Back to Journals » The Application of Clinical Genetics » Volume 10
Maple syrup urine disease: mechanisms and management
Authors Blackburn PR, Gass JM, Vairo FP, Farnham KM, Atwal HK, Macklin S ![]() , Klee EW, Atwal PS
, Klee EW, Atwal PS ![]()
Received 1 June 2017
Accepted for publication 14 July 2017
Published 6 September 2017 Volume 2017:10 Pages 57—66
DOI https://doi.org/10.2147/TACG.S125962
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Patrick R Blackburn,1,2,* Jennifer M Gass,1,* Filippo Pinto e Vairo,3,4,* Kristen M Farnham,5 Herjot K Atwal,6 Sarah Macklin,5 Eric W Klee,3,4,7,8 Paldeep S Atwal1,5
1Center for Individualized Medicine, 2Department of Health Sciences Research, Mayo Clinic, Jacksonville, FL, 3Center for Individualized Medicine, 4Department of Health Sciences Research, Mayo Clinic, Rochester, MN, 5Department of Clinical Genomics, Mayo Clinic, Jacksonville, FL, 6Department of Pharmacy, Mayo Clinic, Rochester, MN, 7Department of Clinical Genomics, Mayo Clinic, Jacksonville, FL, 8Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, USA
*These authors contributed equally to this work
Abstract: Maple syrup urine disease (MSUD) is an inborn error of metabolism caused by defects in the branched-chain α-ketoacid dehydrogenase complex, which results in elevations of the branched-chain amino acids (BCAAs) in plasma, α-ketoacids in urine, and production of the pathognomonic disease marker, alloisoleucine. The disorder varies in severity and the clinical spectrum is quite broad with five recognized clinical variants that have no known association with genotype. The classic presentation occurs in the neonatal period with developmental delay, failure to thrive, feeding difficulties, and maple syrup odor in the cerumen and urine, and can lead to irreversible neurological complications, including stereotypical movements, metabolic decompensation, and death if left untreated. Treatment consists of dietary restriction of BCAAs and close metabolic monitoring. Clinical outcomes are generally good in patients where treatment is initiated early. Newborn screening for MSUD is now commonplace in the United States and is included on the Recommended Uniform Screening Panel (RUSP). We review this disorder including its presentation, screening and clinical diagnosis, treatment, and other relevant aspects pertaining to the care of patients.
Keywords: maple syrup urine disease, BCKDHA, BCKDHB, DBT, newborn screening, alloisoleucine, branched-chain amino acids
Introduction
Maple syrup urine disease (MSUD, MIM #248600) is an autosomal recessive disease characterized by disruption of the normal activity of the branched-chain α-ketoacid dehydrogenase (BCKAD) complex, the second step in the catabolic pathway for the branched-chain amino acids (BCAAs) that include leucine, isoleucine, and valine. Pathogenic homozygous or compound heterozygous variants in BCKDHA (MIM #608348), BCKDHB (MIM #248611), DBT (MIM #248610), or DLD (MIM #238331), which form the catalytic subunits of BCKAD, can result in MSUD, which is characterized by neurological and developmental delay, encephalopathy, feeding problems, and a maple syrup odor to the urine. Patients with this disorder have elevations of branch chain ketoacids in the urine in addition to elevated BCAAs in the plasma. MSUD is amenable to treatment through dietary restriction of BCAAs, and with early treatment, patients typically have good clinical outcomes. MSUD is therefore included on the Recommended Uniform Screening Panel (RUSP), a list of actionable, early onset disorders for which screening is recommended for all newborns in the United States. The use of tandem mass spectrometry (MS/MS) in newborn screening (NBS) has helped facilitate early detection of and timely medical intervention for patients with MSUD, thus improving clinical outcomes in affected individuals. In this review, we will discuss the pathophysiology, clinical presentation, screening and diagnosis, as well as treatment of patients with MSUD with a particular focus on the management of adult patients.
