Back to Archived Journals » Research and Reports in Neonatology » Volume 8
Managing neonatal bowel obstruction: clinical perspectives
Authors Desoky SM, Kylat RI ![]() , Udayasankar U
, Udayasankar U ![]() , Gilbertson-Dahdal D
, Gilbertson-Dahdal D
Received 28 July 2017
Accepted for publication 7 November 2017
Published 5 February 2018 Volume 2018:8 Pages 19—32
DOI https://doi.org/10.2147/RRN.S125760
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Robert Schelonka
Sarah M Desoky,1 Ranjit I Kylat,2 Unni Udayasankar,1 Dorothy Gilbertson-Dahdal1
1Department of Medical Imaging, University of Arizona College of Medicine, Tucson, AZ, USA; 2Division of Neonatal-Perinatal Medicine, Department of Pediatrics, University of Arizona College of Medicine, Tucson, AZ, USA
Abstract: Neonatal intestinal obstruction is a common surgical emergency and occurs in approximately 1 in 2,000 live births. The causes of obstruction are diverse with varied embryological origins, and some underlying etiologies are not yet well described. Some findings of neonatal bowel obstruction can be detected prenatally on ultrasound imaging. The obstruction is classified as “high” when the level of obstruction is proximal to the ileum, and “low” when the level of obstruction is at the ileum or colon. Early diagnosis of the type of intestinal obstruction and localization of the obstructive bowel segment guides timely and appropriate management of the underlying pathologic entity. Neonatal bowel obstructions are ideally managed at specialized centers with a large volume of neonatal surgery and dedicated pediatric surgical and anesthesia expertise. Although surgical intervention is necessary in most cases, initial management strategies often target underlying metabolic, cardiac, or respiratory abnormalities. Imaging plays a key role in early and accurate diagnosis of the abnormalities. When bowel obstruction is suspected clinically, initial imaging workup usually involves abdominal radiography, which may direct further evaluation with fluoroscopic examination such as upper gastrointestinal (UGI) contrast study or contrast enema. This article provides a comprehensive review of clinical and radiological features of common and less common causes of intestinal obstruction in the neonatal age group, including esophageal atresia, enteric duplication cysts, gastric volvulus, congenital microgastria, hypertrophic pyloric stenosis, duodenal atresia, intestinal malrotation, intussusception, intestinal atresia, meconium ileus, functional immaturity of the colon, Hirschsprung disease, and anorectal malformation. The embryology, epidemiology, clinical presentation, and brief medical and surgical management of each is described.
Keywords: neonatal, bowel, intestinal, obstruction
Introduction
Intestinal obstruction is a common surgical emergency in the neonate and occurs in approximately 1 in 2,000 live births.1 The diagnosis may be suspected prior to the delivery of the infant due to prenatal ultrasound findings, including polyhydramnios and nonvisualization of normally visible fluid-filled structures, such as the stomach in esophageal atresia, or dilation of structures, such as the stomach and duodenum (the fetal double-bubble sign) in duodenal atresia. Other abnormalities which may be seen on prenatal ultrasound in the setting of bowel obstruction include bowel wall thickness greater than 3 mm, echogenic bowel, intraluminal and/or abdominal calcifications, as well as other abnormalities seen in the VACTERL (vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula, renal anomalies, and limb abnormalities) association.
Intestinal obstruction is classified as “high” when the level of obstruction is proximal to the ileum and “low” when the level of obstruction is at the ileum or colon. Prenatally, proximal obstruction is more readily diagnosed than distal obstruction because bowel dilatation and/or polyhydramnios are more likely to be present. Early prenatal diagnosis provides an opportunity for parental counseling and preparation, screening for associated anomalies, pregnancy termination, or delivery in an appropriate setting.
Although radiological appearances may vary, the clinical features and initial management of both types of obstruction are similar.
