Back to Journals » International Journal of Nanomedicine » Volume 11
Manipulating the NF-κB pathway in macrophages using mannosylated, siRNA-delivering nanoparticles can induce immunostimulatory and tumor cytotoxic functions
Authors Ortega R, Barham W, Sharman K, Tikhomirov O, Giorgio T ![]() , Yull F
, Yull F
Received 31 July 2015
Accepted for publication 12 November 2015
Published 18 May 2016 Volume 2016:11 Pages 2163—2177
DOI https://doi.org/10.2147/IJN.S93483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Ryan A Ortega,1–3 Whitney Barham,3 Kavya Sharman,4 Oleg Tikhomirov,3 Todd D Giorgio,1–3 Fiona E Yull3
1Department of Biomedical Engineering, Vanderbilt University, 2Vanderbilt Institute for Nanoscale Science and Engineering, 3Department of Cancer Biology, Vanderbilt-Ingram Cancer Center, 4Department of Neuroscience, Vanderbilt University, Nashville, TN, USA
Abstract: Tumor-associated macrophages (TAMs) are critically important in the context of solid tumor progression. Counterintuitively, these host immune cells can often support tumor cells along the path from primary tumor to metastatic colonization and growth. Thus, the ability to transform protumor TAMs into antitumor, immune-reactive macrophages would have significant therapeutic potential. However, in order to achieve these effects, two major hurdles would need to be overcome: development of a methodology to specifically target macrophages and increased knowledge of the optimal targets for cell-signaling modulation. This study addresses both of these obstacles and furthers the development of a therapeutic agent based on this strategy. Using ex vivo macrophages in culture, the efficacy of mannosylated nanoparticles to deliver small interfering RNA specifically to TAMs and modify signaling pathways is characterized. Then, selective small interfering RNA delivery is tested for the ability to inhibit gene targets within the canonical or alternative nuclear factor-kappaB pathways and result in antitumor phenotypes. Results confirm that the mannosylated nanoparticle approach can be used to modulate signaling within macrophages. We also identify appropriate gene targets in critical regulatory pathways. These findings represent an important advance toward the development of a novel cancer therapy that would minimize side effects because of the targeted nature of the intervention and that has rapid translational potential.
Keywords: nanotechnology, targeted nanoparticles, cancer immunology, RNAi
Introduction
Many therapeutic strategies for cancer immunotherapy, such as adoptive T-cell transfer for prostate cancer, focus on activating or enhancing adaptive immunity against cancer cells.1,2 However, cells of the innate immune system are also an attractive target for cancer therapies. Tumor-associated macrophages (TAMs) have been implicated as one of the most prevalent and impactful types of immune cells in tumor-related stroma.4–6 In most cases, interactions between the tumor cells and the resident or infiltrating macrophage population cause the macrophages to adopt a phenotype characterized by the constant, low-level production of inflammatory cytokines.7 This produces a state of smoldering inflammation in the tumor and surrounding tissue. This type of inflammation is insufficient to induce apoptosis or other mechanisms of tumor cell death but is significant enough to cause survivable DNA damage in the genetic material of tumor cells as well as activate survival signals in the nearby cells.8,9 TAMs also contribute to the immunosuppressive microenvironment by producing cytokines, such as interleukin (IL)-10, which inhibit the ability of resident immune cells to act against tumor cells and prevent the recruitment of CD8+ T-cells, natural killer cells (NK cells), and other cytotoxic or proinflammatory immune cells.10–12 Furthermore, TAMs produce trophic cytokines, can degrade the surrounding connective tissue, and induce angiogenesis, allowing the primary tumor to grow.13–15 Finally, TAMs participate in the metastatic process by creating pathways that aid in tumor cell intravasation.16,17
It has been reported that the tumorigenic and metastatic effects generated by TAMs can be reduced or removed by ablating macrophages with liposomal clodronate in mouse models of human cancer.17–19 Though TAMs have adopted a protumor phenotype, we and others have shown that they retain the potential to produce cytotoxic levels of inflammation, lyse surrounding cells, and coordinate an immune response from cells of the innate and adaptive immune system.21–24 The ability to recapitulate these cytotoxic and immunostimulatory functions in TAMs, thus creating an antitumor phenotype, would be a powerful therapeutic tool for treating tumors and metastases with a significant macrophage population. An antitumor macrophage phenotype could potentially be produced by strategically manipulating the nuclear factor-kappaB (NF-κB) signaling pathway in TAMs. The two arms of the NF-κB pathway (classical and alternative) are potent controllers of the macrophage phenotype and regulate many of their inflammatory and trophic functions (Figure 1).25–28
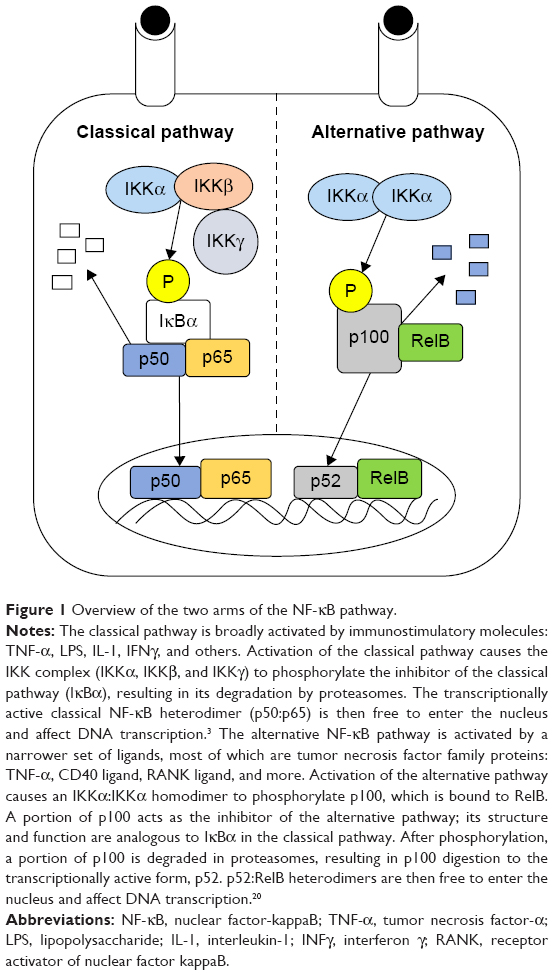 | Figure 1 Overview of the two arms of the NF-κB pathway. |
Previous work supports the idea of therapeutically targeting TAMs with the intention of reeducating them to become tumor killing rather than tumor supportive. In addition, NF-κB signaling may be a phenotypic fulcrum that could promote this switch if manipulated appropriately. However, little progress has been made toward translating this knowledge into viable, clinically relevant therapeutic agents. This study addresses two of the major hurdles that must be overcome to move this type of therapy forward: 1) developing a means to achieve macrophage-specific and pathway-specific cell-signaling modulation and 2) gaining additional knowledge about which components of the NF-κB pathway would be the most advantageous to target.
A possible mechanism to change NF-κB signaling in TAMs is the use of small interfering RNAs (siRNAs). These are short (20–25 bp), double-stranded RNA sequences that can activate RNA interference in cells, resulting in the degradation of intracellular messenger RNA (mRNA) sequences that are bound by the guide strand of the siRNA.29 This effect targets a specific mRNA sequence, and the strength and duration of the effect can be tuned by altering siRNA dosing. To facilitate delivery of NF-κB siRNA to TAMs, we have developed a targeted polymeric nanoparticle capable of encapsulating and protecting siRNA sequences. The particle is designed such that the pH-responsive core disrupts the endosomal compartment upon cellular uptake, rupturing the organelle as the internal pH drops and releasing the functional siRNA into the cytoplasm.30–32 The particle has a modular surface that can be modified with targeting ligands using facile, copper-catalyzed “click” chemistry.33 We have functionalized the surface of these particles with mannose as a targeting ligand in order to facilitate uptake of the particles in TAMs, which have increased amounts of mannose receptor (CD206) on their surface.34,35 The mannose receptor is an endosomal pattern-recognition receptor, which, when present at high levels, is associated with suppression of the classical immune response.36 We have shown previously that uptake of these mannosylated endosomal escape nanoparticles (MnNPs) by macrophages is mannose dependent and that uptake of the MnNPs is enhanced in macrophages compared to uptake of untargeted, hydroxyl-capped nanoparticles (OHNPs).37 MnNPs are designed to condense and shield siRNA in the interior of the particle for optimal systemic transport, enter the tumor vasculature via the enhanced permeability and retention effect, specifically target TAMs in the tumor microenvironment, and escape the low pH of the late endosome to deliver functional siRNA into the cytoplasm. We recently determined that MnNPs are biocompatible in vivo and that the targeting moiety enhances uptake in TAMs in vivo.38 The present study takes this work forward by validating the efficacy of MnNPs as transfection agents for delivering functional siRNA to macrophages and measuring siRNA-mediated modulation of NF-κB activity resulting from suppression of signal transduction proteins.