Pathophysiology of MSUD
MSUD is a metabolic disorder caused by decreased function of the BCKAD enzyme complex. Biallelic pathogenic variants in the catalytic components of BCKAD decrease its activity thereby increasing BCAA levels and causing toxicity within skeletal muscle and brain tissue.1,2 BCAA catabolism is essential for normal physiological functions.3 The first step involves the conversion of leucine, isoleucine, and valine into their relevant α-ketoacids by branch-chain aminotransferase within the mitochondria (Figure 1). Unlike most other amino acid metabolism, the majority of this process does not take place in the liver but rather in skeletal muscle.3–5 BCAAs can be found in protein-rich diets and are among the nine amino acids essential for human life, playing important roles in protein synthesis and function, cellular signaling, and glucose metabolism.6,7
 | Figure 1 Overview of BCAA catabolic pathway. The BCAAs undergo transamination that is catalyzed by the branched-chain aminotransferase (BCAT) and requires α-ketoglutarate, leading to the production of the α-ketoacids KIC, KMV, and KIV. These intermediates then undergo oxidative decarboxylation, catalyzed by the BCKAD complex. Abbreviations: KIC, α-ketoisocaproic acid; KIV, α-ketoisovaleric acid; KMV, α-keto-β-methylvaleric acid; Glu, glutamate; BCAAs, branched-chain amino acids; BCKAD, branched-chain α-ketoacid dehydrogenase. |
During the second step in BCAA catabolism, the BCKAD complex initiates oxidative decarboxylation of α-ketoacids.4 This process results in the conversion of α-ketoacids into acetoacetate, acetyl-CoA, and succinyl-CoA as illustrated in Figure 1. The BCKAD complex is made up of several components, including subunits E1α and E1β, E2, and E3. Increased BCAA levels within the body due to pathogenic defects in these components cause MSUD, leading to a variety of symptoms mentioned above, including dysfunction of the immune system, skeletal muscle, and central nervous system (CNS).2–4 The generation of various mouse models has been used to study MSUD and the effects of dietary BCAA restriction.8,9 In these models, as well as MSUD patients, excessive amounts of BCAAs build up and can cause severe tissue damage if left untreated.10 Within the brain, BCAA metabolism functions to maintain glutamate levels.2,11,12 Glutamate serves as a neurotransmitter within the CNS and plays important roles in brain development and cognitive functions such as learning and memory. Disorders of BCAA metabolism can cause abnormalities in glutamate synthesis leading to various neurological problems in patients.13 Controlling plasma concentrations of BCAA levels is key to preventing these symptoms. Furthermore, the accumulation of leucine is highly neurotoxic.2 Elevated levels of leucine can affect water homeostasis within the subcortical gray matter causing swelling within the brain, alter nitrogen homeostasis further depleting glutamate levels, increase oxidative stress, and compete with other important amino acids within the CNS such as tyrosine, which is involved in protein signaling. In addition, there is evidence that α-ketoisocaproic acid, an intermediate in the metabolism of leucine, is a major neurotoxin contributing to the encephalopathic syndrome.
Although the specific outcomes of BCAA abnormalities have not been fully characterized, it is apparent that proper metabolism is critical for human health.4 While genetic variants in certain components of this pathway lead to MSUD, several other disorders have been associated with abnormal BCAA metabolism, including insulin resistance and type 2 diabetes mellitus, liver disease, and certain types of cancer, broadening its respective roles in human health.3,4
Clinical presentation of MSUD
Clinical presentation
There are five distinct clinical phenotypes of MSUD, with no good genotype–phenotype correlation. However, the MSUD forms can be categorized based on age at onset, severity of symptoms, response to thiamine supplementation, and biochemical findings (Table 1). Classic and E3-deficient MSUD typically present in the neonatal period, while the intermediate, intermittent, and thiamine-responsive forms may present at any time of life, with decompensations occurring during periods of illness or stress. The clinical presentation of MSUD depends on the BCKAD residual activity, although the phenotypic classification relies on the leucine tolerance and metabolic response to illness.
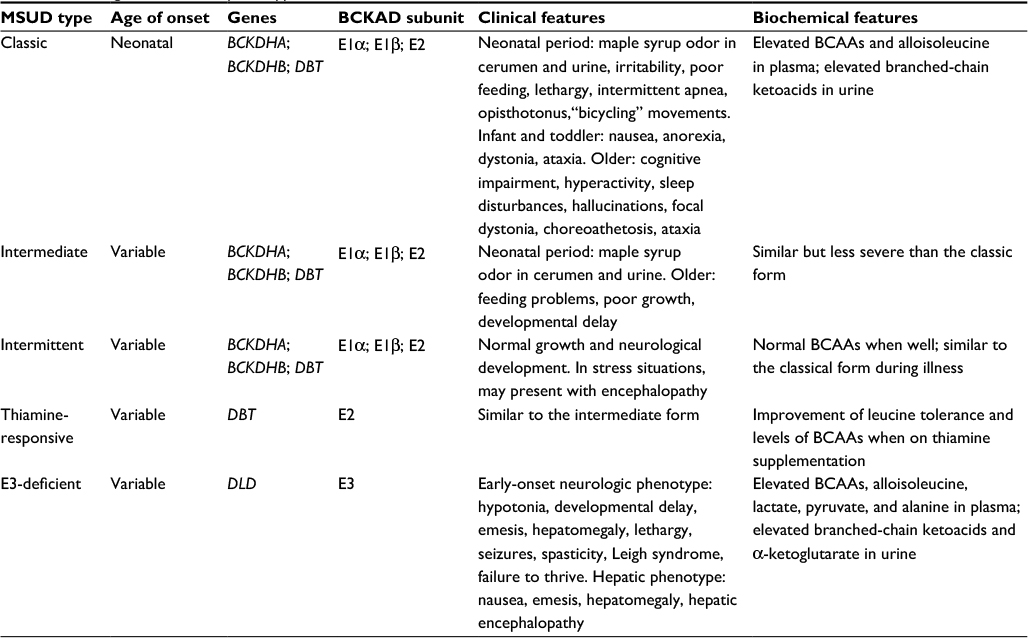 | Table 1 MSUD genetics, clinical phenotype, and biochemical features Abbreviations: BCKAD, branched-chain α-ketoacid dehydrogenase complex; BCAAs, branched-chain amino acids; MSUD, maple syrup urine disease. |
Individuals with the classic neonatal form have <2% of BCKAD enzymatic activity and present with maple syrup odor in cerumen shortly after birth and in urine during the first week of life. If untreated, the neonate may develop irritability, lethargy, poor feeding, apnea, opisthotonus, “bicycling” movements, followed by coma and early death due to brain edema.14,15 In older individuals, increased levels of leucine (leucinosis) causes epigastric pain, anorexia, vomiting, muscle fatigue, altered level of consciousness, psychiatric symptoms, movement disorders, and ataxia.16 Furthermore, clinical features similar to Wernicke encephalopathy have been reported in patients with acute decompensation.17 During periods of catabolism of endogenous protein, such as fever, infections, exercise, trauma, or surgery, individuals with MSUD can develop neurological deterioration due to acute leucine intoxication.18
The intermediate form of MSUD is characterized by up to 30% of BCKAD residual activity. These individuals may appear healthy during the neonatal period, although maple syrup odor in cerumen may be present. During the first years of life, they may experience feeding problems, poor growth, and intellectual disability, and are susceptible to similar neurologic features as individuals with the classic form.19
In a third form, individuals with intermittent MSUD are completely asymptomatic, having normal growth and neurological development, even on an unrestricted diet. During catabolic states, clinical and biochemical features of the classic form may arise in patients, and should be controlled similarly to individuals with the severe form.