Clinical presentation in neonatal bowel obstruction may include bilious emesis, failure to pass meconium, and abdominal distension; the sequence of appearance of symptoms varies depending on the level of the obstruction.1 When a bowel obstruction is suspected in a neonate, the initial imaging study of choice is an abdominal radiograph, which may direct further imaging or clinical workup. If less than four dilated loops of bowel are noted, then the obstruction is likely proximal, in which case an upper gastrointestinal (UGI) contrast study may be conducted. If four or more loops of bowel are dilated, then the obstruction is more likely distal, in which case it is best to obtain a contrast enema.
When neonatal intestinal obstruction is suspected, management at a tertiary care center with a large volume of neonatal surgery and dedicated pediatric surgical and anesthesia expertise is ideal. A critically ill neonate requiring surgery should be transported emergently by a specialized neonatal transport team after initial resuscitative measures, which may include elective intubation. Most patients also need intravenous fluid and dextrose administration, gastrointestinal decompression with a large-gauge nasogastric suction catheter (replogle), as well as withholding of oral feeds until further evaluation and assessment. Although routine preoperative gastrointestinal decontamination is unnecessary in neonates, many surgical centers use perioperative broad-spectrum antibiotics. Maintenance of temperature is vital in this population, especially if the infant is premature. Constant monitoring of perfusion, blood pressure, blood glucose, electrolytes, and acid–base status should occur, with corrective measures instituted as needed. Measures to prevent negative nitrogen balance with early parenteral nutrition should be part of the management protocol for ideal postoperative care. If the infant is ill and intestinal malrotation with midgut volvulus has been excluded, immediate surgery for intestinal atresia is not necessary. The operation can be postponed until metabolic, cardiac, and/or respiratory abnormalities have been diagnosed and treated, and the patient is in optimal condition. If surgical treatment is deferred for a prolonged period of time, however, there is increasing risk of emesis and aspiration, sepsis, and other complications.
Esophageal atresia
Esophageal atresia occurs embryologically when there is a problem with the longitudinal ridge that separates the trachea and the esophagus. If tracheal dominance occurs, there is esophageal atresia. If there is a failure of separation of the longitudinal ridge, a tracheoesophageal fistula occurs, often in association with esophageal atresia.2
Esophageal atresia occurs in approximately 1 in 3,000 live births and is commonly associated with other abnormalities such as vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal fistula, renal anomalies, and limb anomalies, making up the VACTERL association.
There are multiple types of esophageal atresia that occur with or without tracheoesophageal fistula (Figures 1 and 2). The most common type is an esophageal atresia with a tracheoesophageal fistula involving the distal esophageal segment, which occurs in 75% of cases. In 10%–15% of cases, there is no fistula. In 5% there is no atresia, but there is an “H-type” fistula between the trachea and the esophagus, and these may present later in life.2
 | Figure 1 Schematic diagram depicting types of esophageal atresia. Abbreviations: EA, esophageal atresia, TEF, tracheoesophageal fistula. |
 | Figure 2 Esophageal atresia in newborns with failure to pass a nasogastric tube. (A) Supine chest radiograph shows nasogastric tube tip in dilated air-filled proximal esophageal pouch. Distal bowel gas indicates presence of tracheoesophageal fistula. (B) Supine chest and abdominal radiograph shows nasogastric tube coiled in dilated air-filled proximal esophageal pouch. Lack of distal bowel gas indicates absence of tracheoesophageal fistula. (C) Supine chest radiograph shows findings of esophageal atresia and tracheoesophageal fistula along with vertebral and cardiac anomalies seen in VACTERL association. Abbreviation: VACTERL, vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula, renal anomalies, and limb abnormalities. |
Esophageal atresia usually presents with a history of maternal polyhydramnios and postnatal inability to feed, with failure to pass a nasogastric tube. Chest radiography often reveals a nasogastric tube tip in a dilated air-filled proximal esophageal pouch. There may or may not be air in the stomach and bowel depending on the presence or absence of a distal tracheoesophageal fistula.
