")
Back to Journals » Journal of Blood Medicine » Volume 8
Many factor VIII products available in the treatment of hemophilia A: an embarrassment of riches?
Authors Lieuw K
Received 16 February 2017
Accepted for publication 17 May 2017
Published 15 June 2017 Volume 2017:8 Pages 67—73
DOI https://doi.org/10.2147/JBM.S103796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Kenneth Lieuw1,2
1Department of Pediatrics, Walter Reed National Military Medical Center, 2Department of Pediatrics, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
Abstract: Hemophilia A (HA) is a common bleeding disorder caused by the deficiency of factor VIII (FVIII) with an incidence of ~1 in 5000 male births. Replacement of FVIII is necessary to prevent and treat bleeding episodes. However, with multiple new drugs in addition to old standards, choosing among the different FVIII treatment options is harder than ever. There are FVIII products that are plasma derived or recombinant, FVIII products designed to extend the half-life of FVIII, and the first single-chain FVIII product, recombinant factor VIII single chain (rFVIII-SC). As development of inhibitors to FVIII continues to be a major problem in the care of HA patients, recent studies showing lower rates of inhibitor development with plasma-derived FVIIII products versus recombinant FVIII products have made choosing among the many options now available even more complex. Although still unproven, extended half-life (EHL) products may provide the hope of decreased immunogenicity but need further testing in previously untreated patients (PUPs). This review highlights some of the differences between FVIII products currently available and hopefully assists the clinician to decide which FVIII product to choose for their patients.
Keywords: hemophilia, hemophilia A, factor VIII, recombinant factor VIII single chain, extended half-life factor VIII, immunogenicity, bleeding disorder, inhibitor development
Introduction
Hemophilia A (HA) is an X-linked bleeding disorder, first reported in medical literature from the 1800s, in which the deficiency of factor VIII (FVIII) results in problems with hemostasis.1 Before therapy with FVIII infusions became available, hemophilia patients suffered from prolonged bruising and bleeding with manifestations ranging from severe and recurrent joint hemorrhages to life-threatening bleeding episodes and had a decreased life expectancy. HA is defined as mild when endogenous FVIII levels are >5%, moderate when between 1% and 5%, and severe when <1%. Treatment for HA with replacement factor first started with the use of fresh frozen plasma in the 1950s followed by cryoprecipitate in the 1960s. Finally, in the 1970s, lyophilized FVIII derived from pooled plasma became available and revolutionized treatment for HA, even allowing home infusion therapy. The HA community then suffered from the contamination of factor supplies with the HIV in the 1980s. This tragedy stimulated research and technical advances such as heat inactivation and solvent detergent treatment to generate safer plasma-derived FVIII products. The cloning of the gene for FVIII in 1984 resulted in recombinant FVIII preparations becoming available in the 1990s. Currently, the standard of care in developed countries is to offer primary prophylactic FVIII infusions to patients with severe HA and has led to dramatic increase in the quality of life for these patients.2,3
Now, a child born with HA is expected to have a normal lifespan as prophylactic infusions of FVIII permit normal activity without restrictions albeit with great financial burden ($150000–$300000 per year in the US4). However, due to the short half-life of FVIII, frequent infusions are required in order to maintain a trough level >1%. In addition, the development of inhibitors in 25–30% of HA patients leads to increased risk for bleeding and the need for expensive treatment regimens to induce immune tolerance or the use of bypassing agent treatments to control bleeding episode. Therefore, the development of factor products with decreased immunogenicity and longer half-lives is an important unmet need for the HA community.
