well as the amplification of neuroinflammation through gut-brain axis imbalances and Th17/Treg immune deviations. Furthermore, the translational prospects of targeting immune regulation (eg, IL-17A antagonists, microbiota transplantation) and neuroprotective strategies are discussed.
MIA and Neurodevelopmental Disorders
Epidemiological Evidence Linking MIA to Neurodevelopmental Disorders
The association between environmental factors, MIA, and NDDs is well-established. Epidemiological studies demonstrate significant correlations between gestational infections and elevated risks of SCZ, ASD, and epilepsy in offspring. Historical cohort analyses reveal marked increases in schizophrenia and ASD incidence following the 1918–1919 influenza pandemic and 1964 rubella epidemic.22,23 Second-trimester influenza infection confers a 2- to 8-fold increased schizophrenia risk,24 while prenatal exposure to Toxoplasma gondii,25 herpes simplex virus type 2,26 and urinary/respiratory infections27–30 similarly elevates neuropsychiatric vulnerability.
Notably, the California Department of Developmental Services documented a “winter birth phenomenon”, wherein infants conceived during winter months exhibit heightened ASD susceptibility.31 A 1990–2002 pediatric cohort study suggests this seasonal risk may correlate with increased respiratory infections in colder months.31 A Danish nationwide study of 1.6 million children further confirmed that hospitalization-requiring maternal infections or inflammation significantly elevates offspring psychiatric risks, underscoring MIA’s pathogen-agnostic immunopathic effects. The Kaiser Permanente Northern California Birth Cohort study also found that fever or infections (including viral and bacterial) during the second trimester (4–6 months of gestation) were no Significantly increased risk of ASD, especially fever lasting ≥1 week32 Influenza vaccination during pregnancy can reduce the risk of ASD associated with MIA.33 Male offspring with high levels of HSV-2 IgG antibodies in maternal plasma during the second trimester have also been found to have an increased risk of autism spectrum disorders34–37 The COVID-19 pandemic has intensified concerns regarding prenatal viral impacts, with SARS-CoV-2 infection linked to fetal growth restriction and neurodevelopmental sequelae.38 Maternal COVID-19 infection may be associated with fetal neurodysplasia.39 Analysis of 460 infants whose mothers were infected with OVID 19 (COVID+) from January 2020 to September 2021 found that reduced levels of certain immune molecules in newborns born to COVID+ mothers may increase the risk of NDDs.40 Globalization-driven resurgent infections and autoimmune disease proliferation may synergistically fuel the NDD pandemic, necessitating urgent dissection of MIA-specific immune pathways and gene-environment interplay to inform early intervention strategies. Beyond infections, maternal autoimmune conditions (eg, asthma, allergies), acute stress, environmental pollutants, and nutritional imbalances amplify ASD risk through immune hyperactivation.35–37 For example, a meta-analysis of the effects of maternal diabetes on neurodevelopmental disorders in offspring showed that maternal diabetes increased the risk of neurodevelopmental disorders in offspring by 28%, especially maternal pregestational diabetes was more strongly associated and may lead to impaired neurodevelopmental capacity.41 Antibiotic use during pregnancy may alter the maternal microbiome, affecting the colonization of microbes in the fetal gastrointestinal system, and maternal and early antibiotic use has been confirmed to be associated with a higher risk of ASD and ADHD.42
In conclusion, the epidemiological evidence consistently demonstrates that the risk for ASD is substantially heightened specifically when maternal immune activation results from severe infectious or inflammatory insults. It is not just any case of infection, but rather the intensity and duration of the immune response that determines neurodevelopmental outcomes. This crucial distinction underscores the need for particular clinical attention to pregnancies experiencing significant inflammatory events, while reassuring that routine infections do not appear to carry the same level of risk.
Experimental Animal Evidence of MIA
Mounting evidence from animal models has robustly validated neurodevelopmental abnormalities induced by MIA.43 Experimental paradigms typically involve artificial immune system activation during gestation to assess offspring neurodevelopmental outcomes. Widely used MIA induction methods include exposure to human influenza virus, viral mimetic polyinosinic: polycytidylic acid (Poly(I:C)), bacterial endotoxin lipopolysaccharide (LPS), localized inflammatory agents (eg, turpentine), or specific proinflammatory cytokines.16,44 Amodeo et al demonstrated that gestational Poly(I:C) exposure impairs offspring learning and social interaction capabilities, inducing autism- and schizophrenia-like behavioral phenotypes. These effects correlate with persistent disruptions in glutamatergic neurotransmission, mTOR signaling, and potassium channel activity.45 In murine models, O’Loughlin et al observed upregulated amygdalar proinflammatory cytokine expression alongside chronic microglial activation and astrogliosis following intraperitoneal LPS administration during pregnancy.46 Rat studies by Fortunato et al revealed that LPS-induced MIA elevates stereotyped repetitive behaviors and reduces social engagement in offspring—phenotypes reversible through ω-3 polyunsaturated fatty acid intervention.47
Complementing rodent studies, non-human primate (NHP) models—with closer physiological parallels to human pregnancy—are increasingly employed in MIA research.48 Seminal work by Bauman et al in rhesus macaques demonstrated that mid-gestation Poly(I:C) exposure exacerbates abnormal behaviors (eg, tremors, tail clasping, self-clasping) during weaning, with adults exhibiting heightened stereotypies and impaired social reciprocity.49
Abnormal Levels of Cytokines in Neurodevelopmental Disorders
Inflammatory Mediators in MIA-Induced Neurodevelopmental Abnormalities
The developing brain exhibits heightened sensitivity to environmental signals, with MIA -driven cytokine cascades and inflammatory responses serving as pivotal disruptors of fetal neurodevelopment. Gilmore and Jarskog first proposed in 1997 that inflammatory mediators might underlie infection-associated fetal brain developmental disruptions,50 a hypothesis now substantiated by extensive epidemiological studies and experimental MIA models delineating cytokine network dysregulation as central to MIA pathophysiology. Herein, we comprehensively review the mechanistic roles of key inflammatory mediators in MIA-induced neurodevelopmental perturbations.
Prenatal inflammatory exposure triggers maternal-fetal upregulation of cytokines and chemokines—including interferon-gamma (IFN-γ), IFN-β, tumor necrosis factor-alpha (TNF-α), interleukins (IL-4, IL-5, IL-6, IL-8, IL-1α, IL-1β, IL-17), granulocyte-macrophage colony-stimulating factor (GM-CSF), and C-reactive protein (CRP)—that induce fetal endogenous cytokine production, thereby altering cerebral ontogeny.51–53 Maternal-fetal cytokine profiling demonstrates MIA-induced dysregulation critically contributes to ASD pathogenesis. Clinical detection of interleukin-6 (IL-6), interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ) in blood, cerebrospinal fluid or brain tissue of patients with autism spectrum disorder and other diseases Increased levels of Interleukin-17 (IL-17),54,55 Interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) may be reduced or relatively insufficient.56,57
Analysis of 60 inflammatory mediators and growth factors in mid-pregnancy maternal serum and umbilical cord blood from the Norwegian Mother, Father and Child Cohort Study (MoBa) identified TNF-α, Serpin E1, VCAM1, and IL-1β as key predictors of neurodevelopmental trajectories and ASD diagnosis.53 A longitudinal birth cohort study on ADHD revealed that elevated umbilical cord blood IL-6 and TNF-α levels—when integrated with polygenic risk scores—predicted increased ADHD symptom severity in 8- to 9-year-olds via structural equation modeling, implicating perinatal inflammation in ADHD pathogenesis through early neural circuit modulation.58 Similarly, fetal exposure to IL-8 shows significant schizophrenia risk association.59
IL-6 has been shown that its level is increased in ASD subjects and autistic mice and its concentration may be positively correlated with the severity of symptoms in ASD patients.60 IL-6 not only inhibits neurogenesis in neural stem cells (NSCs) via the JAK2/STAT3 pathway but also promotes Th17 cell differentiation, leading to IL-17 production.61 The concentration of IL-17 can be detected in the serum, cerebrospinal fluid or cerebral cortex of D children, which is significantly higher than that of the healthy control group61,62 The increase of IL-17A in ASD patients with gastrointestinal symptoms is more significant.62 Studies have found that the expression of TNF-α in the amygdala and dorsal-lateral prefrontal cortex of ASD children is increased. Serum levels of proinflammatory factors such as TNF-α are also significantly elevated in individuals with ASD compared to healthy controls,63 The risk of ADHD is increased in children of mothers with inflammation and immune system disorders64 Gustafsson’s team was the first to link inflammation to defects in brain development/behavior. It is suggested that the imbalance of maternal serum IL-6, TNF-α and MCP-1 levels may be closely related to the risk of ADHD.65
Interleukin-6 (IL-6)
Dysregulated IL-6 levels critically disrupt fetal brain development, driving neurobehavioral abnormalities in MIA-exposed offspring66 (Figure 1). Under physiological conditions, IL-6 remains minimally detectable in the brain, but its production escalates during pathological states. Severe peripheral or central inflammatory challenges—such as bacterial meningitis—trigger IL-6 accumulation in serum and cerebrospinal fluid (CSF).67 Elevated IL-6 levels are observed in the prefrontal cortex, CSF, and cerebellum of autistic children. In MIA models, maternal serum IL-6 peaks 3 hours post-challenge before returning to baseline within 24 hours.68 Strikingly, recombinant IL-6 administration alone induces prepulse inhibition (PPI) deficits and ASD/schizophrenia-like behaviors in adult offspring.69,70 Conversely, IL-6 antibody-treated or IL-6 knockout dams fail to transmit behavioral or transcriptional anomalies to progeny following immune challenge.66 IL-6 has further emerged as a prognostic biomarker for neuropsychiatric disorders including depression, schizophrenia, bipolar disorder, and ASD.71 In rodent MIA models, maternal IL-6 activates placental decidual immune cells, stimulating localized IL-6 production.72 This placental IL-6 signaling initiates pathogenic cascades by: Downregulating placental growth hormone (GH) and insulin-like growth factor 1 (IGF-1)—critical neurodevelopmental regulators linked to ASD; Inducing cerebellum-specific Purkinje cell deficits in lobule VII; Driving neuronal loss and behavioral impairments reversible via IL-6 receptor α (IL-6Rα) blockade.66 Mechanistically, maternal IL-6 traverses the placental barrier during mid-late gestation, directly stimulating fetal IL-6 synthesis and establishing feedforward inflammatory loops.73,74 It synergizes with IL-1β and TGF-β to disrupt chorionic tight junctions (TJ), compromising placental integrity.75 Placental JAK/STAT3 pathway activation—particularly in spongiotrophoblasts—upregulates proinflammatory genes while altering neurodevelopmental factor secretion.76 IL-6/JAK2/STAT3 signaling further suppresses neurogenesis via DNMT1/TET3-mediated epigenetic remodeling, with JAK2 inhibition rescuing hippocampal neurogenesis and cognitive deficits in APP/PS mice.77 In cortical neurons differentiated from induced pluripotent stem cells (iPSCs), exposure to interleukin-6 (IL-6) resulted in upregulated gene expression within the chemokine signaling pathway and Toll-like receptor (TLR) signaling pathway categories. Concurrently, IL-6-induced oxidative stress elevated basal respiration, ATP production, and non-mitochondrial respiration in differentiated neurons, suggesting a marked increase in metabolic activity and energy demand.78 Notably, Hyper-IL-6 treatment of dorsal forebrain organoids (DFOs) recapitulates MIA’s early corticogenic effects,79 activating JAK/STAT cascades and inducing transcriptional shifts mirroring human neurodevelopmental perturbations.80
