Back to Journals » The Application of Clinical Genetics » Volume 9
Marfan syndrome: current perspectives
Authors Pepe G, Giusti B, Sticchi E, Abbate R, Gensini G, Nistri S
Received 12 September 2015
Accepted for publication 27 January 2016
Published 9 May 2016 Volume 2016:9 Pages 55—65
DOI https://doi.org/10.2147/TACG.S96233
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Guglielmina Pepe,1,2 Betti Giusti,1,2 Elena Sticchi,1,2 Rosanna Abbate,1,2 Gian Franco Gensini,1–3 Stefano Nistri2,4
1Department of Experimental and Clinical Medicine, Section of Critical Medical Care and Medical Specialities, DENOTHE Center, University of Florence, 2Cardiothoracovascular Department, Marfan Syndrome and Related Disorders Regional Referral Center, Careggi Hospital, 3Santa Maria agli Ulivi, Fondazione Don Carlo Gnocchi, Onlus, Institute for Cancer Research and Treatment, Florence, 4Cardiology Service, CMSR Veneto Medica, Altavilla Vicentina, Italy
Abstract: Marfan syndrome (MFS) is a pleiotropic connective tissue disease inherited as an autosomal dominant trait, due to mutations in the FBN1 gene encoding fibrillin 1. It is an important protein of the extracellular matrix that contributes to the final structure of a microfibril. Few cases displaying an autosomal recessive transmission are reported in the world. The FBN1 gene, which is made of 66 exons, is located on chromosome 15q21.1. This review, after an introduction on the clinical manifestations that leads to the diagnosis of MFS, focuses on cardiovascular manifestations, pharmacological and surgical therapies of thoracic aortic aneurysm and/or dissection (TAAD), mechanisms underlying the progression of aneurysm or of acute dissection, and biomarkers associated with progression of TAADs. A Dutch group compared treatment with losartan, an angiotensin II receptor-1 blocker, vs no other additional treatment (COMPARE clinical trial). They observed that losartan reduces the aortic dilatation rate in patients with Marfan syndrome. Later on, they also reported that losartan exerts a beneficial effect on patients with Marfan syndrome carrying an FBN1 mutation that causes haploinsufficiency (quantitative mutation), while it has no significant effect on patients displaying dominant negative (qualitative) mutations. Moreover, a French group in a 3-year trial compared the administration of losartan vs placebo in patients with Marfan syndrome under treatment with beta-receptor blockers. They observed that losartan decreases blood pressure but has no effect on aortic diameter progression. Thus, beta-receptor blockers remain the gold standard therapy in patients with Marfan syndrome. Three potential biochemical markers are mentioned in this review: total homocysteine, serum transforming growth factor beta, and lysyl oxidase. Moreover, markers of oxidative stress measured in plasma, previously correlated with clinical features of Marfan syndrome, may be explored as potential biomarkers of clinical severity.
Keywords: Marfan syndrome, thoracic aortic aneurysm, fibrillin 1, cardiovascular manifestations, diagnosis, therapy
Introduction
Marfan syndrome (MFS; Online Mendelian Inheritance in Man #154700) is an autosomal dominant inherited connective tissue disorder (CTD) mostly caused by mutations in FBN1, the gene encoding fibrillin 1, a structural component of the extracellular matrix (ECM) also involved in the regulation of transforming growth factor β (TGF-β) bioavailability.1 MFS is a rare pleiotropic disease (1:5,000) characterized by three clinical criteria (thoracic aortic aneurysm and/or dissection [TAAD], ectopia lentis [EL], and systemic features [SFs, multisystemic manifestations] with score ≥7) and two genetic criteria (the presence of a first-grade relative with MFS diagnosed according to revised Ghent-2 criteria and presence of a pathogenic mutation in FBN1 gene in presence of TAAD or EL) (Table 1).2 The detection of two of the three clinical criteria or one of the clinical criteria plus a family history or the presence of TAAD with causal FBN1 mutation or EL with a causal FBN1 mutation allows for the diagnosis of MFS.2 An exhaustive comparison among Beighton,3 Ghent-1,4 and Ghent-22 nosologies has been already performed, reaching the conclusion that Ghent-1 and Ghent-2 criteria improve the detection of patients with MFS compared to Beighton nosology, highlighting the most common and specific mutations and giving a higher score to the more specific manifestations, and that Ghent-2 nosology is easier to use compared to Ghent-1 criteria.5,6 The latter issue is due to the simplification of the diagnostic criteria in Ghent-2 nosology by removing the minor criteria, by stating five criteria, and by the increased importance of detection of pathogenic FBN1 mutations.5,6 It is now common opinion that MFS is associated with mutations in FBN1 gene, although mutations in this gene underlie several other diseases such as myopia or mitral valve prolapse, mild aortic dilation, skeleton, and skin (MASS) syndrome, acromelic dysplasia, Weill–Marchesani syndrome, and stiff skin syndrome (SSS).2,6 Ghent-2 nosology highlights the importance of performing FBN1 mutation screening analysis to confirm the clinical diagnosis because a wide time onset of clinical manifestations, a high inter- and intrafamilial clinical phenotype variability, and a wide spectrum of CTDs in differential diagnosis always make a precocious definite clinical diagnosis more difficult.2 Early clinical diagnosis allows more appropriate clinical follow-ups, with great advantage to the quality of life of patients and partly reducing the cost for Public Health Service.2,6 In this review, we will focus on the perspectives regarding some old and new clinical manifestations and on the recent advancements in biomarkers and molecular genetics.