Various approaches have been used for increasing the half-life of recombinant FVIII, which is a glycoprotein that is normally bound by its chaperone protein, Von Willebrand factor (VWF), which is also important in achieving hemostasis and influences the half-life of FVIII.3,5 Once FVIII is synthesized from the gene located on the X chromosome, it undergoes post-translational modification such as N-glycosylation and sulfation that are important for protein–protein interactions. Activation of FVIII is induced by thrombin and results in proteolysis of FVIII into its active form that is rapidly inactivated and cleared. The most common methods of prolonging the half-life of FVIII have been conjugation of FVIII with larger molecules such as polyethylene glycol (PEG), albumin, or to the human immunoglobulin Fc. Conjugation with albumin takes advantage of the natural half-life of albumin, whereas PEGylation protects FVIII from proteolytic enzymes. Fc conjugation takes advantage of the existence of the neonatal Fc receptor that allows protection and recirculation of the FVIII via this receptor pathway.5,6 Recently, a novel single-chain recombinant FVIII (Afstyla®) has been approved by the US Food and Drug Administration (US FDA) in May 2016. Recombinant factor VIII single chain (rFVIII-SC) has increased stability and affinity for VWF, extending its half-life with the possibility of decreased immunogenicity.7–11
Current factors available and extended half-life (EHL) factor development
Numerous plasma-derived FVIII products and recombinant FVIII products are now available (Table 1). First-generation recombinant FVIII products such as Recombinate® were generated from the full FVIII gene and contained both human albumin and animal proteins in the production process. FVIII gene protein production results in the production of a single polypeptide that is modified and cleaved to form light and heavy chains. These light and heavy chains are held together by a labile metal ion bridge that is required for proper functioning of the protein.3 It was found that deleting most of the B domain of FVIII actually improved secretion from the cell in the recombinant process,12 and so B-domain-deleted (BDD) FVIII products such as ReFacto® were developed. Because of the concern about unknown pathogens, second-generation factors such as Kogenate® removed human albumin as a stabilizing agent. Third-generation products, introduced over the last 5 years, no longer use any animal or human products in the cell culture or production of FVIII products. The recent availability of EHL products adds to the choices of agents available to the HA community (Table 1). With the many choices now available, it is becoming more difficult and complicated to determine which FVIII product to use for which patients. This review focuses on the many new FVIII products now available to help in choosing between the different options available.
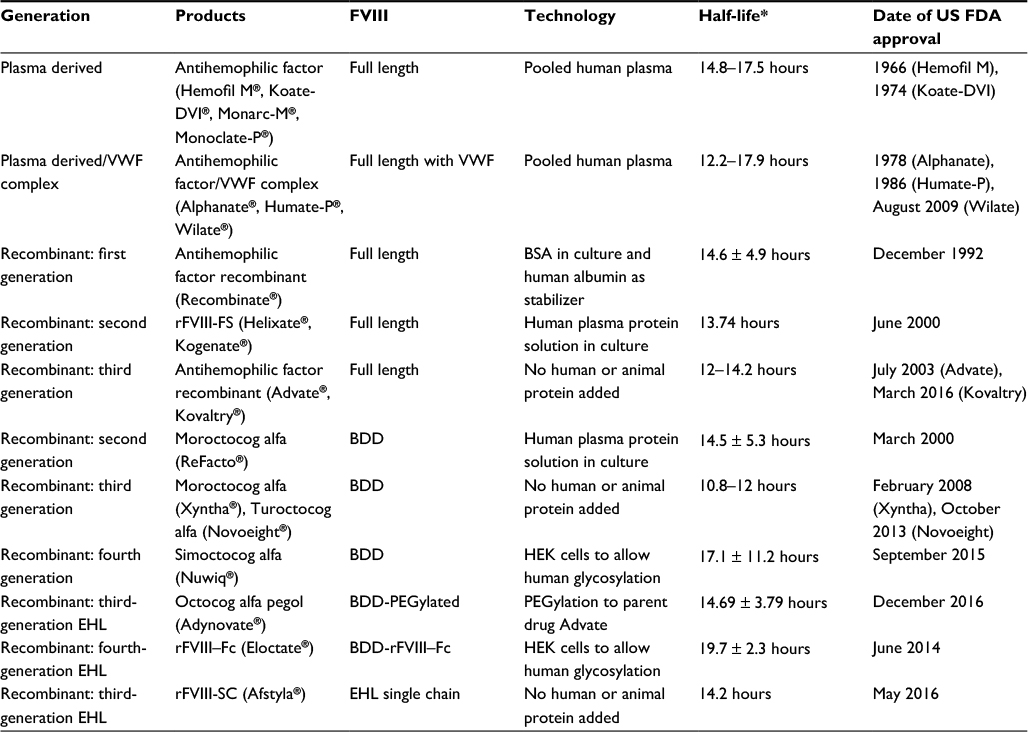 | Table 1 Available FVIII products Note: *The half-life of the different factors was taken from the product brochures from the manufacturers and differs in how it was determined. Abbreviations: FVIII, factor VIII; US FDA, US Food and Drug Administration; VWF, Von Willebrand factor; BSA, bovine serum albumin; rFVIII, recombinant factor VIII; BDD, B domain deleted; HEK, human embryonic kidney; EHL, extended half-life; PEG, polyethylene glycol; SC, single chain; rFVIII-FS, recombinant FVIII formulated with sucrose; rFVIII-Fc, antihemophilic factor (recombinant), Fc fusion protein; rFVIII-SC, antihemophilic factor (recombinant), single chain. |