 |
Figure 1 Mechanisms of IL-6–Mediated Disruption of Fetal Neurodevelopment through Placental and Neural Pathways. The schematic illustrates the cascade by which maternally derived IL-6 impairs fetal brain development via placental and direct neural mechanisms. Upper panel: Production and placental transfer of maternal IL-6. Maternal immune activation triggers release of inflammatory factors, which activate placental immune cells leading to IL-6 production. This maternal IL-6 either crosses the placental barrier directly or stimulates placental intrinsic immune cells to generate additional IL-6, which then enters fetal circulation. Lower panel: Multilevel impacts on fetal neurodevelopment. In the fetal brain, IL-6 synergizes with IL-1β and TGF-β to disrupt placental barrier integrity; activates JAK/STAT3 signaling to regulate angiogenesis and uterine growth; directly inhibits fetal neuronal production; increases neuronal basal respiration via oxidative stress; and activates microglia and other immune cells, ultimately affecting neuronal synaptic pruning. These coordinated pathways collectively contribute to neurodevelopmental abnormalities associated with maternal immune activation. |
Interleukin-17A
Interleukin-17A (IL-17A), a proinflammatory cytokine predominantly expressed by TH17 cells,81 plays pivotal roles in autoimmune disorders, inflammatory responses, tumorigenesis, and host defense against bacterial and fungal infections. Notably, IL-17A critically regulates neural function during cortical development and modulates neuroinflammatory processes. Genome-wide copy number variation (CNV) analysis in ASD patients identified IL-17A as one of the enriched genes.82 Elevated IL-17A levels were observed in the serum of children with ASD and in neutrophils isolated from ASD patients,83,84 with its concentration positively correlating with autism severity.83 These alterations were accompanied by upregulated phosphorylated nuclear factor-κB (p-NF-κB), IL-6, and NADPH oxidase 2 (NOX2)/reactive oxygen species (ROS) signaling. Murine studies confirmed that increased maternal IL-17A levels in MIA models induce elevated IL-17 receptor A (IL-17RA) expression in fetal brains, establishing IL-17A as a key pathogenic factor in ASD. IL-17A is primarily secreted by TH17 cells, and IL-6 serves as a critical factor for TH17 cell differentiation. Elevated IL-6 levels in the placenta or fetal brain following MIA may drive IL-17A upregulation. This upregulation further stimulates the production of multiple cytokines, including IL-6, IL-1β, tumor necrosis factor-α (TNF-α), granulocyte-macrophage colony-stimulating factor (GM-CSF), and granulocyte colony-stimulating factor (G-CSF). MIA induces aberrant expression of cortical layer-specific marker genes, including SATB2, TBR1, and CTIP2, in the cortical cells of offspring mice. However, offspring from MIA dams pretreated with IL-17A-blocking antibodies exhibit normal expression of these markers.85 In miR-301a knockdown mice, suppression of the SOCS3/STAT3 pathway and subsequent reduction in downstream cytokines IL-6 and IL-17A significantly ameliorate poly(I:C)-induced ASD-like behaviors, including deficits in social interaction and anxiety-related behaviors.86
IL-17A signaling influences fetal brain development through multiple mechanisms (Figure 2). Maternal IL-17A accumulates in the placenta and directly crosses the placental barrier to impact fetal development.87 Concurrently, maternal IL-17A enhances IL-17 receptor (IL-17R) expression at both mRNA and protein levels in placental trophoblasts, amplifying signal transduction.88 Elevated IL-17A does not alter placental permeability but binds to placental IL-17RA receptors and traverses the placenta.87 Evidence indicates that MIA downregulates microglial Gpr56 expression in fetal brains in an IL-17A-dependent manner, leading to parvalbumin-positive (PV+) interneuron deficits and autism-like behavioral phenotypes in offspring.89 Placental IL-17A signaling modulates fetal brain microglia; IL-17A administration activates microglia, enhancing their phagocytic activity without increasing cellular density.90 Moreover, integrated stress response (ISR) activation in MIA offspring is IL-17A-dependent and necessary for the emergence of core behavioral phenotypes. Genetic knockdown of ISR components alleviates ASD-like phenotypes in male MIA offspring, revealing a novel mechanism by which IL-17A mediates MIA effects.91 Additionally, the TRAF6/MMP9 pathway under IL-17A signaling is implicated in ASD pathophysiology. Modulation of the IL-17A/TRAF6/MMP9 axis ameliorates MIA-induced ASD-like behaviors in adulthood.92 As a gut barrier cytokine, IL-17A is critical for maintaining intestinal microbiota homeostasis. MIA exacerbates IL-17A-driven immune responses, altering gut microbial composition and inducing immune sensitization in offspring, thereby increasing susceptibility to bacterial infection-triggered intestinal inflammation.93
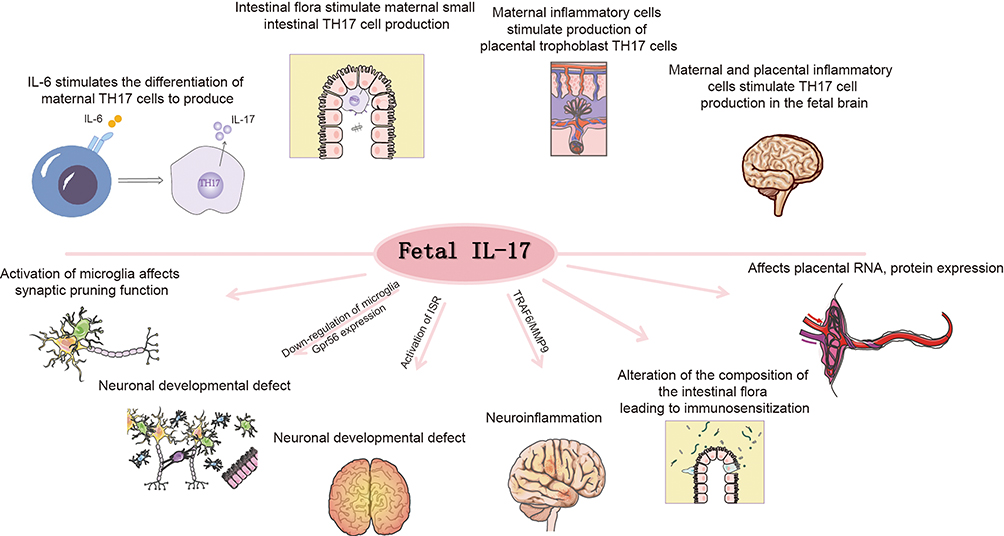 |
Figure 2 Mechanisms of IL-17A-Mediated Neurodevelopmental Impairment through Maternal-Fetal Immune Activation. This schematic illustrates the pathways through which IL-17A contributes to fetal neurodevelopmental abnormalities. Upper panel: Sources and production of IL-17A. Multiple pathways contribute to IL-17A production: IL-6 stimulates the differentiation of maternal TH17 cells; intestinal flora activate maternal small intestinal TH17 cells; maternal inflammatory cells trigger placental trophoblast TH17 cell production; and maternal-placental inflammatory cells induce TH17 cell production in the fetal brain. Lower panel: Neurodevelopmental consequences of IL-17A signaling. IL-17A exerts multiple detrimental effects including: activation of microglia leading to impaired synaptic pruning function; down-regulation of microglial Gpr56 expression causing neuronal developmental defects; induction of neuroinflammation; alteration of intestinal flora composition resulting in immune sensitization; and modulation of placental RNA and protein expression. These interconnected mechanisms collectively disrupt normal neurodevelopment and contribute to the pathogenesis of neurodevelopmental disorders. |
Interleukin-1β (IL-1β)
Interleukin-1β (IL-1β) is one of the key cytokines elevated in ASD.94 In MIA models mimicking viral infection, fetal brain IL-1β levels peaked 24 hours post-injection and continued to rise, exhibiting the highest concentration among cytokines analyzed.95 Offspring of LPS-exposed MIA dams also showed increased IL-1β mRNA levels in fetal brains.96 Multiple studies confirm that viral mimetics significantly elevate fetal brain IL-1β expression for at least 24 hours post-MIA induction.95 The association between ASD pathogenesis and IL-1 signaling, including its receptor family, has been experimentally validated,97 with IL-1β gene polymorphisms being more prevalent in children with ASD.98,99 Disrupted IL-1β signaling during early development leads to synaptic homeostasis dysregulation and ASD-like behavioral deficits,99 as neuronal function critically depends on tightly regulated IL-1β levels.100 IL-1β likely mediates MIA effects through microglial modulation: IL-1 receptor type 1 (IL-1R1) deficiency reduces microglial synaptic phagocytosis, while IL-1R1 antagonism in wild-type microglia similarly impairs synaptic engulfment capacity.99 Furthermore, IL-1β selectively enhances downstream mTOR pathway activity, influencing synaptic pruning and phagocytosis. Dysregulated IL-1β expression is implicated in the etiology of multiple neurodevelopmental and neuropsychiatric disorders.99 Maternal inflammation during pregnancy can indirectly damage placental integrity via IL-1 signaling, resulting in fetal mortality, abnormal brain development, or offspring neuropathology. Notably, IL-1 receptor antagonist administration effectively rescues maternal inflammation-induced placental and neurodevelopmental defects.21
TNF-α and IFNs
In MIA models, male offspring of dams exhibit anxiety-like and obsessive-compulsive behavioral deficits, which correlate with reduced hypothalamic concentrations of granulocyte colony-stimulating factor (G-CSF), interleukin-4 (IL-4), interleukin-7 (IL-7), interferon-gamma (IFNγ), and tumor necrosis factor-alpha (TNFα).101 Among these cytokines, TNF-α—primarily released by macrophages and microglia—modulates brain development and function.102,103 Late-gestation immune challenges specifically activate IL-10 and TNF-α responses in fetal brains, which are linked to increased postnatal apoptosis.104 Furthermore, offspring-derived TNF-α may impair fetal development by suppressing MAP2+ dendritic growth. In a human induced pluripotent stem cell (hiPSC)-derived neuron-macrophage co-culture system, MAP2+ dendritic growth was inhibited by TNF-α and IL-1α secreted by GM-CSF-primed macrophages (GM-CSFMΦ), with this suppression being more pronounced in GM-CSFMΦ from ASD individuals compared to typically developing (TD) controls.105 Inflammation also induces microglial activation in the medial prefrontal cortex (mPFC) of offspring mice, triggering proinflammatory cytokines such as IL-1α and TNF-α. This leads to dendritic shortening, reduced branching, and subsequent social avoidance behaviors.106,107
In murine and zebrafish models, type I interferons (IFN-I) promote microglial phagocytosis of neurons during cortical developmental remodeling, thereby limiting the accumulation of DNA-damaged neurons.108 Within MIA mechanisms affecting fetal brain development, IFNγ may regulate synaptic refinement. This immunomodulatory cytokine induces prolonged synaptic modifications to modulate synaptic activity while disrupting glutamate receptor clustering on dendrites.109 Cortical neurons differentiated from iPSCs and exposed to IFNγ during differentiation exhibit aberrant neurite outgrowth and altered dendritic spine density.78
Given the association between MIA and diverse neurodevelopmental disorders, developing interventions targeting inflammatory mediators is critical. For instance, blocking specific cytokines (eg, IL-17A) or their signaling pathways may effectively mitigate MIA-induced adverse effects on fetal neurodevelopment.