In order to address the second hurdle and determine the optimal strategy for modulation of NF-κB signaling, we have screened several siRNA sequences specific for NF-κB pathway proteins using both the commercial transfection agent Lipofectamine and our MnNPs. The prevailing dogma has been that suppression of NF-κB signaling reduces protumor TAM effects.39 However, our recent work in mice with activatable classical NF-κB signaling in macrophages has shown that strategic activation of the classical pathway reduces metastatic tumor burden in a tail vein-injected mouse model of metastatic breast cancer and in a model of metastatic melanoma.23,40 In line with these studies, we discovered that MnNP-delivered siRNA against the inhibitor protein of the classical pathway was capable of activating the NF-κB pathway and inducing a proinflammatory, immunogenic phenotype in the transfected macrophages. This result highlights a novel strategy to generate antitumor functions in TAMs and indicates that the combination of our novel MnNP with IκBα siRNA could represent the first step toward harnessing TAM-targeted siRNA strategies for cancer therapy.
Materials and methods
Cell culture
Unless otherwise stated, all primary cells and cell lines used in this study were maintained in Dulbecco’s Modified Eagle’s Medium (Corning Incorporated, Corning, NY, USA; MT-10-13-CV) with the addition of 10% (v/v) fetal bovine serum and 1% Pen Strep (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C in a 5% CO2 humidified atmosphere.
Bone marrow-derived macrophage culture
All animal work was approved by the Vanderbilt University Institutional Animal Care and Use Committee. Bone marrow-derived macrophages (BMDMs) were made by harvesting bone marrow from wild-type and NF-κB green fluorescent protein (GFP)-luciferase (NGL) reporter transgenic mice on an FVB background.41 Cells from NGL mice produce a luciferase/GFP fusion protein as a readout of total NF-κB activation: activity of either the classical or alternative pathway induces expression of a GFP-luciferase reporter protein. The medium for these cells contained 10% (v/v) fetal bovine serum, 1% Pen Strep (Thermo Fisher Scientific), 5% heat-inactivated horse serum (Thermo Fisher Scientific), 1% MEM nonessential amino acid mixture (Sigma-Aldrich Co., St Louis, MO, USA), and 50 μM 2-mercaptoethanol (Sigma-Aldrich Co.) and was supplemented with media from L-129 fibroblasts as a source of macrophage colony stimulating factor (M-CSF).24 The bone marrow was cultured for 6 days in the M-CSF-supplemented medium, and the resultant BMDMs were scraped from their plates and replated as necessary for further experiments.
Luciferase activity readout in reporter cells
For cells producing luciferase as a readout of total NF-κB activation, the Promega luciferase assay system with reporter lysis buffer was used to measure luciferase activity. Plated cells were frozen in 1× reporter lysis buffer, then subjected to a thaw–freeze–thaw cycle before being scraped from the plates, and centrifuged at 13,000 rpm for 4 minutes. Twenty microliters of the supernatant was added to 100 μL of the luciferin substrate, and the resulting luminescence was measured to determine luciferase activity.
Effects of tumor necrosis factor-α stimulation on BMDMs
In order to induce NF-κB activation in macrophages, NGL BMDMs were stimulated with 0.01–1,000 ng/mL of tumor necrosis factor-α (TNF-α) (PeproTech, Rocky Hill, NJ, US) for 6 hours. After 6 hours, NF-κB activation was assessed by the luciferase assay. For subsequent experiments, including TNF-α stimulation, 10 ng/mL concentrations were used. To assess mRNA changes following TNF-α stimulation, wild-type BMDMs were plated in six-well plates at a density of 2,000,000 cells/well (~210,000 cells/cm2). Each sample was taken from a different mouse, for a total of three biological replicates, with three experimental replicates per biological replicate. The cells were stimulated for 6 hours with TNF-α (10 ng/mL). After stimulation, mRNA was collected from the cells for quantitative real-time polymerase chain reaction (qRT-PCR). The effect of TNF-α stimulation on the macrophages was analyzed by determining the changes in mRNA for the NF-κB pathway proteins IKKβ and p100/p52 as well as the mRNA for CCL3, an inflammatory cytokine, and the mRNA for IL-10, an immunoinhibitory cytokine (qRT-PCR procedure and primer sequences are detailed later).
RNA preparation and qRT-PCR
mRNA was extracted from cells using RNeasy kit (Qiagen NV, Venlo, the Netherlands), and DNA was removed from the samples using DNA-free kit (Thermo Fisher Scientific). cDNA was prepared from the isolated mRNA using dNTPs from Hoffman-La Roche Ltd., Basel, Switzerland, and random hexamers and superscript II reverse transcriptase were from Thermo Fisher Scientific. qRT-PCR was performed with SYBR green real-time PCR master mix (Thermo Fisher Scientific) using Step One Plus real-time PCR systems hardware and software (Thermo Fisher Scientific). The Step One Plus software was used to automatically calculate optimized baseline and threshold values. Differences in cDNA levels were calculated using the ΔΔCt method with glyceraldehyde 3-phosphate dehydrogenase as an internal control. The sequences used for qRT-PCR are summarized in Table 1.
 | Table 1 qRT-PCR primer sequences |
Screening effective siRNA sequences against NF-κB with Lipofectamine
Several siRNA sequences against NF-κB proteins were purchased from Thermo Fisher Scientific. The siRNA targets and their serial numbers are summarized in Table 2. The siRNAs were complexed with Lipofectamine and delivered to NGL BMDMs (12-well plates, 300,000 cells/well [~80,000 cells/cm2], four samples per condition) for 24 hours at an siRNA concentration of 10 nM. The most successful sequences were then used at 50 nM and delivered to NGL BMDMs using both Lipofectamine and MnNPs.
 | Table 2 siRNA targets and serial number of siRNA sequences purchased from Thermo Fisher Scientific (Waltham, MA, USA) |
DNA and siRNA delivery to macrophages by MnNPs
Wild-type BMDMs were cultured in normal media or in media containing IL-4 (10 ng/mL) (PeproTech) for 72 hours. IL-4 stimulation has been reported to increase mannose receptor presence on the cell surface.42 The cells were then plated in 96-well plates at a density of 50,000 cells/well (~350,000 cells/cm2). A 21-base pair, Cy3-labled DNA sequence was purchased from Sigma-Aldrich Co. for complexation with MnNPs. The sequence was designed to have the same base pair order and charge characteristics as the scrambled siRNA sequence from Thermo Fisher Scientific. To form nanoparticle–siRNA complexes, mannosylated endosomal escape polymers were synthesized as previously described (4 mg/mL in aqueous solution).37,38 For complexation with the polymer, all nucleotides were diluted to 50 μM in sterile nuclease-free water. The nucleotides were mixed with the polymer solution at a ratio of 160 ng of MnNP polymer per pmol of siRNA and allowed to form complexes at room temperature for 1 hour prior to use. For both studies, the effects of the siRNA on total NF-κB activity were assessed by luciferase assay following TNF-α stimulation of the cells. The Cy3_DNA (50 μM solution) was complexed with MnNPs or OHNPs, and the complexes were delivered to BMDMs. Cy3 fluorescence was measured at 0.5 hour, 1 hour, 3 hours, 5 hours, 10 hours, and 18 hours by washing the cells with phosphate-buffered saline (PBS) three times and then measuring fluorescence with a Tecan Infinite M1000-Pro plate reader as a measure of the particle uptake. Another set of BMDMs were plated as described earlier, and the Cy3_DNA sequence was delivered to the cells using either Lipofectamine or MnNPs with a 20-hour transfection time. These samples were measured for fluorescence with the Tecan Infinite M1000-Pro plate reader and imaged using a Nikon Eclipse Ti microscope. The total number of Cy3-positive cells was counted, and the percentage of Cy3-positive cells was determined.