Thiamine-responsive MSUD is a rare phenotype associated with pathogenic variants in DBT that encodes the BCKAD E2 subunit. Affected individuals present with symptoms similar to the intermediate form and usually require a combination of BCAA dietary restriction and thiamine supplementation. However, there has been one case of full responsiveness to thiamine without dietary restriction.20
Dihydrolipoamide dehydrogenase acts as the E3 subunit for three mitochondrial enzyme complexes: branched chain alpha-ketoacid dehydrogenase (BCKAD) complex, α-ketoglutarate dehydrogenase (αKGDH) complex, and pyruvate dehydrogenase (PDH) complex, so individuals with the E3-deficient MSUD form may present with a wide phenotypic spectrum, ranging from early-onset neurologic manifestations to adult-onset isolated liver disease. The most frequent is the severe form characterized by metabolic acidosis, encephalopathy, feeding difficulties, liver failure, and early death. Besides the biochemical hallmarks of MSUD, patients have increased levels of lactate, alanine, and α-ketoglutarate, which are related to mitochondrial dysfunction.21,22 Individuals with hepatic presentation may present with signs and symptoms at any age, and experience recurring episodes of hepatopathy that decrease with age and are often triggered by hypercatabolic states.23 The different clinical variants of MSUD are summarized in Table 1.
Medical management
NBS
Historically, MSUD has had a worldwide incidence, occurring in one in 185,000 live births,19 but is much more frequent in certain founder populations including the Old Order Mennonites of Pennsylvania, where it may be observed in as many as one in every 200 live births.24 Since MSUD is amenable to treatment through dietary restriction of BCAAs (or treatment with thiamine in the responsive forms) and outcomes are generally very good if it is detected and treated within the first few days of life, routine NBS for the disorder is recommended. MSUD screening is currently incorporated in NBS programs throughout the United States, five Canadian provinces, 22 European countries, two Latin American countries (Costa Rica and Uruguay), and eight countries in the Asia Pacific region.25
NBS for MSUD has been performed since 1964 when the bacterial inhibition assay for leucine on dried blood spots (Guthrie specimen) was introduced.26 Today, NBS for inborn errors of BCAA metabolism typically involves quantitative plasma amino acid profiling by MS/MS (Figure 2).27,28 If elevations of BCAAs are detected, the laboratory may perform additional studies including urine organic acid analysis by gas chromatography (GC)-MS/MS, dinitrophenylhydrazine (DNPH) test, quantitative plasma amino acids by liquid chromatography (LC)-MS/MS, and confirmatory molecular testing in suspected cases. Diagnosis of MSUD relies upon the presence of clinical, molecular, and biochemical features, including elevations of BCAAs and alloisoleucine in plasma as well as the presence of branched-chain α-hydroxyacids and branched-chain α-ketoacids (BCKAs) in urine. MS/MS testing for MSUD examines the leucine–isoleucine concentration plus its ratio with other amino acids including alanine, glutamate, glutamine, tryptophan, methionine, histidine, phenylalanine, and tyrosine.15,29 MS/MS cannot distinguish isobaric amino acids (ions with the same mass) including leucine, isoleucine, alloisoleucine, and hydroxyproline and MS/MS-positive cases may require second-tier testing such as LC-MS/MS (Figure 2).30
 | Figure 2 Overview of MSUD testing algorithm in NBS Abbreviations: BCAAs, branched-chain amino acids; MSUD, maple syrup urine disease; NBS, newborn screening |
The milder variant forms of MSUD are often missed by NBS due to normal leucine levels in the newborn period.31 Individuals with mild forms may have greater residual enzyme activities and they typically appear after the neonatal period, usually before the second year of life.19 All classic MSUD patients have pathognomonic elevations of alloisoleucine; however, even second-tier testing can miss variant cases, which may only be detectable during acute illness or metabolic crises.31 Second-tier testing can also distinguish MSUD from benign hydroxyprolinemia (4-hydroxy-l-proline oxidase deficiency, MIM #237000), which has no known clinical phenotype aside from elevated hydroxyproline. Since newborns with benign hydroxyprolinemia test positive for MSUD, they represent a false-positive result on NBS.