Currently, most of our patients with HA are offered prophylactic infusions with third-generation products, with some patients transitioning to the newer third-generation or EHL products now available. Because third-generation products do not use any human or animal proteins in the production of their products, these are often requested by the families. Two newly approved third-generation FVIII products with standard half-life are now available: turoctogog alfa (Novoeight®) is a BDD FVIII produced in the Chinese hamster ovary (CHO) cell line and octocog alfa (Kovaltry®) is a full-length FVIII product that is produced in the baby hamster kidney (BHK) cell line. Octocog alfa (Kovaltry) is reportedly improved from its predecessor, rFVIII-FS (Kogenate), in the consistency of glycosylation and co-expression with protein chaperone HSP70 that may enhance the protein folding of FVIII. Both turoctogog alfa and octocog alfa showed effectiveness in clinical trials and are expected to replace the second-generation FVIII products.13–16 Finally, simoctogog alfa, human-cl rhFVIII (Nuwiq®), is a fourth-generation BDD FVIII product made in the human embryonic kidney (HEK) cell line. Because it has the full human post-translational modifications, it has been speculated that it may also provide decreased immunogenicity.17,18
EHL FVIII products have brought exciting new options for the HA community, providing the possibility of fewer scheduled infusions. Despite the various reported half-lives found in the product brochures of FVIII products, it is commonly understood that the half-life of standard FVIII products is between 8 and 12 hours with individual variation seen with different brands. There have been several strategies for extending FVIII half-life such as attaching a PEG moiety, albumin, or the Fc fragment of immunoglobulin to the FVIII protein.6,19 PEGylation protects FVIII from proteolytic degradation, while both Fc fragment and albumin conjugation use the recycling of the endocytosed fusion protein through the neonatal Fc receptor pathway.6 Although both immunoglobulins and albumin normally have very long half-lives, these approaches have only resulted in a modest increase in the half-life of FVIII, likely due to the dependence on VWF that stabilizes FVIII.5 Even with only a modest gain in half-life, the decrease in the frequency of infusions can significantly increase the quality of life for HA patients. Recently, the US FDA approved both PEGylated FVIII (Adynovate®) and the Fc–FVIII fusion product (Eloctate®). Clinical studies thus far showed that rFVIII–Fc fusion product (Eloctate) has the longest half-life among the EHL products currently available with a half-life of 19.7 hours in adolescents and adults, although it decreased to 14.6 in patients 6–12 years old.20,21 Interestingly, when the same BDD FVIII gene is expressed using the HEK cell line allowing for human-like post-translational modifications, the resultant FVIII protein’s half-life also appears to be slightly increased. Simoctocog alfa (Nuwiq) was developed with the hope to generate a less immunogenic FVIII product by eliminating potentially immunogenic glycosylation patterns found in non-human cell lines, and it appears that the humanized post-translational modifications may have also prolonged the half-life.22 Further studies, however, are needed to determine if these new products are indeed less immunogenic in previously untreated patients (PUPs).