Effects of IκBα knockout macrophages on cocultured tumor cells
Immortalized murine macrophages from IκBα knockout mice were incubated with GFP-expressing ID8 murine ovarian tumor cells on glass slides.39,43,44 The coculture was fixed and stained after 3 days of incubation. The samples were fixed in 4% paraformaldehyde for 30 minutes and permeabilized in 0.4% Triton X-100 for 10 minutes. Next, the slides were washed for 10 minutes two times in Tris-buffered saline (TBS) with 0.05% Tween 20. They were then washed with TBS for 5 minutes and blocked for 1 hour in blocking buffer (0.01 M Tris–HCl, pH 7.4, 2% bovine serum albumin, 2% normal goat serum). The samples were then incubated with primary tubulin antibodies in blocking buffer overnight at 4°C, washed three times in TBST (TBS + Tween), and incubated for 2 hours with secondary antibodies in the blocking buffer. After staining with secondary antibodies, the slides were washed three times with TBS containing 0.05% Tween 20, then washed once in TBS, and then stained with Molecular Probes TO-PRO-3 (Thermo Fisher Scientific) for 15 minutes (1 μM in TBS). The slides were washed with TBS and the coverslips mounted using Molecular Probes ProlongGold antifade reagent (Thermo Fisher Scientific). Images were acquired using an LSM 510 Meta confocal microscope in the Vanderbilt University Medical Center Imaging Core.
Effect of IκBα siRNA on total NF-κB activation in macrophages
To investigate the possibility of activating the classical NF-κB pathway by knocking down the inhibitor of the classical pathway IκBα, siRNA against IκBα was delivered to NGL BMDMs in 12-well plates at 50 nM for 24 hours using MnNPs. Free siRNA and empty MnNPs were also delivered separately as controls. Total NF-κB activation was assessed at 6 hours, 12 hours, and 24 hours by luciferase assay. Wild-type BMDMs were plated in six-well plates at a density of 2,000,000 cells/well (~210,000 cells/cm2). Each sample was taken from a different mouse, for a total of three biological replicates, with three experimental replicates per biological replicate. The cells were transfected using MnNPs for 24 hours with IκBα or scrambled siRNA, with and without TNF-α stimulation. After transfection, mRNA was collected from the cells for qRT-PCR (as earlier).
Statistical analysis
Data are reported as the sample mean, and error bars represent one SD from the mean. Paired comparisons were made using a two-tailed t-test to determine significant differences between samples. For datasets where multiple experimental groups were compared to the control group, Dunnett’s test was used to determine significant differences. For samples with multiple comparisons, analysis of variance was used to confirm significant differences within the groups; then post hoc analysis was performed using two-tailed t-tests. The probability for type I error in the post hoc analysis was reduced by minimizing the number of paired comparisons using a priori knowledge of the relationships between groups and selecting only the most pertinent comparisons.
Results and discussion
TNF-α stimulation of BMDMs
It has been reported that BMDMs are a viable in vitro model for TAMs.7 BMDMs are made from myeloid progenitor cells harvested from femoral bone marrow and matured in media supplemented with M-CSF. In vivo, M-CSF is a potent macrophage-recruiting and maturation cytokine and is known to play a role in the recruitment of myeloid progenitors into tumors and maturing these cells into macrophages.45 After 6 hours of incubation of BMDMs in TNF-α-treated media, total NF-κB activity increased in a dose-dependent manner as measured by luciferase readout as a proxy measure of NF-κB activity (Figure 2A). These results were used to select a sensitive baseline of NF-κB activation for subsequent assessments of activation suppression resulting from siRNA treatments. TNF-α stimulation of BMDMs was also confirmed by qRT-PCR, which shows the expected increase in the inflammatory cytokine CCL3 and a decrease in the immunoinhibitory IL-10 (Figure 2B). While mRNA levels of IKKβ were not found to increase after stimulation, mRNA for the transcriptionally active p100/p52 protein significantly increased.
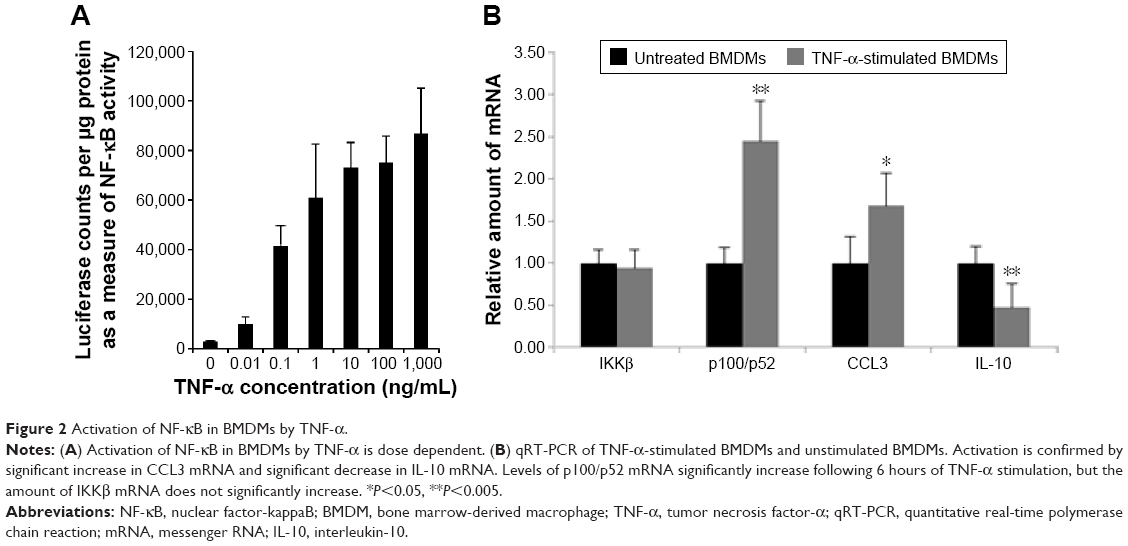 | Figure 2 Activation of NF-κB in BMDMs by TNF-α. |
Screening NF-κB-specific siRNA sequences for efficacy using Lipofectamine and MnNPs
In order to confirm that siRNA could knockdown NF-κB activity in macrophages, several sequences for IKKβ, p52/p100, and p65 were screened in NGL BMDMs using Lipofectamine as the transfection agent. Luciferase siRNA was used as an additional control for the effect of direct knockdown of the reporter protein. Efficacious siRNA sequences were confirmed for the NF-κB-related proteins shown in Figure 3A. Transfection with IKKβ siRNA significantly reduced the total NF-κB activity by ~40% for sequences B and C. Similar significant reduction in activation (40%–45%) was measured following transfection with p65 siRNA sequences B and C. This level of knockdown is consistent with siRNA knockdown of a single pathway, which would leave the unaffected pathway free to normally respond to stimuli. It is interesting to note that knockdown of the alternative pathway with p52/p100-specific siRNA sequences (B and C) resulted in a 70%–80% decrease in total NF-κB activity following TNF-α stimulation. This result is consistent with the existence of crosstalk between the two NF-κB signaling pathways, or that TNF-α stimulation of NF-κB activation in these cells was driven largely by the alternative pathway, and the siRNA sequences for this pathway were very effective.