 | Table 1 Revised Ghent criteria for Marfan syndrome diagnosis |
Cardiovascular manifestations
TAAD and TGF-β in MFS
Cardiovascular manifestations represent the major morbidity and mortality factors requiring main clinical focus while managing Marfan (MF) patients (Figure 1).7,8
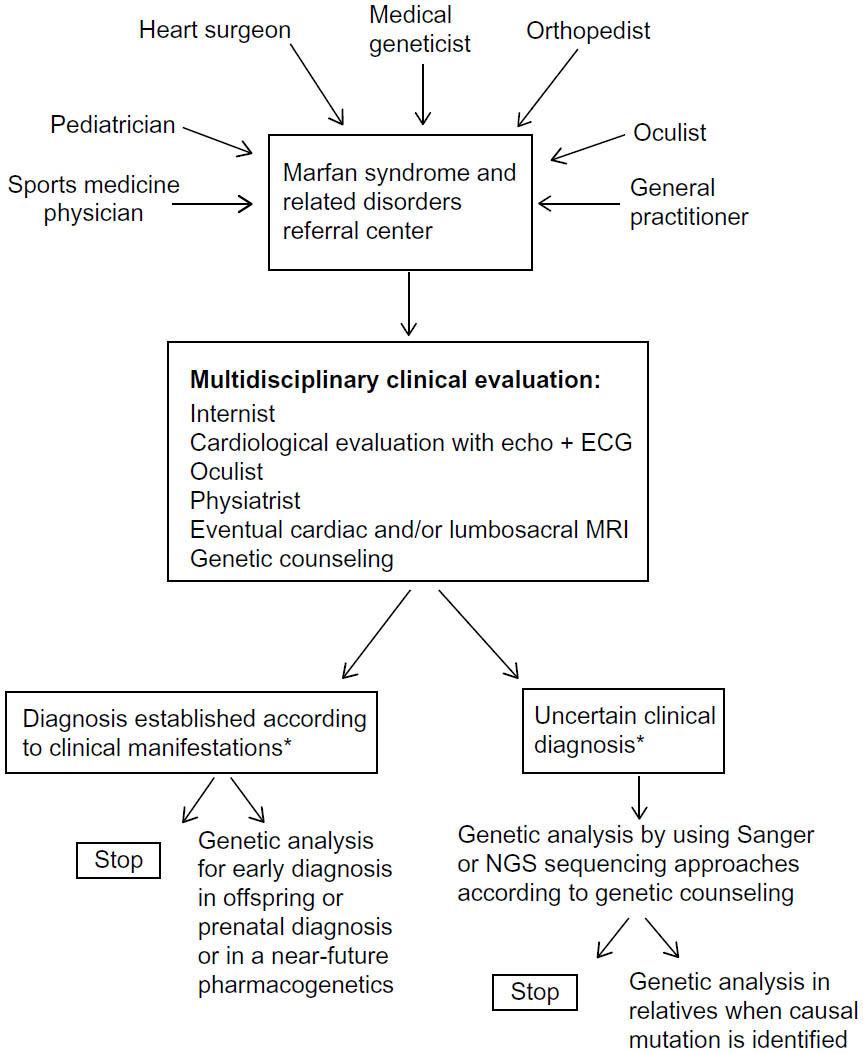 | Figure 1 Marfan syndrome and related disorders: flow chart of clinical and genetic management. |
MFS displays a number of abnormalities of the thoracic and abdominal aorta, ranging from abnormal aortic stiffness9,10 to aortic aneurysm and dissection. Histopathological changes are represented by accelerated vessel aging and maladaptive remodeling;11 tissue histologic analyses display elastic lamellae fragmentation, disorganization of the aortic architecture with excessive collagen and mucopolysaccharide accumulation, and a relative decrease of vascular smooth muscle cells.12 Thoracic aortic aneurysms (TAAs) can occur early in life and can be also detected in the fetus by echocardiography (hereinafter abbreviated as echo). Because an echo does not allow the visualization of the entire thoracic aorta, periodic magnetic resonance imaging (MRI) or computed tomography (CT) scan, as appropriate, is necessary. However, the rate of progression and risk of dissection are unpredictable on a single-patient basis. The clinical standardized follow-up of patients with MF syndrome includes limitation in sports, administration of beta-receptor blockers,13,14 and an echo test (every 1 year or 2 years), with subsequent prophylactic aortic root surgery in patients with an aortic diameter >50 mm. However, in patients with a family history of aortic dissection, a rapid increase of the aortic diameter (>3 mm/y on repeated measurements using the same imaging technique, at the same aorta level, with side-by-side comparison and confirmation by another technique), or presence of severe aortic or mitral regurgitation (or if a pregnancy is planned), aortic surgery should be performed when the diameter is >45 mm.15 Because the aortic diameter in MF males and females still increases during life, reaching sizes at high risk for rupture or dissection, future prospects in this field are the improvement of drug therapy and the search for biomarkers able to indicate predisposition to dissection or early aortic wall rupture.16
Soon after the discovery of perturbation of TGF-β signaling, as part of the pathophysiological mechanism underlying MFS pleiotropic manifestations, an FBN1 mutation knock-in mouse model (Fbn1C1039G/+) was treated with an oral angiotensin receptor blocker (ARB), losartan, which totally inhibited dilatation and also showed a regression of aortic ectasia.17 Thus, an ARB seems to lower the excessive activation of TGF-β signaling in mice. Clinicians over the world started clinical trials, some of which are still in progress, to test the hypothesis that losartan therapy or combined therapy including beta-receptor blockers and losartan may act more strongly against aortic dilatation in MFS, and probably also in other inherited TAAs.14,18–23
In the past 3 years, contrasting data came out from the first results of trials regarding the comparative effect of beta-receptor blocker (atenolol) vs ARB (losartan) over 3 years of follow-up. Losartan seems to reduce aortic root dilatation rate (AoDR) in a small retrospective cohort of pediatric patients with a severe MF phenotype and in adult patients with MF syndrome.18 Moreover, in patients with replacement of the aortic root, losartan reduces the dilatation rate of the aortic arch.19 Instead, no significantly different effect between atenolol and losartan was detected by Lacro et al20 in children and young adults. Finally, Milleron et al14 carried out a randomized, large-scale, double-blind, placebo-controlled trial with losartan. They concluded that although losartan is able to decrease blood pressure in patients with MF syndrome older than 10 years of age, it is not able to limit aortic dilatation during a 3-year period. Moreover, in normotensive patients, the vasodilator properties of losartan are limited. This study did not show any benefit of losartan in MF patients. Thus, according to these findings, beta-receptor blocker therapy remains the first-line therapy that should be proposed to these patients and, according to Milleron et al,14 also to patients with TAA and abdominal aortic aneurysms derived from other etiologies.