Biochemical workup
MSUD in a newborn may be suspected due to the presence of illness and/or an abnormal neonatal screening test result. The maple syrup-like odor characteristic of MSUD is usually present in the cerumen and may be detected as early as 12 hours after birth. Expanded NBS by MS/MS can show elevations of BCAAs on dried blood samples, but is unable to distinguish the isobaric amino acids as described above. In MSUD patients, plasma leucine, isoleucine, and valine are usually elevated, but may range from normal to slightly reduced. Elevations of BCAAs may be accompanied by decreased concentrations of other amino acids, leading to elevated concentration ratios (Figure 2).15,29 Subsequent quantitative plasma amino acid profiling that includes alloisoleucine detection provides the strongest evidence supporting a diagnosis of MSUD. Urine organic acid analysis by GC-MS/MS to detect BCKA (including α-ketoisocaproate, α-keto-β-methylisovalerate, α-ketoisovalerate) also provides supporting evidence for a diagnosis of MSUD. Elevations of BCKAs can be detected 48–72 hours after birth and correspond with massive elevations of BCAAs. The DNPH test, a nonquantitative screening test, can be performed in lieu of urine organic acid testing and detects α-ketoacids in urine. Ketonuria can serve as a surrogate marker suggestive of underlying metabolic instability and can be detected using urine test strips, particularly in resource poor settings that do not have access to the DNPH test or other analytic methods.
Monitoring in adult patients
The DNPH test reagent can also be used in the outpatient setting for detection of urine BCKAs during episodes of metabolic decompensation. If detected early, many MSUD patients can be treated at home by dietary restriction of BCAAs (leucine in particular) and the administration of high-calorie “sick day” formulas accompanied by frequent follow-up testing. Treatment details are outlined in the subsequent sections.
Molecular genetics of MSUD
Previous genetic studies have determined that MSUD is an autosomal recessive disease caused by pathogenic variants in genes encoding the E1α, E1β, E2, and E3 components of BCKAD. In 1989, the first genetic variants linked to MSUD were discovered in the E1α subunit (BCKDHA) of the BCKAD complex.32 Analysis of BCKDH activity in cultured fibroblasts showed that both the father and mother had levels that were 50% of the normal, while the patient’s levels were about 5% of normal.32 DNA sequencing then confirmed that each parent was a carrier for different pathogenic variants in BCKDHA and that the affected proband was compound heterozygous.32
Since then, over 190 different pathogenic or likely pathogenic variants have been identified in E1α and the other BCKAD components including E1β (BCKDHB), E2 (DBT), and E3 (DLD). All pathogenic variants that have been identified are homozygous or compound heterozygous variants within the same gene.33–35 Genetic testing is essential for a clinical diagnosis of MSUD and to determine which subunit is deficient, which may be helpful in the future for determining individualized therapies.36,37
Genetic counseling
Since MSUD is inherited in an autosomal recessive pattern, both parents of an individual with MSUD are most often unaffected carriers of the condition. A carrier for MSUD has one pathogenic variant in one of the previously described genes. When two carrier parents with mutations in the same gene reproduce, there is a 25% chance that any child will have MSUD. There is a 50% chance that any offspring will be a carrier and a 25% chance that they will be neither a carrier nor affected. There are many reproductive options available for couples who are both carriers of the same MSUD gene. Parents can choose to pursue natural conception with no genetic testing, consider adoption, or sperm or egg donation. Additionally, when carrier status has been molecularly confirmed, parents can also consider prenatal testing through chorionic villus sampling or amniocentesis or preimplantation genetic diagnosis with in vitro fertilization.