Contrast studies are generally not needed as they carry the risk of aspiration and usually add little diagnostic value to radiographs.3
Treatment for esophageal atresia is surgical and involves anastomosis of the proximal and distal esophageal segments and closure of the fistula. Surgery may be delayed depending on birth weight, presence of other anomalies, and if the proximal and distal esophageal segments are too far apart. In these rare instances, a gastric tube is placed until the esophagus can be repaired.4
Duplication anomalies
Duplication anomalies may be found anywhere along the alimentary tract and can be cystic or tubular. Embryologically, they are classified as foregut, midgut, or hindgut and their exact origin is unclear, with numerous theories including luminal recanalization, intrauterine vascular accident, abortive or incomplete twinning, embryonic diverticula and recanalization defect, and split notochord.5,6 They are associated with other anomalies including spinal and gastrointestinal tract abnormalities.5,7
Whereas esophageal duplication cysts are a rare cause of obstruction, they are the second most common gastrointestinal duplication anomaly, accounting for 15%–20% of all duplications.6 Esophageal duplication cysts are twice as common in males. Embryologically, an esophageal duplication cyst results due to an abnormal vacuolization process or defective separation of the developing foregut. These cysts are usually asymptomatic and diagnosis is often made when an imaging study is performed for an unrelated symptom. Rarely, feeding difficulty, tachypnea, or gastrointestinal hemorrhage can be presenting symptoms. A diagnosis may be suspected by a contrast esophagram; however, definitive diagnosis is made with computed tomography (CT) or magnetic resonance imaging (MRI). The treatment is complete excision, and as these cysts may communicate with the vertebrae, an MRI should be done prior to surgical resection of the cyst and repair of an esophageal defect, to delineate the spinal cord so as to prevent potential cord injury.5,6
Gastric duplication anomalies are usually cystic and do not communicate with the gastric lumen.5,6 The rarer form of tubular duplication often communicates with the gastric lumen. Both cystic and tubular duplications are seen on the greater curvature of the stomach. Neonates present with emesis and poor weight gain secondary to inadequate feeding.5,6 Gastrointestinal bleeding also can also occur due to ulceration.5,6 Treatment for gastric duplication anomalies is resection, which can be achieved by enucleation or by a limited gastric resection, and laparoscopic resection is ideal for smaller lesions.5,6
Duodenal duplication anomalies occur in the second or third part of the duodenum. They do not communicate with the gastrointestinal tract.5,6 Common symptoms are generally vague and include abdominal pain, failure to thrive, or jaundice. Rarely, patients can present with pancreatitis caused by compression of the pancreatic duct, hemorrhage, or perforation. These cases can often be confused with choledochal cysts.5,6 The treatment options are complete excision, marsupialization, or a Roux-en-Y internal drainage to the duodenum or jejunum.5,6
Gastroduodenal duplication anomalies may be detected by prenatal ultrasound. Postnatal UGI contrast study, abdominal ultrasound, and CT aid in diagnosis. Technetium-99m scans may be used to identify ectopic gastric mucosa within a duplication anomaly.