Finally, using recombinant DNA technology, CSL Behring created a novel recombinant FVIII to modestly extend the half-life by increasing the affinity of FVIII to VWF, which stabilizes the protein and prevents it from degradation. Normally, FVIII is composed of a heavy and a light chain held together by a labile metal ion bridge. CSL Behring developed a BDD recombinant FVIII molecule in which the heavy and light chains are covalently linked that has a twofold increased affinity to VWF.7,23 The increased stability provided by the single-chain covalent bond permits a higher efficiency manufacturing of the full-length factor and results in a more stable protein after reconstitution.24 Moreover, the enhanced affinity for VWF may accelerate the formation of VWF:FVIII complex that not only increases the half-life but also may result in decreased immunogenicity by inhibiting endocytosis of FVIII by dendritic cells.25,26 These modifications resulted in a FVIII molecule with an increased half-life that is comparable to the PEGylated EHL product (Adynovate).8,10,27,28 When FVIII is normally activated, the heavy and light chains are cleaved and released from VWF. Because these key thrombin cleavage sites in the single-chain FVIII are intact, once activated, rFVIII-SC becomes structurally identical to FVIII.
Preclinical testing in animals confirmed that recombinant rFVIII-SC had equal potency compared to other full-length and BDD FVIII products.11 In the AFFINITY clinical trial program, rFVIII-SC showed excellent hemostatic efficacy with a median annualized bleeding rate (ABR) of 1.14 that is comparable to other recently approved factor products.8 For example, rFVIII-Fc (Eloctate) reported a median ABR of 1.6, and simoctocog alfa (Nuwiq) reported a median ABR of 0.9. Furthermore, no participants (previously treated patients [PTPs] with >150 exposure days to other FVIII products) in the AFFINITY study developed an inhibitor, and the immunogenicity of rFVIII-SC is currently being studied in PUPs (#NCT02172950).8
Inhibitor development and immunogenicity of new EHL FVIII
The most feared complication in the treatment of HA is the development of neutralizing anti-FVIII antibodies or “inhibitors” that occur in up to 25–30% of HA patients.29 The development of inhibitors results in the need for new treatments with bypassing agents and cumbersome and expensive immune tolerance induction protocols. Why the immunogenicity of FVIII products is so high is unclear, but it seems to be a combination of ethnicity, genetic mutation, and environmental factors.2 Also important are the differences in the post-translational modification patterns of FVIII – dependent on the cell line used, whether non-mammalian cell lines (e.g., CHO or BHK) or the human cell line (HEK) – and the role of VWF in both increasing the half-life of FVIII and reducing the endocytosis of FVIII by antigen-presenting cells.5
With regard to ethnicity, Black HA patients have twice the rates of inhibitor development than White HA patients. It has been thought that this is due to the differences in genetic haplotypes of the patient’s FVIII gene versus the haplotypes that are found in various plasma-derived and recombinant FVIII products.30 Gunasekera et al31 reported that African Americans with intron-22 inversion had two- to threefold higher risk of inhibitor development than White HA patients with the same inversion. However, recent studies looking at the inhibitor risk in severe HA patients and ethnic haplotypes did not find a correlation between the haplotypes and inhibitor formation.31,32 Thus, whether inhibitor risk is increased due to ethnic differences in the FVIII gene compared to commercially available FVIII products remains unanswered.
