Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Marketed nonsteroidal anti-inflammatory agents, antihypertensives, and human immunodeficiency virus protease inhibitors: as-yet-unused weapons of the oncologists’ arsenal
Authors Papanagnou P, Baltopoulos P, Tsironi M
Received 2 February 2015
Accepted for publication 25 March 2015
Published 18 May 2015 Volume 2015:11 Pages 807—819
DOI https://doi.org/10.2147/TCRM.S82049
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Panagiota Papanagnou,1 Panagiotis Baltopoulos,2 Maria Tsironi1
1Department of Nursing, Faculty of Human Movement and Quality of Life Sciences, University of Peloponnese, Sparta, 2Department of Sports Medicine and Biology of Physical Activity, Faculty of Physical Education and Sport Science, National and Kapodistrian University of Athens, Athens, Greece
Abstract: Experimental data indicate that several pharmacological agents that have long been used for the management of various diseases unrelated to cancer exhibit profound in vitro and in vivo anticancer activity. This is of major clinical importance, since it would possibly aid in reassessing the therapeutic use of currently used agents for which clinicians already have experience. Further, this would obviate the time-consuming process required for the development and the approval of novel antineoplastic drugs. Herein, both pre-clinical and clinical data concerning the antineoplastic function of distinct commercially available pharmacological agents that are not currently used in the field of oncology, ie, nonsteroidal anti-inflammatory drugs, antihypertensive agents, and anti-human immunodeficiency virus agents inhibiting viral protease, are reviewed. The aim is to provide integrated information regarding not only the molecular basis of the antitumor function of these agents but also the applicability of the reevaluation of their therapeutic range in the clinical setting.
Keywords: repositioning, tumorigenesis, pleiotropy, exploitation
Introduction
Research advances have largely “molecularized” medicine and other life sciences, not only at the theoretical but also at the practical level.1 Inevitably, pharmacology has been transformed into molecular pharmacology, a basic medical science that continuously surprises researchers with data being accrued daily opening novel perspectives. Consistent with this, numerous pharmacological agents that are already available in the market and have gained approval for the management of diseases other than neoplasia are being characterized as potent anticancer compounds.2,3 Therefore, the possible expansion of the therapeutic uses of already prescribed pharmaceuticals could be harnessed in the field of cancer therapeutics, in order to save time and money from bench to bedside. Moreover, this would be advantageous over launching newly characterized agents, due to the preexisting clinical experience. From a theoretical point of view, however, this also highlights functional pleiotropy as a prominent feature in intracellular signaling routes and their components.4–6
This review aims to present the current knowledge regarding the anticancer function of certain non-antineoplastic agents gathered from pre-clinical and clinical experimentation. These include nonsteroidal anti-inflammatory drugs (NSAIDs), human immunodeficiency virus (HIV) agents falling into the category of protease inhibitors (PIs), and finally different types of antihypertensive drugs. Although many other non-antineoplastic agents available in the market are also known to exhibit anticancer properties,7–9 the aforementioned pharmaceuticals were chosen for many purposes: first, NSAIDs have attracted appreciable scientific interest as potent anticancer agents.10,11 Second, antihypertensive and anti-HIV medication is primarily prescribed to elderly people and to HIV patients, respectively, with both of these population categories being at high risk of developing neoplasia. In fact, cancer is considered an age-related pathology,12,13 while HIV patients commonly develop such tumors as Kaposi’s sarcoma and non-Hodgkin’s lymphoma.14,15 Considerations for using and repositioning them are also presented here in an attempt to compel the reassessment of the clinical utility of the aforementioned agents. This could possibly enable the better management of tumorigenesis by virtue of novel therapeutic schemes that are less toxic and more efficacious than conventional chemotherapy.
NSAIDs
Current clinical use
NSAIDs are commonly prescribed pharmacological agents that principally serve as selective or non-selective inhibitors of cyclooxygenase (COX)-mediated pathway(s).16,17 Prostaglandins are the main COX-derived pro-inflammatory, tumor-promoting eicosanoids that act via binding to their cognate G protein-coupled receptors, most importantly EP1–EP4.
The most widely known and the oldest NSAID in use is aspirin. Depending on its concentration, low (≤100 mg/day) or high, aspirin inhibits COX-1 and both COX-1/2, respectively, thereby blocking the formation of prostanoids. COX-2, however, can be selectively targeted by a specific class of NSAIDs including celecoxib and etoricoxib, the so-called coxibs. Etodolac and meloxicam also display selectivity toward COX-2, whereas NSAIDs, such as sulindac and ibuprofen, mediate non-selective COX-1/2 blockage.17,18 Currently, the main therapeutic indication of NSAIDs is for the management of different types of acute or chronic pain and inflammation.16,19,20 In addition, such NSAIDs as aspirin and sulindac find application in the prevention of thrombosis and preeclampsia21,22 or as antipyretic and tocolytic agents.23,24 This further emphasizes the versatility of NSAIDs as therapeutics aside their potency as antitumor drugs, as presented in the following section.
The possible therapeutic repositioning of NSAIDs: mechanistic basis, pre-clinical, clinical, and epidemiological data
Although NSAIDs have been approved for the treatment of the aforementioned pathological conditions (ie, inflammation and pain), the existence of a mechanistic link among inflammation/COX signaling and tumorigenesis and pre-clinical data points to the future harnessing of NSAIDs in oncology.16 More importantly, the ample clinical as well as epidemiological evidence that is presented in the next paragraphs encourages the use of COX inhibitors in tumor therapeutics or even in the prevention of tumorigenesis.
In fact, it is well established that chronic inflammation and aberrant COX-2 expression is causally linked to cancer,25 while NSAID-mediated inhibition of COX-1/2 is known to exert protective effects against the development of several malignancies, especially the gastrointestinal ones.26,27 Additionally, the pro-tumorigenic function of prostaglandin E2 (PGE2) and its receptors EP1–EP4 has been firmly demonstrated in animal models of colon carcinogenesis.28–30 Intriguingly, in colon cancer cell lines, experimental data suggest crosstalk between PGE2- and β-catenin-dependent pathways, strongly arguing for a mechanistic interconnection among the APC- and the PGE2-driven carcinogenesis. Apart from colon cancer, the COX-2/PGE2/EP1–EP4 signaling axis, fostering angiogenesis, tumor growth and metastasis, involving both tumor and stromal cells, has been incriminated in multiple types of solid tumors, such as non-small cell lung cancer (NSCLC), head and neck squamous cell carcinoma, and breast cancer.31–34
The COX-2 inhibitor etodolac has shown remarkable anti-metastatic function in animal models by virtue of its ability to downregulate the expression of matrix metalloproteinase (MMP)-9 as well as to interfere with the formation and the tone of lymphatic vessels.35–37 The COX-2 selective inhibitor meloxicam has been shown to trigger apoptosis in both COX-2-dependent and COX-2-independent routes in hepatocellular carcinoma cells.38 Similarly, meloxicam can act either in a COX-2-dependent or COX-2-independent fashion in osteosarcoma at multiple levels.39
The chemopreventive role of the coxibs against the emergence, malignant progression, or recurrence of colorectal polyps has been clinically confirmed, and clinical data regarding combinational schemes of COX-2 inhibitors with conventional chemotherapy for the treatment of various solid tumors are quite encouraging. In fact, APC mutation-positive familial adenomatous polyposis (FAP) patients receiving COX-2 inhibitors experience a marked numerical reduction and shrinkage of adenomas, thereby being less likely to develop colorectal cancer.25,40 Similar data for marketed NSAIDs that non-selectively inhibit COX-1/2, such as aspirin and ibuprofen, point to the chemopreventive role of these agents in smoking-induced lung cancer and skin cancer.41,42
Interestingly, the chemopreventive and/or antitumor therapeutic potency of non-selective COX inhibitors, such as aspirin, sulindac, and ibuprofen, may go beyond the realm of COX regulation. This notion is supported by in vitro experimentation, epidemiological data, or data from clinical studies.43–48 Aspirin or other NSAIDs results in the reduction of the risk of developing prostate cancer, as well as the risk of high-grade prostate cancer in men, irrespectively of the concurrent use of the antiandrogen agent dutasteride. This was evidenced by a study in which there were enrolled subjects with cancer-negative biopsies prior to initiation of the experimentation.49 Moreover, aspirin may not only serve as an adjuvant agent in FAP patients who have been subjected to prophylactic colectomy but also in patients suffering from Lynch syndrome. The latter are highly prone to develop malignancies in the gastrointestinal tract, as well as in other organs. In fact, Lynch syndrome patients who are chronic users of aspirin (>10 years) are significantly protected from developing cancers related to their genetic disorder.50 Hopefully, there is evidence that long-term usage of sulindac (≥5 months) can also act in a prophylactic manner, even in FAP patients who have not been colectomized for prophylactic purposes.51 Several ongoing clinical trials (eg, NCT01187901 and NCT00468910) will shed more light on this issue. Of note, there have been designed phosphoderivatives of various NSAIDs that display marked anticancer function.52–54 However, the antitumor potency of these agents is not discussed here, because these agents are not commercially available, at least so far.