A challenge in the management of patients with MF syndrome is whether molecular genetics may help in the selection of pharmaceutical therapy (Figure 1). Franken et al,24 in an article that includes among the authors the Dutch clinicians who performed the COMPARE clinical trial19 mentioned earlier, reported that losartan reduces AoDR only in patients carrying an FBN1 mutation resulting in a haploinsufficiency (such as splicing mutations or one nucleotide insertions/deletions causing frame shifts). Apparently, patients carrying dominant negative FBN1 mutations (mainly missense mutations) do not gain any advantage from this drug. An explanation of this important difference may be the fact that a dominant negative FBN1 mutation (qualitative defect) may alter functions or foldings of the protein, affecting interactions with fibrillin 1 and/or other proteins, leading to a disorganized ECM.24 As a result, both the strength of the fibrillin matrix and the release of binding proteins, such as TGF-β, may be altered. Noteworthy, plasma levels of TGF-β turned out to be increased in MFS and related to TAADs. Furthermore, a decrease in fibrillin 1 results in an increase of plasma TGF-β levels due to the quantitative reduction of TGF-β binding and, in turn, its serum level increases. The various mechanisms of haploinsufficiency (quantitative defect) all lead to a decreased level of normal fibrillin 1 protein, probably resulting in a thinner fibrillin 1 matrix, which reduces aortic wall strength.24 A recent article, based on analysis using new techniques, demonstrates that dominant negative FBN1 mutations among different fibrillinopathies, MFS, SSS, and acromelic dysplasias act through different mechanisms.25 More precisely, substitutions in fibrillin 1 domains TB4 and TB5 associated with SSS and acromelic dysplasias do not interfere with secretion or assembly into microfibrils; instead, the mutation results in a loss of recombinant proteins in the culture medium and no association with microfibrils.25
Why does losartan act only on patients with haploinsuffiiency? A possible explanation is that the aortic wall, being thinner because of the decreased fibrillin matrix, may suffer from hypertension, which as a consequence, alone or with aortic stretch, may directly damage the aorta by activating angiotensin II (AngII) receptor type 1 (AT1R), which responds to the damage by producing TGF-β (Figure 2). Local AngII has been associated with aneurysm formation. Patients with MF syndrome with haploinsufficiency have a more beneficial effect from losartan because they have more AngII in the aortic wall.24 Losartan, by blocking AT1R, decreases TGF-β production and blood pressure, as well as increases proinflammatory responses, myofibroblast differentiation, and production of reactive oxygen species (all AngII-mediated detrimental processes in the aortic wall) (Figure 2).26,27
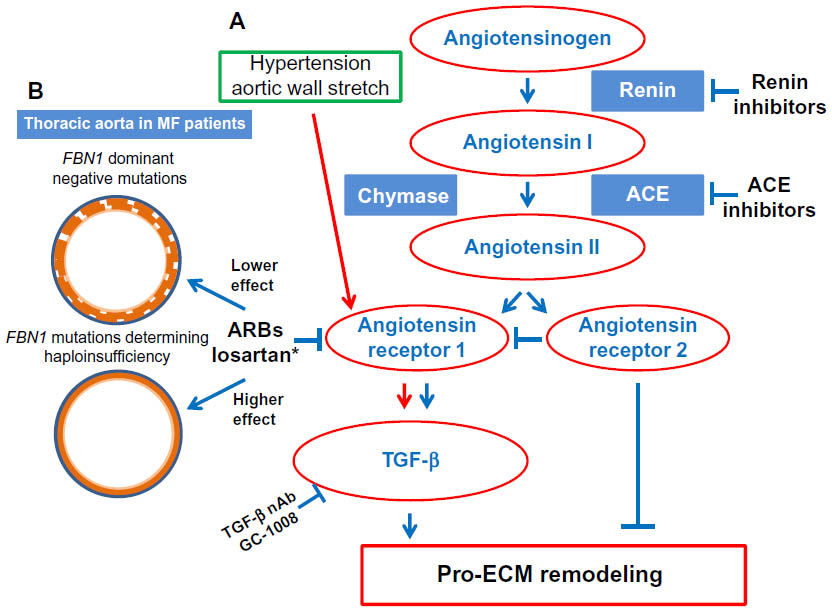 | Figure 2 Effects of drug therapies on thoracic aortic wall of MF patients. |
Our group28 reported plasmatic signs of oxidative stress in patients with MF syndrome, with a well-defined and statistically significant positive correlation between plasma protein carbonyl content as an expression of reactive oxygen species and clinical involvement (according to Ghent-1 criteria), thus suggesting that in MFS, the intensity of oxidative stress may be a marker of clinical severity. These data may be now partly associated with the blocking of AT1R. Other data will be required to confirm this hypothesis. It would be interesting to know whether each single MF patient with haploinsufficiency has beneficial effects from losartan and what are the results of the French trial based on the two groups of patients separated on the base of qualitative vs quantitative mutations (Figure 2).
Thoracic aortic dissection (TAD) may occur as a result of TAA; nonetheless, aortic diameter is an unreliable predictor of TAA rupture or dissection because a significant proportion of TAD happens at diameters well below the suggested threshold for prophylactic aortic surgery. Thus, future prospective studies should include a search for potential predictors of TAD.
Détaint et al29 reported that, by the age of 60 years, ~100% of patients with MF syndrome will have developed aortic root dilatation at varying degrees and three quarters of them would have undergone aortic root replacement on the basis of increased aortic diameter to critical levels and/or symptomatic aortic valve insufficiency (or Stanford type “A” dissection). Regarding life expectancy and quality of life of patients with MF syndrome, an optimal timing of these operations is highly required to avoid complications. Moreover, cardiac surgical interventions performed unreasonably early (with smaller aortic diameters than that stated by the European Society of Cardiology and the American College of Cardiology) in young adulthood or childhood may lead, in certain cases, to an increase in the probability of a reoperation. Furthermore, when a mechanical valve is implanted, lifelong anticoagulation therapy is required, thereby worsening the quality of life further. TAD may also develop in patients with the aorta having normal or smaller diameters than those reported in the current surgical criteria. These data suggest that good indicators or biomarkers of TAADs are not available at the moment.30 Recently, increased total serum levels of TGF-β1 have been investigated as a potential marker for TAAD in MFS. Franken et al26 analyzed 99 patients with MF syndrome carrying an FBN1 mutation. After a follow-up of 38 months, they concluded that elevated TGF-β levels correlate with 1) larger aortic root diameters, 2) faster aortic root growth, and 3) earlier aortic root surgery when TGF-β1 level is >140 pg/mL.