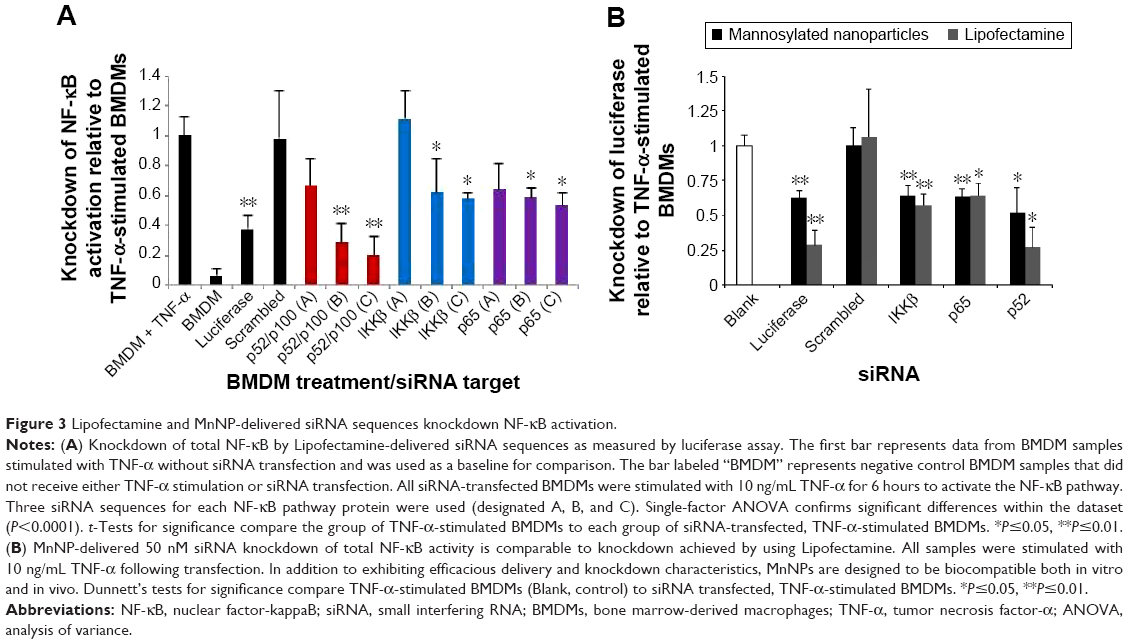 | Figure 3 Lipofectamine and MnNP-delivered siRNA sequences knockdown NF-κB activation. |
It is known that products of one NF-κB pathway can modulate another, in particular, the TNF family of proteins.3 Knocking down activity of the alternative pathway may also affect the classical pathway via inhibition of cytokine production that could crosstalk to stimulate that pathway. NF-κB activation can be accomplished with protein dimers other than p50:p65 and p52:RelB. Since it is possible that knockdown of p52 suppresses the formation of p52:p65 heterodimers, it is also possible that reduction of p52 activates a compensatory mechanism and results in more production of p105, which is degraded to p50, although there is currently no direct evidence for this mechanism. An increase in p50 could result in the formation of p50 homodimers, which are known to inhibit classical activation.46
To compare the transfection ability of MnNPs with Lipofectamine, 50 nM siRNA was delivered to NGL BMDMs for 24 hours by MnNPs or Lipofectamine; knockdown of total NF-κB activity was analyzed by luciferase assay following TNF-α stimulation. The most successful sequence for each protein from Figure 3A was used. MnNP-mediated siRNA knockdown of total NF-κB activity was similar to Lipofectamine-mediated knockdown (Figure 3B). Total NF-κB activity was significantly decreased upon MnNP delivery of siRNA sequences. The average knockdown of total NF-κB activity by Lipofectamine-delivered siRNA was less than the knockdown from 10 nM siRNA shown in Figure 3A. It is possible that this decrease in efficacy is due to an increase in cytotoxicity associated with increasing the amounts of Lipofectamine combined with TNF-α stimulation, as suggested previously.38
Comparing the uptake of MnNPs with untargeted particles and Lipofectamine
We have previously shown that uptake of MnNPs can be reduced by introducing free mannose during transfection, which competes with the MnNPs for uptake by the macrophage mannose receptor.37 To further investigate the potential for specific delivery to TAMs, untargeted OHNPs or MnNPs loaded with Cy3-labeled dsDNA were delivered to BMDMs for 18 hours. Two different populations of BMDMs were used: untreated BMDMs and BMDMs stimulated with IL-4 to increase mannose receptor production.42 Particle uptake by BMDMs was measured at various time points by measuring Cy3 fluorescence in the BMDMs. In IL-4-stimulated macrophages, uptake of MnNPs is significantly increased as compared to that of MnNPs in unstimulated macrophages (Figure 4A and B). Uptake of OHNPs was not significantly different between the two groups of cells. This result indicates that the mannose ligand on the surface of the MnNPs is mediating enhanced uptake, which is specific for macrophages that have undergone phenotypic differentiation by IL-4.
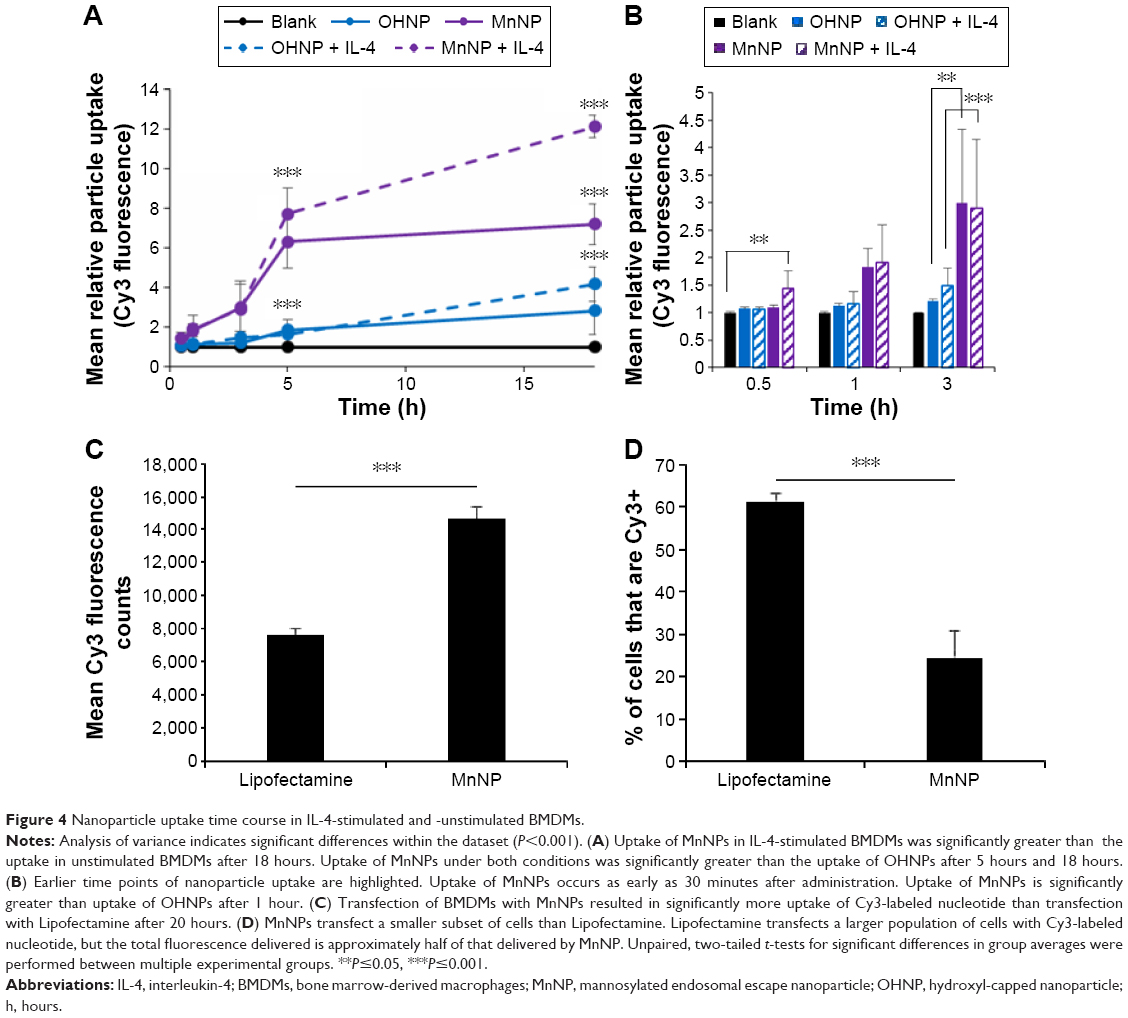 | Figure 4 Nanoparticle uptake time course in IL-4-stimulated and -unstimulated BMDMs. |
Although IL-4-stimulated BMDMs are reported to have increased mannose receptor production, unstimulated BMDMs also express this receptor at their surface. This explains why, even at earlier time points, MnNP uptake is enhanced relative to OHNP uptake in both IL-4-stimulated and -unstimulated BMDMs. Significant uptake of MnNP occurs as early as 30 minutes after administration to IL-4-stimulated BMDMs. This rapid uptake is consistent with endosomal uptake of the particles, presumably mediated by the mannose receptor. Some modest nonspecific uptake is also evident. It is likely that some particles are phagocytized by the macrophages or taken up via other nonspecific endosomal pathways.