Considerations for using and repositioning NSAIDs
Although coxibs seem promising anticancer agents, gastrointestinal side effects (ranging from irritation of the gastrointestinal mucosa to gastrointestinal bleeding), renal toxicity, and cardiovascular complications (thrombotic events, myocardial infraction, and ischemic episodes) raised serious concerns about the safety of this class of drugs, which is still a point of controversy, culminating in the withdrawal of distinct coxibs from the market.43,55–61 Consequently, there is much skepticism regarding the applicability of coxibs in the field of oncology.
Theoretically, cancer patients would suffer more than non-cancer subjects from the unwanted effects of these NSAIDs, given the hypercoagulable state associated with malignancy, as well as the cardiotoxicity of some anticancer drugs that they might receive.62,63 Hypersensitivity reactions to NSAIDs and NSAID-induced central nervous system toxicity are additional issues of concern,64,65 given that commonly used chemotherapeutic drugs may also be “provocative” to the immune system and induce hypersensitivity reactions or they may exhibit neurotoxicity.66,67
On the other hand, a severe limitation of the potential usage of NSAIDs in the field of oncology is that their antiplatelet function might increase the risk of hemorrhage.68 Notably, there are tumors such as gastrointestinal stromal tumors, which are commonly associated with gastrointestinal bleeding.69 In addition, one should also consider the probability of intratumoral hemorrhage. Certain types of tumors, eg, intracranial tumors, are not associated with a high risk for intratumoral bleeding, at least spontaneously.70,71 In any case, given that bleeding is a severe, life-threatening complication, ongoing and future clinical trials evaluating the antitumor function of NSAIDs in histologically different types of malignancies are necessary to address this issue.
Another issue is that non-selective COX inhibitors exemplified by aspirin can cause gastrointestinal irritation. Therefore, these drugs should cautiously be administered in subjects suffering from peptic ulcers. However, in the latter case, these unwanted effects can be satisfactorily managed by drugs protecting the digestive tract mucosa, such as the proton pump inhibitor omeprazole.72 Another promising option to avoid toxicity is nanoparticle formulation, something that allows successful usage of much lower concentrations, at least in the case of some NSAIDs.73 Using NSAIDs in the field of oncology without causing major health problems is a big future challenge.
Drugs blocking the renin–angiotensin system
Current clinical use
Agents targeting the renin–angiotensin system (RAS) are common active ingredients of combination drugs used for the management of hypertension and other health issues that may arise for hypertensive individuals, such as proteinuria.74,75 They are typically combined with a thiazide diuretic or even both with a thiazide and a calcium channel blocker into a single formulation. The vasoconstrictory effects of angiotensin II are mediated by the angiotensin II type 1 receptor (AT1R).76 Consequently, pharmacological agents targeting RAS interfere either with AT1R-dependent signaling or with the cleavage of angiotensin I into angiotensin II. Antihypertensives acting as AT1R blockers are termed sartans, whereas ACE inhibitors (ACEIs) prevent the formation of angiotensin II.
But why is the pharmacological control of RAS so important? The answer lies in the physiological significance of this system. RAS is a signaling circuit that critically modulates the volume of extracellular fluids and arterial blood pressure. It is a hormonal route that principally relies on the sensory function of the juxtaglomerular cells in kidneys, which secrete the aspartyl protease renin into the bloodstream in response to various stimuli, including decreased blood pressure or signals deriving from the sympathetic nervous system. Consequently, renin converts angiotensinogen into angiotensin I. The latter is eventually cleaved into the vasoactive peptide angiotensin II via the ACE.76,77 More importantly, the RAS is molecularly linked to the pathogenesis of cancer, as referred to in the following section.
Possible therapeutic repositioning of drugs blocking the renin–angiotensin system
Overview
The theoretical cornerstone of redirecting agents that block the RAS into tumor therapeutics is the fact that the RAS has been incriminated in carcinogenesis. In addition, angiotensin II exerts pleiotropic cellular effects. It acts not only as a vasoactive peptide but also as a powerful mitogenic and as an angiogenic agent.78,79 In fact, angiotensin II receptors are commonly expressed in human cancers,80–82 while AT1R blockage gains ground as a novel antitumor approach.83,84 Moreover, it is well-documented that ACEI-receiving hypertensive patients are somehow protected from developing cancer, and clinical data also support the beneficial effects of sartans in the performance status of hormone-refractory prostate cancer patients.85,86
Taking into account that some sartans exhibit antiangiogenic activity, as is discussed in the following sections, such a future application would possibly help in obviating the necessity of administering antiangiogenic drugs, such as anti-VEGF agents, which may exacerbate hypertension.87 Since hypertension and cancer commonly coexist in aged subjects, the therapeutic reevaluation of sartans and ACEIs would be feasible. The expansion of the clinical use of these antihypertensive drugs in the field of oncology is further corroborated by numerous pre-clinical and clinical data, as presented in the following sections.
Pre-clinical data regarding sartans
The AT1R blocker olmesartan was found to mitigate the growth of tumors developed in nude mice upon the concurrent injection of a pancreatic cancer cell line and pancreatic stellate cells.88 The latter cells constitute a specific pancreatic cell subpopulation that exhibits profibrotic effects and fuels pancreatic cancer, due to interactions both with malignant cells and the cancer-promoting stromal elements in pancreas.89
Candesartan has been demonstrated to exert remarkable in vivo antiangiogenic activity. In fact, it reduces the occurrence of renal cancer lung metastases and inhibits VEGF production in androgen-independent human prostate cancer mouse xenografts, along with suppression of tumor growth and reduction of serum prostate-specific antigen (PSA).90 In urogenital cancers, inhibition of angiogenesis is considered the predominant antitumor mechanism of AT1R blockage.82 Accordingly, a clinically relevant dosing scheme of candesartan exhibits antitumor activity in human bladder cancer murine xenografts via suppressing angiogenesis without a direct pro-apoptotic effect.91
Candesartan also suppresses the production of VEGF and the invasiveness of AT1R-positive SKOV-3 human ovarian carcinoma cells. Additionally, candesartan reduces tumor angiogenesis in vivo and the ability of SKOV-3 cells to disseminate into the peritoneum in mice. Therefore, given that in clinical samples from patients with invasive ovarian cancer, AT1R immunoreactivity is associated with increased VEGF expression and microvascular density, candesartan is a candidate pharmacological tool not only for male-specific neoplasias (prostate cancer) but also for gynecological cancers (ovarian lesions).81 In xenografted prostate tumors, candesartan also acts as an antiangiogenic agent.92 Further, candesartan displays antifibrotic and antiproliferative activity in gastric cancer.93
Losartan has been shown to cause tumor shrinkage and apoptosis in rats with C6 glioma.80,94 Moreover, losartan exerts synergistic cancer cell-killing or antiproliferative effects when it is coadministered with other potent anticancer agents, specifically the angiotensin II type 2 receptor agonist CGP42112A in ovarian carcinoma95 and anti-miR-155 in endometrial cancer cells, respectively.96 Therefore, losartan may find use in the treatment of neurological as well as gynecological tumors. Of note, another possible application of this AT1R blocker is its ability to potentiate the therapeutic value of tumor-targeting nanoparticles; either these are oncolytic herpes viruses or liposomal doxorubicin conjugated with polyethylene glycol. This is achieved owing to the ability of losartan to impair the deposition of collagen in various different models of desmoplasia.97 Conceivably, losartan may not only be a valuable antitumor agent per se but may also open the road for increasing the efficacy of other antitumor factors.
The high-affinity AT1R antagonist telmisartan, which is known to exhibit the longest plasma half-life among all the sartans, triggers apoptosis in prostate cancer cells in a concentration-dependent fashion without affecting normal prostate stromal cells. Still, it is postulated that the cytotoxic activity of telmisartan is attributed to its PPARγ agonism rather than its AT1R-blocking activity.98 In addition, telmisartan restrains the EGFR-dependent proliferation of colon cancer cells in response to the tumor promoter 12-O-tetradecanoylphorbol-13-acetate by preventing the nuclear translocation of the C-terminal fragment of the EGF family member HB-EGF.99
Pre-clinical data regarding ACEIs
On the basis of the structural similarity of ACE with MMPs, the ACEI captopril inhibits MMP-mediated gelatinolysis like MMP inhibitors do. In this way, captopril suppresses the invasiveness of HT1080 fibrosarcoma cells and T98G glioma cells in vitro and impedes the growth of human gastric adenocarcinoma cells in a mouse xenograft model, either when it is administered alone or in synergy with cisplatin.100,101 A short-term clinically relevant dosing scheme (2.8 mg) of captopril displays remarkable antitumor activity against lung cancer growth and lymph node metastasis in mouse xenografts though evoking an apoptotic response without impinging on tumor neoangiogenesis,102 although there is evidence that this ACEI negatively regulates the chemotactic behavior of capillary endothelial cells and neovascularization.103 Moreover, captopril has been shown to serve as a free sulfhydryl-group donor for the plasmin-mediated generation of angiostatin and to display synergistic anticancer properties in human melanoma xenografts with tissue-type plasminogen activator (a protease participating in the conversion of plasminogen to plasmin).104 Captopril also attenuates renal cancer cell growth, possibly via sensitizing renal cancer cells to the cytostatic activities of TGFβ105 and inhibits renal carcinogenesis in a mouse model.106 The in vitro antiproliferative properties of captopril in human mammary ductal carcinoma are possibly attributed to its ability to interfere with the expression of sex-steroid receptors and key biosynthetic routes (ribonucleic acid/protein synthesis),107 further perplexing its biological activity.