Hillebrand et al31 have extended the analysis of total serum TGF-β1 to genetic aortic syndromes. They found that TGF-β1 levels were elevated in the following groups: 1) patients with MF syndrome with causative FBN1 mutation and bicuspid aortic valve (BAV) patients with causative NOTCH1 mutation vs patients without mutations; 2) patients with BAV with causative NOTCH1 mutations vs patients with ACTA2/MYH11 mutations with tricuspid aortic valves; 3) patients with MFS or BAV vs patients without genetic aortic syndromes; 4) patients with MF syndrome with FBN1 in-frame mutations.
Recently, TGF-β has been used as a therapeutic biomarker for effectiveness of losartan on the AoDR in patients with MF syndrome.27 Baseline plasma TGF-β levels in 22 healthy controls and in 99 patients with MF syndrome (121 pg/mL vs 54 pg/mL; P=0.006) were measured. After 1 month of treatment with losartan, TGF-β levels were remeasured in 42 patients with MF syndrome. AoDR was assessed by MRI at baseline and after 3-year follow-up. As a result, 15 out of 42 patients with higher baseline TGF-β levels (189 pg/mL vs 54 pg/mL in 27/42 patients) were found to respond to losartan therapy with a decrease of the plasma TGF-β level and an increase of AoDR.26 These data suggest that increased AngII, but not elevated TGF-β, is the initiator of aorta dilatation. TGF-β now seems to be a marker of aortic damage instead of being considered the initial cause of aortic dilatation.27
Other cardiovascular manifestations
Mitral valve elongation and myxomatous thickening usually lead to mitral valve prolapse (MVP) and regurgitation. MVP is the most common valvular abnormality in MFS, with a highly variable reported prevalence of 28%–75% in comparison with an overall prevalence of 2.4% in the general population.32 Noteworthy, in children, MVP can be complicated by severe mitral regurgitation and myocardial dysfunction, resulting in heart failure.33 Surgical repair of MVP can be performed in patients with MF syndrome who develop severe regurgitation; however, they have an increased risk of developing a TAD, secondary to the rapid postoperative increase in cardiac output.34 Neonatal MF patients may present valvular insufficiency, atrial and ventricular septal defects, and congestive heart failure.35,36 Left ventricular (LV) systolic and diastolic dysfunction in adult MF patients is usually considered to be secondary to valvular insufficiency and LV volume overload. Increased arterial stiffness may contribute to cardiac dysfunction by altering the hemodynamic load on the LV. Moreover, researchers37 have identified primary heart disease in the absence of MVP or TAA, suggesting an intrinsic problem with myocardial function in MFS. In asymptomatic patients with MF syndrome, early primary LV dysfunction can be detected by new echo techniques.38 The LV dysfunction has been found to be related to the severity of gene mutation, suggesting possible primary cardiomyopathy in patients with MF syndrome. Follow-up studies with regard to the prognostic value of these observations are warranted.38
Ocular manifestations
EL is one of the five criteria used to diagnose MFS, while myopia >3 diopters (D) belongs and contributes to the SF criteria (Table 1). They may both trigger complications such as retinal detachment, glaucoma, and amaurosis.2,6
Systemic features
The SFs include chest, vertebral, and feet skeletal deformities; excessive elongation of upper and lower limbs; and altered ratios among the body’s segments (Table 1). Moreover, apical pulmonary blebs predisposing to spontaneous pneumothorax, striae distensae, MVP, myopia >3 D, and dural ectasia (DE) are also SF indicators (Table 1).2 The presence of SFs with an increasing score characterize the following clinical phenotypes: ≤4: nonspecific CTDs; 5–6: MASS syndrome; ≥7: potential Marfan; ≤4+ MVP: MVP syndrome.
Dural ectasia
Among the diagnostic features, DE is now (Ghent-2 nosology) considered only one manifestation, a part of the SFs2 with a score of two, while earlier (Ghent-1 criteria),4 it was considered a major criterion in the diagnosis of MFS. This change is due to the fact that DE may also be present in neurofibromatosis type 1 (NF1), Ehlers–Danlos syndrome (EDS), Loeys–Dietz syndrome (LDS), and probably is present in other CTDs. DE usually is localized in the lumbar or sacral spine in MFS, while in EDS, it is present also in the dorsal spine. It is characterized by a widening of the spinal canal, a posterior scalloping of the vertebral body, an increased thinning of the cortex of pedicles and laminae, a widening of the neural foramina, or the presence of a meningocele.4 Anterior sacral meningocele is known to be a consequence of DE in MFS. Two cases initially described as nongynecological pelvic masses highlight the clinical difficulty in both the diagnosis and interpretation of the classic radiological findings.39 Although, at present, there is no standardized method for the diagnosis of DE, two qualitative4,40 and one quantitative41 methods have been reported. DE prevalence has been investigated by MRI in MFS, whereby 92% (76 out of 82 patients) had DE starting from the young age of 12 years.40 A higher capacity for DE detection was attributed to MRI compared to CT.4,42 Recently, DE prevalence in LDS has also been investigated with one of these three methods by comparing LDS to MFS and healthy controls. The qualitative method applied to patients with LDS showed 40% of DE, the quantitative method 1 showed up 50%, and quantitative method 2 identified 70%. The corresponding prevalences in patients with MF syndrome were 50%, 75%, and 85% and prevalences in controls were 0%, 0%, and 5%. Both patients with LDS and MFS had a significantly wider dura compared to controls.43 DE is also reported to be aspecific and abundantly present: 58% (19/33 patients) even in patients with MF syndrome in whom FBN1/TGFBR1 and TGFBR2 mutations were excluded and who did not reach the diagnosis of MFS.44 In our experience, it was still useful because its presence allowed us to make an MFS diagnosis at a younger age than usual.45
Classic clinical manifestations of DE in MFS are represented by low back pain, headache, proximal leg pain, weakness and numbness above and below the knee, and genital/rectal pain. Their daily/several-times-per-week expression ranges between moderate and severe, is exacerbated by upright posture, and is not always relieved by recumbency.46
Old and new emerging genetic and biochemical information
MFS is due to mutations in the FBN1 gene, which at the moment is known to be the only major gene.47 More than 2,000 mutations in this gene have been published.48–50 In addition to dominant mutations, some recessive mutations in the FBN1 gene have been detected,51,52 therefore, autosomal recessive transmission has to be always kept in mind, especially in sporadic cases.