Small bowel duplication anomalies are the most common among all duplications. Duplication anomalies of the small or large intestine can be cystic or tubular. Cystic duplications may cause obstruction, volvulus, or hemorrhage. Simple cystic duplications of the small or large intestine are managed by resection and primary anastomosis. Long tubular duplications may need mucosal stripping. Cystic rectal duplications must be distinguished from sacrococcygeal teratomas and anterior meningoceles.5,6,8
Gastric volvulus
Gastric volvulus is defined as twisting of all or a portion, of the stomach for at least 180° about an axis that causes an obstruction of the foregut.9 In contrast, twisting of less than 180° is termed gastric torsion. The stomach is normally anchored to the abdominal cavity by the gastrocolic, gastrohepatic, gastrophrenic, and gastrosplenic ligaments. If there is agenesis, elongation, or disruption of these ligaments, primary idiopathic gastric volvulus may result. Volvulus is organoaxial or mesenteroaxial due to ligament laxity of the gastrohepatic and gastrosplenic or the gastrophrenic and gastrocolic attachments, respectively. It can be associated with intestinal malrotation, asplenia, congenital abnormalities of the diaphragm, repair of congenital diaphragmatic hernia, gastropexy, gastrostomy tube placement, and hypertrophic pyloric stenosis. The obstruction may be acute or chronic and intermittent in nature. The incidence is rare in infants and neonates and, if intermittent, it is rarely diagnosed in infancy.9 When presenting acutely, gastric volvulus carries significant mortality if not recognized and treated immediately. Sudden onset of persistent nonbilious emesis, retching, acute pain, and abdominal distention and, less commonly, respiratory distress and hematemesis are the presenting symptomatology. There could also be difficulty in advancing a nasogastric tube into the stomach.9
An abdominal radiograph reveals gastric dilation at or above the level of the diaphragm lying in either a horizontal (organoaxial) or vertical (mesenteroaxial) plane, with a distinct incisura pointing toward the right upper quadrant, and bowel gas may be displaced inferiorly. UGI contrast study confirms the diagnosis by demonstrating an abnormal position of the stomach.9 Incomplete filling of the stomach with contrast is indicative of acute gastric volvulus. Treatment involves operative reduction of the volvulus with either open or laparoscopic techniques and repair of a diaphragmatic defect if found.9
Microgastria
Congenital microgastria occurs as a result of arrested embryogenesis of the dorsal mesogastrium that occurs during the fifth week of fetal development, leading to a stomach that has a small volume, a tubular shape, and an abnormal fixation. The incidence is extremely rare and less than 100 cases have been reported. Known associations include intestinal malrotation, situs inversus, asplenia, skeletal defects, as well as esophageal and biliary anomalies.10,11
Infants usually have persistent emesis, diarrhea, gastroesophageal reflux (GER), and failure to thrive. UGI contrast study is diagnostic, which classically demonstrates a small transverse stomach and, often, an abnormal esophagus.10,11
The initial management includes nutritional rehabilitation with continuous enteral feeding and supplemental parenteral nutrition. In many cases, continuous feedings can eventually be transitioned to small, frequent bolus feeds. In patients with persistent symptoms, surgical management includes gastric augmentation with a Roux-en-Y jejunal reservoir (Hunt–Lawrence pouch) and, more recently, using a modified (Bianchi) gastric dissociation procedure.10,11
Lactobezoar
Bezoars are stony concretions that can form in the gastrointestinal tract. Lactobezoars form when undigested milk compacts within the gastric lumen in the setting of high-caloric density formulas, but can also be seen in infants who are fed cow’s milk or given human milk fortifiers.12,13 Gastric lactobezoars are the most common form of bezoars, but the incidence is extremely rare.13 Lactobezoars can infrequently cause obstructive symptoms in infants and can mimic pyloric stenosis with abdominal distention, nonbilious emesis, diarrhea, and a palpable abdominal mass.
Radiographs may show an opacity with frothy appearance in the gastric lumen (Figure 3). Abdominal ultrasound and UGI contrast study are the diagnostic techniques of choice. Conservative management or gastric lavage are sufficient to overcome the obstruction, but protein-cleaving enzyme N-acetylcysteine has been used for faster disintegration of the bezoars.12,13
 | Figure 3 Abdominal radiograph in 19-day-old neonate shows nasogastric tube tip in stomach which also contains a lactobezoar (L). |
Congenital gastric outlet obstruction
Congenital gastric outlet obstruction is a rare cause of feeding intolerance and can be partial or complete. Pyloric or antral webs and atresia are the common forms. Ectopic pancreatic tissue in the submucosa of the pyloric channel can also cause a partial obstruction.