The process of liver regeneration and tumor recurrence after partial hepatectomy performed in colorectal cancer patients with liver metastases is associated with an increase in the intrahepatic levels of ACE in mouse models. On the contrary, administration of captopril hinders tumor angiogenesis and triggers tumor apoptotic death.108 Aside from metastatic colorectal cancer, this ACEI has been suggested to be effective for the pharmacological management of recurrent glioblastoma in a mixture with other medicines, collectively termed CUSP9*.109
Of note, captopril is able to exert remarkable antimitotic activity in cancer cells that are devoid of functional RAS110 and to counteract endothelial cell migration irrespectively of ACE inhibition.103 Therefore, there is evidence that this ACEI can impede carcinogenesis irrespective of the link among RAS and cancer and its mechanistic basis. This warrants further investigation.
The ACEI perindopril has been shown to inhibit hepatocellular carcinogenesis in mice and to suppress tumor neovascularization at the clinically achievable dose of 2 mg/kg/day. The profound antiangiogenic activity of perindopril in vivo and its biologically active metabolite perindoprilat in vitro, as evidenced by the impediment of the formation of endothelial cell-derived tubular structures and the reduction of CD31 immunoreactivity within tumors, are possibly attributed to its ability to shut off VEGF gene transcription. The antitumor activity of perindopril does not seem to be dependent on the blockage of AT1R signaling, since neither losartan nor candesartan, even at higher doses, could suppress hepatocellular carcinoma development.111,112 Given the fact that perindopril displays virtually no cytotoxicity112 and is generally well tolerated,113 it is a promising drug against liver cancer. Of note, in rodent models of hepatocellular carcinoma, perindopril exhibits a remarkable synergism in the suppression of tumorigenesis and chemoprevention with IFN-β and vitamin K2, respectively.114–116 This further consolidates the notion of its anticancer exploitation alone or in combination with other agents already clinically used. In addition, perindopril is a potent antiangiogenic agent in head and neck squamous cell carcinoma, as evidenced by in vitro and in vivo experimentation.117
Clinical/epidemiological data regarding drugs blocking the renin–angiotensin system
A pilot clinical study assessing the benefits of 8 mg candesartan (total daily clinically relevant dose to treat hypertension ranges from 8 mg to 32 mg) in combination with antiandrogens in PSA expression and performance status in hormone-refractory prostate cancer patients118 yielded encouraging results. In addition, there is clinical evidence supporting that the antihypertensive treatment with ACEIs or sartans prolongs life expectancy of advanced lung cancer patients receiving conventional platinum drugs.119 A Phase II clinical trial indicated remarkable benefits of receiving low-dose (4 mg) candesartan or perindopril in combination with IFN-α, the COX-2 inhibitor meloxicam, and cimetidine, namely the “I-CCA therapy”, as first-line treatment in advanced renal cell carcinoma patients, with trifling toxicity.120 Though oral administration of candesartan (16 mg) in combination with intravenously infused gemcitabine was reported to be well tolerated in advanced pancreatic cancer patients, it does not seem to be an effective combinational therapeutic scheme for this type of malignancy.121,122
Noteworthy, according to epidemiological data the ACE genotype DD, which is associated with high ACE enzymatic activity, both predisposes carriers to breast cancer development and increases their responsiveness to the antitumor function of ACEIs or sartans.123 Ongoing clinical trials assessing the antitumor activity of antihypertensive agents that target RAS components (eg, NCT00077064) will aid in the repositioning of these drugs beyond the field of cardiovascular therapeutics. Taking into account the polymorphisms in the enrolled patients at loci which are critical for RAS and its targeting, would be of great predictive importance.
Considerations for using and repositioning drugs blocking the renin–angiotensin system
In general, sartans are devoid of major side effects, and they are well tolerated by the majority of hypertensive patients.124 However, considerable caution should be taken regarding the putative application of captopril or other ACEIs in oncology, given the pulmonary toxicity of this class of drugs: ACEIs may lead to bronchospasm, dyspnea, or the provocation of persistent dry cough, due to the drug-induced increase in bradykinin levels. This happens because ACE is also responsible for the catabolism of bronchoconstrictive kinins.125–127 This is a significant limiting factor, especially in cancer patients concomitantly suffering from respiratory diseases, lung cancer patients with compromised respiratory function, or even cancer patients with irradiation-induced pulmonary fibrosis and lung malfunction.
Patients suffering from advanced cancer may experience hypotension. This could be ascribed to cancer-associated deregulated function of the autonomous nervous system.128 Alternatively, drop in blood pressure may be iatrogenic.129 This can be the case when patients are treated with IFN, given that a decrease in blood pressure is a well-known adverse effect of IFN.130 Hopefully, however, most of the available clinical data stem from studies assessing the antitumor potency of drugs targeting the RAS in cancer in which there were enrolled patients with advanced-stage solid tumors.118–120 In these studies, agents blocking the RAS were well tolerated by the recruited patients, even when a sartan or ACEI was combined with IFN-α.120 However, in the case that agents targeting the RAS would induce hypotension upon the concomitant administration of cytokines, this could be managed either with melatonin129 or with conventional medication that is indicated for the treatment of hypotension, such as etilefrine. Importantly, the administration of this sympathomimetic agent would possibly yield multiple beneficial effects in cancer patients with concomitant hypotension and chylothorax.131,132
HIV protease inhibitors
Current clinical use
HIV aspartyl PIs gained US Food and Drug Administration (FDA) approval and entered the anti-HIV cocktail market in the early 1990s.133 Actually, saquinavir was the first drug of this class to be approved by the FDA in 1995 through a relatively rapid process of only 3 months. Saquinavir and other PIs gained approval for the control of HIV infection as well as to offer HIV-infected individuals a better quality of life and increased survival rates. PIs target viral protease, an enzyme that is crucial for HIV replication.134 This class of antiviral drugs comprises both peptidomimetic agents, like the prototype drug saquinavir, and non-peptidic drugs, such as nelfinavir. The latter was launched in the late 1990s and was the first PI to be approved for pediatric use.135
The possible therapeutic repositioning of HIV PIs: pre-clinical and clinical data
PIs are currently employed only in the management of HIV infection. Surprisingly, however, there is ample evidence highlighting their antitumor function. As a matter of fact, PI-receiving HIV patients are less likely to develop infection-associated tumors, such as non-Hodgkin’s lymphomas and Kaposi’s sarcoma (KS),136 or may even experience KS regression.137 Further, there are numerous in vitro and in vivo experiments clearly demonstrating that PIs inhibit the growth of many non-HIV-related human cancer models. Let us note that aside from PIs, the anti-HIV nucleoside analog reverse-transcriptase inhibitor azidothymidine has also been reported to exert antitumor activity. Still, only the antitumor properties of PIs are presented in the following paragraphs, since the in vitro anticancer potency of azidothymidine does not correlate with in vivo evidence,138 thereby attracting no more research interest.
The antiretroviral agent nelfinavir was found to exhibit a wide range of antitumor activities in several cancer cell lines, including chemoresistant ones, as well as in NSCLC mouse xenografts, at clinically attainable doses. Mechanistically, the cytotoxic effects of nelfinavir are associated with both caspase-dependent apoptotic and non-apoptotic cell death that are overall mitigated by a prosurvival autophagic response that coincides with Akt inhibition.139 Similar antigrowth and pro-apoptotic function of nelfinavir along with Akt-pathway inhibition by this drug has been reported by other researchers as well. In fact, there has been observed a chemosensitizing effect of nelfinavir in NSCLC cells and IL-6/STAT3 and androgen receptor (AR) signaling in prostate cancer cells, which hinders their proliferation. Suppression of the IL-6/STAT3 pathway occurs either at the level of STAT3 phosphorylation stimulated by IL-6 or at the level of STAT3 binding to deoxyribonucleic acid (DNA).140,141 AR blockage results from the fact that Akt and STAT3 function as coactivators for AR. Yang et al combined the in vitro evidence of nelfinavir’s activity against prostate cancer with in vivo data in LNCaP-xenografted mice that received small short-term doses of nelfinavir (60 mg/kg five times a week) with excellent tolerability.140 The therapeutic potency of nelfinavir in prostate cancer is also supported by a more recent publication, wherein this PI triggered ER stress and apoptosis in castration-resistant prostate cancer cells. In this case, apoptosis was actually triggered due to blockage of site-2 protease, which mediates the transcriptional activation of SREBP-1 and ATF6 through regulated intramembrane proteolysis.142 A similar anticancer mechanism has been reported in liposarcoma cells also.143
Another study underscores the possible utility of nelfinavir in multiple myeloma, where this anti-HIV agent displayed anti-proteasomal and pro-apoptotic activity.144 Further, nelfinavir may also exert anti-glioblastoma activity by virtue of its property to block catalysis mediated by MMP-2 and -9.145 In melanoma cells, nelfinavir triggers apoptosis and ceases the cell cycle via decreasing CDK2 activity through stimulating the proteasomal degradation of CDC25A.146 Another study demonstrating the pro-apoptotic and cytostatic activity of nelfinavir in ovarian cancer cells offers further impetus toward the rapid clinical testing of this PI for oncological purposes.147
Interestingly, nelfinavir also blunts the transcriptional upregulation of VEGF by Sp1 and HIF-1α under normoxic and hypoxic conditions, respectively, presumably via inhibiting Akt (Figure 1). VEGF downregulation is functionally associated with in vitro and in vivo perturbation of angiogenesis. In addition, nelfinavir exhibits in vivo radiosensitizing effects, through inducing tumor reoxygenation via an as-yet-unidentified mechanism.148 Since pO2 critically determines radiosensitivity, this would be of major clinical importance, particularly in hypoxic solid tumors that are resistant to radiotherapy.