MFS displays a high interfamilial and intrafamilial phenotype variability that suggests the presence of important modifying genes. At present, we know that in MFS, mainly transmitted in an autosomal dominant manner, the following conditions may act as modifier allele/gene: a) the normal allele of FBN1 gene,53 b) mild mutations in FBN1, TGFBR1 and TGFBR2 alleles,54,55 c) mild mutations in other genes that may involve/affect a single organ/apparatus or more, or a single clinical feature.56,57
In particular, our group studied patients subdivided into three subgroups based on the severity of cardiovascular manifestations: subgroup A: no involvement; subgroup B: mild involvement; and subgroup C: aortic dilatation or aortic dissection.56 Total homocysteine (tHcy) was significantly higher in subgroup C than in subgroup B. In subgroup C patients with dissection, tHcy was higher than in those without dissection. In subgroup C, the prevalence of 677T homozygotes was higher, but not significantly so, than in the subgroup B. In patients with dissection, the prevalence of 677T homozygotes was significantly higher than in those without dissection and higher than in subgroup B. In the logistic regression analysis, severe cardiovascular manifestations and aortic dissection in patients with MF syndrome were associated with plasma levels of tHcy. These data indicate an association between the severity of the cardiovascular manifestations, in particular aortic dissection, and elevated tHcy levels. This suggests an important role for tHcy in determining phenotypic variability in patients with MF syndrome.56
Recently, Radonic et al58 showed that both TGF-β and inflammation are upregulated in patients with MFS. They analyzed transcriptome-wide gene expression in patients with MF syndrome and levels of TGF-β and various cytokines in their plasma. Within the MF population, increased plasma levels of TGF-β were found, especially in patients with MF syndrome with aortic root dilatation, when compared to patients with MF syndrome with normal aorta. Microarray data showed that increased expression of inflammatory genes was associated with major clinical features within the patients with MF syndrome group, namely, severity of the aortic root dilatation (HLA-DRB1 and HLA-DRB5 genes), ocular lens dislocation (RAET1L, CCL19, and HLA-DQB2), and specific skeletal features (HLA-DRB1, HLA-DRB5, and GZMK). Patients with progressive aortic disease had higher levels of macrophage colony stimulating factor in blood. When comparing MF aortic root vessel wall with non-MF aortic root wall, increased number of CD4+ T-cells was found in the media and increased number of CD8+ T-cells in the adventitia of the patients with MF syndrome.58
Other potential causes of aortic wall damage and potential biomarkers are lysyl oxidase (LOX) enzyme and LOX isoforms (LOXLs), important in ECM overstructure formation and, in particular, for the correct structure of the aortic wall. Recently, studies on LOX, an enzyme involved in the formation of covalent cross-linking of collagens and elastin essential for ECM maturation, have highlighted its importance in the correct formation and maintenance of the aortic wall.59 LOX catalyzes the conversion of lysine and hydroxylysine groups of collagen and elastin into highly reactive aldehydes, which eventually condense with other aldehyde groups or intact lysine residues to form a variety of inter- and intrachain cross-linkages.59 The cross-links provide the tensile strength and elastic properties for various ECMs, including vascular walls. Interestingly, both genetic and pharmacological inactivation of LOX enzymatic activity is linked to TAA, cardiovascular dysfunction, perinatal death in mice, and abdominal aortic aneurysms.59,60 Recently, Busnadiego et al61 showed elevated LOX expression levels in aorta of patients with MF syndrome. Their work supported a protective role for some LOX enzymes in the development of aneurysms in MFS, as shown in the tissues of an MFS mouse model (Fbn1C1039G/+). LOX isoforms may act as potential modifiers of the MFS phenotype. In support of these results, a case report62 described a spontaneous arterial wall dissection associated with ECM disorganization and highly reduced LOX expression. Noteworthy, LOX decrease/inhibition affects mainly the ascending aorta compared to the aortic root. Ascending aorta ectasia is highly associated with BAV, wherein hemodynamic derangements are thought to cause alterations in the ascending aorta, suggesting that both collagen and collagen cross-linking play an important role in providing the ascending aorta with the mechanical properties required to sustain the hemodynamic load. Moreover, the ascending aorta in mice treated with LOX inhibitors, such as AngII, goes through aneurysm and rupture only in presence of hypertension. This observation confirms that the formation of aortic ascending ectasia requires a concomitant structural alteration of the aortic wall, associated with an increment of the hemodynamic loading.63 Taken together, this model may be conceived as an association of MFS and BAV, a human pathology first reported by our group,64 which becomes a more severe aortic involvement, with a higher and earlier requirement of surgical intervention. A higher overlap exists between MFS and BAV because some MFS/BAV and two patients with BAV in whom MFS was clinically excluded display mutations in the FBN1 gene.64–66
High levels of LOX and LOX-like 1 (LOXL1) expression might stabilize aneurysms, while low levels may be associated with increased risk of aortic rupture. LOX isoforms are secreted out of the cells; therefore, they may be measured in serum samples as markers of tissue expression and as a prognostic biomarker in MFS. Studies are in progress to verify this hypothesis.67
Regarding the still-discussed utility of losartan in MFS, analyzed through clinical trials,14,18–20 the Dutch group in a recent article shows that LOX inhibition results in an overactivation of both canonical (SMAD) and noncanonical (ERK) TGF-β-signaling pathways. They also show that AT1R antagonism with losartan prevents β aminopropionitrile (a compound shown to irreversibly inhibit LOX activity in vitro and in vivo)-mediated aorta deterioration while restoring basal levels of activation of TGF-β-dependent pathways. The fact that losartan shows effectiveness in preventing aortic disease in MF mice, with or without inhibition of LOX activity, positions the AT1R as a central receptor in the pathogenetic route leading to aortic aneurysms. Current hypotheses on the pathogenesis of aortic aneurysm in MFS invoke mechanosignaling-dependent pathways, such as AngII/AT1R, coupled to downstream TGF-β signaling as a major factor responsible for disease progression.68–70
According to this model, LOX inhibition may aggravate the aortic pathology by worsening the aortic phenotype through weakening of the ECM; this, in turn, activates the hemodynamic stress that increases TGF-β signaling and TGF-β-dependent aortic responses. Properly cross-linked collagen protects from events leading to pathological remodeling.71 Alternatively, recent in vitro and in vivo evidences suggest a direct role for LOX in the control of active TGF-β through a regulatory TGF-β/LOX feedback loop.72
We also know that mutations in many other genes, coding for proteins of TGF-β signaling (TGFBRs, SMADs), vascular smooth muscle cell contractility (MYH11, ACTA2), and other ECM proteins (COL3, FBN2) cause clinical phenotypes overlapping with MFS. For these reasons (and others), many groups are switching from the analysis of single genes to the analysis of multiple genes, also considering that the time and cost of the analysis will be lower, although the cost of personnel will be higher. Recently, two papers reported the setting up of a next-generation sequencing (NGS) panel for MF and MF-like syndromes’ assay73 and a second one for syndromic and nonsyndromic TAAs.74 Wooderchak–Donahue et al73 used a panel of ten genes: ACTA2 (TAAD), MYH11 (TAD with patent ductus arteriosus), COL3A1 (EDS), FBN1 (MFS, MASS, and TAAD), FBN2 (CCA), SMAD3 (LDS type 3 with osteoarthritis), TGFBR1, TGFBR2 (LDS type 1 and 2), MYLK (TAAD), and SLC2A10 (ATS). NGS results were analyzed and variants validated by Sanger sequencing. Of a total of 175 patients, 18 showed a pathogenic mutation and 32 had a variant of uncertain significance. Most (72%) of the pathogenic variants were detected in the FBN1 gene. A novel large SMAD3 duplication and FBN1 deletion were detected. The mutation detection displays a low rate, but the authors propose to add other genes; moreover, revision of the uncertain mutations will allow for the detection of further 10% of mutations.