A final experiment investigating the mechanism of MnNP delivery to macrophages compared 20-hour MnNP delivery to Lipofectamine delivery. Both transfection agents were labeled with Cy3_dsDNA. After 20 hours of incubation, Cy3 fluorescence was measured as an indicator of particle uptake, and the percentage of Cy3-positive cells was determined from fluorescent and bright-field images of the cells. Transfection with MnNPs resulted in significantly more uptake of Cy3-labeled nucleotide than transfection with Lipofectamine (Figure 4C). Furthermore, Lipofectamine transfects a larger population of cells with Cy3-labeled nucleotides, but the total fluorescence delivered is approximately half of that delivered by MnNPs (Figure 4D). Compared to Lipofectamine, MnNPs strongly transfect a smaller subset of macrophages. This is consistent with the mechanism of MnNP transfection as a more targeted delivery to cells expressing the corresponding surface receptor. This result, combined with our previous report that the uptake of MnNP is mannose dependent, is consistent with MnNPs targeting a subset of macrophages with greater amounts of mannose receptor. Macrophages (and TAMs) with a tissue remodeling/immunoinhibitory phenotype have increased mannose receptor production. This effect is so pronounced that the presence of high levels of mannose receptor in macrophages is a commonly used indicator for this macrophage phenotype. Therefore, this result enhances our previous work showing enhanced uptake of MnNPs by TAMs in vivo.38
Target protein selection for strategic manipulation of NF-κB to produce therapeutically relevant changes in TAM phenotype
Our screen of siRNA sequences for NF-κB proteins confirms knockdown of total NF-κB activity by MnNP-delivered siRNA. Although siRNA sequences for IKKβ, p65, and p52 all resulted in significant knockdown, it is important to establish whether these proteins will be efficacious targets or not. Although p65 is implicated in producing smoldering inflammation in the tumor microenvironment, it is also responsible for survival signaling in macrophages. Therefore, p65 may not be an optimum target because knocking down p65 could potentially induce apoptosis in the macrophages being targeted for phenotypic modification.47,48
Viable targets for therapeutic siRNA knockdown should ideally also express increased amounts of the target mRNA while in the pathological state. qRT-PCR of TNF-α-stimulated BMDMs indicates that levels of p52 mRNA are increased upon exposure to TNF-α, making p52 a viable target for knocking down alternative NF-κB activity in TAMs. However, levels of IKKβ do not significantly increase (Figure 2B). It is known that the classical NF-κB pathway is rapidly activated within minutes of stimulation, while alternative activation is more gradual.27,49,50 If there is an increase in IKKβ mRNA, it is possible that the increase is rapidly induced and then rapidly degraded. Another alternative is that there is already enough IKKβ protein active in the cell to activate enough p50:p65 homodimers and induce a classical NF-κB response; increased production of IKKβ may not be necessary. Regardless of the reason, this lack of increase in IKKβ mRNA makes it a poor target for siRNA-mediated knockdown. In addition, there are significant non-NF-κB functions of IKKβ, and knocking down this protein could potentially have off-target effects.3
As discussed earlier, neither IKKβ nor p65 is an attractive target for siRNA-mediated knockdown to achieve a reduction in the smoldering inflammation produced by TAMs. However, decreasing the smoldering inflammation from these cells is not the only therapeutic option. If the full inflammatory potential of the TAMs could be reactivated, these cells could promote a potent immune response at the site of the tumor by releasing inflammatory cytokines and inducing apoptosis as well as by recruiting cytotoxic immune cells of innate and adaptive immune system. This effect could be achieved by strongly activating the classical NF-κB pathway in TAMs.
Although the strategy to deliver siRNA with MnNPs was initially designed to knockdown NF-κB activity, it should theoretically be able to activate the classical pathway as well. Activation of transcriptional pathways by siRNA has been reported before, but only as an undesirable, off-target side effect. Because the IκBα protein inhibits activation of the classical pathway, knocking down this protein could potentially increase classical NF-κB activation. This proposed activation of a key transcriptional pathway is a novel approach to manipulating cell phenotype for immunoengineering. To test this possibility, NGL BMDMs were transfected with MnNP-delivered siRNA against IκBα for 24 hours, and total NF-κB activation was measured at multiple time points during transfection. Delivery of IκBα siRNA by MnNPs induced significant activation of NF-κB without any other source of stimulation (Figure 5). The particle-delivered siRNA increased the total NF-κB activity in the macrophage population 2.3-fold; however, this measurement was taken from the combined lysate of the entire macrophage population and, as Figure 4D shows, the MnNPs transfect ~25% of the macrophages in the population. The effect of transfection in the targeted cells is presumably diluted by the presence of untransfected cells. When the 75% untransfected population is taken into account in the luciferase assay results reported in Figure 5, the actual increase in total NF-κB activity solely in the transfected population is estimated to be approximately ninefold.
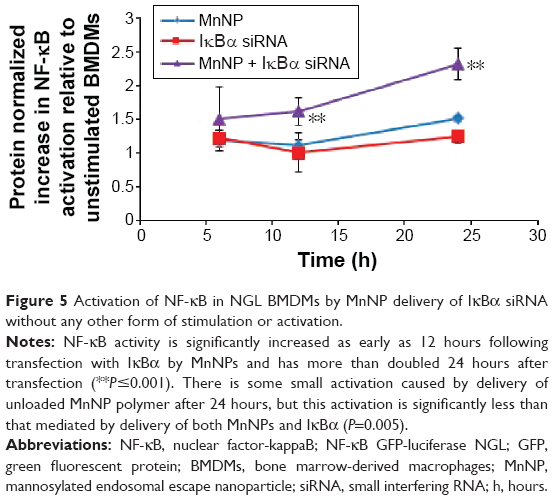 | Figure 5 Activation of NF-κB in NGL BMDMs by MnNP delivery of IκBα siRNA without any other form of stimulation or activation. |
This work identifies a novel approach to therapeutic RNAi: using protein knockdown to suppress expression of an inhibitor and increase activation of a transcriptionally active pathway. Knocking down the inhibitor of the classical NF-κB pathway increases total NF-κB activity, but it is important to understand whether this knockdown is physiologically and therapeutically relevant. In order to understand what effect IκBα knockdown in macrophages might have on tumor cells, an IκBα knockout (IκBα k/o) macrophage line was cocultured with GFP expressing ovarian tumor cells (ID8 cell line) for 3 days, and the coculture was imaged at day 1 and day 3. The IκBα k/o macrophage/ID8 tumor cell coculture is compared to a wild-type macrophage line, also at 3 days cocultured with ID8 cells (Figure 6). After 3 days, the wild-type macrophages exist in stable coculture with the tumor cells and there are no overt morphological signs of cytotoxicity in either cell type. However, the IκBα k/o macrophages appear to mediate tumor cell cytotoxicity presumably via extracellular lysis of the tumor cells such that only fragments of the tumor cells are left.51,52 This result suggests that activation of NF-kappaB via deletion of the IκBα inhibitor can induce tumor cytotoxic functions.