Recently, the Research of Determinants of Inhibitor Development (RODIN) study group reported that second-generation FVIII products had higher inhibitor rates than third-generation products. This finding was confirmed by Calvez et al in their report of a French cohort of HA patients that also showed higher rates of inhibitor development (37.5%) with second-generation FVIII products33,34 and an adjusted hazard ratio of 1.58 when they compared the rates of inhibitor formation between second-generation and third-generation products. The authors speculated that this might be related to the cell line used for the production of the product: BHK cells (second generation) vs CHO cells (third generation) as the degree of product modifications such as sulfation and glycosylation differ between the two cell types.34
Another potential benefit of the EHL FVIII products is the possibility of decreased immunogenicity of these new factor products. It must be emphasized that this is not yet proven and requires further studies in PUPs. However, it has long been demonstrated that conjugation of immunogenic peptides to a non-immunogenic protein carrier can induce tolerance and that immunoglobulin conjugation was superior to other protein carriers such as albumin.35 Recent case reports using the recombinant FVIII–Fc fusion protein in immune tolerance induction for HA patients with inhibitors also support this notion.36,37 Both Fc and albumin conjugations can potentially decrease the immunogenicity of FVIII by what is called antigen shielding, where the fusion moiety inhibits uptake of the FVIII product by the antigen-presenting cells needed to generate an immune response.38 Conjugation of PEG to a molecule not only prolongs the half-life but also is thought to make it less immunogenic by inhibiting the binding of protein to endocytic receptors on antigen-presenting cells.39 This strategy has been utilized to increase the half-life of multiple drugs now. For example, it is known that PEGylated asparaginase, which is used for the treatment of leukemia, causes fewer allergic reactions than the native asparaginase enzyme.40
With the advent of various EHL FVIII products now approved or in development utilizing the Fc fusion, albumin fusion, or PEGylation, further clinical trials are needed to determine if they also result in decreased immunogenicity. While animal studies support the notion that these new products have decreased immunogenicity,39,41 clinical studies with PUPs will be especially important. In addition, the effects of long-term exposure to a molecule such as PEG in HA patients still need to be evaluated.
Immunogenicity of plasma-derived FVIII products versus recombinant FVIII products
Recently reported results from the clinical trial called Study on Inhibitors in Plasma-Product Exposed Toddlers (SIPPET) trial are that plasma-derived FVIII products had lower rates of inhibitor formation than recombinant FVIII products in that population, complicating what we thought we knew was best for the treatment of patients with HA.42 While the use of recombinant FVIII has become the standard in most US and European clinics, the results of the SIPPET trial now further confuses the choice of factor that is best for each patient. What can be the reasons behind the decreased immunogenicity of plasma-derived FVIII products? One possible answer is that most recombinant FVIII products are produced using a non-human cell line and contain differences in the post-translational modifications of the resulting FVIII protein.5,43 Another potential reason is that preformed complexes containing VWF result in decreased immunogenicity, which is supported by animal studies.25 If human glycosylation decreases the immunogenicity of FVIII, use and studies of the newly approved FVIII products such as simoctocog alfa (Nuwiq) and rFVIII-Fc (Eloctate) should help to prove this point, as these are made using HEK cell line. It should be noted that pharmacokinetic studies showed that simoctocog alfa (Nuwiq) has increased half-life compared to Advate®, which is the same recombinant product made in non-human cell lines.22 It is thus tempting to speculate that human glycosylation pattern increases the affinity of FVIII to VWF, which not only increases the half-life but also decreases the immunogenicity of the FVIII.5 It should also be mentioned that plasma-derived FVIII products contain many other proteins as well that may influence FVIII immunogenicity.
With regard to the SIPPET trial, the medical and scientific advisory council (MASAC) of the National Hemophilia Foundation has highlighted the fact that the SIPPET trial differed from previous prospective studies comparing plasma-derived FVIII to recombinant FVIII such as RODIN and the European Pediatric Network trial (PedNet), which found no significant differences.44 Because the ethnicities in the SIPPET trial (mostly from Egypt, India, and Iran) differed significantly from the ethnicities of the patients in the other studies, they concluded that the results may not be applicable to the US HA community that are mostly Caucasian. In addition, they pointed out that in the US, majority of patients receive prophylactic infusions and that third-generation products are in use that differed from the study population. They recommended that the SIPPET study results may not be applicable to the US population and that the risk of inhibitor development must be weighed against the potential infectious risk of emerging viruses, which has not been tested with the current manufacturing processes.44
Incorporating the EHL FVIII products into clinical practice
Based on in vitro and animal studies done thus far, EHL FVIII products may also have a potential benefit of decreased immunogenicity and inhibitor development in addition to prolonged half-life and are very attractive to implement into clinical practice.39 However, it is worth noting that individualized dosing must be done using observed pharmacokinetic values, as the half-life of each product can differ between patients, and in individual cases, the use of EHL FVIII products may not prolong the half-life enough to justify the increased cost of these new factors. For example, despite the longest half-life advertised of any of the factors available, the actual half-life is much shorter in younger patients, and we have been unable to go to less than thrice-weekly infusions of rFVIII–Fc (Eloctate) in order to maintain a trough level >1%. Nevertheless, it is exciting to have so many options in coming up with individualized treatment programs for HA patients.