Special attention and care should be provided to young adults with MSUD who are beginning to take on more responsibilities regarding their health.38 The new pressure of managing MSUD without assistance from a parent is added to what can already be a challenging time. Young adults with MSUD may feel isolated from peers based on their diagnosis and may struggle with how to discuss the diagnosis in new relationships. Typical activities, like going out to a restaurant with friends, can be complicated by dietary restrictions. It can also be uncomfortable for some young adults to continue to receive care through a facility for children. Young adults may face more concrete barriers, like difficulty obtaining various insurance coverages as well. Not all individuals will have the same experiences, but incorporation of a mental health professional into the care of these adolescents may help with the transitional period.39
Medical nutrition management
Basics of management
One of the primary goals in treating MSUD is to manage diet by reducing BCAAs and provide adequate macronutrients to prevent catabolism and help maintain plasma BCAAs within targeted treatment ranges. Initiation of dietary management usually begins in the newborn period following a positive NBS result and clinical confirmation. Amounts of BCAAs are titrated in the diet by close monitoring of biochemical lab values and growth throughout infancy, childhood, and into adulthood (Table 2).40 Long-term treatment requires careful manipulation of calories, restriction of dietary BCAAs, and supplementation using a BCAA-free amino acid mixture (Table 3 shows a list of available products) to provide non-BCAAs and other nutrients for protein synthesis.41
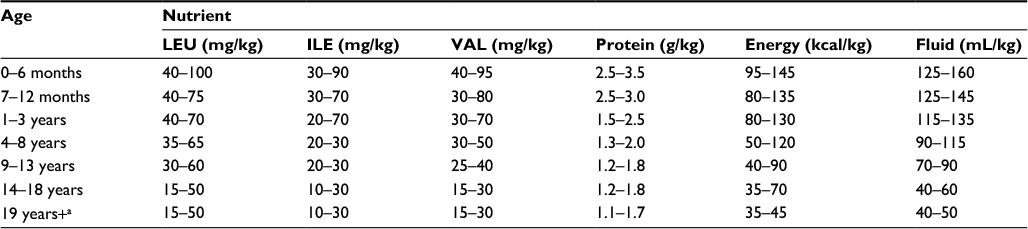 | Table 2 Recommended daily nutrient intake of BCAA, PRO, energy, and fluids for non-symptomatic individuals with MSUD Note: aMales and nonpregnant, non-lactating females. Abbreviations: BCAA, branched-chain amino acids; MSUD, maple syrup urine disease; PRO; protein; LEU, leucine; ILE, isoleucine; VAL, valine. |
 | Table 3 Selected medical foods Notes: aContains L-amino acids (without BCAA), as well as fat, carbohydrate, vitamins, and minerals; bcontains L-amino acids (without BCAA) low in or devoid of fat, carbohydrate, vitamins, or minerals; cNutricia North America, Rockville, MD, USA; dAbbott Nutrition, Abbott Park, IL, USA; eMead Johnson Nutrition, Glenview, IL, USA; fCambrooke Therapeutics, Ayer, MA, USA; gVitaflo USA, Alexandria, VA, USA. Abbreviations: BCAA, branched-chain amino acids; MSUD, maple syrup urine disease. |
Nutrition management consists of BCAA-free medical food specially formulated to provide 80%–90% of protein needs, and a majority of energy and micronutrient necessities throughout life.42 Leucine requirements are usually achieved using breast milk or infant formula. Leucine from breast milk or infant formula is replaced by solid foods when the infant is developmentally ready. The dietary protein restriction is guided by leucine requirements. Valine and isoleucine need to be supplemented, as the content of these tend to be lower than leucine in medical foods. Micronutrient intake should be monitored and supplemented as needed.42
Acute management
Individuals with MSUD are at risk for metabolic decompensation. Families and individuals should be equipped with an emergency protocol provided by the managing physician, such as a biochemical geneticist, for both the family and medical professionals caring for the patient. The goal in acute management is to suppress catabolism and promote protein anabolism. Patients must have a “sick day” prescription in order to manage illness at home, although this practice is not universal. Sick day protocol usually entails guidelines on increasing BCAA-free amino acid formula intake to 120% of the usual intake, decreasing leucine intake by 50%–100%, and providing small but frequent feedings throughout a 24 hour period. Monitoring BCAA plasma concentrations is necessary to guide appropriate diet adjustments during the illness.42 The sick day protocol is usually initiated upon the first signs of an illness and for minor illnesses can be managed at home. In serious cases, more aggressive approaches should be taken (e.g., dialysis, hemofiltration, parenteral nutrition, and/or tube feedings).40 Acute dietary treatment needs to be aggressive and include sufficient energy (up to 150% of the normal energy consumption), based on BCAA-free formula and fluid administration (up to 150 mL/kg).40 In cases where gastrointestinal delivery is not tolerated, BCAA-free formulations exist (Coram Specialty Infusion Services).42
Special considerations for management in adults
Acute metabolic management in adults
Adult patients in metabolic crisis can be difficult to manage. Typically, most expertise for management of inborn errors of metabolism rests in academic pediatric metabolic centers. Furthermore, most biochemical geneticists are trained in pediatrics and lack experience in treating adult patients. Finally, the treating physicians in intensive care and the hospital floors may not be familiar with the condition or how to manage it. All of these factors create a very challenging situation and underline the need for extreme vigilance in monitoring these patients.
Management strategies in adults are similar to those in children in that the primary goals are to 1) stop protein intake for 24–72 hours, 2) provide hydration and calorific support, 3) correct any metabolic abnormalities, 4) eliminate toxic metabolites, 5) address the underlying cause of the metabolic crisis, 6) consider cofactor supplementation, and 7) minimize associated clinical sequelae.
Once the patient shows clinical improvement, protein can be gradually reintroduced back in the range of 25%–50% of normal intake initially using total parenteral nutrition and then increased to 100% orally over the course of 3–5 days depending on the patient’s clinical course.
Calorific support is crucial to supporting protein anabolism. In the pediatric setting, the ratio of body weight/body surface is used to guide fluid infusion rate. This is not common practice in the adult setting and may well lead to confusion among treating physicians if these guidelines are given. Typical maintenance fluid rates for adults are 2–3 L of fluid per day. The 24 hour rates of 4.5 L per day would equal around 1.5 times maintenance fluid rates and represent a high fluid infusion rate of 187.5 mL/hour. We have seen successful treatment with infusion rates of 150–200 mL/hour of fluid for adult patients which should be titrated depending on the body size of the individual. Ideally, fluids should be given as 10%–12.5% dextrose. Most hospitals do not have high concentration dextrose infusions readily available. IV insulin should be considered to promote anabolism and should also be used to control the increase in glucose levels after the use of IV dextrose. A common error in the management of acute decompensated patients with MSUD is that treating physicians reduce infusion rates of IV dextrose if the blood sugar is significantly elevated, for example above 150–200 mg/dl. In this scenario, rather than decreasing the fluid rate, the rate of insulin infusion should be increased.