Proost et al74 set up the analysis of 14 genes for TAA: ACTA2, COL3A1, EFEMP2, FBN1, FLNA, MYH11, MYLK, NOTCH1, SKI, SLC2A10, SMAD3, TGFB2, TGFBR1, and TGFBR2. They first analyzed 100 patients with MF syndrome and detected FBN1 mutations, 44 of which were novel, in 90 patients. By multiplex ligation-dependent probe amplification analysis, they found six other large deletions in the FBN1 gene. They excluded the presence of FBN1, TGFBR1, and TGFBR2 by direct Sanger sequencing analysis in the remaining four patients. Then, they investigated 55 patients with TAA and detected 15 mutations: seven in FBN1, two in NOTCH1, and one in each of the following genes: ACTA2, COL3A1, TGFBR1, MYLK, SMAD3, and SLC2A10 (homozygous). These last six variants were rare and all were classified as rare. These results are very encouraging, and due to the rapid improvement of high-throughput technologies and their methods of data analysis, they represent a big challenge in increasing the knowledge and improving the genetic diagnosis of MFS and related disorders. Overall, the negative aspect in these articles is that there are no complete and clear clinical data that allow the interpretation of the genetic results, and this is a problem common to many articles that do not allow performing a genotype–phenotype correlation. The same problem seems to affect the MFS databases.75
Conclusion and perspectives
On the whole, there are exciting and interesting data regarding the correlation between losartan therapy and different types of FBN1 mutations. The increase of disorders in differential diagnosis with MFS required the increase of FBN1 mutation analysis because the detection of FBN1 gene in a patient highly suspected to be affected by MFS may allow an early diagnosis, which in turn permits preventive therapies and more accurate clinical follow-up. New data will come out in the next few years regarding new major/modifier genes associated with MFS and related disorders, aided by improved techniques such as NGS, which will allow a better clinical follow-up and a more precise surgery timing. In addition, identification of new biochemical and genetic markers, as well as the extensive characterization of the biomarkers already detected, will help in a more precise prediction of some clinical events and their progression.
The detection of other pleiotropic clinical manifestations that can trigger complications, such as retinal detachment, glaucoma, or amaurosis caused by myopia or EL or intrapelvic meningocele, and dural leak with postural headache caused by DE,6 as well as the highly variable onset of clinical manifestations (0–60 years of age) and their unpredictable progression, require a wide clinical follow-up of patients with MF syndrome and of patients with related disorders such as MASS phenotype, MVP syndrome, and EL syndrome.76,77
Last but not the least, at present, it is desirable to frame the patients with MF syndrome in a more general psychosocial context. For example, many patients are anxious to reach a final diagnosis; therefore, the detection of a pathogenic FBN1 mutation or the exclusion of an FBN1 mutation may help in reducing anxiety. Moreover, more attention should be given to correct lifestyles. Thus, the choice of the right physical exercise and/or the correct diet also depending on the clinical manifestations may help in improving prevention and a long-term enhancement of quality of life.