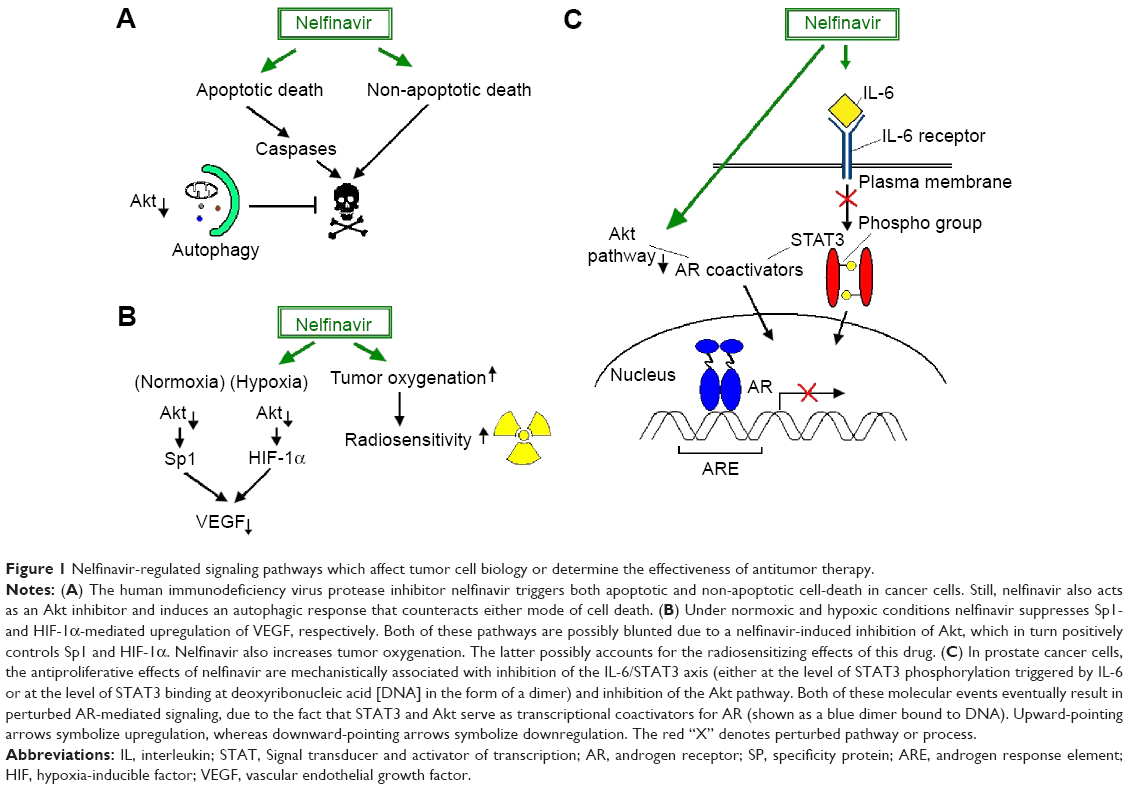 | Figure 1 Nelfinavir-regulated signaling pathways which affect tumor cell biology or determine the effectiveness of antitumor therapy. |
Apart from nelfinavir, other PIs also exhibit antitumor activity, including ritonavir in breast cancer cells, ritonavir and saquinavir in ovarian cancer cells,149–151 lopinavir in cervical cancer cells,152 and amprenavir in hepatocarcinoma xenografts.153 In breast cancer cells, ritonavir-mediated growth inhibition partially depends on disrupting the assembly of the Akt/Hsp90 complex,149 which had been previously shown to dampen ASK1-dependent apoptosis.154 Noticeably, although ER stress response has been reported to induce autophagy that eventually counteracts nelfinavir-induced cell death, the ability of atazanavir and nelfinavir to kill malignant glioma cells seems to rely on an active ER stress response/caspase 4 pathway.155 This is not surprising, given that ER stress is known to result in apoptosis via multiple pathways.156 Saquinavir and indinavir display in vitro antiangiogenic properties comparable to those of taxol and promote the regression of KS-like lesions in murine disease models.137 Saquinavir, like nelfinavir, inhibits the proteasome.157 The latter molecular event is functionally associated with the induction of apoptosis and potentiation of the cytotoxic effects of ionizing radiation.
In a small, Phase I clinical trial, the clinically relevant dose of 1,250 mg twice daily of nelfinavir combined with conventional chemotherapy (gemcitabine and cisplatin) and radiotherapy yielded satisfying antitumor response, as evidenced with positron emission tomography in pancreatic cancer patients. In detail, there was a complete response and stabilization of disease progression in five and two of nine patients, respectively, with minor toxicity.158 Interestingly, in glioblastoma multiforme patients, a recent Phase I study reported that 1,250 mg twice daily was the maximally tolerated dose of nelfinavir when combined with radiotherapy and temozolomide.159 Hopefully, nelfinavir is well tolerated even at a dosing scheme that exceeds the dose that has gained FDA approval for the management of HIV infection by 2.5 times, as a Phase I trial reported.160 Another Phase I study showed that 750 mg of nelfinavir twice daily yields encouraging preoperative results in locally advanced rectal cancer patients when combined with chemotherapy and radiation therapy.161 A Phase I trial demonstrated that up to 1,250 mg of nelfinavir twice a day combined with chemotherapy and radiotherapy was well tolerated in patients suffering from advanced, unresectable lung cancer.162 More importantly, the results regarding the clinical response of the patients enrolled in the latter study were satisfactory enough. However, nelfinavir at 1,250 mg twice daily does not significantly impact on progression-free survival of patients with recurrent adenoid cystic carcinoma, as a Phase II trial showed.163 Further clinical evaluation of HIV PIs in cancer therapeutics is needed.
Considerations for using and repositioning HIV PIs
Given the multi-year clinical experience of nelfinavir administration in HIV patients and its broad-spectrum anticancer activity, scientists have envisioned the introduction of this drug in the field of oncology as a promising cancer-fighting strategy. This is also the case for other PIs as well, such as ritonavir.150 Unfortunately, PIs commonly cause disturbances in the glycemic and lipidemic profile. However, these unwanted effects can be pharmacologically managed in acquired immunodeficiency syndrome (AIDS) patients. In fact, there are certain medications that are recommended for patients receiving antiviral therapy. It is also important for clinicians to take into account each patient’s individual physiology before prescribing a glucose- and/or lipid-lowering agent to AIDS patients.164–166 Such a cautious, individualized management of the aforementioned clinical conditions would allow the safe use of PIs in oncology. In turn, this could hopefully pave the road for the design of more efficacious anticancer modalities, circumventing the laggard process of new drug approval.139,147,167
Conclusion and future prospects
The non-antitumor pharmaceuticals with anticancer properties presented here reflect the functional redundancy characterizing the molecules and/or signaling pathways targeted by these drugs. Moreover, the potent multiple utility of a given pharmacological agent is not a novel phenomenon. For instance, the lysosomotropic agent hydroxychloroquine is currently used both as an antimalarial drug and in the treatment of various inflammatory diseases, including rheumatoid arthritis and systemic lupus erythematosus.168 Additionally, experimental evidence indicates that hydroxychloroquine could also be therapeutically used in the field of oncology.169 In general, repositioning of drugs is an emerging concept that gains ground in light of novel data.170,171
Herein, there was provided evidence for the antitumor activity of three different categories of non-antineoplastic drugs that are already commercially available. The fact that only data stemming from pre-clinical experimentation and that Phase I or II clinical trials were reviewed should be considered as a limitation. To the best of our knowledge, no Phase III clinical trials have been conducted in order to assess the clinical value of the aforementioned marketed drugs in cancer therapeutics so far. However, the evidence presented herein could achieve its goal, ie, the conceptual corroboration of the repositioning of these marketed drugs, the compelling of the prioritization of basic or clinical research toward this direction, and the instigation of further experimentation.