MIA-Associated Cellular Mechanisms in Neurodevelopmental Abnormalities
Peripheral Immune Cells
Aberrant T cell activation is closely linked to neurodevelopmental disorders. CD4+ T cell dysregulation has been observed in children with ASD, characterized by an elevated proportion of Th17 cells and reduced regulatory T cells (Tregs).110,111 The Th17/Treg ratio exhibits a significant positive correlation with ASD severity, while Treg abundance inversely correlates with symptom severity.112
Using MIA animal models, Choi et al demonstrated that maternal CD4+ T cells expressing RORγt are essential for the emergence of ASD-like phenotypes in offspring. In poly(I:C)-induced MIA offspring, CD4+ T cells exhibit enhanced differentiation into Th17 cells upon stimulation,113–115 constitutive upregulation of Toll-like receptor (TLR) expression,115, and systemic Treg deficiencies. Elevated CD4+ T cell reactivity in the spleen and mesenteric lymph nodes further reflects compromised immune homeostasis in MIA offspring.116 Bioinformatic and experimental analyses revealed that activation of the PI3K/Akt/NF-κB pathway in offspring upregulates STAT3 signaling, driving Treg/Th17 imbalance.117 Similarly, in MIA models induced by Toxoplasma gondii infection, no significant differences in T or B cell percentages were observed in offspring at 2 or 4 weeks of age. However, granulocyte counts nearly doubled by 4 weeks, and TH1/TH17 subsets expanded dramatically in T cells by 10–12 weeks, accompanied by declining Treg and TH2 populations. This T cell dysregulation exhibited time-dependent exacerbation,118 representing a major contributor to neurodevelopmental impairments. Intravenous administration of purified Tregs to MIA offspring ameliorates Th1/Th17 skewing119 and reverses behavioral deficits. Notably, Tregs isolated from MIA dams can remodel inflammatory microenvironments in offspring brains.118 Parasite-specific T lymphocytes release high interferon-gamma (IFNγ) levels, promoting Th1 effector differentiation and macrophage activation—critical for antimicrobial functions such as nitric oxide release.120
Lipopolysaccharide (LPS), a widely used MIA inducer, paradoxically increases natural regulatory CD4+Foxp3+ T cell numbers and proportions in offspring by weeks 8–10. Gene Ontology (GO) analysis identified upregulation of 18 proteins in LPS-treated CD4+ T cells, 13 of which are metabolism-associated.121 LPS-induced MIA also shifts offspring immune development toward Th1-biased responses, disrupting Th1/Th2 balance. Additionally, offspring T cells display impaired IL-10 secretion in vitro, indicating MIA-induced dysregulation of immunomodulatory cytokine production.122 In SARS-CoV-2-associated MIA offspring, CD14+ monocytes exhibit upregulated interferon-stimulated gene (ISG) expression, downregulated IFNAR2, and elevated TLR transcripts (TLR2, TLR4, TLR5) alongside increased FOS and reduced NF-κB inhibitor levels. These changes likely reflect prenatal IFN exposure. Fetal bone marrow CD14+ monocytes also show reduced antigen-presenting capacity and transcriptional signatures of B cell dysfunction.123
Beyond offspring alterations, maternal T cells critically mediate intergenerational effects. Maternal antibodies and immune cells can cross the placenta to influence fetal brain development. Post-poly(I:C) administration, MIA dams exhibit pronounced M1 macrophage polarization (placenta, spleen, peritoneum) with concomitant M2 reduction. Macrophages are essential for MIA-induced placental maldevelopment and inflammation. Depleting maternal macrophages via chlorophosphate liposomes (CL) rescues anxiety-like phenotypes and autism-associated behaviors in adult offspring.124 Helminth-derived Sjp90α protein induces Treg production of IL-10, modulating macrophage activity.125 MIA rapidly elevates maternal plasma IL-17A levels within 12–24 hours post-poly(I:C) injection at E12.5. Pre-existing Th17 cells—not de novo differentiated populations—constitute the primary IL-17A source in inflamed pregnant mice.126
Microglia
Microglia, a critical component of the central nervous system (CNS) glial population, play pivotal roles in CNS development and homeostasis maintenance.127 These cells sense neuronal activity and regulate neurodevelopment through the release of immunomodulatory factors, including phagocytosis of neural progenitor cells, guidance of dopaminergic axon extension, neuronal positioning,128 structural integrity maintenance, and selective synaptic pruning.129–131 Microglia can polarize into M1 or M2 phenotypes.132 The M1 phenotype produces pro-inflammatory cytokines (eg, IL-6, TNF-α, IL-1β) and chemokines, exerting pathogen clearance and tissue repair functions, yet excessive inflammatory mediator release may impair neuronal function. Conversely, the M2 phenotype secretes anti-inflammatory factors (eg, IL-10, TGF-β) to promote tissue repair and neuroprotection. Microglial priming—a major consequence of MIA—serves as a key mechanism underlying age-dependent neuronal dysfunction and behavioral phenotypes in offspring (Figure 3). Distinct MIA induction methods differentially alter microglial phenotypes in postnatal and adult offspring brains.133 For instance, prenatal valproic acid (VPA) exposure increases microglial soma size, while prenatal LPS exposure enhances hippocampal microglial activation in MIA offspring.134 MIA also induces microglial hyperplasia in brain regions like the hippocampus (HPC). Although MIA does not alter microglial morphology, its timing may subtly influence postnatal microglial differentiation.135 Activated microglia in MIA exhibit functional alterations, including upregulated inflammatory mediator gene expression, enhanced proliferation, phagocytic activity, and release of cytotoxic cytokines, chemokines, and reactive oxygen species (ROS),136,137 collectively disrupting fetal brain development.19
 |
Figure 3 Central Role of Microglial Dysregulation in Maternal Immune Activation-Induced Neurodevelopmental Impairments. Maternal infection-induced immune activation triggers a cascade of microglial-mediated pathological events in the fetal brain. Microglia undergo significant activation characterized by population expansion and functional transformation, serving as the central effector in neurodevelopmental disruption. The activated microglia exhibit multiple pathological manifestations: compromised blood-brain barrier integrity; enhanced phagocytic activity with aberrant synaptic pruning; increased secretion of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6); impaired myelin formation through disrupted oligodendrocyte differentiation; and mitochondrial damage via oxidative stress pathways. These microglial dysfunctions collectively drive neurodevelopmental impairments through disrupted neural circuitry, compromised white matter integrity, and sustained neuroinflammatory responses, establishing microglia as the pivotal cellular mediator linking maternal immune activation to fetal brain abnormalities. |
During development, microglia regulate neural progenitor cell (NPC) pool size by stimulating NPC proliferation138 and subsequently phagocytosing excess NPCs.139 MIA disrupts these microglia-mediated neurodevelopmental processes.128,140,141 CX3CR1 deficiency impairs microglial signaling and function by altering developmental trajectories.142 LPS- or poly(I:C)-induced MIA perturbs microglia-neuron communication in young male offspring via CX3CL1-CX3CR1 and/or CD200-CD200R signaling dysregulation, contributing to neurodevelopmental deficits.143 LPS-MIA offspring show reduced CX3CR1 mRNA expression at postnatal day 15.144 Beyond providing neurotrophic support (eg, insulin-like growth factor-1),145,146 microglia dynamically remodel synapses through phagocytosis and trogocytosis,129 modulating synaptic plasticity and strength via activity-dependent interactions.147 MIA exacerbates microglial synaptic pruning and phagocytosis,19 with dysfunctional phagocytosis implicated in ASD and schizophrenia (SCZ). Excessive NPC phagocytosis and defective synaptic pruning disrupt neuronal connectivity and brain circuitry.148 Altered microglial homeostasis impedes NPC self-renewal/differentiation, predisposing to autism-like behaviors.149 Microglia may regulate MIA-induced interneuron deficits via G protein-coupled receptor 56 (GPR56).89 Aberrant microglial synaptic elimination and autophagy underlie MIA-associated circuit phenotypes;150 mTOR inhibitor rapamycin rescues dendritic pruning defects and ASD-like behaviors in Tsc2± mice,151 while activating microglial autophagy ameliorates SCZ-related abnormalities in MIA offspring.152 Activated microglia release excess ROS,137 damaging mitochondria. Microglial immune receptors can specifically recognize mitochondrial components—such as mitochondrial DNA, transcription factor A (TFAM), and cardiolipin—released during mitochondrial dysfunction. This recognition activates pro-inflammatory processes and triggers mitochondrion-dependent inflammation,153 subsequently inducing intracellular cascades that regulate mitochondrial metabolism and function154 Microglia can help restore blood-brain barrier (BBB) permeability by limiting the penetration of peripheral factors into the CNS parenchyma. On the other hand, activated microglia increase BBB permeability and vascular leakage through the release of cytokines or reactive oxygen species, adversely affecting BBB integrity.155,156 Chronic MIA-induced inflammation promotes microglial phagocytosis of astrocytic end-feet, further compromising the BBB.157
Contrary to the prevailing view of microglial hyperactivation post-MIA, Akira Sawa et al propose that prenatal stress induces long-term microglial hyporesponsiveness. In MIA offspring, microglia exhibit sustained immune hypoactivation across development, accompanied by chromatin remodeling, altered transcription factor occupancy, and dysregulated gene expression—factors increasing schizophrenia risk. These “primed” microglia underlie ventral striatal circuit dysfunction, which prenatal microglial replacement can rescue alongside IL-1β, TNFα, and IL-6 normalization.158 MIA-induced microglial alterations represent a pathophysiological bridge linking maternal inflammation to neurodevelopmental disorder (NDD) risk, positioning microglia as novel therapeutic targets. Future research should dissect interactions among diverse cell types (T cells, microglia, astrocytes, neural stem cells) in MIA mechanisms and their collective impact on neurodevelopment.