Acknowledgment
We thank Ms Stella Sagaria for careful and extensive English revision of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Ramirez F, Dietz HC. Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev. 2007;17(3):252–258. | |
Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476–485. | |
Beighton P, de Paepe A, Danks D, et al. International nosology of heritable disorders of connective tissue, Berlin, 1986. Am J Med Genet. 1988;29(3):581–594. | |
De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62(4):417–426. | |
Hoffjan S. Genetic dissection of Marfan syndrome and related connective tissue disorders: an update 2012. Mol Syndromol. 2012;3(2):47–58. | |
von Kodolitsch Y, De Backer J, Schüler H, et al. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl Clin Genet. 2015;8:137–155. | |
Pyeritz RE. The Marfan syndrome. Annu Rev Med. 2000;51:481–510. | |
Pyeritz RE. Marfan syndrome and related disorders. Ann Thorac Surg. 2008;86(1):335–336. | |
Aldrich HR, Labarre RL, Roman MJ, Rosen SE, Spitzer MC, Devereux RB. Color flow and conventional echocardiography of the Marfan syndrome. Echocardiography. 1992;9(6):627–636. | |
Dormand H, Mohiaddin RH. Cardiovascular magnetic resonance in Marfan syndrome. J Cardiovasc Magn Reson. 2013;15:33. | |
Summers KM, Nataatmadja M, Xu D, et al. Histopathology and fibrillin-1 distribution in severe early onset Marfan syndrome. Am J Med Genet A. 2005;139(1):2–8. | |
Wu D, Shen YH, Russell L, Coselli JS, LeMaire SA. Molecular mechanisms of thoracic aortic dissection. J Surg Res. 2013;184(2):907–924. | |
Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med. 1994;330(19):1335–1341. | |
Milleron O, Arnoult F, Ropers J, et al. Marfan Sartan: a randomized, double-blind, placebo-controlled trial. Eur Heart J. 2015;36(32):2160–2166. | |
Baumgartner H, Bonhoeffer P, De Groot NM, et al; Task Force on the Management of Grown-up Congenital Heart Disease of the European Society of Cardiology (ESC); Association for European Paediatric Cardiology (AEPC); ESC Committee for Practice Guidelines (CPG). ESC guidelines for the management of grown-up congenital heart disease (new version 2010). Eur Heart J. 2010;31(23):2915–2957. | |
Detaint D, Aegerter P, Tubach F, et al. Rationale and design of a randomized clinical trial (Marfan Sartan) of angiotensin II receptor blocker therapy versus placebo in individuals with Marfan syndrome. Arch Cardiovasc Dis. 2010;103(5):317–325. | |
Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–121. | |
Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358(26):2787–2795. | |
Groenink M, den Hartog AW, Franken R, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J. 2013;34(45):3491–3500. | |
Lacro RV, Dietz HC, Sleeper LA, et al; Pediatric Heart Network Investigators. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014;371(22):2061–2071. | |
Chiu HH, Wu MH, Wang JK, et al. Losartan added to beta-blockade therapy for aortic root dilation in Marfan syndrome: a randomized, open-label pilot study. Mayo Clin Proc. 2013;88(3):271–276. | |
Jost CHA, Greutmann M, Connolly HM, et al. Medical treatment of aortic aneurysms in Marfan syndrome and other heritable conditions. Curr Cardiol Rev. 2014;10(2):161–171. | |
Loeys BL. Angiotensin receptor blockers: a panacea for Marfan syndrome and related disorders? Drug Discov Today. 2015;20(2):262–266. | |
Franken R, den Hartog AW, Radonic T, et al. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ Cardiovasc Genet. 2015;8(2):383–388. | |
Jensen SA, Iqbal S, Bulsiewicz A, Handford PA. A microfibril assembly assay identifies different mechanisms of dominance underlying Marfan syndrome, stiff skin syndrome and acromelic dysplasias. Hum Mol Genet. 2015;24(15):4454–4463. | |
Franken R, den Hartog AW, de Waard V, et al. Circulating transforming growth factor-β as a prognostic biomarker in Marfan syndrome. Int J Cardiol. 2013;168(3):2441–2446. | |
Franken R, Radonic T, den Hartog AW, et al; COMPARE Study Group. The revised role of TGF-β in aortic aneurysms in Marfan syndrome. Neth Heart J. 2015;23(2):116–121. | |
Fiorillo C, Becatti M, Attanasio M, et al. Evidence for oxidative stress in plasma of patients with Marfan syndrome. Int J Cardiol. 2010;145(3):544–546. | |
Détaint D, Faivre L, Collod-Beroud G, et al. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur Heart J. 2010;31(18):2223–2229. | |
Song HK, Kindem M, Bavaria JE, et al; Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions Consortium. Long-term implications of emergency versus elective proximal aortic surgery in patients with Marfan syndrome in the Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions Consortium registry. J Thorac Cardiovasc Surg. 2012;143(2):282–286. | |
Hillebrand M, Millot N, Sheikhzadeh S, et al. Total serum transforming growth factor-β1 is elevated in the entire spectrum of genetic aortic syndromes. Clin Cardiol. 2014;37(11):672–679. | |
Pyeritz RE, Wappel MA. Mitral valve dysfunction in the Marfan syndrome. Clinical and echocardiographic study of prevalence and natural history. Am J Med. 1983;74(5):797–807. | |
Morse RP, Rockenmacher S, Pyeritz RE, et al. Diagnosis and management of infantile Marfan syndrome. Pediatrics. 1990;86(6):888–895. | |
Schroeyers P, Wellens F, Degrieck I, et al. Aggressive primary treatment for poststernotomy acute mediastinitis: our experience with omental- and muscle flaps surgery. Eur J Cardiothorac Surg. 2001;20(4):743–746. | |
Stheneur C, Faivre L, Collod-Béroud G, et al. Prognosis factors in probands with an FBN1 mutation diagnosed before the age of 1 year. Pediatr Res. 2011;69(3):265–270. | |
Fujiseki Y, Okuno K, Tanaka M, Shimada M, Takahashi M, Kawanishi K. Myocardial involvement in the Marfan syndrome. Jpn Heart J. 1985;26(6):1043–1050. | |
Alpendurada F, Wong J, Kiotsekoglou A, et al. Evidence for Marfan cardiomyopathy. Eur J Heart Fail. 2010;12(10):1085–1091. | |
Abd El Rahman M, Haase D, Rentzsch A, et al. Left ventricular systolic dysfunction in asymptomatic Marfan syndrome patients is related to the severity of gene mutation: insights from the novel three dimensional speckle tracking echocardiography. PLoS One. 2015;10(4):e0124112. | |
Voyvodic F, Scroop R, Sanders RR. Anterior sacral meningocele as a pelvic complication of Marfan syndrome. Aust N Z J Obstet Gynaecol. 1999;39(2):262–265. | |