Taking into account the highly interlocked intracellular pathways, the therapeutic utility of many agents that are currently available in the market is rather underrated. Repositioning of the aforementioned non-antitumor drugs may offer clinicians the opportunity to fight cancer through therapeutic schemes with a safer toxicological profile. The latter is a major challenge, inasmuch as targeted therapeutic agents, such as monoclonal antibodies, were found to have serious adverse effects, such as drug-induced hypertension,172 that raise concerns, especially in elderly people, who are most prone to tumorigenesis. Many clinical studies that are under way (eg, NCT01485731, NCT01729923) will hopefully aid in the exploitation of the antineoplastic function of non-antitumor agents, such as NSAIDs, antihypertensive drugs, and HIV PIs, thereby opening new avenues for the development of safer and perhaps more efficacious alternative anticancer medications.
Disclosure
The authors report no conflicts of interest in this work.
References
Quirke V, Gaudillière JP. The era of biomedicine: science, medicine, and public health in Britain and France after the Second World War. Med Hist. 2008;52(4):441–452. | ||
Bernhard EJ, Brunner TB. Progress towards the use of HIV protease inhibitors in cancer therapy. Cancer Biol Ther. 2008;7(5):636–637. | ||
Kast RE, Boockvar JA, Brüning A, et al. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget. 2013;4(4):502–530. | ||
LaPorte SL, Juo ZS, Vaclavikova J, et al. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. 2008;132(2):259–272. | ||
Behar M, Barken D, Werner SL, Hoffmann A. The dynamics of signaling as a pharmacological target. Cell. 2013;155(2):448–461. | ||
Lang BT, Wang J, Filous AR, Au NP, Ma CH, Shen Y. Pleiotropic molecules in axon regeneration and neuroinflammation. Exp Neurol. 2014;258:17–23. | ||
Fang Z, Tang Y, Fang J, et al. Simvastatin inhibits renal cancer cell growth and metastasis via AKT/mTOR, ERK and JAK2/STAT3 pathway. PLoS One. 2013;8(5):e62823. | ||
Agostini M, Almeida LY, Bastos DC, et al. The fatty acid synthase inhibitor orlistat reduces the growth and metastasis of orthotopic tongue oral squamous cell carcinomas. Mol Cancer Ther. 2014;13(3):585–595. | ||
Hiroshima Y, Maawy A, Hassanein MK, et al. The tumor-educated-macrophage increase of malignancy of human pancreatic cancer is prevented by zoledronic acid. PLoS One. 2014;9(8):e103382. | ||
Huang L, Wong CC, Mackenzie GG, et al. Phospho-aspirin (MDC-22) inhibits breast cancer in preclinical animal models: an effect mediated by EGFR inhibition, p53 acetylation and oxidative stress. BMC Cancer. 2014;14:141. | ||
Bowers LW, Maximo IX, Brenner AJ, et al. NSAID use reduces breast cancer recurrence in overweight and obese women: role of prostaglandin-aromatase interactions. Cancer Res. 2014;74(16):4446–4457. | ||
Ahmad A, Banerjee S, Wang Z, Kong D, Majumdar AP, Sarkar FH. Aging and inflammation: etiological culprits of cancer. Curr Aging Sci. 2009;2(3):174–186. | ||
Djansugurova LB, Perfilyeva AV, Zhunusova GS, Djantaeva KB, Iksan OA, Khussainova EM. The determination of genetic markers of age-related cancer pathologies in populations from Kazakhstan. Front Genet. 2013;4:70. | ||
Gibson TM, Morton LM, Shiels MS, Clarke CA, Engels EA. Risk of non-Hodgkin lymphoma subtypes in HIV-infected people during the HAART era: a population-based study. AIDS. 2014;28(15):2313–2318. | ||
Nelwan EJ, Pramono LA, Lubis AM, Djoerban Z. Kaposi sarcoma of the eye in an HIV patient well-responded to HAART. Acta Med Indones. 2014;46(3):253–255. | ||
Hiľovská L, Jendželovský R, Fedoročko P. Potency of non-steroidal anti-inflammatory drugs in chemotherapy. Mol Clin Oncol. 2015; 3(1):3–12. | ||
Karnezis T, Shayan R, Fox S, Achen MG, Stacker SA. The connection between lymphangiogenic signalling and prostaglandin biology: a missing link in the metastatic pathway. Oncotarget. 2012;3(8):893–906. | ||
Baigent C, Patrono C. Selective cyclooxygenase 2 inhibitors, aspirin, and cardiovascular disease: a reappraisal. Arthritis Rheum. 2003; 48(1):12–20. | ||
Lynch ME, Watson CP. The pharmacotherapy of chronic pain: a review. Pain Res Manag. 2006;11(1):11–38. | ||
Chou R, Huffman LH. Medications for acute and chronic low back pain: a review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Ann Intern Med. 2007;147(7):505–514. | ||
Bujold E, Roberge S, Lacasse Y, et al. Prevention of preeclampsia and intrauterine growth restriction with aspirin started in early pregnancy: a meta-analysis. Obstet Gynecol. 2010;116(2 Pt 1):402–414. | ||
Floyd CN, Ferro A. Mechanisms of aspirin resistance. Pharmacol Ther. 2014;141(1):69–78. | ||
Bachert C, Chuchalin AG, Eisebitt R, Netayzhenko VZ, Voelker M. Aspirin compared with acetaminophen in the treatment of fever and other symptoms of upper respiratory tract infection in adults: a multicenter, randomized, double-blind, double-dummy, placebo-controlled, parallel-group, single-dose, 6-hour dose-ranging study. Clin Ther. 2005;27(7):993–1003. | ||
Park S, Lee NR, Lee KE, Park JY, Kim YJ, Gwak HS. Effects of single-nucleotide polymorphisms of FMO3 and FMO6 genes on pharmacokinetic characteristics of sulindac sulfide in premature labor. Drug Metab Dispos. 2014;42(1):40–43. | ||
Brown JR, DuBois RN. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 2005;23(12):2840–2855. | ||
Harris RE, Beebe-Donk J, Doss H, Burr Doss D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: a critical review of non-selective COX-2 blockade. Oncol Rep. 2005;13(4):559–583. | ||
Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010;62(2):233–244. | ||
Sonoshita M, Takaku K, Sasaki N, et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(delta 716) knockout mice. Nat Med. 2001;7(9):1048–1051. | ||
Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, Whelan J. Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in Apc(Min/+) mice. Cancer Res. 2002;62(2):403–408. | ||
Hull MA, Ko SC, Hawcroft G. Prostaglandin EP receptors: targets for treatment and prevention of colorectal cancer? Mol Cancer Ther. 2004;3(8):1031–1039. | ||
Hida T, Yatabe Y, Achiwa H, et al. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res. 1998;58(17):3761–3764. | ||
Liu CH, Chang SH, Narko K, et al. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem. 2001;276(21):18563–18569. | ||
Chang SH, Ai Y, Breyer RM, Lane TF, Hla T. The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia. Cancer Res. 2005;65(11):4496–4499. | ||
Mazhar D, Ang R, Waxman J. COX inhibitors and breast cancer. Br J Cancer. 2006;94(3):346–350. | ||
Ishizaki T, Katsumata K, Tsuchida A, et al. Etodolac, a selective cyclooxygenase-2 inhibitor, inhibits liver metastasis of colorectal cancer cells via the suppression of MMP-9 activity. Int J Mol Med. 2006;17(2):357–362. | ||
Iwata C, Kano MR, Komuro A, et al. Inhibition of cyclooxygenase-2 suppresses lymph node metastasis via reduction of lymphangiogenesis. Cancer Res. 2007;67(21):10181–10189. | ||
Karnezis T, Shayan R, Caesar C, et al. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell. 2012;21(2):181–195. | ||
Dong X, Li R, Xiu P, et al. Meloxicam executes its antitumor effects against hepatocellular carcinoma in COX-2-dependent and -independent pathways. PLoS One. 2014;9(3):e92864. | ||
Naruse T, Nishida Y, Hosono K, Ishiguro N. Meloxicam inhibits osteosarcoma growth, invasiveness and metastasis by COX-2-dependent and independent routes. Carcinogenesis. 2006;27(3):584–592. | ||
Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1(1):11–21. | ||
Harris RE, Beebe-Donk J, Schuller HM. Chemoprevention of lung cancer by non-steroidal anti-inflammatory drugs among cigarette smokers. Oncol Rep. 2002;9(4):693–695. | ||
Muranushi C, Olsen CM, Pandeya N, Green AC. Aspirin and non-steroidal anti-inflammatory drugs can prevent cutaneous squamous cell carcinoma: a systematic review and meta-analysis. J Invest Dermatol. 2014;135(4):975–983. | ||