Astrocyte, Neuron and NSC
The density of microglia in the brain (especially in the prefrontal cortex) of ASD patients is increased,159 and microglia are activated.160 The secretion of proinflammatory cytokines such as IL-1β and TNF-α is increased, while synaptic pruning is enhanced, the complement C3-CR3 pathway is activated, and the phagocytosis of dendritic spines is abnormal. In addition to microglia, multiple brain regions such as the cortex also show activation of astrocytes.160 The expression of glutamate transporter GLT-1 in astrocytes is reduced, resulting in glutamate accumulation in synaptic cleft and subsequent excitotoxicity. May be closely related to the occurrence of ASD,161 ASD patients can show brain endothelial cell damage and dysfunction,162 increased permeability of the blood-brain barrier, and inflammatory cytokines can also cross the barrier to activate microglia. There are abnormalities in the number and function of neuronal synapses in the brain of patients with ASD, and the connection mode and strength between neurons may also change.163,164 At the same time, the number of microglia in the brain increases, and the activation of microglia triggers a variety of inflammatory cytokines and chemokines, and the generation of a large number of reactive oxygen species, which affects neurogenesis and synapse formation. ADHD patients have decreased expression of histamine H2 receptors on PV neurons in the substantia nigra pars reticulata (SNr), resulting in hyperactivity of dopaminergic neurons.,165 ADHD patients show higher temporal variability and lower spatial stability in neural responses in the salience and frontoparietal network regions.166 Mental retardation (ID) and fragile X syndrome (FXS) also show abnormalities in neuronal number and dendritic morphology, and synaptic development. Maternal immune activation (MIA) can alter the regulation of protein translation and phosphorylation of microtubule-associated protein 2 (MAP2) in neural stem cells, leading to defects in synaptic connectivity during neuronal maturation. It can also affect the neuromaturation and function of offspring166 and induce abnormal synaptic development and pruning during neurodevelopment, destroying the excitation-inhibition balance of neurons (such as hippocampal neurons).167 Maternal immune activation (MIA) has also been shown to interfere with the development and behavior of offspring microglia, thereby disrupting the coordinated neurodevelopment process of microglia,168 MIA can also damage the integrity of the blood-cerebrospinal fluid barrier,169 change the composition of cerebrospinal fluid, inhibit the concentration of neurotrophins such as insulin-like growth factor-I (IGF-I),169 and destroy the brain microenvironment for neurodevelopment. It is noteworthy that maternal immune stimulation of the fetal immune system leads to increased levels of inflammatory cytokines in the offspring brain.170 Cytokines such as IL-6 and IL-17A can interfere with the neural development program in the embryonic period and early postnatal period, and have a significant impact on the proliferation, differentiation, migration and survival of neural stem cells. It has been shown that H-IL-6 promotes the differentiation of neural stem cells (NSCs) into glutamate-responsive neurons, oligodendrocytes, and distinct glial subtypes, an effect that is highly dependent on the presence of the soluble IL-6 receptor (sIL-6R).171 Using a brain organoid model, Hyper-IL-6 treatment slightly increases the proportion of radial glial cells, which exhibit the most pronounced transcriptional alterations, including downregulation of protein translation–related genes.80 Notably, even transient increases in systemic IL-6 can trigger short-term proliferation followed by lasting reductions in adult NSC pools, suggesting that IL-6 signaling is essential for NSC maintenance but that its dysregulation may contribute to neurodevelopmental and psychiatric disorders.172 IL-17 activation in neural stem cells (NSCs) leads to a notable reduction in cell numbers and impaired neurosphere formation, without causing cytotoxicity or apoptosis. While IL-17 inhibits NSC proliferation, it also disrupts their differentiation into astrocytes and oligodendrocyte precursor cells (OPCs). These effects are partially mitigated by blocking the p38 MAPK pathway.173 A recent study examined the impact of MIA using neural stem cells (NSCs), which demonstrated significant changes in protein expression and phosphorylation, particularly in proteins involved in neuronal function. Transcriptomic, proteomic, and phosphoproteomic analyses revealed alterations in proteins related to cytoskeletal dynamics and synaptic activity. Of particular note, changes in the phosphorylation of MAP family proteins, especially MAP2, were found to influence neuronal plasticity and synapse formation. These findings underscore the critical role of NSCs in studying the molecular mechanisms of MIA and highlight the importance of MAP2 phosphorylation in regulating neuronal structure and function.174
Potential Mechanism of MIA-Induced Neurodevelopmental Disorders: Gut Microbiota Dysbiosis
The gut microbiota serves as a critical modulator of core symptoms in various neurological disorders,175 exerting profound effects on the central nervous system (CNS). The gut-brain axis—a bidirectional communication network involving neurons, hormones, immune cells, and microbial metabolites—mediates intimate crosstalk between the gut and brain.176,177 Through this axis, gut microbes regulate host behaviors associated with neurodevelopmental dysfunction.178 MIA compromises intestinal integrity. Offspring from poly(I:C)-induced MIA dams exhibit reduced expression of zonula occludens-1 (ZO-1), a scaffold protein critical for tight junction (TJ) stabilization, in adult small and large intestines. Bacteroides fragilis supplementation restores intestinal permeability and upregulates genes governing gut barrier integrity in MIA offspring,179 while also promoting stem cell regeneration and enhancing mucus secretion.180 MIA alters maternal gut microbiota composition and function, inducing dysbiosis that may impact maternal-fetal health through multiple pathways. Prenatal valproic acid (VPA) exposure induces ASD-like behaviors in offspring,181 accompanied by gut microbial compositional shifts. Lactobacillus reuteri treatment rescues social deficits in ASD mice via vagus nerve-dependent mechanisms, restoring social interaction-induced synaptic plasticity in the ventral tegmental area.177
Elevated IL-17A levels during MIA are influenced by maternal gut microbiota. The presence of segmented filamentous bacteria (SFB) in the maternal microbiome drives increased maternal serum IL-17A. In SFB-deficient dams, even under immune challenge, IL-17A remains unchanged, and offspring behavioral abnormalities are absent.182 Preemptive eradication of vancomycin-sensitive SFB prior to MIA induction prevents offspring behavioral deficits.182 Accumulating evidence highlights the maternal and offspring microbiomes as key regulators of microglial development, homeostasis, and MIA-associated behavioral outcomes. Adult germ-free mice or those with microbiota complexity limitations exhibit global microglial defects and impaired innate immune responses.183 Prenatally, maternal Lactobacillus murinus deficiency disrupts embryonic microglial transcriptomes, alters brain microglial colonization (eg, increased density and hyper-ramification), and elevates chromatin accessibility.184,185 Microbiota-targeted interventions in MIA offspring partially ameliorate gut alterations, stereotyped behaviors, anxiety-like phenotypes, and sensorimotor gating deficits.186
While the gut microbiome modulates neurological outcomes via the microbiota-gut-brain axis, mechanisms underlying microbial rescue of neuronal dysfunction remain incompletely understood. Clinical interventions targeting gut microbiota to ameliorate neurobehavioral disorders represent a promising frontier.
Alterations in Synaptogenesis and Pruning
Synaptogenesis and synaptic pruning are critical processes during brain development, governing the formation of new synaptic connections and the refinement of neuronal networks. Impairments in synaptic structure and function are central to neurodevelopmental disorders such as ASD and attention-deficit/hyperactivity disorder (ADHD).
MIA disrupts synaptic development, maturation, and pruning through multiple pathways, including inflammatory cytokine-mediated interference with signaling cascades and microglial activation. Maternal inflammatory signals traverse the placental barrier, perturbing neurodevelopmental pathways in offspring. For instance, IL-6 inhibits neurogenesis via JAK2/STAT3 signaling,77 while offspring-derived TNF-α suppresses MAP2+ dendritic growth, impairing synaptic development.105 Poly(I:C)-induced MIA significantly reduces expression of presynaptic (eg, synaptophysin) and postsynaptic (eg, PSD95) proteins in offspring hippocampi, compromising basal synaptic transmission (field potential [FV] and excitatory postsynaptic potential [fEPSP]) and plasticity (paired-pulse facilitation [PPF] and long-term potentiation [LTP]).187 Additionally, MIA downregulates Shank3 expression, disrupting neuronal connectivity, synaptogenesis, and dendritic spine maturation.
Beyond synaptogenesis, aberrant synaptic pruning profoundly impacts neurodevelopment.188 In male offspring of LPS-induced MIA dams, dendritic density in dentate gyrus granule cells remains elevated during peak pruning stages (postnatal day 15),144 a phenotype linked to persistent network hyperconnectivity and ASD-like behaviors.189 IL-17A signaling enhances microglial phagocytic activity in fetal brains,90 while poly(I:C)-exposed MIA offspring exhibit upregulated hippocampal IL-1β expression in adulthood.144 IL-1β selectively amplifies mTOR pathway activity, further dysregulating synaptic pruning and phagocytosis.99
MIA disrupts fetal synaptogenesis and pruning primarily via microglial activation. Targeted interventions to modulate these pathways may mitigate long-term neurodevelopmental risks in offspring.
Other Mechanisms
MIA induces neurodevelopmental disorders through multiple mechanisms, including interference with critical signaling pathways, disruption of the blood-brain barrier (BBB), placental dysfunction, and activation of the hypothalamic-pituitary-adrenal (HPA) axis. The crosstalk between inflammatory and neurodevelopmental signaling pathways constitutes a complex regulatory network. MIA disrupts fetal neurodevelopmental processes by impairing key pathways such as Notch,190 NF-κB,117 and JAK/STAT,191 which modulate the expression of neurodevelopmental genes (eg, BDNF, GSK-3β, and components of Notch/Wnt pathways). These perturbations inhibit neural stem cell proliferation/differentiation and neuronal migration/synaptogenesis, ultimately altering cortical circuit architecture and function.192
Notably, MIA increases BBB permeability, compromising its structural integrity and allowing immune cells, cytokines, and neurotoxic molecules to infiltrate the brain parenchyma, thereby amplifying neuroinflammation. The placenta serves as a physical and immunological interface during MIA. Animal studies demonstrate that maternal inflammation transmits to the fetus via both direct (eg, altered mRNA expression of placental amino acid transporters like leucine transporters) and indirect mechanisms, disrupting fetal brain growth trajectories.193
Mechanistically, MIA dysregulates the neuroendocrine system through HPA axis hyperactivation—a hallmark of ADHD pathophysiology. Chronic stress-induced HPA dysfunction elevates corticotropin and cortisol levels, Promote catabolic states via lipolysis/gluconeogenesis. Inhibit neuronal growth and dendritic arborization; Impair synaptic plasticity and neurocognitive functions. This endocrine-metabolic cascade exacerbates both ADHD symptoms and metabolic disturbances.58
Summary and Perspectives
Accumulating evidence underscores the pivotal role of MIA in neurodevelopmental disorders (NDDs). However, MIA lacks standardized diagnostic criteria and is broadly defined as prenatal exposure to infection, aberrant inflammation, or immune dysregulation. The interplay of environmental, genetic, and immunological factors complicates precise mechanistic delineation. While animal models and epidemiological studies have established cytokine-driven immune responses as central to MIA-induced neuropathology, the developmental mechanisms underlying human MIA-associated NDD phenotypes remain incompletely resolved.
Accumulating evidence underscores the pivotal role of Maternal Immune Activation (MIA) as a key environmental risk factor for neurodevelopmental disorders (NDDs). The core pathogenesis hinges on a maternal immune response triggered by infection or inflammation during pregnancy, leading to a surge in pro-inflammatory cytokines such as IL-6, IL-17A, IL-1β, and TNF-α. These cytokines disrupt fetal brain developmental programs directly or indirectly via the placental interface. IL-6 has been identified as a critical initiator, propagating downstream pathology by impairing placental function, altering epigenetic regulation via the JAK/STAT pathway, and directly suppressing neurogenesis. IL-17A, primarily derived from maternal Th17 cells, can not only cross the placenta but also bind to IL-17RA receptors in the fetal brain, provoking microglial dysregulation and the Integrated Stress Response (ISR), which ultimately disrupts cortical neuron differentiation and interneuron function. In tandem, cytokines like IL-1β and TNF-α act synergistically to compromise neural circuit formation and plasticity by modulating microglial phagocytosis and synaptic pruning, as well as directly impairing neuronal dendrite development. This collaborative interplay among cytokines forms the immunopathological foundation of MIA-induced neurodevelopmental damage, culminating in the core behavioral phenotypes of NDDs such as ASD and ADHD.
Substantial research gaps remain in this field. Current mechanistic studies have primarily concentrated on a limited repertoire of cytokines and microglial responses, while paying inadequate attention to other significant pathogens like Toxoplasma gondii and crucial cellular components such as vascular endothelial cells. Moreover, the long-term pathophysiological processes through which maternal immune activation persistently disrupts neuronal circuitry—particularly the specific interactions between primed microglia and interneurons—are not yet fully elucidated. A more comprehensive investigation into the complex interplay between environmental triggers like MIA and individual genetic susceptibilities is essential to unravel the determinants of phenotypic heterogeneity in NDDs.
Successful MIA modeling holds translational potential for developing therapeutic or preventive strategies against NDDs. Leveraging diverse MIA models enables dynamic tracking of disease progression and symptom-specific biomarker quantification, facilitating early identification of high-risk populations. A deeper understanding of MIA mechanisms—particularly immune-neuroinflammatory crosstalk—will inform targeted interventions. Strategies modulating immune pathways (eg, cytokine blockade, microglial replacement) represent promising therapeutic avenues, warranting validation in preclinical models. Given the central role of maternal immune activation (MIA) in neurodevelopmental disorders (NDDs), intervention strategies targeting its associated immune signaling pathways have become a major research focus. At the pharmacological level, studies indicate that bumetanide can ameliorate autism-like behaviors through immuno-behavioral covariation mechanisms by modulating the expression of inflammatory factors such as IFN-γ.194 In more targeted receptor blockade strategies, animal studies have demonstrated that specifically blocking the signaling transduction of key cytokines like IL-17A85,195 or IFN-γ169 with antibodies significantly alleviates abnormal behavioral phenotypes in MIA offspring models.