So, what is the best approach to incorporate these new FVIII products into clinical practice? It has been suggested that the EHL products may be helpful in patients who have decreased compliance since these may decrease the frequency of infusions. In addition, in a patient who is having breakthrough bleeding with an already aggressive prophylactic regimen, it is reasonable to try an EHL product in an attempt to decrease breakthrough bleeds.4 Although there have been concerns about potential inhibitor development in switching FVIII products, there is little evidence that inhibitor rates increase with switching FVIII products. However, immunologically, recombinant products do have varying levels of post-translational protein modifications due to production in different cell lines that may provide potential epitopes for immune reaction.38 Therefore, further studies are needed to verify whether these new EHL products indeed decrease immunogenicity. Unfortunately, it is likely that new clinical studies in PUPs will be underpowered to detect a difference in immunogenicity and will make it difficult to have convincing scientific basis to declare the least immunogenic product. For PTPs, although it may make sense to switch to a product that is generated in the same cell line so that glycosylation patterns remain the same, the overwhelming evidence from clinical studies shows that PTPs hardly ever develop an inhibitor after a FVIII product switch.
Conclusion
Treatment options for HA have never been better with the approval of several new recombinant FVIII products and the new EHL FVIII products that are now even safer and may offer the potential of decreased immunogenicity and longer half-lives. Additional factor products that are still in development (Table 2) will add to the choices that we will have in the treatment of HA patients.4,19 However, there remains much work to be done in terms of satisfying the unmet needs for HA patients. The rate of inhibitor development is still the scourge of HA treatment, and improvements are needed to minimize the development of inhibitors while optimizing immune tolerance induction protocols. Novel products currently in development continue to offer exciting possibilities. New products such as rFVIII-VWF-XTEN in animal studies allow subcutaneous dosing with fourfold increase in half-life, and the development of a humanized bispecific antibody that provides FVIII-mimetic function provides exciting new possibilities in the treatment of HA.5,45,46 Perhaps, with the introduction of many new EHL products to the market, the cost of FVIII infusions will decrease enough to allow more children in developing countries to receive prophylactic therapy. Moreover, the recent success with gene therapy trials for factor IX deficiency provides hope that we will soon overcome the obstacles in developing an engineered gene therapy construct for FVIII that will result in a cure.47
 | Table 2 FVIII products in development Abbreviations: FVIII, factor VIII; EHL, extended half-life; BDD, B domain deleted; PEG, polyethylene glycol. |
Acknowledgments
I would like to acknowledge Anne Warwick, Gary Crouch, and David Scott for helpful reading of the manuscript and suggestions. I would also like to thank Michele Mason-Coles for helping with the references.
Disclosure
The views expressed in this article are those of the author and do not reflect the official policy or position of the Walter Reed National Military Medical Center, Uniformed Services University, United States Army, the Department of Defense, or the US Government. The identification of specific products, scientific instrumentation, or organization is considered an integral part of the scientific endeavor and does not constitute endorsement or implied endorsement on the part of the author, DoD, or any component agency. The author reports no conflicts of interest in this work.