Wong CC, Cheng KW, Rigas B. Preclinical predictors of anticancer drug efficacy: critical assessment with emphasis on whether nanomolar potency should be required of candidate agents. J Pharmacol Exp Ther. 2012;341(3):572–578. | ||
Matos P, Kotelevets L, Gonçalves V, et al. Ibuprofen inhibits colitis-induced overexpression of tumor-related Rac1b. Neoplasia. 2013;15(1): 102–111. | ||
Su B, Xu B, Wan J. Correlation between long-term aspirin use and F-fluorodeoxyglucose uptake in colorectal cancer measured by PET/CT. PLoS One. 2014;9(10):e109459. | ||
Liggett JL, Choi CK, Donnell RL, et al. Nonsteroidal anti-inflammatory drug sulindac sulfide suppresses structural protein nesprin-2 expression in colorectal cancer cells. Biochim Biophys Acta. 2014;1840(1): 322–331. | ||
Liggett JL, Min KW, Smolensky D, Baek SJ. A novel COX-independent mechanism of sulindac sulfide involves cleavage of epithelial cell adhesion molecule protein. Exp Cell Res. 2014;326(1):1–9. | ||
Liggett JL, Zhang X, Eling TE, Baek SJ. Anti-tumor activity of non-steroidal anti-inflammatory drugs: cyclooxygenase-independent targets. Cancer Lett. 2014;346(2):217–224. | ||
Vidal AC, Howard LE, Moreira DM, Castro-Santamaria R, Andriole GL, Freedland SJ. Aspirin, NSAIDs, and risk of prostate cancer: results from the REDUCE study. Clin Cancer Res. 2015;21(4):756–762. | ||
Chan AT. Aspirin and familial adenomatous polyposis: coming full circle. Cancer Prev Res (Phila). 2011;4(5):623–627. | ||
Kim KY, Jeon SW, Park JG, et al. Regression of colonic adenomas after treatment with sulindac in familial adenomatous polyposis: a case with a 2-year follow-up without a prophylactic colectomy. Ann Coloproctol. 2014;30(4):201–204. | ||
Xie G, Sun Y, Nie T, et al. Phospho-ibuprofen (MDC-917) is a novel agent against colon cancer: efficacy, metabolism, and pharmacokinetics in mouse models. J Pharmacol Exp Ther. 2011;337(3):876–886. | ||
Cheng KW, Wong CC, Alston N, et al. Aerosol administration of phospho-sulindac inhibits lung tumorigenesis. Mol Cancer Ther. 2013;12(8):1417–1428. | ||
Huang L, Wong CC, Cheng KW, Rigas B. Phospho-aspirin-2 (MDC-22) inhibits estrogen receptor positive breast cancer growth both in vitro and in vivo by a redox-dependent effect. PLoS One. 2014;9(11):e111720. | ||
Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286(8):954–959. | ||
Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352(11):1092–1102. | ||
Baron JA, Sandler RS, Bresalier RS, et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006;131(6):1674–1682. | ||
Fujimura T, Ohta T, Oyama K, Miyashita T, Miwa K. Cyclooxygenase-2 (COX-2) in carcinogenesis and selective COX-2 inhibitors for chemoprevention in gastrointestinal cancers. J Gastrointest Cancer. 2007;38(2–4):78–82. | ||
Lanas A, Baron JA, Sandler RS, et al. Peptic ulcer and bleeding events associated with rofecoxib in a 3-year colorectal adenoma chemoprevention trial. Gastroenterology. 2007;132(2):490–497. | ||
Patterson SL, Colbert Maresso K, Hawk E. Cancer chemoprevention: successes and failures. Clin Chem. 2013;59(1):94–101. | ||
Cooper DL, Murrell DE, Conder CM, et al. Exacerbation of celecoxib-induced renal injury by concomitant administration of misoprostol in rats. PLoS One. 2014;9(2):e89087. | ||
Kawasaki A, Suzuki K, Takekawa H, et al. Co-occurrence of multiple cerebral infarctions due to hypercoagulability associated with malignancy and meningeal carcinomatosis as the initial manifestation of gastric cancer. BMC Neurol. 2014;14:160. | ||
Tian S, Hirshfield KM, Jabbour SK, et al. Serum biomarkers for the detection of cardiac toxicity after chemotherapy and radiation therapy in breast cancer patients. Front Oncol. 2014;4:277. | ||
Auriel E, Regev K, Korczyn AD. Nonsteroidal anti-inflammatory drugs exposure and the central nervous system. Handb Clin Neurol. 2014; 119:577–584. | ||
Saff RR, Banerji A. Management of patients with nonaspirin-exacerbated respiratory disease aspirin hypersensitivity reactions. Allergy Asthma Proc. 2015;36(1):34–39. | ||
Sioka C, Kyritsis AP. Central and peripheral nervous system toxicity of common chemotherapeutic agents. Cancer Chemother Pharmacol. 2009;63(5):761–767. | ||
Bruchim I, Goldberg A, Fishman A, Confino-Cohen R. Carboplatin hypersensitivity: evaluation and successful desensitization protocol. Immunotherapy. 2014;6(8):905–912. | ||
Bak S, Andersen M, Tsiropoulos I, et al. Risk of stroke associated with nonsteroidal anti-inflammatory drugs: a nested case-control study. Stroke. 2003;34(2):379–386. | ||
Cruz RJ Jr, Vincenzi R, Ketzer BM, Cecilio AL, Cepeda LA. Spontaneous intratumoral bleeding and rupture of giant gastric stromal tumor (>30 cm) in a young patient. World J Surg Oncol. 2008;6:76. | ||
Kwon Y, Ahn JS, Jeon SR, et al. Intratumoral bleeding in meningioma after gamma knife radiosurgery. J Neurosurg. 2002;97(5 Suppl):S657–S662. | ||
Lakshmi Prasad G, Ramdurg SR, Suri A, Mahapatra AK. A rare association of meningioma with intratumoral bleed and acute subdural hematoma. Neurol India. 2010;58(6):977–978. | ||
Chen WC, Li YD, Chiang PH, et al. Comparison of proton pump inhibitor and histamine-2 receptor antagonist in the prevention of recurrent peptic ulcers/erosions in long-term low-dose aspirin users: a retrospective cohort study. Biomed Res Int. 2014;2014:693567. | ||
Bonelli P, Tuccillo FM, Federico A, et al. Ibuprofen delivered by poly(lactic-co-glycolic acid) (PLGA) nanoparticles to human gastric cancer cells exerts antiproliferative activity at very low concentrations. Int J Nanomedicine. 2012;7:5683–5691. | ||
Mourad JJ. The evolution of systolic blood pressure as a strong predictor of cardiovascular risk and the effectiveness of fixed-dose ARB/CCB combinations in lowering levels of this preferential target. Vasc Health Risk Manag. 2008;4(6):1315–1325. | ||
Kalaitzidis RG, Bakris GL. The current state of RAAS blockade in the treatment of hypertension and proteinuria. Curr Cardiol Rep. 2009; 11(6):436–442. | ||
Vajapey R, Rini D, Walston J, Abadir P. The impact of age-related dysregulation of the angiotensin system on mitochondrial redox balance. Front Physiol. 2014;5:439. | ||
Schweda F. Salt feedback on the renin-angiotensin-aldosterone system. Pflugers Arch. 2014;467(3):565–576. | ||
Tamarat R, Silvestre JS, Durie M, Levy BI. Angiotensin II angiogenic effect in vivo involves vascular endothelial growth factor- and inflammation-related pathways. Lab Invest. 2002;82(6):747–756. | ||
Ager EI, Neo J, Christophi C. The renin-angiotensin system and malignancy. Carcinogenesis. 2008;29(9):1675–1684. | ||
Arrieta O, Guevara P, Escobar E, García-Navarrete R, Pineda B, Sotelo J. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br J Cancer. 2005;92(7):1247–1252. | ||
Suganuma T, Ino K, Shibata K, et al. Functional expression of the angiotensin II type 1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin Cancer Res. 2005; 11(7):2686–2694. | ||
Miyajima A, Kikuchi E, Kosaka T, Oya M. Angiotensin II type 1 receptor antagonist as an angiogenic inhibitor in urogenital cancer. Rev Recent Clin Trials. 2009;4(2):75–78. | ||
Wasa J, Sugiura H, Kozawa E, Kohyama K, Yamada K, Taguchi O. The tumor suppressive effect of angiotensin II type 1 receptor antagonist in a murine osteosarcoma model. Anticancer Res. 2011;31(1):123–127. | ||
Shirotake S, Miyajima A, Kosaka T, et al. Regulation of monocyte chemoattractant protein-1 through angiotensin II type 1 receptor in prostate cancer. Am J Pathol. 2012;180(3):1008–1016. | ||
Uemura H, Nakaigawa N, Ishiguro H, Kubota Y. Antiproliferative efficacy of angiotensin II receptor blockers in prostate cancer. Curr Cancer Drug Targets. 2005;5(5):307–323. | ||
Uemura H, Ishiguro H, Kubota Y. Pharmacology and new perspectives of angiotensin II receptor blocker in prostate cancer treatment. Int J Urol. 2008;15(1):19–26. | ||