Microglia play a central role in MIA-induced neurodevelopmental disorders, and microglia replacement therapy has emerged as a promising strategy with significant advances in treating neurological diseases. Compelling evidence from multiple disease models demonstrates the therapeutic potential of this approach. In CSF1R-related leukoencephalopathy (ALSP), researchers successfully replaced pathogenic microglia in the central nervous system, effectively blocking disease progression in animal models.196 This strategy also showed promising results in a small-scale clinical trial involving eight patients, where it stabilized both motor and cognitive functions over a two-year follow-up period.196 In Sandhoff disease models, two independent studies utilizing bone marrow transplantation and direct intracerebral injection of in vitro-cultured cells respectively confirmed the efficacy of microglia replacement. These interventions not only significantly improved neurological function but also remarkably extended the lifespan of treated mice from 135 to 250 days.197,198 Furthermore, in Krabbe disease models, perinatal microglia replacement completely normalized transcriptional signatures, rescued histopathological abnormalities, and achieved a two-fold extension in average survival.199 These collective findings strongly support the broad applicability of microglia replacement strategies across various neurological disease model.
However, the field still faces important challenges. Studies have found that early microglia depletion may exacerbate seizure severity in adulthood, and transplanted cells from different sources (such as monocyte-derived versus authentic microglia) exhibit distinct functional characteristics in neural circuits. RNA sequencing revealed that the developmental origin of cells and the transplantation process significantly influence their response to excitotoxic brain environments.200 Additionally, traditional bone marrow transplantation methods suffer from low efficiency and require radiation preconditioning, while the novel “tricyclic microglial depletion for transplantation (TCMDT)” strategy enables efficient transplantation without preconditioning through CSF1R inhibitors,201 demonstrating therapeutic potential in multiple disease models.
In conclusion, interventional strategies for neurodevelopmental disorders are continuously evolving, ranging from pharmacological modulation and antibody blockade to innovative cell replacement therapies. Future research should focus on optimizing cell source selection, improving replacement safety and efficiency, and deeply exploring the interaction mechanisms between transplanted cells and neural circuits, thereby advancing the clinical management of neurodevelopmental disorders into a new era of precision medicine.
This review has systematically elucidated the multi-level mechanisms through which MIA contributes to NDDs, encompassing inflammatory cytokine cascades at the maternal-fetal interface, microglia-mediated synaptic pruning deficits, gut-brain axis dysregulation, and Th17/Treg immune deviations. Evidence strongly supports the crucial role of specific cytokines including IL-6, IL-17A, and IL-1β in disrupting neural circuit formation through various signaling pathways. Current intervention strategies have evolved from pharmacological modulation to more targeted approaches, with antibody blockade of specific cytokine pathways and emerging microglia replacement therapy showing particular promise for resetting the neuroimmune environment.
Looking forward, several research priorities demand urgent attention. The intricate interactions between genetic susceptibility and environmental factors in determining NDD phenotypes require deeper investigation through integrated multi-omics approaches. Standardized protocols for cell replacement therapies need development to address critical challenges in transplantation safety and efficiency. Furthermore, the translation of immune-based interventions necessitates careful consideration of temporal windows, target specificity, and individual patient characteristics. The evolving landscape of NDD interventions—spanning immunomodulation, targeted blockade, and cellular replacement—heralds a new era in precision medicine for neurodevelopmental disorders. Future research integrating fundamental mechanistic insights with innovative therapeutic approaches will ultimately enable more effective prevention and treatment strategies for these complex conditions.
Acknowledgments
Research on application of brain organoids in neurodevelopmental disorders in Kun Xia lab is supported by MOE Key laboratory of Rare Pediatric Diseases; National Natural Science Foundation of China (82130043, 82330035 and 82361138573 to K.X., 82471198 to L.X.), Natural Science Foundation of Guangdong Province, China (2022A1515111190 to T.Z.).
Disclosure
The authors declare no competing interests in this work.
References
1. Schaffer LS, Breunig S, Lawrence JM, Foote IF, Grotzinger AD. Characterizing genetic pathways unique to autism spectrum disorder at multiple levels of biological analysis. Mol Autism. 2024;15(1):46. doi:10.1186/s13229-024-00624-2
2. Tick B, Bolton P, Happé F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry. 2016;57(5):585–595. doi:10.1111/jcpp.12499
3. Bai D, Yip BHK, Windham GC, et al. Association of genetic and environmental factors with autism in a 5-country cohort. JAMA psychiatry. 2019;76(10):1035–1043. doi:10.1001/jamapsychiatry.2019.1411
4. Minakova E, Warner BB. Maternal immune activation, central nervous system development and behavioral phenotypes. Birth Defects Res. 2018;110(20):1539–1550. doi:10.1002/bdr2.1416
5. Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, Ginty DD. Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell. 2016;166(2):299–313. doi:10.1016/j.cell.2016.05.033
6. Gardner Z, Holbrook O, Tian Y, Odamah K, Man HY. The role of glia in the dysregulation of neuronal spinogenesis in Ube3a-dependent ASD. Exp Neurol. 2024;376:114756. doi:10.1016/j.expneurol.2024.114756
7. Wang Z, Lin X, Luo X, et al. Pleiotropic association of CACNA1C variants with neuropsychiatric disorders. Schizophr Bull. 2023;49(5):1174–1184. doi:10.1093/schbul/sbad073
8. Peça J, Feliciano C, Ting JT, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472(7344):437–442. doi:10.1038/nature09965
9. Sun F, Chen Y, Gao Q, Zhao Z. Abnormal gray matter structure in children and adolescents with high-functioning autism spectrum disorder. Psychiatry Res Neuroimaging. 2022;327:111564. doi:10.1016/j.pscychresns.2022.111564
10. Norman LJ, Carlisi C, Lukito S, et al. Structural and functional brain abnormalities in attention-deficit/hyperactivity disorder and obsessive-compulsive disorder: a comparative meta-analysis. JAMA psychiatry. 2016;73(8):815–825. doi:10.1001/jamapsychiatry.2016.0700
11. Castellanos FX, Proal E. Large-scale brain systems in ADHD: beyond the prefrontal-striatal model. Trends Cogn Sci. 2012;16(1):17–26. doi:10.1016/j.tics.2011.11.007
12. Greven CU, Bralten J, Mennes M, et al. Developmentally stable whole-brain volume reductions and developmentally sensitive caudate and putamen volume alterations in those with attention-deficit/hyperactivity disorder and their unaffected siblings. JAMA psychiatry. 2015;72(5):490–499. doi:10.1001/jamapsychiatry.2014.3162
13. Xiong Y, Chen J, Li Y. Microglia and astrocytes underlie neuroinflammation and synaptic susceptibility in autism spectrum disorder. Front Neurosci. 2023;17:1125428. doi:10.3389/fnins.2023.1125428
14. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167(3):261–280. doi:10.1176/appi.ajp.2009.09030361
15. Bauman MD, Van de Water J. Translational opportunities in the prenatal immune environment: promises and limitations of the maternal immune activation model. Neurobiol Dis. 2020;141. doi:10.1016/j.nbd.2020.104864
16. Meyer U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry. 2014;75(4):307–315. doi:10.1016/j.biopsych.2013.07.011
17. Han VX, Patel S, Jones HF, Dale RC. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat Rev Neurol. 2021;17(9):564–579. doi:10.1038/s41582-021-00530-8
18. Knuesel I, Chicha L, Britschgi M, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol. 2014;10(11):643–660. doi:10.1038/nrneurol.2014.187
19. Guma E, Bordeleau M, González Ibáñez F, et al. Differential effects of early or late exposure to prenatal maternal immune activation on mouse embryonic neurodevelopment. Proc Natl Acad Sci U S A. 2022;119(12):e2114545119. doi:10.1073/pnas.2114545119
20. van Rooij D, Anagnostou E, Arango C, et al. Cortical and subcortical brain morphometry differences between patients with autism spectrum disorder and healthy individuals across the lifespan: results from the ENIGMA ASD working group. Am J Psychiatry. 2018;175(4):359–369. doi:10.1176/appi.ajp.2017.17010100
21. Girard S, Tremblay L, Lepage M, Sébire G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J Immunol. 2010;184(7):3997–4005. doi:10.4049/jimmunol.0903349
22. Kendell RE, Kemp IW. Maternal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. 1989;46(10). doi:10.1001/archpsyc.1989.01810100020004
23. C S. Autism in children with congenital rubella. J Autism Child Schizophr. 1971;1(1). doi:10.1007/BF01537741
24. B M, A E, B B, E WW, M PB. Obstetric conditions and risk of first admission with schizophrenia: a Danish national register based study. Schizophr Res. 2007;97(1–3). doi:10.1016/j.schres.2007.07.018
25. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167(3). doi:10.1176/appi.ajp.2009.09030361
26. Mortensen PB, Nørgaard-Pedersen B, Waltoft BL, et al. Toxoplasma gondii as a risk factor for early-onset schizophrenia: analysis of filter paper blood samples obtained at birth. Biol Psychiatry. 2007;61(5). doi:10.1016/j.biopsych.2006.05.024
27. Mortensen PB, Pedersen CB, Hougaard DM, et al. A Danish National Birth Cohort study of maternal HSV-2 antibodies as a risk factor for schizophrenia in their offspring. Schizophr Res. 2010;122(1–3). doi:10.1016/j.schres.2010.06.010
28. Sørensen HJ, Mortensen EL, Reinisch JM, Mednick SA. Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophr Bull. 2009;35(3). doi:10.1093/schbul/sbn121
29. Clarke MC, Tanskanen A, Huttunen M, Whittaker JC, Cannon M. Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. Am J Psychiatry. 2009;166(9):1025–30. doi:10.1176/appi.ajp.2009.08010031
30. Buka SL, Cannon TD, Torrey EF, Yolken RH. Maternal exposure to herpes simplex virus and risk of psychosis among adult offspring. Biol Psychiatry. 2008;63(8). doi:10.1016/j.biopsych.2007.09.022
31. Z O, I AM, D L, W C, I H-P. Month of conception and risk of autism. Epidemiology. 2011;22(4). doi:10.1097/EDE.0b013e31821d0b53