Pyloric atresia constitutes less than 1% of all gastrointestinal atresias.1 Complete obstruction presents early in life with nonbilious vomiting. Maternal history of polyhydramnios is likely, and prenatal ultrasound reveals a single fluid-filled bubble.1 Postnatal diagnosis is made on the basis of a large gastric air bubble, with absence of gas distally. UGI contrast study shows poor visualization of the duodenum. Preoperative gastric decompression, fluid resuscitation, and correction of the hypochloremic hypokalemic alkalosis are necessary prior to corrective surgery, which involves excision of webs and transverse closure or primary anastomosis of the atresia.1,14,15
Hypertrophic pyloric stenosis
Hypertrophic pyloric stenosis is relatively common with an incidence of 2.4 per 1,000 Caucasians, and is more common in males. This acquired pathology can be seen as early as 2 weeks of life, but is generally thought to occur between 3 and 12 weeks. The exact cause is unknown, although a genetic component is established. The circumferential muscle of the pylorus becomes hypertrophied as well as the mucosa.16 The patient presents with nonbilious projectile vomiting, which, if left untreated, can lead to dehydration and electrolyte imbalance.
Abdominal ultrasound is the initial and optimal diagnostic test. The abnormal pyloric muscle will be thickened and elongated, measuring greater than 3–4 mm in thickness and greater than 12–16 mm in length, depending on the reference (Figure 4A). The sonographer should observe the pylorus for up to 10 minutes to identify passage of gastric contents through the pyloric channel to differentiate between pyloric spasm, which is a transient phenomenon, and pyloric stenosis. Feeding the infant during ultrasound evaluation can be useful to aid visualization of gastric emptying. It is also useful to simultaneously evaluate the relationship of the superior mesenteric artery (SMA) and superior mesenteric vein (SMV), as the differential diagnosis for a vomiting infant includes intestinal malrotation (Figure 4B). If the SMA–SMV relationship is reversed, an immediate UGI contrast study should be undertaken to evaluate for intestinal malrotation and midgut volvulus.
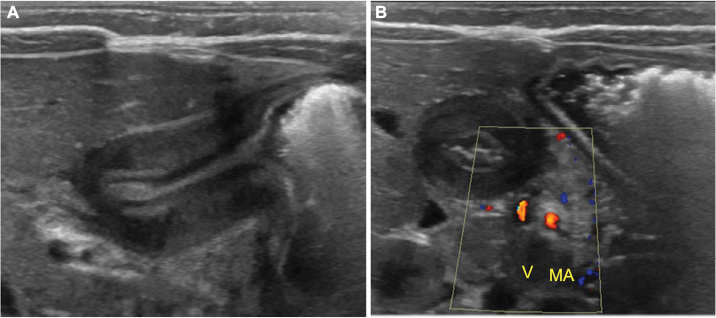 | Figure 4 Pyloric stenosis in infant with vomiting. (A) Abdominal ultrasound shows thickened and elongated pyloric muscle in longitudinal plane. (B) Abdominal ultrasound with color Doppler shows normal relationship of the superior mesenteric artery (MA) and superior mesenteric vein (V). |
Sometimes, vomiting infants are sent for UGI contrast study before ultrasound examination. Signs of hypertrophic pyloric stenosis on fluoroscopic examination include the caterpillar sign where abnormal gastric peristalsis is present, as well as abnormal narrowing of the pyloric canal as evidenced by the string, teat, and mushroom signs.