Metabolic abnormalities should be corrected as needed. Intervenous sodium bicarbonate should be considered when blood acidosis is below pH 7.2 or serum bicarbonate <14 Meq/L. Plasma sodium should be monitored closely, due to hyponatremia enhancing the risk of brain edema, and kept in the range of 140–145 Meq/L. Frequent monitoring of amino acids is essential and ideally daily amino acids should be obtained to guide management. Often, this is not possible due to the availability of a biochemical genetics lab or long turnaround time, and therefore, it is important to reintroduce protein after a maximum of 72 hours to guard against catabolism.
If symptoms are very severe and the patient is comatose with associated abnormalities such as refractory acidosis or electrolyte abnormalities, hemodialysis should be considered. This has been performed successfully in pediatric populations and detailed information on the dialysis technique has been described previously.43
Normally, there is a precipitating metabolic stressor that causes acute decompensation in adults and children. The most common causes are infection, surgery, injury, or significant dietary change. These should be treated as appropriate – infection should be treated with antibiotics; before surgical procedures, the patient should receive IV fluid with 10% dextrose which should be continued during and after surgery; and dietary changes should be addressed promptly by the treating metabolic team.
Pregnancy and lactation
Unlike other inborn errors of metabolism, there are insufficient data concerning the potential harmful effects of elevated BCAAs on the developing fetus.44 Careful attention and planning is important in managing pregnant patients with MSUD. During pregnancy, the mother must maintain plasma BCAA levels while increasing protein to support maternal changes and fetal health.40 It is reported that there are increases in leucine tolerance during pregnancy and lactation.44,45 Calorie and protein needs are calculated based on the requirements for pregnancy. Supplemental vitamins and minerals may be necessary to meet pregnancy needs that are not met through medical foods. Wessel et al published a successful case management by providing adequate energy and BCAA-free protein during pregnancy, delivery, and the postpartum period.45 The use of BCAA-free parenteral nutrition can be obtained through Coram Specialty Infusion Services during pre- and postpartum emergencies. This treatment will provide adequate energy and BCAA-free protein preventing elevated plasma leucine. Successful lactation in the same woman with MSUD was also reported. Management of pregnancy in an MSUD patient requires thorough observations between obstetrics and laboratory nutrition services. Careful planning and monitoring, such as reduced fasting time, is necessary for optimal outcomes during any surgical procedure.42
Treatment options
Liver transplantation
Orthotopic liver transplantation in pediatric patients with classic MSUD has been a very successful treatment. In a study of 52 individuals with classic MSUD who underwent liver transplant, all were able to achieve an unrestricted diet posttransplant.46 Interestingly, the risks associated with surgery and lifelong immunosuppression were similar to other pediatric liver transplant populations, with no reversal of cognitive deficit or progression of neurocognitive impairment.47,48 To date, there are no major studies looking at liver transplantation in an adult population. While orthotopic liver transplant is not a universal recommendation for classic MSUD, studies have shown it to be an effective therapy and should be considered in difficult to manage patients.
The most frequent transplant approach is to use liver from unrelated deceased individuals, but when the availability of deceased donor tissue is limited, living related donor liver can be used. It is important to point out that neither deceased unrelated or living related livers can restore the enzyme activity to normal levels, so acute metabolic intoxication can still occur posttransplantation.49 In general, weight-adjusted leucine tolerance is more favorable in young patients. One hypothesis for this finding is that adolescent and adult recipients tend to consume more protein and typically have less control over their daily amino acid intake. Another reason could be related to the decrease in dietary leucine tolerance from childhood (when the need matches the demand for normal growth) to adulthood.50
Sodium phenylbutyrate
Sodium phenylbutyrate (NaPBA), a nitrogen scavenging medication, is commonly used for the treatment of patients with urea cycle disorders (UCDs). It has been noted in these patients that NaPBA lowers BCAA amino acid levels. Burrage et al examined a cohort of 533 patients with UCDs, confirmed this BCAA reduction, and suggested follow-up studies to determine whether it could be a therapeutic option for MSUD.51 There is no clinical role for NaPBA at present; however, ongoing studies are assessing its efficacy.
Summary
MSUD is an organic acidemia with a varying presentation and clinical course. Dietary management is key to successful long-term treatment. As treatment has improved, a new subfield of adult management is emerging which presents new challenges. Finally, new treatments are needed to better manage difficult cases and timely clinical diagnosis remains a challenge in nonclassical patients.
Acknowledgment
The authors would like to thank the Mayo Clinic Center for Individualized Medicine for their support.
Disclosure
The authors report no conflicts of interest in this work.