32. Zerbo O, Qian Y, Yoshida C, Grether JK, Van de Water J, Croen LA. Maternal infection during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2015;45(12):4015–4025. doi:10.1007/s10803-013-2016-3
33. Zerbo O, Qian Y, Yoshida C, Fireman BH, Klein NP, Croen LA. Association between influenza infection and vaccination during pregnancy and risk of autism spectrum disorder. JAMA Pediatr. 2017;171(1):e163609. doi:10.1001/jamapediatrics.2016.3609
34. Mahic M, Mjaaland S, Bøvelstad HM, et al. Maternal immunoreactivity to herpes simplex virus 2 and risk of autism spectrum disorder in male offspring. mSphere. 2017;2(1). doi:10.1128/mSphere.00016-17
35. Atladóttir HO, Thorsen P, Østergaard L, et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010;40(12). doi:10.1007/s10803-010-1006-y
36. P S, M A, D RC, et al. Social impairments in autism spectrum disorder are related to maternal immune history profile. Mol Psychiatry. 2018;23(8). doi:10.1038/mp.2017.201
37. Croen LA, Grether JK, Yoshida CK, Odouli R, Van de Water J. Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: a case-control study. Arch Pediatr Adolesc Med. 2005;159(2). doi:10.1001/archpedi.159.2.151
38. Dashraath P, Wong JL, Lim MX, et al. Coronavirus disease 2019 (COVID-19) pandemic and pregnancy. Am J Obstet Gynecol. 2020;222(6). doi:10.1016/j.ajog.2020.03.021
39. Duan L, Yin H, Liu J, et al. Maternal COVID-19 infection associated with offspring neurodevelopmental disorders. Mol Psychiatry. 2025;30(5):2108–2118. doi:10.1038/s41380-024-02822-z
40. Kim DH, Croen LA, Iosif AM, et al. The association of maternal COVID-19-infection during pregnancy on the neonatal immune profile and associations with later diagnosis of neurodevelopmental disorders. Brain Behav Immun. 2025;123:1071–1080. doi:10.1016/j.bbi.2024.11.014
41. Ye W, Luo C, Zhou J, et al. Association between maternal diabetes and neurodevelopmental outcomes in children: a systematic review and meta-analysis of 202 observational studies comprising 56·1 million pregnancies. Lancet Diabetes Endocrinol. 2025;13(6):494–504. doi:10.1016/s2213-8587(25)00036-1
42. Njotto LL, Simin J, Fornes R, et al. Maternal and early-life exposure to antibiotics and the risk of autism and attention-deficit hyperactivity disorder in childhood: a swedish population-based cohort study. Drug Safety. 2023;46(5):467–478. doi:10.1007/s40264-023-01297-1
43. Kentner AC, Bilbo SD, Brown AS, et al. Maternal immune activation: reporting guidelines to improve the rigor, reproducibility, and transparency of the model. Neuropsychopharmacology. 2019;44(2):245–58. doi:10.1038/s41386-018-0185-7
44. Bergdolt L, Dunaevsky A. Brain changes in a maternal immune activation model of neurodevelopmental brain disorders. Prog Neurobiol. 2019;175:1–19. doi:10.1016/j.pneurobio.2018.12.002
45. Amodeo DA, Lai CY, Hassan O, Mukamel EA, Behrens MM, Powell SB. Maternal immune activation impairs cognitive flexibility and alters transcription in frontal cortex. Neurobiol Dis. 2019;125:211–218. doi:10.1016/j.nbd.2019.01.025
46. O’Loughlin E, Pakan JMP, Yilmazer-Hanke D, McDermott KW. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J Neuroinflammation. 2017;14(1):212. doi:10.1186/s12974-017-0981-8
47. da Rosa N, de Medeiros FD, de Oliveira J, et al. 6-shogaol exerts a neuroprotective factor in offspring after maternal immune activation in rats. Dev Neurosci. 2022;44(1):13–22. doi:10.1159/000519992
48. Ryan AM, Bauman MD. Primate models as a translational tool for understanding prenatal origins of neurodevelopmental disorders associated with maternal infection. Biol Psychiatry Cogn Neurosci Neuroimaging. 2022;7(5):510–523. doi:10.1016/j.bpsc.2022.02.012
49. Bauman MD, Iosif AM, Smith SE, Bregere C, Amaral DG, Patterson PH. Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol Psychiatry. 2014;75(4):332–341. doi:10.1016/j.biopsych.2013.06.025
50. Gilmore JH, Jarskog LF. Exposure to infection and brain development: cytokines in the pathogenesis of schizophrenia. Schizophr Res. 1997;24(3):365–367. doi:10.1016/s0920-9964(96)00123-5
51. Wang R, Wu Z, Huang C, Hashimoto K, Yang L, Yang C. Deleterious effects of nervous system in the offspring following maternal SARS-CoV-2 infection during the COVID-19 pandemic. Transl Psychiatry. 2022;12(1):232. doi:10.1038/s41398-022-01985-z
52. Meltzer A, Van de Water J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology. 2017;42(1):284–298. doi:10.1038/npp.2016.158
53. Meyer U. Neurodevelopmental resilience and susceptibility to maternal immune activation. Trends Neurosci. 2019;42(11):793–806. doi:10.1016/j.tins.2019.08.001
54. Than UTT, Nguyen LT, Nguyen PH, et al. Inflammatory mediators drive neuroinflammation in autism spectrum disorder and cerebral palsy. Sci Rep. 2023;13(1):22587. doi:10.1038/s41598-023-49902-8
55. Croonenberghs J, Bosmans E, Deboutte D, Kenis G, Maes M. Activation of the inflammatory response system in autism. Neuropsychobiology. 2002;45(1):1–6. doi:10.1159/000048665
56. Tamayo JM, Rose D, Church JS, Schwartzer JJ, Ashwood P. Maternal allergic asthma induces prenatal neuroinflammation. Brain Sci. 2022;12(8):1041. doi:10.3390/brainsci12081041
57. Masi A, Breen EJ, Alvares GA, et al. Cytokine levels and associations with symptom severity in male and female children with autism spectrum disorder. Mol Autism. 2017;8:63. doi:10.1186/s13229-017-0176-2
58. Takahashi N, Nishimura T, Harada T, et al. Interaction of genetic liability for attention deficit hyperactivity disorder (ADHD) and perinatal inflammation contributes to ADHD symptoms in children. Brain Behav Immun. 2023;30:100630. doi:10.1016/j.bbih.2023.100630
59. Ellman LM, Deicken RF, Vinogradov S, et al. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr Res. 2010;121(1–3):46–54. doi:10.1016/j.schres.2010.05.014
60. Nadeem A, Ahmad SF, Attia SM, Al-Ayadhi LY, Al-Harbi NO, Bakheet SA. Dysregulation in IL-6 receptors is associated with upregulated IL-17A related signaling in CD4+ T cells of children with autism. Prog Neuropsychopharmacol Biol Psychiatry. 2020;97:109783. doi:10.1016/j.pnpbp.2019.109783
61. Ullah MA, Revez JA, Loh Z, et al. Allergen-induced IL-6 trans-signaling activates γδ T cells to promote type 2 and type 17 airway inflammation. J Allergy Clin Immunol. 2015;136(4):1065–1073. doi:10.1016/j.jaci.2015.02.032
62. Butera A, De Simone R, Potenza RL, et al. Effects of a gut-selective integrin-targeted therapy in male mice exposed to early immune activation, a model for the study of autism spectrum disorder. Brain Behav Immun. 2024;115:89–100. doi:10.1016/j.bbi.2023.09.024
63. Tsilioni I, Patel AB, Pantazopoulos H, et al. IL-37 is increased in brains of children with autism spectrum disorder and inhibits human microglia stimulated by neurotensin. Proc Natl Acad Sci U S A. 2019;116(43):21659–21665. doi:10.1073/pnas.1906817116
64. Instanes JT, Halmøy A, Engeland A, Haavik J, Furu K, Klungsøyr K. Attention-deficit/hyperactivity disorder in offspring of mothers with inflammatory and immune system diseases. Biol Psychiatry. 2017;81(5):452–459. doi:10.1016/j.biopsych.2015.11.024
65. Gustafsson HC, Sullivan EL, Battison EAJ, et al. Evaluation of maternal inflammation as a marker of future offspring ADHD symptoms: a prospective investigation. Brain Behav Immun. 2020;89:350–356. doi:10.1016/j.bbi.2020.07.019
66. Wu WL, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun. 2017;62. doi:10.1016/j.bbi.2016.11.007
67. Kępa L, Oczko-Grzesik B, Boroń-Kaczmarska A. Cerebrospinal fluid interleukin-6 concentration in patients with purulent, bacterial meningitis - own observations. Przegl Epidemiol. 2014;68(4):645–649.
68. Murray KN, Edye ME, Manca M, et al. Evolution of a maternal immune activation (mIA) model in rats: early developmental effects. Brain Behav Immun. 2019;75:48–59. doi:10.1016/j.bbi.2018.09.005
69. Liang ZK, Xiong W, Wang C, et al. Resolving neuroinflammatory and social deficits in ASD model mice: dexmedetomidine downregulates NF-κB/IL-6 pathway via α2AR. Brain Behav Immun. 2024;119:84–95. doi:10.1016/j.bbi.2024.03.040
70. Smith SE, Li J, Garbett K, Mirnics K, Patterson P. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27(40). doi:10.1523/JNEUROSCI.2178-07.2007
71. García-Juárez M, Camacho-Morales A. Defining the role of anti- and pro-inflammatory outcomes of interleukin-6 in mental health. Neuroscience. 2022. 492:32–46.doi:10.1016/j.neuroscience.2022.03.020
72. Osman HC, Moreno R, Rose D, Rowland ME, Ciernia AV, Ashwood P. Impact of maternal immune activation and sex on placental and fetal brain cytokine and gene expression profiles in a preclinical model of neurodevelopmental disorders. J Neuroinflammation. 2024;21(1):118.doi:10.1186/s12974-024-03106-7
73. D J, S AM, J T, H A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res. 2006;60(2). doi:10.1203/01.pdr.0000230026.74139.18
74. Zaretsky MV, Alexander JM, Byrd W, Bawdon RE. Transfer of inflammatory cytokines across the placenta. Obstetr Gynecol. 2004;103(3). doi:10.1097/01.AOG.0000114980.40445.83
75. Tossetta G, Paolinelli F, Avellini C, et al. IL-1β and TGF-β weaken the placental barrier through destruction of tight junctions: an in vivo and in vitro study. Placenta. 2014;35(7). doi:10.1016/j.placenta.2014.03.016
76. Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25(4). doi:10.1016/j.bbi.2010.12.017
77. K X, G Z, Z L, et al. JAK2/STAT3 signaling mediates IL-6-inhibited neurogenesis of neural stem cells through DNA demethylation/methylation. Brain Behav Immun. 2019;79. doi:10.1016/j.bbi.2019.01.027
78. K A, K -L-L, R JL, K R. Distinct effects of interleukin-6 and interferon-γ on differentiating human cortical neurons. Brain Behav Immun. 2022;103. doi:10.1016/j.bbi.2022.04.007
79. Velasco S, Kedaigle AJ, Simmons SK, et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 2019;570(7762):523–527. doi:10.1038/s41586-019-1289-x
80. Sarieva K, Kagermeier T, Khakipoor S, et al. Human brain organoid model of maternal immune activation identifies radial glia cells as selectively vulnerable. Mol Psychiatry. 2023;28(12):5077–5089. doi:10.1038/s41380-023-01997-1
81. Rosine N, Miceli-Richard C. Innate cells: the alternative source of IL-17 in axial and peripheral spondyloarthritis? Front Immunol. 2020;11:553742. doi:10.3389/fimmu.2020.553742
82. vdZ B, F L, P M, et al. Gene-network analysis identifies susceptibility genes related to glycobiology in autism. PLoS One. 2009;4(5). doi:10.1371/journal.pone.0005324
83. Al-Ayadhi LY, Mostafa GA. Elevated serum levels of interleukin-17A in children with autism. J Neuroinflammation. 2012;9(1). doi:10.1186/1742-2094-9-158
84. S K, M H, I K, et al. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PLoS One. 2011;6(5). doi:10.1371/journal.pone.0020470
85. Choi GB, Yim YS, Wong H, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351(6276):933–939. doi:10.1126/science.aad0314