Izzedine H, Ederhy S, Goldwasser F, et al. Management of hypertension in angiogenesis inhibitor-treated patients. Ann Oncol. 2009;20(5): 807–815. | ||
Masamune A, Hamada S, Kikuta K, et al. The angiotensin II type I receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scand J Gastroenterol. 2013;48(5):602–609. | ||
Wilson JS, Pirola RC, Apte MV. Stars and stripes in pancreatic cancer: role of stellate cells and stroma in cancer progression. Front Physiol. 2014;5:52. | ||
Kosaka T, Miyajima A, Takayama E, et al. Angiotensin II type 1 receptor antagonist as an angiogenic inhibitor in prostate cancer. Prostate. 2007;67(1):41–49. | ||
Kosugi M, Miyajima A, Kikuchi E, Horiguchi Y, Murai M. Angiotensin II type 1 receptor antagonist candesartan as an angiogenic inhibitor in a xenograft model of bladder cancer. Clin Cancer Res. 2006;12(9):2888–2893. | ||
Alhusban A, Al-Azayzih A, Goc A, Gao F, Fagan SC, Somanath PR. Clinically relevant doses of candesartan inhibit growth of prostate tumor xenografts in vivo through modulation of tumor angiogenesis. J Pharmacol Exp Ther. 2014;350(3):635–645. | ||
Okazaki M, Fushida S, Harada S, et al. The angiotensin II type 1 receptor blocker candesartan suppresses proliferation and fibrosis in gastric cancer. Cancer Lett. 2014;355(1):46–53. | ||
Rivera E, Arrieta O, Guevara P, Duarte-Rojo A, Sotelo J. AT1 receptor is present in glioma cells; its blockage reduces the growth of rat glioma. Br J Cancer. 2001;85(9):1396–1399. | ||
Park YA, Choi CH, Do IG, et al. Dual targeting of angiotensin receptors (AGTR1 and AGTR2) in epithelial ovarian carcinoma. Gynecol Oncol. 2014;135(1):108–117. | ||
Choi CH, Park YA, Choi JJ, et al. Angiotensin II type I receptor and miR-155 in endometrial cancers: synergistic antiproliferative effects of anti-miR-155 and losartan on endometrial cancer cells. Gynecol Oncol. 2012;126(1):124–131. | ||
Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U S A. 2011;108(7):2909–2914. | ||
Funao K, Matsuyama M, Kawahito Y, et al. Telmisartan is a potent target for prevention and treatment in human prostate cancer. Oncol Rep. 2008;20(2):295–300. | ||
Ozeki K, Tanida S, Morimoto C, et al. Telmisartan inhibits cell proliferation by blocking nuclear translocation of ProHB-EGF C-terminal fragment in colon cancer cells. PLoS One. 2013;8(2):e56770. | ||
Nakagawa T, Kubota T, Kabuto M, Kodera T. Captopril inhibits glioma cell invasion in vitro: involvement of matrix metalloproteinases. Anticancer Res. 1995;15(5B):198–199. | ||
Williams RN, Parsons SL, Morris TM, Rowlands BJ, Watson SA. Inhibition of matrix metalloproteinase activity and growth of gastric adenocarcinoma cells by an angiotensin converting enzyme inhibitor in in vitro and murine models. Eur J Surg Oncol. 2005;31(9):1042–1050. | ||
Attoub S, Gaben AM, Al-Salam S, et al. Captopril as a potential inhibitor of lung tumor growth and metastasis. Ann N Y Acad Sci. 2008;1138:65–72. | ||
Volpert OV, Ward WF, Lingen MW, et al. Captopril inhibits angiogenesis and slows the growth of experimental tumors in rats. J Clin Invest. 1996;98(3):671–679. | ||
de Groot-Besseling RR, Ruers TJ, van Kraats AA, et al. Anti-tumor activity of a combination of plasminogen activator and captopril in a human melanoma xenograft model. Int J Cancer. 2004;112(2):329–334. | ||
Miyajima A, Asano T, Hayakawa M. Captopril restores transforming growth factor-beta type II receptor and sensitivity to transforming growth factor-beta in murine renal cell cancer cells. J Urol. 2001;165(2):616–620. | ||
Hii SI, Nicol DL, Gotley DC, Thompson LC, Green MK, Jonsson JR. Captopril inhibits tumour growth in a xenograft model of human renal cell carcinoma. Br J Cancer. 1998;77(6):880–883. | ||
Small W Jr, Molteni A, Kim YT, Taylor JM, Chen Z, Ward WF. Captopril modulates hormone receptor concentration and inhibits proliferation of human mammary ductal carcinoma cells in culture. Breast Cancer Res Treat. 1997;44(3):217–224. | ||
Koh SL, Ager EI, Costa PL, Malcontenti-Wilson C, Muralidharan V, Christophi C. Blockade of the renin-angiotensin system inhibits growth of colorectal cancer liver metastases in the regenerating liver. Clin Exp Metastasis. 2014;31(4):395–405. | ||
Kast RE, Karpel-Massler G, Halatsch ME. CUSP9* treatment protocol for recurrent glioblastoma: aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget. 2014;5(18):8052–8082. | ||
Reddy MK, Baskaran K, Molteni A. Inhibitors of angiotensin-converting enzyme modulate mitosis and gene expression in pancreatic cancer cells. Proc Soc Exp Biol Med. 1995;210(3):221–226. | ||
Yoshiji H, Kuriyama S, Kawata M, et al. The angiotensin-I-converting enzyme inhibitor perindopril suppresses tumor growth and angiogenesis: possible role of the vascular endothelial growth factor. Clin Cancer Res. 2001;7(4):1073–1078. | ||
Yoshiji H, Kuriyama S, Fukui H. Perindopril: possible use in cancer therapy. Anticancer Drugs. 2002;13(3):221–228. | ||
Ogilvie RI, Anand S, Roy P, De Souza S. Perindopril for control of blood pressure in patients with hypertension and other cardiovascular risk factors: an open-label, observational, multicentre, general practice-based study. Clin Drug Investig. 2008;28(11):673–686. | ||
Noguchi R, Yoshiji H, Kuriyama S, et al. Combination of interferon-beta and the angiotensin-converting enzyme inhibitor, perindopril, attenuates murine hepatocellular carcinoma development and angiogenesis. Clin Cancer Res. 2003;9(16 Pt 1):6038–6045. | ||
Yoshiji H, Noguchi R, Kuriyama S, Yoshii J, Ikenaka Y. Combination of interferon and angiotensin-converting enzyme inhibitor, perindopril, suppresses liver carcinogenesis and angiogenesis in mice. Oncol Rep. 2005;13(3):491–495. | ||
Yoshiji H, Kuriyama S, Noguchi R, et al. Combination of vitamin K2 and the angiotensin-converting enzyme inhibitor, perindopril, attenuates the liver enzyme-altered preneoplastic lesions in rats via angiogenesis suppression. J Hepatol. 2005;42(5):687–693. | ||
Yasumatsu R, Nakashima T, Masuda M, et al. Effects of the angiotensin-I converting enzyme inhibitor perindopril on tumor growth and angiogenesis in head and neck squamous cell carcinoma cells. J Cancer Res Clin Oncol. 2004;130(10):567–573. | ||
Uemura H, Hasumi H, Kawahara T, et al. Pilot study of angiotensin II receptor blocker in advanced hormone-refractory prostate cancer. Int J Clin Oncol. 2005;10(6):405–410. | ||
Wilop S, von Hobe S, Crysandt M, Esser A, Osieka R, Jost E. Impact of angiotensin I converting enzyme inhibitors and angiotensin II type 1 receptor blockers on survival in patients with advanced non-small-cell lung cancer undergoing first-line platinum-based chemotherapy. Cancer Res Clin Oncol. 2009;135(10):1429–1435. | ||
Tatokoro M, Fujii Y, Kawakami S, et al. Phase-II trial of combination treatment of interferon-α, cimetidine, cyclooxygenase-2 inhibitor and renin-angiotensin-system inhibitor (I-CCA therapy) for advanced renal cell carcinoma. Cancer Sci. 2011;102(1):137–143. | ||
Nakai Y, Isayama H, Ijichi H, et al. Phase I trial of gemcitabine and candesartan combination therapy in normotensive patients with advanced pancreatic cancer: GECA1. Cancer Sci. 2012;103(8): 1489–1492. | ||
Nakai Y, Isayama H, Ijichi H, et al. A multicenter phase II trial of gemcitabine and candesartan combination therapy in patients with advanced pancreatic cancer: GECA2. Invest New Drugs. 2013;31(5):1294–1299. | ||
van der Knaap R, Siemes C, Coebergh JW, van Duijn CM, Hofman A, Stricker BH. Renin-angiotensin system inhibitors, angiotensin I-converting enzyme gene insertion/deletion polymorphism, and cancer: the Rotterdam Study. Cancer. 2008;112(4):748–757. | ||
Siragy HM. A current evaluation of the safety of angiotensin receptor blockers and direct renin inhibitors. Vasc Health Risk Manag. 2011;7:297–313. | ||
Lunde H, Hedner T, Samuelsson O, et al. Dyspnoea, asthma, and bronchospasm in relation to treatment with angiotensin converting enzyme inhibitors. BMJ. 1994;308(6920):18–21. | ||