Treatment for hypertrophic pyloric stenosis is gastric decompression by nasogastric tube and fluid resuscitation, followed by open or laparoscopic surgical release of the hypertrophied pyloric muscle.17–19
Duodenal atresia
Congenital obstruction of the duodenum can be complete or partial, and intrinsic or extrinsic. Intrinsic obstruction is often caused by duodenal atresia and occurs in approximately 1 in 5,000 to 1 in 10,000 live births.1,20 Extrinsic obstruction can be caused by intestinal malrotation, annular pancreas, gastroduodenal duplication anomalies, and pseudocysts of the pancreas and biliary tree.1
During the sixth and seventh weeks of gestation, portions of the intestinal tract normally become occluded as the endodermal epithelium proliferates. Subsequently, recanalization restores the patency of the lumen between the eighth and tenth weeks of gestation. Duodenal atresia is thought to result from failure of the bowel to recanalize during this period, and is most commonly postampullary in location.1,20
Duodenal atresia can be classified into three types. Type I is the most common, accounting for 90% of cases, and is due to webs or membranes, some of which may be fenestrated.20 A windsock anomaly may result if the intraluminal membrane causing the obstruction distends and prolapses distally within the lumen, and can cause an appearance of bowel dilation far beyond the actual location of the obstruction.20 Type II duodenal atresia has a grossly dilated proximal segment and a smaller distal segment. In type III duodenal atresia, the two bowel segments are completely discontinuous. Duodenal atresia can be associated with other intrinsic duodenal and pancreaticobiliary anomalies, including annular pancreas which occurs when the ventral pancreatic bud fails to rotate behind the duodenum, resulting in pancreatic tissue encircling the second portion of the duodenum. The incidence of associated anomalies with duodenal atresia varies between 50% and 80%. Congenital heart disease (30%), Trisomy 21 (30%), intestinal malrotation (20%), esophageal atresia, imperforate anus (10%–20%), thoracoabdominal heterotaxia, and gallbladder agenesis are common associations (Figure 5).20–22
 | Figure 5 Duodenal atresia in newborns with abnormality on prenatal ultrasound. (A) Supine abdominal radiograph shows classic “double bubble” sign. (B) Intraoperative fluoroscopic image following contrast administration through a gastric tube in a different patient shows findings of duodenal atresia as well as esophageal atresia with dilated contrast-filled distal esophageal pouch. |
On prenatal ultrasound examination, maternal polyhydramnios and a double-bubble sign of two dilated fluid-filled structures in the fetal abdomen aid in the diagnosis of duodenal atresia. Postnatally, clinical features of duodenal atresia include bilious emesis with a lack of abdominal distension. Abdominal radiography shows the classic double-bubble sign of two distinct gas collections or air-fluid levels representing a markedly dilated stomach and duodenal bulb (Figure 5). In the classic double-bubble sign, the duodenum appears distended and rounded because of chronic obstruction in utero. When a distended stomach is associated with a normal-caliber duodenum, the diagnosis is likely to be intestinal malrotation with duodenal obstruction secondary to Ladd’s bands. If a double-bubble sign is seen on abdominal radiograph and the diagnosis is presumed to be duodenal atresia, it may be prudent to obtain an UGI contrast study for confirmation if surgery is delayed for any reason, as malrotation with midgut volvulus can mimic duodenal atresia.23
In the preoperative period, nasogastric decompression; fluid, electrolyte and nutritional management; prophylactic antibiotics; and evaluation for associated anomalies are necessary. The surgical approach depends upon the site of the atresia. In every case, the possibility of a second or multiple atresias should be considered. With a proximal obstruction, distal atresias may be overlooked at surgery, especially if they are not associated with a gap in the bowel or mesentery.
Duodenal atresia is approached through a transverse right upper quadrant incision and can also be repaired laparoscopically; however, this approach has not been widely adopted. The procedure of choice is an end-to-side or side-to-side duodenoduodenostomy. A duodenojejunostomy is also acceptable. During surgery, a small catheter should be passed into the distal bowel. Either air or saline should be injected through the entire small bowel to rule out additional stenoses or atresias.
In cases of duodenal stenosis or atresia due to an intrinsic duodenal web, a vertical duodenotomy, web resection, and transverse closure along with enteroplasty to optimize the duodenal lumen can be undertaken. Alternatively, duodenoduodenostomy is an operative choice where the proximal and distal duodenal pouches are opened and joined, as resection of the obstructing web or membrane may result in damage to the common bile duct or pancreatic duct.