86. L X, F Q, Z M, et al. Quantitative proteomics of the miR-301a/SOCS3/STAT3 axis reveals underlying autism and anxiety-like behavior. Mol Ther Nucleic Acids. 2024;35(1). doi:10.1016/j.omtn.2024.102136
87. K H. Visualization of maternal IL-17a across the placental membrane. Sci Prog. 2023;106(3). doi:10.1177/00368504231195500
88. Lee JY, Kim H. IL-17A cytokine-regulated Glut1 expression in placenta cells. Curr Issues Mol Biol. 2024;46(7). doi:10.3390/cimb46070438
89. Yu D, Li T, Delpech JC, et al. Microglial GPR56 is the molecular target of maternal immune activation-induced parvalbumin-positive interneuron deficits. Sci Adv. 2022;8(18):eabm2545. doi:10.1126/sciadv.abm2545
90. S T, T S, T Y. Intraventricular IL-17A administration activates microglia and alters their localization in the mouse embryo cerebral cortex. Mol Brain. 2020;13(1). doi:10.1186/s13041-020-00635-z
91. Kalish BT, Kim E, Finander B, et al. Maternal immune activation in mice disrupts proteostasis in the fetal brain. Nat Neurosci. 2021;24(2):204–213. doi:10.1038/s41593-020-00762-9
92. F L, Z X, J Y, et al. Yigansan ameliorates maternal immune activation-induced autism-like behaviours by regulating the IL-17A/TRAF6/MMP9 pathway: network analysis and experimental validation. Phytomedicine. 2024;128. doi:10.1016/j.phymed.2024.155386
93. Kim E, Paik D, Ramirez RN, et al. Maternal gut bacteria drive intestinal inflammation in offspring with neurodevelopmental disorders by altering the chromatin landscape of CD4(+) T cells. Immunity. 2022;55(1):145–158.e7. doi:10.1016/j.immuni.2021.11.005
94. Estes ML, McAllister AK. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat Revi Neurosci. 2015;16(8). doi:10.1038/nrn3978
95. Arrode-Brusés G, Brusés JL. Maternal immune activation by poly I:C induces expression of cytokines IL-1β and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. J Neuroinflammation. 2012;9:83. doi:10.1186/1742-2094-9-83
96. B P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun. 2010;24(6). doi:10.1016/j.bbi.2010.03.005
97. O’Roak BJ, Deriziotis P, Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6). doi:10.1038/ng.835
98. S K, A AM, AA A-R, et al. Polymorphism of interleukin-1β and interleukin-1 receptor antagonist genes in children with autism spectrum disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2020;103. doi:10.1016/j.pnpbp.2020.109999
99. B A, M C, D G, et al. Loss of interleukin 1 signaling causes impairment of microglia- mediated synapse elimination and autistic-like behaviour in mice. Brain Behav Immun. 2024;117. doi:10.1016/j.bbi.2024.01.221
100. K A, V E, N Y, et al. Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1 beta on long term potentiation in rat dentate gyrus. J Biol Chem. 2003;278(21). doi:10.1074/jbc.M301938200
101. Church JS, Tamayo JM, Ashwood P, Schwartzer JJ. Repeated allergic asthma in early versus late pregnancy differentially impacts offspring brain and behavior development. Brain Behav Immun. 2021;93. doi:10.1016/j.bbi.2020.12.014
102. G R, L K, F B. Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2000;20(1). doi:10.1097/00004647-200001000-00009
103. T D, K L, S EH, S JG, T PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117(2). doi:10.1016/j.pharmthera.2007.10.001
104. M U, N M, E A, et al. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26(18). doi:10.1523/JNEUROSCI.0099-06.2006
105. T R, T M, Y T, et al. Granulocyte macrophage colony-stimulating factor-induced macrophages of individuals with autism spectrum disorder adversely affect neuronal dendrites through the secretion of pro-inflammatory cytokines. Mol Autism. 2024;15(1). doi:10.1186/s13229-024-00589-2
106. McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79(1):16–29.doi:10.1016/j.neuron.2013.06.028
107. N X, K S, T K, et al. The innate immune receptors TLR2/4 mediate repeated social defeat stress-induced social avoidance through prefrontal microglial activation. Neuron. 2018;99(3). doi:10.1016/j.neuron.2018.06.035
108. Escoubas CC, Dorman LC, Nguyen PT, et al. Type-I-interferon-responsive microglia shape cortical development and behavior. Cell. 2024;187(8). doi:10.1016/j.cell.2024.02.020
109. Vikman KS, Owe-Larsson B, Brask J, Kristensson KS, Hill RH. Interferon-gamma-induced changes in synaptic activity and AMPA receptor clustering in hippocampal cultures. Brain Res. 2001;896(1–2). doi:10.1016/s0006-8993(00)03238-8
110. Ahmad SF, Zoheir KM, Ansari MA, et al. Dysregulation of Th1, Th2, Th17, and T regulatory cell-related transcription factor signaling in children with autism. Mol Neurobiol. 2017;54(6). doi:10.1007/s12035-016-9977-0
111. Mostafa GA, Al Shehab A, Fouad NR. Frequency of CD4+CD25high regulatory T cells in the peripheral blood of Egyptian children with autism. J Child Neurol. 2010;25(3). doi:10.1177/0883073809339393
112. Moaaz M, Youssry S, Elfatatry A, Abd El Rahman M. Th17/Treg cells imbalance and their related cytokines (IL-17, IL-10 and TGF-β) in children with autism spectrum disorder. J Neuroimmunol. 2019;337. doi:10.1016/j.jneuroim.2019.577071
113. M M, M AC, D R, P NM. Preferential development of Th17 cells in offspring of immunostimulated pregnant mice. J Reproduct Immunol. 2010;87(1–2). doi:10.1016/j.jri.2010.06.156
114. M M, M AC, D R, P NM. Maternal immune stimulation during pregnancy affects adaptive immunity in offspring to promote development of TH17 cells. Brain Behav Immun. 2011;25(5). doi:10.1016/j.bbi.2010.09.011
115. Ponzio NM, Mandal M, Elkabes S, et al. Pro-inflammatory phenotype induced by maternal immune stimulation during pregnancy. In: Recent Advances in Autism Spectrum Disorders - Volume I. Intechopen; 2013. doi:10.5772/53990
116. Hsiao EY, McBride SW, Chow J, Mazmanian M, Patterson P. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc Natl Acad Sci U S A. 2012;109(31). doi:10.1073/pnas.1202556109
117. L Y, Y G, W R, et al. Maternal immune activation mediated prenatal chronic stress induces Th17/Treg cell imbalance may relate to the PI3K/Akt/NF-κB signaling pathway in offspring rats. Int Immunopharmacol. 2024;126. doi:10.1016/j.intimp.2023.111308
118. X Z, Z X, C H, et al. Rescue of maternal immune activation-induced behavioral abnormalities in adult mouse offspring by pathogen-activated maternal Treg cells. Nat Neurosci. 2021;24(6). doi:10.1038/s41593-021-00837-1
119. Xu Z, Zhang X, Chang H, et al. Rescue of maternal immune activation-induced behavioral abnormalities in adult mouse offspring by pathogen-activated maternal T(reg) cells. Nat Neurosci. 2021;24(6):818–830. doi:10.1038/s41593-021-00837-1
120. Denkers EY, Gazzinelli RT. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev. 1998;11(4):569–588. doi:10.1128/cmr.11.4.569
121. L R, C H, L L, et al. Maternal lipopolysaccharide exposure promotes immunological functional changes in adult offspring CD4+ T cells. Am J Reprod Immunol. 2015;73(6). doi:10.1111/aji.12364
122. Z A, P ML, F-d-P V, R A, J P-N. Increased cell-mediated immunity in male mice offspring exposed to maternal immune activation during late gestation. Int Immunopharmacol. 2013;17(3). doi:10.1016/j.intimp.2013.08.007
123. Matute JD, Finander B, Pepin D, et al. Single-cell immunophenotyping of the fetal immune response to maternal SARS-CoV-2 infection in late gestation. Pediatr Res. 2022;91(5). doi:10.1038/s41390-021-01793-z
124. Shen C, Zhu X, Chang H, et al. The rebalancing of the immune system at the maternal-fetal interface ameliorates autism-like behavior in adult offspring. Cell Rep. 2024;43(10):114787. doi:10.1016/j.celrep.2024.114787
125. S C, Z X, C H, et al. The rebalancing of the immune system at the maternal-fetal interface ameliorates autism-like behavior in adult offspring. Cell Rep. 2024;43(10). doi:10.1016/j.celrep.2024.114787
126. Kim S, Kim H, Yim YS, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549(7673):528–532. doi:10.1038/nature23910
127. C M, B O. Microglia function in the central nervous system during health and neurodegeneration. Ann Rev Immunol. 2017;35(1). doi:10.1146/annurev-immunol-051116-052358
128. Squarzoni P, Oller G, Hoeffel G, et al. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014;8(5):1271–1279. doi:10.1016/j.celrep.2014.07.042
129. Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi:10.1126/science.1202529
130. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi:10.1016/j.neuron.2012.03.026
131. Gunner G, Cheadle L, Johnson KM, et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci. 2019;22(7):1075–1088. doi:10.1038/s41593-019-0419-y
132. Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76(2):77–98. doi:10.1016/j.pneurobio.2005.06.004
133. Smolders S, Notter T, Smolders SMT, Rigo JM, Brône B. Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav Immun. 2018;73:51–65. doi:10.1016/j.bbi.2018.06.001
134. Luo L, Chen J, Wu Q, et al. Prenatally VPA exposure is likely to cause autistic-like behavior in the rats offspring via TREM2 down-regulation to affect the microglial activation and synapse alterations. Envir Toxicol Pharmacol. 2023;99:104090. doi:10.1016/j.etap.2023.104090
135. Ozaki K, Kato D, Ikegami A, et al. Maternal immune activation induces sustained changes in fetal microglia motility. Sci Rep. 2020;10(1):21378. doi:10.1038/s41598-020-78294-2
136. G H, E D, M A, P R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J Neuroimmunol. 2014;274(1–2). doi:10.1016/j.jneuroim.2014.07.012
137. Sousa NA, Oliveira GAL, de Oliveira AP, et al. Novel ocellatin peptides mitigate lps-induced ros formation and NF-kB activation in microglia and hippocampal neurons. Sci Rep. 2020;10(1):2696. doi:10.1038/s41598-020-59665-1
138. Antony JM, Paquin A, Nutt SL, Kaplan DR, Miller FD. Endogenous microglia regulate development of embryonic cortical precursor cells. J Neurosci Res. 2011;89(3). doi:10.1002/jnr.22533
139. Cunningham CL, Martínez-Cerdeño V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33(10). doi:10.1523/JNEUROSCI.3441-12.2013
140. Cunningham CL, Martínez-Cerdeño V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33(10):4216–4233. doi:10.1523/jneurosci.3441-12.2013
141. Thion MS, Mosser CA, Férézou I, et al. Biphasic impact of prenatal inflammation and macrophage depletion on the wiring of neocortical inhibitory circuits. Cell Rep. 2019;28(5):1119–1126.e4. doi:10.1016/j.celrep.2019.06.086
142. Matcovitch-Natan O, Winter DR, Giladi A, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353(6301). doi:10.1126/science.aad8670
143. Chamera K, Kotarska K, Szuster-Głuszczak M, et al. The prenatal challenge with lipopolysaccharide and polyinosinic:polycytidylic acid disrupts CX3CL1-CX3CR1 and CD200-CD200R signalling in the brains of male rat offspring: a link to schizophrenia-like behaviours. J Neuroinflammation. 2020;17(1):247. doi:10.1186/s12974-020-01923-0
144. FdC L, G A, vdV S, L GN. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav Immun. 2017;63. doi:10.1016/j.bbi.2016.09.028