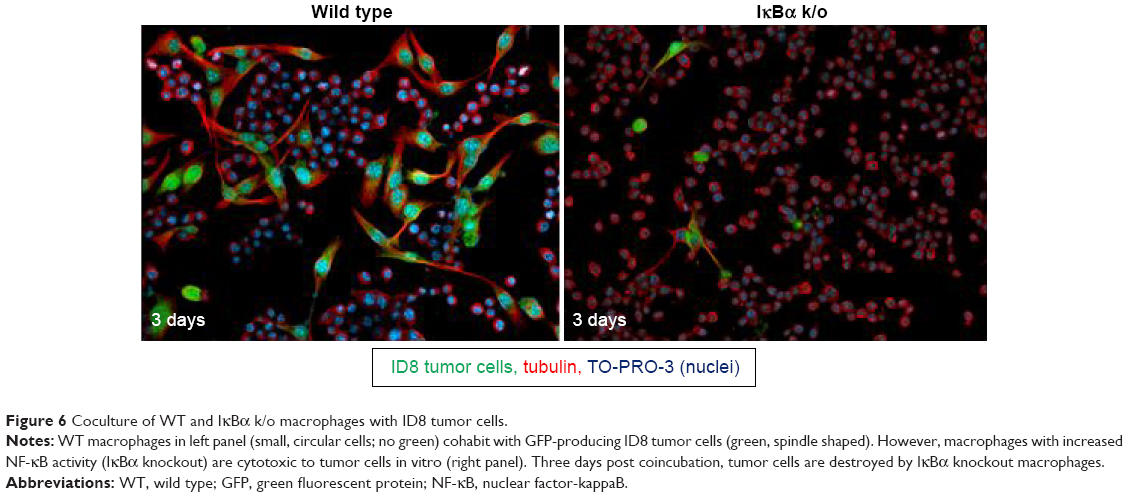 | Figure 6 Coculture of WT and IκBα k/o macrophages with ID8 tumor cells. |
qRT-PCR of BMDM transfected with NF-κB-specific siRNA sequences by MnNPs
To determine whether interaction with the MnNP transfection agent and delivery of control siRNA by MnNPs results in any significant expression of relevant genes, MnNPs were used to transfect BMDMs with a scrambled siRNA sequence. Figure 7 shows that there are no significant differences between macrophages transfected with scrambled siRNA sequences and untransfected macrophages for mRNA for IκBα, p52, IL-10, or CXCL9 (a T-cell recruiting cytokine). There is a significant increase in CD206 (mannose receptor) upon uptake of the MnNPs. This is consistent with an expected increase in receptor production following CD206 binding events and activation that contributes to the rapid recycling of this receptor.53,54
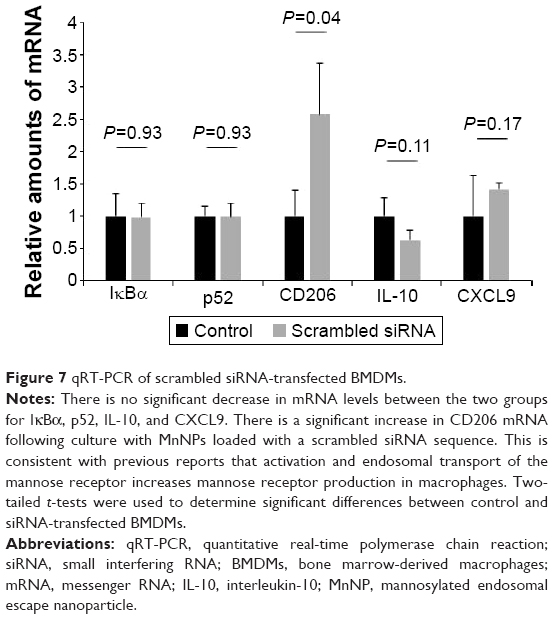 | Figure 7 qRT-PCR of scrambled siRNA-transfected BMDMs. |
To assess the potential for delivery of IκBα siRNA by MnNPs to induce immunostimulatory and antitumor effects, mRNA from transfected BMDMs was analyzed by qRT-PCR. mRNA levels for IκBα are significantly decreased following transfection with IκBα siRNA, indicating successful knockdown at the mRNA level of IκBα (Figure 8A). Effects on the mRNA levels of relevant cytokines in the cancer microenvironment that are produced by macrophages were also quantified. IκBα RNAi increases the amount of CXCL9 mRNA in macrophages to a level that is equivalent to levels of CXCL9 mRNA in TNF-α-stimulated macrophages (Figure 8B). Furthermore, the combination of IκBα siRNA and TNF-α acts in an additive manner, resulting in significantly more CXCL9 mRNA than by either treatment alone. CXCL9 is a chemotactic signal that recruits CD8+ T-cells and NK cells, and increasing CXCL9 in the tumor microenvironment has been shown to enhance antitumor immunity in murine breast cancer models.55,56 It has also been reported that intermediate CXCL9 signaling is necessary for T-cells to respond to other recruitment signals, such as IL-12.57 Therefore, the increase in expression of CXCL9 indicates that knockdown of IκBα in TAMs could result in increased recruitment of activated cytotoxic cells of the innate and adaptive immune systems to the tumor microenvironment. The recruitment of T-cells to solid tumors has been the major focus of many efforts to induce tumor immunity in several types of cancer.1,2,58 Increased recruitment of T-cells by modified macrophages in the tumor would be another potential method of activating adaptive tumor immunity.
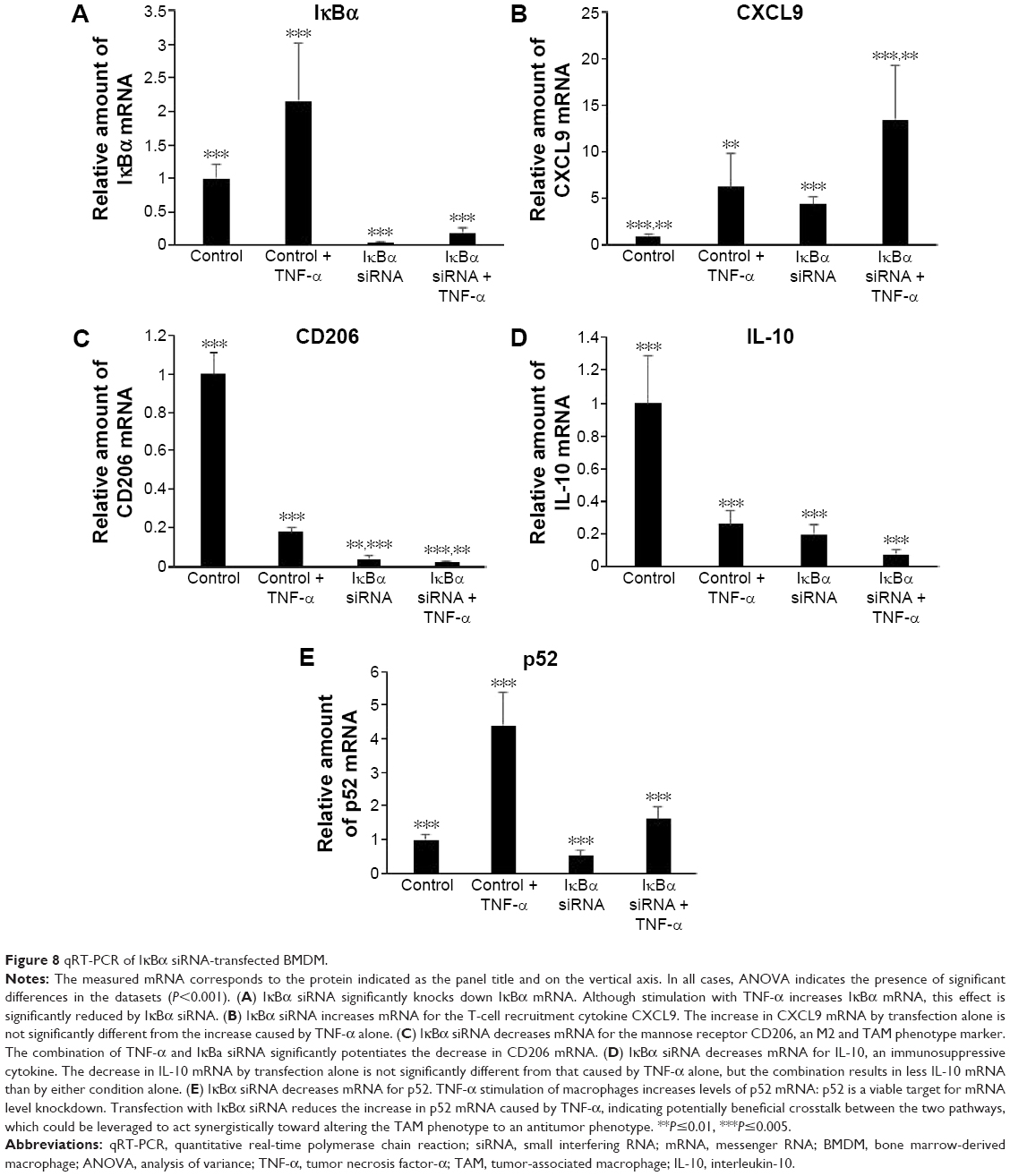 | Figure 8 qRT-PCR of IκBα siRNA-transfected BMDM. |
MnNP-delivered IκBα siRNA also decreases levels of phenotypic markers, which indicates a protumor immunosuppressive macrophage phenotype. Increased levels of the mannose receptor CD206 in macrophages indicate an alternatively activated phenotype and/or a TAM phenotype.34,35 Both TNF-α and IκBα siRNA decrease mRNA for CD206 in macrophages (Figure 8C). The combination of the two treatments also significantly decreases CD206 mRNA. This effect is particularly important because the mannose receptor is the target of the mannose ligand on the MnNP surface. Transfecting macrophages with IκBα siRNA effectively decreases the amount of the target on the macrophage surface. This effect could ensure that only macrophages with a strong TAM phenotype, cells with more CD206 on their surface, have enhanced endosomal uptake of the particles. This self-limiting uptake contributes to preferential targeting of siRNA only in alternatively activated macrophages, potentially suppressing off-target effects. Transfection would then alter the phenotype of the macrophage toward classical immune activity, reducing the amount of CD206 on the cell surface and preventing further unnecessary transfection. The MnNPs would then be free to transfect the remaining resident macrophages with high levels of CD206 or recently recruited macrophages with a TAM phenotype. MnNPs could also act as a surveillance mechanism if there are particles retained extracellularly in the tumor, retransfecting macrophages if they begin to readopt a TAM phenotype and express CD206 at their surface.