Fattori R, Nienaber CA, Descovich B, et al. Importance of dural ectasia in phenotypic assessment of Marfan’s syndrome. Lancet. 1999;354(9182):910–913. | |
Ahn NU, Sponseller PD, Ahn UM, et al. Dural ectasia in the Marfan syndrome: MR and CT findings and criteria. Genet Med. 2000;2(3):173–179. | |
De Paepe A. Dural ectasia and the diagnosis of Marfan’s syndrome. Lancet. 1999;354(9182):878–879. | |
Kono AK, Higashi M, Morisaki H, Morisaki T, Naito H, Sugimura K. Prevalence of dural ectasia in Loeys-Dietz syndrome: comparison with Marfan syndrome and normal controls. PLoS One. 2013;8(9):e75264. | |
Sheikhzadeh S, Rybczynski M, Habermann CR, et al. Dural ectasia in individuals with Marfan-like features but exclusion of mutations in the genes FBN1, TGFBR1 and TGFBR2. Clin Genet. 2011;79(6):568–574. | |
Attanasio M, Pratelli E, Porciani MC, et al. Dural ectasia and FBN1 mutation screening of 40 patients with Marfan syndrome and related disorders: role of dural ectasia for the diagnosis. Eur J Med Genet. 2013;56(7):356–360. | |
Foran JR, Pyeritz RE, Dietz HC, Sponseller PD. Characterization of the symptoms associated with dural ectasia in the Marfan patient. Am J Med Genet A. 2005;134A(1):58–65. | |
Cook JR, Carta L, Galatioto J, Ramirez F. Cardiovascular manifestations in Marfan syndrome and related diseases; multiple genes causing similar phenotypes. Clin Genet. 2015;87(1):11–20. | |
Faivre L, Collod-Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81(3):454–466. | |
Faivre L, Masurel-Paulet A, Collod-Béroud G, et al. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics. 2009;123(1):391–398. | |
Attanasio M, Lapini I, Evangelisti L, et al. FBN1 mutation screening of patients with Marfan syndrome and related disorders: detection of 46 novel FBN1 mutations. Clin Genet. 2008;74(1):39–46. | |
de Vries BB, Pals G, Odink R, Hamel BC. Homozygosity for a FBN1 missense mutation: clinical and molecular evidence for recessive Marfan syndrome. Eur J Hum Genet. 2007;15(9):930–935. | |
Hilhorst-Hofstee Y, Rijlaarsdam ME, Scholte AJ, et al. The clinical spectrum of missense mutations of the first aspartic acid of cbEGF-like domains in fibrillin-1 including a recessive family. Hum Mutat. 2010;31(12):E1915–E1927. | |
Hutchinson S, Furger A, Halliday D, et al. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet. 2003;12(18):2269–2276. | |
Lucarini L, Evangelisti L, Attanasio M, et al. May TGFBR1 act also as low penetrance allele in Marfan syndrome? Int J Cardiol. 2009;131(2):281–284. | |
Lucarini L, Sticchi E, Sofi F, et al. ACE and TGFBR1 genes interact in influencing the susceptibility to abdominal aortic aneurysm. Atherosclerosis. 2009;202(1):205–210. | |
Giusti B, Porciani MC, Brunelli T, et al. Phenotypic variability of cardiovascular manifestations in Marfan syndrome. Possible role of hyperhomocysteinemia and C677T MTHFR gene polymorphism. Eur Heart J. 2003;24(22):2038–2045. | |
Giusti B, Marcucci R, Lapini I, et al. Role of hyperhomocysteinemia in aortic disease. Cell Mol Biol (Noisy-le-grand). 2004;50(8):945–952. | |
Radonic T, de Witte P, Groenink M, et al. Inflammation aggravates disease severity in Marfan syndrome patients. PLoS One. 2012;7(3):e32963. | |
Mäki JM, Räsänen J, Tikkanen H, et al. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106(19):2503–2509. | |
Remus EW, O’Donnell RE Jr, Rafferty K, et al. The role of lysyl oxidase family members in the stabilization of abdominal aortic aneurysms. Am J Physiol Heart Circ Physiol. 2012;303(8):H1067–H1075. | |
Busnadiego O, Gorbenko Del Blanco D, González-Santamaría J, et al. Elevated expression levels of lysyl oxidases protect against aortic aneurysm progression in Marfan syndrome. J Mol Cell Cardiol. 2015;85:48–57. | |
Sibon I, Sommer P, Lamaziere JM, Bonnet J. Lysyl oxidase deficiency: a new cause of human arterial dissection. Heart. 2005; 91(5):e33. | |
Losenno KL, Goodman RL, Chu MW. Bicuspid aortic valve disease and ascending aortic aneurysms: gaps in knowledge. Cardiol Res Pract. 2012;2012:145202. | |
Nistri S, Porciani MC, Attanasio M, Abbate R, Gensini GF, Pepe G. Association of Marfan syndrome and bicuspid aortic valve: frequency and outcome. Int J Cardiol. 2012;155(2):324–325. | |
Pepe G, Nistri S, Giusti B, et al. Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (BAV) without Marfan syndrome. BMC Med Genet. 2014;15:23. | |
De Cario R, Sticchi E, Giusti B, et al. Bicuspid aortic valve syndrome and fibrillinopathies: potential impact on clinical approach. Int Cardiovasc Forum J. 2014;1(4):167–174. | |
Rimar D, Rosner I, Nov Y, et al. Brief report: lysyl oxidase is a potential biomarker of fibrosis in systemic sclerosis. Arthritis Rheumatol. 2014;66(3):726–730. | |
Cook JR, Carta L, Bénard L, et al; GenTAC Registry Consortium. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest. 2014;124(3):1329–1339. | |
Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Cell biology. Dysfunctional mechanosensing in aneurysms. Science. 2014;344(6183):477–479. | |
Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116(8):1448–1461. | |
Atsawasuwan P, Mochida Y, Katafuchi M, et al. Lysyl oxidase binds transforming growth factor-beta and regulates its signaling via amine oxidase activity. J Biol Chem. 2008;283(49):34229–34240. | |
Kutchuk L, Laitala A, Soueid-Bomgarten S, et al. Muscle composition is regulated by a Lox-TGFβ feedback loop. Development. 2015;142(5):983–993. | |
Wooderchak-Donahue W, VanSant-Webb C, Tvrdik T, et al. Clinical utility of a next generation sequencing panel assay for Marfan and Marfan-like syndromes featuring aortopathy. Am J Med Genet A. 2015;167(8):1747–1757. | |
Proost D, Vandeweyer G, Meester JA, et al. Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Hum Mutat. 2015;36(8):808–814. | |
Levenson D. Marfan syndrome database information unreliable for diagnoses. Am J Med Genet A. 2015;167(7):vii–viii. | |
Pepe G, Lapini I, Evangelisti L, et al. Is ectopia lentis in some cases a mild phenotypic expression of Marfan syndrome? Need for a long-term follow-up. Mol Vis. 2007;13:2242–2247. | |
Faivre L, Collod-Beroud G, Adès L, et al. The new Ghent criteria for Marfan syndrome: what do they change? Clin Genet. 2012; 81:433–442. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.