References
Hoyer LW. Hemophilia A. N Engl J Med. 1994;330(1):38–47. | ||
Coppola A, Di Capua M, Di Minno MN, et al. Treatment of hemophilia: a review of current advances and ongoing issues. J Blood Med. 2010;1:183–195. | ||
Orlova NA, Kovnir SV, Vorobiev II, Gabibov AG, Vorobiev AI. Blood clotting factor VIII: from evolution to therapy. Acta Naturae. 2013;5(2):19–39. | ||
Pipe SW. New therapies for hemophilia. Hematology Am Soc Hematol Educ Program. 2016;2016(1):650–656. | ||
Pipe SW, Montgomery RR, Pratt KP, Lenting PJ, Lillicrap D. Life in the shadow of a dominant partner: the FVIII-VWF association and its clinical implications for hemophilia A. Blood. 2016;128(16):2007–2016. | ||
Shapiro AD. Long-lasting recombinant factor VIII proteins for hemophilia A. Hematology Am Soc Hematol Educ Program. 2013;2013:37–43. | ||
Bensen-Kennedy D. Bringing new therapy options to the hemophilia community. Thromb Res. 2013;131(suppl 2):S15–S18. | ||
Mahlangu J, Kuliczkowski K, Karim FA, et al. Efficacy and safety of rVIII-SingleChain: results of a phase 1/3 multicenter clinical trial in severe hemophilia A. Blood. 2016;128(5):630–637. | ||
Pabinger-Fasching I. The story of a unique molecule in hemophilia A: recombinant single-chain factor VIII. Thromb Res. 2016;141 (suppl 3):S2–S4. | ||
Zollner S, Raquet E, Claar P, et al. Non-clinical pharmacokinetics and pharmacodynamics of rVIII-SingleChain, a novel recombinant single-chain factor VIII. Thromb Res. 2014;134(1):125–131. | ||
Zollner SB, Raquet E, Müller-Cohrs J, et al. Preclinical efficacy and safety of rVIII-SingleChain (CSL627), a novel recombinant single-chain factor VIII. Thromb Res. 2013;132(2):280–287. | ||
Miao HZ, Sirachainan N, Palmer L, et al. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103(9):3412–3419. | ||
Santagostino E. A new recombinant factor VIII: from genetics to clinical use. Drug Des Devel Ther. 2014;8:2507–2515. | ||
Kavakli K, Yang R, Rusen L, et al. Prophylaxis vs. on-demand treatment with BAY 81-8973, a full-length plasma protein-free recombinant factor VIII product: results from a randomized trial (LEOPOLD II). J Thromb Haemost. 2015;13(3):360–369. | ||
Sandberg H, Kannicht C, Stenlund P, et al. Functional characteristics of the novel, human-derived recombinant FVIII protein product, human-cl rhFVIII. Thromb Res. 2012;130(5):808–817. | ||
Viuff D, Barrowcliffe T, Saugstrup T, Ezban M, Lillicrap D. International comparative field study of N8 evaluating factor VIII assay performance. Haemophilia. 2011;17(4):695–702. | ||
Kannicht C, Ramström M, Kohla G, et al. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thromb Res. 2012;131(1):78–88. | ||
Tiede A, Brand B, Fischer R, et al. Enhancing the pharmacokinetic properties of recombinant factor VIII: first-in-human trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost. 2013;11(4):670–678. | ||
Kaufman RJ, Powell JS. Molecular approaches for improved clotting factors for hemophilia. Hematology Am Soc Hematol Educ Program. 2013;2013:30–36. | ||
Chowdary P, Fosbury E, Riddell A, Mathias M. Therapeutic and routine prophylactic properties of rFactor VIII Fc (efraloctocog alfa, Eloctate(R)) in hemophilia A. J Blood Med. 2016;7:187–198. | ||
Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123(3):317–325. | ||
Klukowska A, Szczepański T, Vdovin V, Knaub S, Jansen M, Liesner R. Novel, human cell line-derived recombinant factor VIII (Human-cl rhFVIII, Nuwiq(®)) in children with severe haemophilia A: efficacy, safety and pharmacokinetics. Haemophilia. Epub 2015 Sep 14. | ||
Schmidbauer S, Witzel R, Robbel L, et al. Physicochemical characterisation of rVIII-SingleChain, a novel recombinant single-chain factor VIII. Thromb Res. 2015;136(2):388–395. | ||
Schulte S. Pioneering designs for recombinant coagulation factors. Thromb Res. 2011;128(suppl 1):S9–S12. | ||