IκBα siRNA decreases the amount of IL-10 mRNA in macrophages to a level that is equivalent to levels of IL-10 mRNA in TNF-α-stimulated macrophages, and the combination of IκBα siRNA and TNF-α results in significantly less IL-10 mRNA than by either treatments alone (Figure 8D). IL-10 is a potent immunosuppressive cytokine and is implicated in creating the TAM phenotype by paracrine signaling from tumor cells, by other TAMs, and by autocrine signaling.12,59 IL-10 has been reported to specifically inhibit the classical NF-κB pathway by increasing the amount of cytoplasmic p105 and p50:p50 homodimers and by reducing p65 translocation into the nucleus.60,61 Treatment of macrophages with IL-10 reduces classical NF-κB responsiveness to stimulating cytokines such as lipopolysaccharide and TNF-α by reducing nuclear translocation of p65:p50 homodimers, which are responsible for much of the classic inflammatory response governed by NF-κB and by increasing translocation of p50:p50 homodimers that strongly inhibit the classical immune response.62 In many cases, p50 homodimers inhibit transcription of NF-κB gene targets. However, p50 homodimers are reported to activate the production of IL-10 in macrophages.46,63 Furthermore, IL-10 stimulation of macrophages does not affect the ability of the alternative NF-κB pathway in these cells to respond to TNF-α and other TNF protein family members, many of which are present in the tumor microenvironment.61
These studies indicate that IL-10 creates a powerful feedback loop in TAMs. IL-10 stimulation reduces classical NF-κB responsiveness to TNF-α but does not completely eliminate the effect. This is predicted to result in much lower levels of inflammatory cytokine production in response to the constant presence of inflammatory cytokines in the tumor microenvironment, producing protumor smoldering inflammation. In addition, the alternative pathway is free to respond to a smaller subset of these cytokines, activating the tumor-supportive effect of this pathway. Finally, IL-10 stimulation activates a positive feedback loop of IL-10 production, effectively locking the macrophages in the TAM phenotype, unless something can disrupt this signaling loop. Transfection with IκBα siRNA not only decreases IL-10 mRNA but also increases the ability of transfected macrophages to respond to TNF-α, indicating that it might be an effective therapeutic agent for disrupting the IL-10-positive feedback loop and abrogating the TAM phenotype.
Finally, the results suggest that crosstalk between the two pathways could also be leveraged to reduce the negative effects of the TAM phenotype. The expression of p52 increases following TNF-α stimulation, but this effect is inhibited by transfection with IκBα siRNA (Figure 8E). This effect may be the result of direct or indirect crosstalk and could indicate the potential possibility of knocking down of IκBα having combinatorial effects by decreasing the potentially protumor effects of the alternative NF-κB pathway at the same time as activating the classical pathway.
Conclusion
Modulation of the functions of TAMs represents a therapeutic strategy with great potential, and NF-κB signaling is known to be important for defining macrophage functions, thus suggesting that modulation of NF-κB signaling within TAMs could represent a novel therapeutic strategy. In order to develop this new treatment, two components would be required: the methodology for manipulation of gene expression specifically within TAMs and a knowledge of the appropriate gene targets. The optimal strategy for manipulation of NF-κB in TAMs to generate antitumor outcomes is currently unknown. To develop the methodology for manipulation of gene expression within TAMs, we have developed a targeted polymer nanoparticle that encapsulates and protects siRNA sequences to facilitate delivery of NF-κB-specific siRNA to TAMs. The nanoparticles have a mannosylated surface designed to target the mannose receptor, which is upregulated in TAMs macrophages. In the studies reported here, we have shown that MnNPs could deliver efficacious amounts of functional siRNA for manipulating the NF-κB pathways in macrophages and were more specific than commercial transfection agents. MnNPs are taken up to a greater degree in macrophages with increased mannose receptor presence (induced by IL-4 stimulation) compared to wild-type macrophages. The enhanced uptake of MnNPs by IL-4-stimulated macrophages complements our previous results indicating that MnNP uptake is mannose dependent. Furthermore, uptake of untargeted particles by IL-4-stimulated macrophages was not different from uptake of the same particles by wild-type macrophages. There is greater overall uptake of the MnNPs than the OHNPs, and uptake of MnNPs occurs at earlier time points. A comparison of MnNPs and Lipofectamine revealed that MnNPs strongly transfect a small population of macrophages in the BMDM population while Lipofectamine moderately transfects a larger population of BMDMs. Together, these results indicate that we have developed a methodology that is capable of delivering siRNA with specificity for mannose receptor expressing TAMs.
We have performed studies to determine the appropriate NF-κB gene targets to induce antitumor characteristics in macrophages. Following delivery of IκBα siRNA to macrophages by MnNPs, mRNA for IκBα was decreased, and mRNA for a T-cell recruitment cytokine CXCL9 was increased. Furthermore, mRNA for the mannose receptor and for IL-10 were decreased, indicating a loss of immunosuppressive function and a potential mechanism for disrupting the positive feedback loop between TAMs and tumor cells, which is implicated in sustaining the TAM phenotype. Thus, our results suggest that utilizing MnNP-delivered IκBα siRNA to knockdown the inhibitor of the classical pathway and activating classical NF-κB activity represents an effective strategy. Although the NF-κB pathways have been implicated in the generation of many protumor characteristics in TAMs, strong and acute activation of the classical pathway appears to override the TAM phenotype and activate a classically immunogenic phenotype in these macrophages. There have been reports of siRNA activating transcriptional pathways in cells, but this has always been seen as an undesirable, off-target effect of transfection in general. Deliberate activation of a specific transcriptional pathway by using siRNA to knockdown an inhibitor of the pathway has not been reported and is a novel mechanism for manipulating cell phenotype.
The current studies demonstrate the development of a methodology for delivery of siRNA to TAMs together with the identification of an NF-κB pathway gene target that in combination can induce cytotoxic and immunostimulatory functions in TAMs. In future studies, we plan to test the efficacy of this strategy to modulate NF-κB in TAMs in vivo, and activate tumor immunity in solid tumors.
Acknowledgments
The work performed in this study was funded in part by a Collaborative Idea Award through the Department of Defense CDMRP Breast Cancer Research Program (W81XWH-11-1-0242 and W81XWH-11-1-0344) and by donations from Mr Chris Hill via the Anglo-American Charity. Nanoparticle characterization was performed at the Vanderbilt Institute of Nanoscale Science and Engineering, using facilities renovated under National Science Foundation Grant ARI-R2 DMR-0963361. The authors would like to acknowledge Hongmei Li and Shann Yu for their work in the design and synthesis of mannosylated endosomal escape polymers. They would also like to thank Halina Onishko for instruction and advice regarding bone marrow-derived macrophage generation and handling.
Disclosure
The authors report no conflicts of interest in this work.