References
Lang CH, Lynch CJ, Vary TC. BCATm deficiency ameliorates endotoxin-induced decrease in muscle protein synthesis and improves survival in septic mice. Am J Physiol Regul Integr Comp Physiol. 2010;299(3):R935–R944. | ||
Yudkoff M, Daikhin Y, Nissim I, Horyn O, Luhovyy B, Lazarow A. Brain amino acid requirements and toxicity: the example of leucine. J Nutr. 2005;135(6 Suppl):1531S–1538S. | ||
Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol. 2014;10(12):723–736. | ||
Burrage LC, Nagamani SC, Campeau PM, Lee BH. Branched-chain amino acid metabolism: from rare Mendelian diseases to more common disorders. Hum Mol Genet. 2014;23(R1):R1–R8. | ||
Wahren J, Felig P, Hagenfeldt L. Effect of protein ingestion on splanchnic and leg metabolism in normal man and in patients with diabetes mellitus. J Clin Invest. 1976;57(4):987–999. | ||
Brosnan JT, Brosnan ME. Branched-chain amino acids: enzyme and substrate regulation. J Nutr. 2006;136(1 Suppl):207S–211S. | ||
Harper AE, Miller RH, Block KP. Branched-chain amino acid metabolism. Annu Rev Nutr. 1984;4:409–454. | ||
Joshi MA, Jeoung NH, Obayashi M, et al. Impaired growth and neurological abnormalities in branched-chain alpha-keto acid dehydrogenase kinase-deficient mice. Biochem J. 2006;400(1):153–162. | ||
Vogel KR, Arning E, Wasek BL, McPherson S, Bottiglieri T, Gibson KM. Brain-blood amino acid correlates following protein restriction in murine maple syrup urine disease. Orphanet J Rare Dis. 2014;9:73. | ||
Zinnanti WJ, Lazovic J. Interrupting the mechanisms of brain injury in a model of maple syrup urine disease encephalopathy. J Inherit Metab Dis. 2012;35(1):71–79. | ||
Yudkoff M, Daikhin Y, Grunstein L, Nissim I, Stern J, Pleasure D. Astrocyte leucine metabolism: significance of branched-chain amino acid transamination. J Neurochem. 1996;66(1):378–385. | ||
Yudkoff M, Daikhin Y, Lin ZP, Nissim I, Stern J, Pleasure D. Interrelationships of leucine and glutamate metabolism in cultured astrocytes. J Neurochem. 1994;62(3):1192–1202. | ||
Scaini G, Tonon T, de Souza CF, et al. Serum markers of neurodegeneration in maple syrup urine disease. Mol Neurobiol. Epub 2016 Sep 22. | ||
Levin ML, Scheimann A, Lewis RA, Beaudet AL. Cerebral edema in maple syrup urine disease. J Pediatr. 1993;122(1):167–168. | ||
Strauss KA, Wardley B, Robinson D, et al. Classical maple syrup urine disease and brain development: principles of management and formula design. Mol Genet Metab. 2010;99(4):333–345. | ||
Carecchio M, Schneider SA, Chan H, et al. Movement disorders in adult surviving patients with maple syrup urine disease. Mov Disord. 2011;26(7):1324–1328. | ||
Manara R, Del Rizzo M, Burlina AP, et al. Wernicke-like encephalopathy during classic maple syrup urine disease decompensation. J Inherit Metab Dis. 2012;35(3):413–417. | ||
Thompson GN, Francis DE, Halliday D. Acute illness in maple syrup urine disease: dynamics of protein metabolism and implications for management. J Pediatr. 1991;119(1 Pt 1):35–41. | ||
Chuang DT, Shih VE. Maple syrup urine disease (branched-chain ketoaciduria). In: Scriver CR, Beaudet A, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:1971–2006. | ||
Scriver CR, Mackenzie S, Clow CL, Delvin E. Thiamine-responsive maple-syrup-urine disease. Lancet. 1971;1(7694):310–312. | ||
Munnich A, Saudubray JM, Taylor J, et al. Congenital lactic acidosis, alpha-ketoglutaric aciduria and variant form of maple syrup urine disease due to a single enzyme defect: dihydrolipoyl dehydrogenase deficiency. Acta Paediatr Scand. 1982;71(1):167–171. | ||
Quinonez SC, Leber SM, Martin DM, Thoene JG, Bedoyan JK. Leigh syndrome in a girl with a novel DLD mutation causing E3 deficiency. Pediatr Neurol. 2013;48(1):67–72. | ||
Brassier A, Ottolenghi C, Boutron A, et al. Dihydrolipoamide dehydrogenase deficiency: a still overlooked cause of recurrent acute liver failure and Reye-like syndrome. Mol Genet Metab. 2013;109(1):28–32. | ||
Morton DH, Strauss KA, Robinson DL, Puffenberger EG, Kelley RI. Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics. 2002;109(6):999–1008. | ||
Therrell BL, Padilla CD, Loeber JG, et al. Current status of newborn screening worldwide: 2015. Semin Perinatol. 2015;39(3):171–187. | ||
Naylor EW, Guthrie R. Newborn screening for maple syrup urine disease (branched-chain ketoaciduria). Pediatrics. 1978;61(2):262–266. | ||
Chace DH, Hillman SL, Millington DS, Kahler SG, Roe CR, Naylor EW. Rapid diagnosis of maple syrup urine disease in blood spots from newborns by tandem mass spectrometry. Clin Chem. 1995;41(1):62–68. | ||