Delignat S, Repessé Y, Navarrete AM, et al. Immunoprotective effect of von Willebrand factor towards therapeutic factor VIII in experimental haemophilia A. Haemophilia. 2011;18(2):248–254. | ||
Sorvillo N, Hartholt RB, Bloem E, et al. von Willebrand factor binds to the surface of dendritic cells and modulates peptide presentation of factor VIII. Haematologica. 2016;101(3):309–318. | ||
Brand B, Gruppo R, Wynn TT, et al. Efficacy and safety of pegylated full-length recombinant factor VIII with extended half-life for perioperative haemostasis in haemophilia A patients. Haemophilia. 2016;22(4):e251–e258. | ||
Wynn TT, Gumuscu B. Potential role of a new PEGylated recombinant factor VIII for hemophilia A. J Blood Med. 2016;7:121–128. | ||
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in haemophilia A patients – a review of recent studies of recombinant and plasma-derived factor VIII concentrates. Haemophilia. 1999;5(3):145–154. | ||
Viel KR, Ameri A, Abshire TC, et al. Inhibitors of factor VIII in black patients with hemophilia. N Engl J Med. 2009;360(16):1618–1627. | ||
Gunasekera D, Ettinger RA, Nakaya Fletcher S, et al. Factor VIII gene variants and inhibitor risk in African American hemophilia A patients. Blood. 2015;126(7):895–904. | ||
Miller CH, Benson J, Ellingsen D, et al. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. 2012;18(3):375–382. | ||
Gouw SC, van der Bom JG, Ljung R, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368(3):231–239. | ||
Calvez T, Chambost H, Claeyssens-Donadel S, et al. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood. 2014;124(23):3398–3408. | ||
Borel Y. Induction of immunologic tolerance by a hapten (DNP) bound to a non-immunogenic carrier. Nat New Biol. 1971;230(14):180. | ||
Groomes CL, Gianferante DM, Crouch GD, Parekh DS, Scott DW, Lieuw K. Reduction of factor VIII inhibitor titers during immune tolerance induction with recombinant factor VIII-Fc fusion protein. Pediatr Blood Cancer. 2016;63(5):922–924. | ||
Malec LM, Journeycake J, Ragni MV. Extended half-life factor VIII for immune tolerance induction in haemophilia. Haemophilia. 2016;22(6):e552–e554. | ||
Iorio A, Puccetti P, Makris M. Clotting factor concentrate switching and inhibitor development in hemophilia A. Blood. 2012;120(4):720–727. | ||
Ing M, Gupta N, Teyssandier M, et al. Immunogenicity of long-lasting recombinant factor VIII products. Cell Immunol. 2016;301:40–48. | ||
Hasan H, Shaikh OM, Rassekh SR, Howard AF, Goddard K. Comparison of hypersensitivity rates to intravenous and intramuscular PEG-asparaginase in children with acute lymphoblastic leukemia: a meta-analysis and systematic review. Pediatr Blood Cancer. 2016;64(1):81–88. | ||
Krishnamoorthy S, Liu T, Drager D, et al. Recombinant factor VIII Fc (rFVIIIFc) fusion protein reduces immunogenicity and induces tolerance in hemophilia A mice. Cell Immunol. 2015;301:30–39. | ||
Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374(21):2054–2064. | ||
Ettingshausen CE, Kreuz W. Recombinant vs. plasma-derived products, especially those with intact VWF, regarding inhibitor development. Haemophilia. 2006;12(suppl 6):102–106. | ||
MASAC. MASAC Recommendation on SIPPET (Survey of Inhibitors in Plasma-Product-Exposed Toddlers): REsults and Recommendations for Treatment Products for Previously Untreated Patients with Hemophilia A. MASAC; 2016. | ||
Shima M, Hanabusa H, Taki M, et al. Factor VIII-mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374(21):2044–2053. | ||
Drager D, Patarroyo-White S, Chao H, et al. Recombinant FVIIIFc-VWF-XTEN demonstrates significant bioavailability following subcutaneous administration in hemophilia A mice. Blood. 2015; 126:3492. | ||
Nathwani AC, Reiss UM, Tuddenham EG, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994–2004. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.