145. Ueno M, Fujita Y, Tanaka T, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16(5):543–551. doi:10.1038/nn.3358
146. Fujita Y, Yamashita T. Neuroprotective function of microglia in the developing brain. Neuronal Signal. 2021;5(1):Ns20200024. doi:10.1042/ns20200024
147. C J, S S, H HY, Z M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen Res. 2022;17(4). doi:10.4103/1673-5374.322423
148. Yu CJ, Wang M, Li RY, et al. TREM2 and Microglia Contribute to the Synaptic Plasticity: from Physiology to Pathology. Mol Neurobiol. 2023;60(2):512–523. doi:10.1007/s12035-022-03100-1
149. S L, Z M, J F, et al. Microglia homeostasis mediated by epigenetic ARID1A regulates neural progenitor cells response and leads to autism-like behaviors. Mol Psychiatry. 2024;29(6). doi:10.1038/s41380-022-01703-7
150. Kim HJ, Cho MH, Shim WH, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry. 2017;22(11):1576–1584. doi:10.1038/mp.2016.103
151. T G, G K, K SH, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83(5). doi:10.1016/j.neuron.2014.07.040
152. S X, W G, L S, et al. Autophagy defects at weaning impair complement-dependent synaptic pruning and induce behavior deficits. J Neuroinflammation. 2024;21(1). doi:10.1186/s12974-024-03235-z
153. Newman LE, Shadel GS. Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J Cell Biol. 2018;217(10):3327–3329. doi:10.1083/jcb.201808118
154. Morris G, Maes M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Birth Defects Res. 2014;29(1):19–36. doi:10.1007/s11011-013-9435-x
155. Dudvarski Stankovic N, Teodorczyk M, Ploen R, Zipp F, Schmidt MHH. Microglia-blood vessel interactions: a double-edged sword in brain pathologies. Acta Neuropathol. 2016;131(3):347–363. doi:10.1007/s00401-015-1524-y
156. Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun. 2017;60:1–12. doi:10.1016/j.bbi.2016.03.010
157. Haruwaka K, Ikegami A, Tachibana Y, et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun. 2019;10(1):5816. doi:10.1038/s41467-019-13812-z
158. Hayes LN, An K, Carloni E, et al. Prenatal immune stress blunts microglia reactivity, impairing neurocircuitry. Nature. 2022;610(7931):327–334. doi:10.1038/s41586-022-05274-z
159. Morgan JT, Chana G, Pardo CA, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry. 2010;68(4):368–376. doi:10.1016/j.biopsych.2010.05.024
160. Suzuki K, Sugihara G, Ouchi Y, et al. Microglial activation in young adults with autism spectrum disorder. JAMA psychiatry. 2013;70(1):49–58. doi:10.1001/jamapsychiatry.2013.272
161. Bristot Silvestrin R, Bambini-Junior V, Galland F, et al. Animal model of autism induced by prenatal exposure to valproate: altered glutamate metabolism in the hippocampus. Brain Res. 2013;1495:52–60. doi:10.1016/j.brainres.2012.11.048
162. Ouellette J, Toussay X, Comin CH, et al. Vascular contributions to 16p11.2 deletion autism syndrome modeled in mice. Nat Neurosci. 2020;23(9):1090–1101. doi:10.1038/s41593-020-0663-1
163. Russo FB, Freitas BC, Pignatari GC, et al. Modeling the Interplay Between Neurons and Astrocytes in Autism Using Human Induced Pluripotent Stem Cells. Biol Psychiatry. 2018;83(7):569–578. doi:10.1016/j.biopsych.2017.09.021
164. Casey S, Carter M, Looney AM, et al. Maternal mid-gestation cytokine dysregulation in mothers of children with autism spectrum disorder. J Autism Dev Disord. 2022;52(9):3919–3932. doi:10.1007/s10803-021-05271-7
165. An D, You Y, Ma Q, et al. Deficiency of histamine H(2) receptors in parvalbumin-positive neurons leads to hyperactivity, impulsivity, and impaired attention. Neuron. 2025;113(4):572–589.e6. doi:10.1016/j.neuron.2024.12.002
166. Gao Z, Duberg K, Warren SL, et al. Reduced temporal and spatial stability of neural activity patterns predict cognitive control deficits in children with ADHD. Nat Commun. 2025;16(1):2346. doi:10.1038/s41467-025-57685-x
167. Galván EJ, Zepeda A. The impact of maternal immune activation on the morphology and electrophysiological properties of postnatally-born neurons in the offspring. Neural Regen Res. 2024;19(2):399–400. doi:10.4103/1673-5374.379043
168. Zengeler KE, Lukens JR. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat Rev Immunol. 2021;21(7):454–468. doi:10.1038/s41577-020-00487-7
169. Sun YQ, Huang XX, Guo W, et al. IFN-γ signaling links ventriculomegaly to choroid plexus and ependyma dysfunction following maternal immune activation. J Neuroinflammation. 2025;22(1):83. doi:10.1186/s12974-025-03409-3
170. Collins A, Swann JW, Proven MA, et al. Maternal inflammation regulates fetal emergency myelopoiesis. Cell. 2024;187(6):1402–1421.e21. doi:10.1016/j.cell.2024.02.002
171. Islam O, Gong X, Rose-John S, Heese K. Interleukin-6 and neural stem cells: more than gliogenesis. Mol Biol Cell. 2009;20(1):188–199. doi:10.1091/mbc.e08-05-0463
172. Storer MA, Gallagher D, Fatt MP, Simonetta JV, Kaplan DR, Miller FD. Interleukin-6 Regulates Adult Neural Stem Cell Numbers during Normal and Abnormal Post-natal Development. Stem Cell Rep. 2018;10(5):1464–1480. doi:10.1016/j.stemcr.2018.03.008
173. Li Z, Li K, Zhu L, et al. Inhibitory effect of IL-17 on neural stem cell proliferation and neural cell differentiation. BMC Immunol. 2013;14:20. doi:10.1186/1471-2172-14-20
174. Martin-Guerrero SM, Martin-Estebane M, Lara Ordonez AJ, et al. Maternal immune activation imprints translational dysregulation and differential MAP2 phosphorylation in descendant neural stem cells. Mol Psychiatry. 2025;30(7):2994–3007. doi:10.1038/s41380-025-02905-5
175. Xiao L, Zhang F, Zhao F. Large-scale microbiome data integration enables robust biomarker identification. Nat Comput Sci. 2022;2(5):307–316. doi:10.1038/s43588-022-00247-8
176. Xiao L, Zhou T, Zuo Z, Sun N, Zhao F. Spatiotemporal patterns of the pregnancy microbiome and links to reproductive disorders. Sci Bull. 2024;69(9):1275–1285. doi:10.1016/j.scib.2024.02.001
177. Sgritta M, Dooling SW, Buffington SA, et al. Mechanisms underlying microbial-mediated changes in social behavior in mouse models of autism spectrum disorder. Neuron. 2019;101(2):246–259.e6. doi:10.1016/j.neuron.2018.11.018
178. Buffington SA, Dooling SW, Sgritta M, et al. Dissecting the contribution of host genetics and the microbiome in complex behaviors. Cell. 2021;184(7):1740–1756.e16. doi:10.1016/j.cell.2021.02.009
179. Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451–1463. doi:10.1016/j.cell.2013.11.024
180. Zhou Q, Shen B, Huang R, et al. Bacteroides fragilis strain ZY-312 promotes intestinal barrier integrity via upregulating the STAT3 pathway in a radiation-induced intestinal injury mouse model. Front Nutr. 2022;9:1063699. doi:10.3389/fnut.2022.1063699
181. Kim JW, Seung H, Kim KC, et al. Agmatine rescues autistic behaviors in the valproic acid-induced animal model of autism. Neuropharmacology. 2017;113(Pt A):71–81. doi:10.1016/j.neuropharm.2016.09.014
182. K S, K H, Y YS, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549(7673). doi:10.1038/nature23910
183. E D, HdA AL, J D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7). doi:10.1038/nn.4030
184. Thion MS, Low D, Silvin A, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. 2018;172(3):500–516.e16. doi:10.1016/j.cell.2017.11.042
185. Erny D, Angelis A, Prinz M. Communicating systems in the body: how microbiota and microglia cooperate. Immunology. 2016;150(1):7–15. doi:10.1111/imm.12645
186. Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7). doi:10.1016/j.cell.2013.11.024
187. A O-N, O S, S I, M T, S T. Maternal immune activation by polyriboinosinic-polyribocytidilic acid injection produces synaptic dysfunction but not neuronal loss in the hippocampus of juvenile rat offspring. Brain Res. 2010;1363. doi:10.1016/j.brainres.2010.09.054
188. W J, Z J, C X, et al. Microglial over-pruning of synapses during development in autism-associated SCN2A-deficient mice and human cerebral organoids. Mol Psychiatry. 2024;29(8). doi:10.1038/s41380-024-02518-4
189. Z Y, P RC, S F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17(3). doi:10.1038/nn.3641
190. Baines KJ, Hillier DM, Haddad FL, Rajakumar N, Schmid S, Renaud SJ. Maternal Immune Activation Alters Fetal Brain Development and Enhances Proliferation of Neural Precursor Cells in Rats. Front Immunol. 2020;11. doi:10.3389/fimmu.2020.01145
191. Li X, Fu Q, Zhong M, et al. Quantitative proteomics of the miR-301a/SOCS3/STAT3 axis reveals underlying autism and anxiety-like behavior. Mol Ther Nucleic Acids. 2024;35(1):102136. doi:10.1016/j.omtn.2024.102136
192. Lu J, Fan X, Lu L, et al. Limosilactobacillus reuteri normalizes blood-brain barrier dysfunction and neurodevelopment deficits associated with prenatal exposure to lipopolysaccharide. Gut Microbes. 2023;15(1):2178800. doi:10.1080/19490976.2023.2178800
193. Osman HC, Moreno R, Rose D, Rowland ME, Ciernia AV, Ashwood P. Impact of maternal immune activation and sex on placental and fetal brain cytokine and gene expression profiles in a preclinical model of neurodevelopmental disorders. J Neuroinflammation. 2024;21(1):118. doi:10.1186/s12974-024-03106-7
194. Li Q, Zhang L, Shan H, et al. The immuno-behavioural covariation associated with the treatment response to bumetanide in young children with autism spectrum disorder. Transl Psychiatry. 2022;12(1):228. doi:10.1038/s41398-022-01987-x
195. Zhang W, Mou Z, Zhong Q, et al. Transcutaneous auricular vagus nerve stimulation improves social deficits through the inhibition of IL-17a signaling in a mouse model of autism. Front Psychiatry. 2024;15:1393549. doi:10.3389/fpsyt.2024.1393549
196. Wu J, Wang Y, Li X, et al. Microglia replacement halts the progression of microgliopathy in mice and humans. Science. 2025;389(6756):eadr1015. doi:10.1126/science.adr1015
197. Frosch M, Shimizu T, Wogram E, et al. Microglia-neuron crosstalk through Hex-GM2-MGL2 maintains brain homeostasis. Nature. 2025;646(8086):913–924. doi:10.1038/s41586-025-09477-y
198. Mader MM, Scavetti A, Yoo Y, Chai AT, Uenaka T, Wernig M. Therapeutic genetic restoration through allogeneic brain microglia replacement. Nature. 2025;646(8086):903–912. doi:10.1038/s41586-025-09461-6
199. Aisenberg WH, O’Brien CA, Sangster M, et al. Direct microglia replacement reveals pathologic and therapeutic contributions of brain macrophages to a monogenic neurological disease. Immunity. 2025;58(5):1254–1268e9. doi:10.1016/j.immuni.2025.03.019
200. O’Brien CA, Delcy SAS, Shukla S, et al. Neonatal microglia replacement in mice modulates seizure severity in adulthood. Mol Ther. 2025. doi:10.1016/j.ymthe.2025.08.039
201. Chen D, Wang C, Chen X, et al. Brain-wide microglia replacement using a nonconditioning strategy ameliorates pathology in mouse models of neurological disorders. Sci Transl Med. 2025;17(796):eads6111. doi:10.1126/scitranslmed.ads6111
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.