Chace DH, Kalas TA, Naylor EW. Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin Chem. 2003;49(11):1797–1817. | ||
Strauss KA, Morton DH. Branched-chain ketoacyl dehydrogenase deficiency: maple syrup disease. Curr Treat Options Neurol. 2003;5(4):329–341. | ||
Oglesbee D, Sanders KA, Lacey JM, et al. Second-tier test for quantification of alloisoleucine and branched-chain amino acids in dried blood spots to improve newborn screening for maple syrup urine disease (MSUD). Clin Chem. 2008;54(3):542–549. | ||
Puckett RL, Lorey F, Rinaldo P, et al. Maple syrup urine disease: further evidence that newborn screening may fail to identify variant forms. Mol Genet Metab. 2010;100(2):136–142. | ||
Zhang B, Kuntz MJ, Goodwin GW, Edenberg HJ, Crabb DW, Harris RA. cDNA cloning of the E1 alpha subunit of the branched-chain alpha-keto acid dehydrogenase and elucidation of a molecular basis for maple syrup urine disease. Ann N Y Acad Sci. 1989;573:130–136. | ||
Imtiaz F, Al-Mostafa A, Allam R, et al. Twenty novel mutations in BCKDHA, BCKDHB and DBT genes in a cohort of 52 Saudi Arabian patients with maple syrup urine disease. Mol Genet Metab Rep. 2017;11:17–23. | ||
Abiri M, Karamzadeh R, Mojbafan M, et al. In silico analysis of novel mutations in maple syrup urine disease patients from Iran. Metab Brain Dis. 2017;32(1):105–113. | ||
Li X, Ding Y, Liu Y, et al. Eleven novel mutations of the BCKDHA, BCKDHB and DBT genes associated with maple syrup urine disease in the Chinese population: report on eight cases. Eur J Med Genet. 2015;58(11):617–623. | ||
Couce ML, Ramos F, Bueno MA, et al. Evolution of maple syrup urine disease in patients diagnosed by newborn screening versus late diagnosis. Eur J Paediatr Neurol. 2015;19(6):652–659. | ||
McCabe LL, McCabe ER. Genetic screening: carriers and affected individuals. Annu Rev Genomics Hum Genet. 2004;5:57–69. | ||
Packman W, Mehta I, Rafie S, et al. Young adults with MSUD and their transition to adulthood: psychosocial issues. J Genet Couns. 2012;21:5:692–703. | ||
Enns GM and Packman W. The adolescent with an inborn error of metabolism: medical issues and transition to adulthood. Adolesc Med. 2002;13:2:315–329. | ||
Frazier DM, Allgeier C, Homer C, et al. Nutrition management guideline for maple syrup urine disease: an evidence- and consensus-based approach. Mol Genet Metab. 2014;112(3):210–217. | ||
Servais A, Arnoux JB, Lamy C, et al. Treatment of acute decompensation of maple syrup urine disease in adult patients with a new parenteral amino-acid mixture. J Inherit Metab Dis. 2013;36(6):939–944. | ||
van Calcar S. Nutrition management of maple syrup urine disease. In: Bernstein LE, Rohr F, Helm JR, editors. Nutrition Management of Inherited Metabolic Diseases. Switzerland: Springer International Publishing; 2015:173–183. | ||
Atwal PS, Macmurdo C, Grimm PC. Haemodialysis is an effective treatment in acute metabolic decompensation of maple syrup urine disease. Mol Genet Metab Rep. 2015;4:46–48. | ||
Marriage BJ. Nutrition management of patients with inherited disorders of branched-chain amino acid metabolism. In: Acosta P, editor. Nutrition Management of Patients with Inherited Metabolic Disorders. Sudbury, MA: Jones and Bartlett Publishers; 2010:175–236. | ||
Wessel AE, Mogensen KM, Rohr F, et al. Management of a woman with maple syrup urine disease during pregnancy, delivery, and lactation. JPEN J Parenter Enteral Nutr. 2015;39(7):875–879. | ||
Strauss KA, Mazariegos GV, Sindhi R, et al. Elective liver transplantation for the treatment of classical maple syrup urine disease. Am J Transplant. 2006;6(3):557–564. | ||
Muelly ER, Moore GJ, Bunce SC, et al. Biochemical correlates of neuropsychiatric illness in maple syrup urine disease. J Clin Invest. 2013;123(4):1809–1820. | ||
Mazariegos GV, Morton DH, Sindhi R, et al. Liver transplantation for classical maple syrup urine disease: long-term follow-up in 37 patients and comparative United Network for Organ Sharing experience. J Pediatr. 2012;160(1):116–121. | ||
Feier F, Schwartz IV, Benkert AR, et al. Living related versus deceased donor liver transplantation for maple syrup urine disease. Mol Genet Metab. 2016;117(3):336–343. | ||
Strauss KA, Wardley B, Robinson D, et al. Classical maple syrup urine disease and brain development: Principles of management and formula design. Mol Genet Metab. 2010;99(4): 333–345. | ||
Burrage LC, Jain M, Gandolfo L, Lee BH, Nagamani SC. Sodium phenylbutyrate decreases plasma branched-chain amino acids in patients with urea cycle disorders. Mol Genet Metab. 2014;113(1–2):131–135. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.