In cases of type II duodenal atresia, enteroplasty may be necessary to optimize the duodenal lumen.
Small-volume enteral feeding is initially started and gradually increased when bowel function returns. Dysmotility and dyskinesia of the significantly dilated proximal duodenum is a known feature in some patients, but usually improves with time. The outcome after repair is primarily dependent on associated anomalies.1,20–22
Intestinal malrotation
Abnormal embryological development of bowel may lead to rotational abnormalities, termed intestinal malrotation – which include nonrotation, incomplete rotation, and reversed rotation.1 Normally, physiologic intestinal herniation occurs early in the first trimester followed by a 270° counterclockwise rotation of bowel about the SMA while returning to the peritoneal cavity by approximately 12 weeks gestation.24 Normal fixation of the cecum in the right lower quadrant and the duodenojejunal junction in the left upper quadrant results in a broad mesenteric base, which inhibits volvulus or twisting of bowel about the vascular pedicle.1 Intestinal malrotation may result in an abnormal position of the cecum and duodenojejunal junction in close proximity, thereby creating a narrow mesenteric base which predisposes to twisting of bowel loops around the SMA – known as midgut volvulus.1
The incidence of intestinal malrotation is estimated to be 1 in 500 live births.25,26 Autopsy studies indicate the true incidence may be as high as 1% of the total population, with an increased incidence in certain syndromes including heterotaxy and Trisomy 21, as well as anatomical abnormalities including congenital diaphragmatic hernia, omphalocele, and gastroschisis.1,27 The incidence of intestinal malrotation with midgut volvulus is inversely proportional to age, with 75% of cases presenting in the neonatal period.1
Clinical presentation of intestinal malrotation may be without or with midgut volvulus and can be challenging, with chronic, acute, or absent symptomatology.1 Symptoms include bilious emesis, irritability, as well as an initially scaphoid abdomen that exhibits increasing distention and tenderness.1 Late presentation may result in abdominal wall erythema, hematemesis, or melena due to bowel ischemia.1
Laboratory findings include leukocytosis or leukopenia, hyperkalemia, thrombocytopenia, and evidence of metabolic acidosis.1
Radiologic evaluation may be helpful in the diagnosis; however, should not delay treatment if there is high clinical suspicion for intestinal malrotation with midgut volvulus.1 An abdominal radiograph may be obtained in clinically stable patients to guide further workup; however, radiographs may be normal and cannot exclude the diagnosis of intestinal malrotation.1,29 While bilious emesis may be seen in the setting of other neonatal pathologies, intestinal malrotation with midgut volvulus is a surgical emergency with potentially serious consequences, including peritonitis, sepsis, shock, and death; thus, bilious emesis in the neonate should prompt immediate evaluation for intestinal malrotation with a UGI contrast study.1 The gold standard for diagnosis of intestinal malrotation is a UGI contrast study, where an abnormal course of the duodenum is confirmed.1 A Z-shaped duodenum may be seen due to to extrinsic compression on the duodenum by abnormal fibrous peritoneal bands, termed Ladd’s bands, which may be present in intestinal malrotation.28 UGI contrast study may also demonstrate intestinal malrotation with midgut volvulus, which manifests as duodenal obstruction or delayed passage of contrast through a corkscrew-shaped duodenum.1 The sensitivity of UGI contrast study for detecting intestinal malrotation is 93%–100%, with both false positive and false negatives reported, while the sensitivity for detecting midgut volvulus is only 54–79%.26 The expected normal position of the duodenojejunal junction is to the left of the spine at or above the level of the duodenal bulb on the frontal view, with the duodenum entirely retroperitoneal on the lateral view.24 If a water-soluble contrast enema is carried out for another indication, the cecum may be malpositioned in intestinal malrotation; however, this finding may not be present or may be seen with a highly mobile cecum – a normal variant.29 Ultrasound evaluation may suggest the possibility of intestinal malrotation if there is reversal of the SMA–SMV relationship, but this finding may not always be present. Ultrasound findings of duodenal dilation with distal tapering, fixed midline bowel, duodenal wall thickening, ascites, a swirl/whirlpool sign on color Doppler ultrasound, and dilation of the distal SMV may be seen in intestinal malrotation with midgut volvulus.27,29 CT and MRI are not appropriate in the radiologic evaluation of a vomiting neonate.29