Tschöpe C, Schultheiss HP, Walther T. Multiple interactions between the renin-angiotensin and the kallikrein-kinin systems: role of ACE inhibition and AT1 receptor blockade. J Cardiovasc Pharmacol. 2002;39(4):478–487. | ||
Dicpinigaitis PV. Angiotensin-converting enzyme inhibitor-induced cough: ACCP evidence-based clinical practice guidelines. Chest. 2006;129(1 Suppl):S169–S173. | ||
Stone CA, Kenny RA, Nolan B, Lawlor PG. Autonomic dysfunction in patients with advanced cancer; prevalence, clinical correlates and challenges in assessment. BMC Palliat Care. 2012;11:3. | ||
Lissoni P, Pittalis S, Ardizzoia A, et al. Prevention of cytokine-induced hypotension in cancer patients by the pineal hormone melatonin. Support Care Cancer. 1996;4(4):313–316. | ||
Selcukbiricik F, Tural D, Senel TE, Sarica A, Soyluk O, Serdengecti S. Bilateral ischemic optic neuropathy developed under interferon therapy. Case Rep Ophthalmol Med. 2012;2012:102739. | ||
Guillem P, Papachristos I, Peillon C, Triboulet JP. Etilefrine use in the management of post-operative chyle leaks in thoracic surgery. Interact Cardiovasc Thorac Surg. 2004;3(1):156–160. | ||
Kranzfelder M, Gertler R, Hapfelmeier A, Friess H, Feith M. Chylothorax after esophagectomy for cancer: impact of the surgical approach and neoadjuvant treatment: systematic review and institutional analysis. Surg Endosc. 2013;27(10):3530–3538. | ||
Pokorná J, Machala L, Rezáčová P, Konvalinka J. Current and novel inhibitors of HIV protease. Viruses. 2009;1(3):1209–1239. | ||
Brik A, Wong CH. HIV-1 protease: mechanism and drug discovery. Org Biomol Chem. 2003;1(1):5–14. | ||
Perry CM, Frampton JE, McCormack PL, Siddiqui MA, Cvetković RS. Nelfinavir: a review of its use in the management of HIV infection. Drugs. 2005;65(15):2209–2244. | ||
Monini P, Sgadari C, Barillari G, Ensoli B. HIV protease inhibitors: antiretroviral agents with anti-inflammatory, anti-angiogenic and anti-tumour activity. J Antimicrob Chemother. 2003;51(2):207–211. | ||
Sgadari C, Barillari G, Toschi E, et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi sarcoma. Nat Med. 2002;8(3):225–232. | ||
Chow WA, Jiang C, Guan M. Anti-HIV drugs for cancer therapeutics: back to the future? Lancet Oncol. 2009;10(1):61–71. | ||
Gills JJ, Lopiccolo J, Tsurutani J, et al. Nelfinavir, a lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13(17):5183–5194. | ||
Yang Y, Ikezoe T, Takeuchi T, et al. HIV-1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer Sci. 2005;96(7):425–433. | ||
Yang Y, Ikezoe T, Nishioka C, et al. NFV, an HIV-1 protease inhibitor, induces growth arrest, reduced Akt signalling, apoptosis and docetaxel sensitisation in NSCLC cell lines. Br J Cancer. 2006; 95(12):1653–1662. | ||
Guan M, Fousek K, Chow WA. Nelfinavir inhibits regulated intramembrane proteolysis of sterol regulatory element binding protein-1 and activating transcription factor 6 in castration-resistant prostate cancer. FEBS J. 2012;279(13):2399–2411. | ||
Guan M, Fousek K, Jiang C, et al. Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin Cancer Res. 2011;17(7):1796–1806. | ||
Bono C, Karlin L, Harel S, et al. The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica. 2012;97(7):1101–1109. | ||
Kast RE, Halatsch ME. Matrix metalloproteinase-2 and -9 in glioblastoma: a trio of old drugs – captopril, disulfiram and nelfinavir – are inhibitors with potential as adjunctive treatments in glioblastoma. Arch Med Res. 2012;43(3):243–247. | ||
Jiang W, Mikochik PJ, Ra JH, et al. HIV protease inhibitor nelfinavir inhibits growth of human melanoma cells by induction of cell cycle arrest. Cancer Res. 2007;67(3):1221–1227. | ||
Brüning A, Burger P, Vogel M, et al. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol Ther. 2009;8(3):226–232. | ||
Pore N, Gupta AK, Cerniglia GJ, et al. Nelfinavir down-regulates hypoxia-inducible factor 1 alpha and VEGF expression and increases tumor oxygenation: implications for radiotherapy. Cancer Res. 2006; 66(18):9252–9259. | ||
Srirangam A, Mitra R, Wang M, et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin Cancer Res. 2006;12(6):1883–1896. | ||
Kumar S, Bryant CS, Chamala S, et al. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol Cancer. 2009;8:26. | ||
McLean K, VanDeVen NA, Sorenson DR, Daudi S, Liu JR. The HIV protease inhibitor saquinavir induces endoplasmic reticulum stress, autophagy, and apoptosis in ovarian cancer cells. Gynecol Oncol. 2009;112(3):623–630. | ||
Vici P, Mariani L, Pizzuti L, et al. Emerging biological treatments for uterine cervical carcinoma. J Cancer. 2014;5(2):86–97. | ||
Gantt S, Casper C, Ambinder RF. Insights into the broad cellular effects of nelfinavir and the HIV protease inhibitors supporting their role in cancer treatment and prevention. Curr Opin Oncol. 2013;25(5):495–502. | ||
Zhang R, Luo D, Miao R, et al. Hsp90-Akt phosphorylates ASK1 and inhibits ASK1-mediated apoptosis. Oncogene. 2005;24(24):3954–3963. | ||
Pyrko P, Kardosh A, Wang W, Xiong W, Schönthal AH, Chen TC. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res. 2007;67(22):10920–10928. | ||
Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem. 2004;279(25): 25935–25938. | ||
Pajonk F, Himmelsbach J, Riess K, Sommer A, McBride WH. The human immunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non-HIV-associated human cancer cells. Cancer Res. 2002;62(18):5230–5235. | ||
Brunner TB, Geiger M, Grabenbauer GG, et al. Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J Clin Oncol. 2008;26(16):2699–2706. | ||
Alonso-Basanta M, Fang P, Maity A, Hahn SM, Lustig RA, Dorsey JF. A phase I study of nelfinavir concurrent with temozolomide and radiotherapy in patients with glioblastoma multiforme. J Neurooncol. 2014;116(2):365–372. | ||
Blumenthal GM, Gills JJ, Ballas MS, et al. A phase I trial of the HIV protease inhibitor nelfinavir in adults with solid tumors. Oncotarget. 2014;5(18):8161–8172. | ||
Buijsen J, Lammering G, Jansen RL, et al. Phase I trial of the combination of the Akt inhibitor nelfinavir and chemoradiation for locally advanced rectal cancer. Radiother Oncol. 2013;107(2):184–188. | ||
Rengan R, Mick R, Pryma D, et al. A phase I trial of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: a report of toxicities and clinical response. J Thorac Oncol. 2012;7(4):709–715. | ||
Hoover AC, Milhem MM, Anderson CM, et al. Efficacy of nelfinavir as monotherapy in refractory adenoid cystic carcinoma: results of a phase II clinical trial. Head Neck. Epub 2014 Mar 5. | ||
Penzak SR, Chuck SK. Management of protease inhibitor-associated hyperlipidemia. Am J Cardiovasc Drugs. 2002;2(2):91–106. | ||
Calza L, Manfredi R, Chiodo F. Use of fibrates in the management of hyperlipidemia in HIV-infected patients receiving HAART. Infection. 2002;30(1):26–31. | ||
Kalra S, Kalra B, Agrawal N, Unnikrishnan A. Understanding diabetes in patients with HIV/AIDS. Diabetol Metab Syndr. 2011;3(1):2. | ||
Gills JJ, Lopiccolo J, Dennis PA. Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy. 2008;4(1):107–109. | ||
Yogasundaram H, Putko BN, Tien J, et al. Hydroxychloroquine-induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can J Cardiol. 2014;30(12):1706–1715. | ||
Rahim R, Strobl JS. Hydroxychloroquine, chloroquine, and all-trans retinoic acid regulate growth, survival, and histone acetylation in breast cancer cells. Anticancer Drugs. 2009;20(8):736–745. | ||
Brüning A, Gingelmaier A, Friese K, Mylonas I. New prospects for nelfinavir in non-HIV-related diseases. Curr Mol Pharmacol. 2010; 3(2):91–97. | ||
Padhy BM, Gupta YK. Drug repositioning: re-investigating existing drugs for new therapeutic indications. J Postgrad Med. 2011; 57(2):153–160. | ||
Schneider BP, Li L, Shen F, et al. Genetic variant predicts bevacizumab-induced hypertension in ECOG-5103 and ECOG-2100. Br J Cancer. 2014;111(6):1241–1248. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.