References
Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. 2008;222:357–368. | ||
Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. | ||
Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. | ||
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. | ||
DeNardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. | ||
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. | ||
Ojalvo LS, King W, Cox D, Pollard JW. High-density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. Am J Pathol. 2009;174:1048–1064. | ||
Greten FR, Eckmann L, Greten TF, et al. Ikkbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. | ||
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. | ||
Van Ginderachter JA, Movahedi K, Hassanzadeh Ghassabeh G, et al. Classical and alternative activation of mononuclear phagocytes: picking the best of both worlds for tumor promotion. Immunobiology. 2006;211:487–501. | ||
Baj-Krzyworzeka M, Baran J, Szatanek R, Stankiewicz D, Siedlar M, Zembala M. Prevention and reversal of tumor cell-induced monocyte deactivation by cytokines, purified protein derivative (Ppd), and anti-IL-10 antibody. Cancer Immun. 2004;4:8. | ||
Sica A, Saccani A, Bottazzi B, et al. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J Immunol. 2000;164:762–767. | ||
DeNardo DG, Barreto JB, Andreu P, et al. Cd4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102. | ||
Lin EY, Jones JG, Li P, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. | ||
Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007;67:5064–5066. | ||
Qian B, Deng Y, Im JH, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One. 2009;4:e6562. | ||
Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. | ||
Stathopoulos GT, Sherrill TP, Han W, et al. Host nuclear factor-kappaB activation potentiates lung cancer metastasis. Mol Cancer Res. 2008;6:364–371. | ||
Zaynagetdinov R, Sherrill TP, Polosukhin VV, et al. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J Immunol. 2011;187:5703–5711. | ||
Sun SC. The noncanonical NF-κB pathway. Immunol Rev. 2012;246:125–140. | ||
Nathan C, Brukner L, Kaplan G, Unkeless J, Cohn Z. Role of activated macrophages in antibody-dependent lysis of tumor cells. J Exp Med. 1980;152:183–197. | ||
Nathan C, Cohn Z. Role of oxygen-dependent mechanisms in antibody-induced lysis of tumor cells by activated macrophages. J Exp Med. 1980;152:198–208. | ||
Connelly L, Barham W, Onishko HM, et al. NF-kappaB activation within macrophages leads to an anti-tumor phenotype in a mammary tumor lung metastasis model. Breast Cancer Res. 2011;13:R83. | ||
Connelly L, Jacobs AT, Palacios-Callender M, Moncada S, Hobbs AJ. Macrophage endothelial nitric-oxide synthase autoregulates cellular activation and pro-inflammatory protein expression. J Biol Chem. 2003;278:26480–26487. | ||
Bellas RE, FitzGerald MJ, Fausto N, Sonenshein GE. Inhibition of NF-kappa B activity induces apoptosis in murine hepatocytes. Am J Pathol. 1997;151:891–896. | ||
Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011;12:715–723. | ||
Makarov SS. NF-kappaB as a therapeutic target in chronic inflammation: recent advances. Mol Med Today. 2000;6:441–448. | ||
Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. | ||
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. | ||
Behr JP. The proton sponge: a trick to enter cells that viruses did not exploit. Chimia. 1997;51:34–36. | ||
Convertine AJ, Diab C, Prieve M, et al. pH-responsive polymeric micelle carriers for siRNA drugs. Biomacromolecules. 2010;11:2904–2911. | ||
Evans BC, Nelson CE, Yu SS, et al. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J Vis Exp. 2013;73:e50166. | ||
Boyer C, Bulmus V, Davis TP, Ladmiral V, Liu J, Perrier S. Bioapplications of raft polymerization. Chem Rev. 2009;109:5402–5436. | ||
Allavena P, Chieppa M, Bianchi G, et al. Engagement of the mannose receptor by tumoral mucins activates an immune suppressive phenotype in human tumor-associated macrophages. Clin Dev Immunol. 2010;2010:547179. | ||
Luo Y, Zhou H, Krueger J, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116:2132–2141. | ||
Stahl PD, Ezekowitz RA. The mannose receptor is a pattern recognition receptor involved in host defense. Curr Opin Immunol. 1998;10:50–55. | ||
Yu SS, Lau CM, Barham WJ, et al. Macrophage-specific RNA interference targeting via “click”, mannosylated polymeric micelles. Mol Pharm. 2013;10:975–987. | ||
Ortega RA, Barham WJ, Kumar B, et al. Biocompatible mannosylated endosomal-escape nanoparticles enhance selective delivery of short nucleotide sequences to tumor associated macrophages. Nanoscale. 2015;7:500–510. | ||
Hagemann T, Lawrence T, McNeish I, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–1268. | ||
Yang J, Hawkins OE, Barham W, et al. Myeloid IKKβ promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Res. 2014;74:7274–7284. | ||
Everhart MB, Han W, Sherrill TP, et al. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury. J Immunol. 2006;176:4995–5005. | ||
Porcheray F, Viaud S, Rimaniol AC, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142:481–489. | ||
Han W, Joo M, Everhart MB, Christman JW, Yull FE, Blackwell TS. Myeloid cells control termination of lung inflammation through the NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol. 2009;296:L320–L327. | ||
Roby KF, Taylor CC, Sweetwood JP, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–591. | ||
Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–237. | ||
Cao S, Zhang X, Edwards JP, Mosser DM. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281:26041–26050. | ||
Levkau B, Scatena M, Giachelli CM, Ross R, Raines EW. Apoptosis overrides survival signals through a caspase-mediated dominant-negative NF-kappa B loop. Nat Cell Biol. 1999;1:227–233. | ||
Wang Y, Mo X, Piper MG, et al. M-CSF induces monocyte survival by activating NF-κB p65 phosphorylation at Ser276 via protein kinase C. PLoS One. 2011;6:e28081. | ||
Fuseler JW, Merrill DM, Rogers JA, Grisham MB, Wolf RE. Analysis and quantitation of NF-kappa B nuclear translocation in tumor necrosis factor alpha (TNF-alpha) activated vascular endothelial cells. Microsc Microanal. 2006;12:269–276. | ||
Roos C, Wicovsky A, Muller N, et al. Soluble and transmembrane TNF-like weak inducer of apoptosis differentially activate the classical and noncanonical NF-kappa B pathway. J Immunol. 2010;185:1593–1605. | ||
Chattopadhyay U, Bhattacharyya S, Chakrabarty NG. Tumor associated macrophage mediated lysis of autologous tumor cells. Neoplasma. 1986;33:157–165. | ||
Cox GW. Assay for macrophage-mediated anti-tumor cytotoxicity. Curr Protoc Immunol. 2001;Chapter 14:Unit147. | ||
Fiani ML, Beitz J, Turvy D, Blum JS, Stahl PD. Regulation of mannose receptor synthesis and turnover in mouse J774 macrophages. J Leukoc Biol. 1998;64:85–91. | ||
Ezekowitz RA, Sastry K, Bailly P, Warner A. Molecular characterization of the human macrophage mannose receptor: demonstration of multiple carbohydrate recognition-like domains and phagocytosis of yeasts in Cos-1 cells. J Exp Med. 1990;172:1785–1794. | ||
Müller M, Carter S, Hofer MJ, Campbell IL. Review: the chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity – a tale of conflict and conundrum. Neuropathol Appl Neurobiol. 2010;36:368–387. | ||
Palmer K, Hitt M, Emtage PC, Gyorffy S, Gauldie J. Combined CXC chemokine and interleukin-12 gene transfer enhances antitumor immunity. Gene Ther. 2001;8:282–290. | ||
Tannenbaum CS, Tubbs R, Armstrong D, Finke JH, Bukowski RM, Hamilton TA. The CXC chemokines IP-10 and Mig are necessary for IL-12-mediated regression of the mouse renca tumor. J Immunol. 1998;161:927–932. | ||
Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–675. | ||
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. | ||
Armstrong L, Jordan N, Millar A. Interleukin 10 (IL-10) regulation of tumour necrosis factor alpha (TNF-alpha) from human alveolar macrophages and peripheral blood monocytes. Thorax. 1996;51:143–149. | ||
Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: a role for P50. Clin Exp Immunol. 2004;135:64–73. | ||
Smale ST. Hierarchies of NF-κB btarget-gene regulation. Nat Immunol. 2011;12:689–694. | ||
Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.