Bowel obstruction and associated mesenteric vascular compromise may occur, leading to bowel ischemia and, eventually, infarction if untreated.28 Short-gut syndrome, dependence on total parenteral nutrition, or even death may result due to bowel loss, with a mortality rate of 3%.20 Treatment for intestinal malrotation with midgut volvulus includes fluid resuscitation, intravenous broad-spectrum antibiotics, and immediate surgical exploration.1
Intussusception
Intussusception is telescoping of a proximal bowel loop (the intussusceptum) into itself distally (the intussuscipiens).30 It most commonly occurs at the ileocolic junction, without an identifiable lead point, in children between 6 and 18 months of age.31 Intussusception rarely occurs in the neonatal period in full-term infants, with an incidence of 0.3%–1.3%, and is exceedingly rare in premature infants.31 The pathogenesis of neonatal intussusception remains unclear; however, in full-term infants, it is more commonly associated with a pathological lead point and more commonly involves the colon.31
Clinical presentation of a neonate with intussusception differs from that of older infants, who typically have intermittent abdominal pain, a palpable mass, and bloody stools.30 Full-term neonates with intussusception have a presentation that mimics a bowel obstruction, with abdominal distention and bilious emesis within hours of birth.32 Preterm neonates with intussusception have a presentation that mimics necrotizing enterocolitis, with abdominal distention, bilious emesis, feeding intolerance, and, sometimes, rectal bleeding.30 Intussusception is, therefore, rarely sought as a diagnosis in the neonate.
In neonates being evaluated for bowel obstruction or necrotizing enterocolitis, an abdominal radiograph is obtained and dilated loops of small bowel are seen.32 Pathognomonic findings for necrotizing enterocolitis such as pneumatosis intestinalis and portal venous gas are absent, as they are in more than half of cases of necrotizing enterocolitis.31 Abdominal radiography is, thus, insensitive and nonspecific for the diagnosis of intussusception. In patients with suspected low intestinal obstruction, a contrast enema is undertaken and will demonstrate intussusception if a colonic component is present; however, no colonic component is present in 25%–50% of cases of neonatal intussusception.31,32 Abdominal ultrasound is not as routinely conducted in neonates to diagnose intussusception as it is in older infants because the diagnosis is infrequently considered. It has been suggested for further evaluation of neonates in whom intussusception is a diagnostic consideration.30,32
In neonates with a complete bowel obstruction due to intussusception, surgical exploration is curative and not usually delayed.32 In premature neonates in whom necrotizing enterocolitis is the presumed diagnosis, surgical exploration is usually delayed, with an associated increase in morbidity and mortality.32
Intestinal atresias
Jejunal, ileal, and colonic atresias most often occur due to vascular insult with ischemia to the affected bowel, and may coexist.
Jejunal and ileal atresia occur in 1–3 out of 5,000 births.1,25 Jejunal obstruction is almost always due to atresia or stenosis and usually presents with bilious emesis.28 In jejunal atresia, abdominal radiographs may demonstrate the “triple bubble sign” of three dilated air-filled structures representing the distended stomach, duodenum, and proximal jejunum.28 Prior to surgery, an UGI contrast study is sometimes done as well as a contrast enema to evaluate for other atresias which may be concomitant.28 The contrast enema will show a small and unused microcolon. Ileal atresia may present with vomiting and failure to pass meconium. The initial radiograph shows at least four dilated bowel loops, suggesting that the obstruction is distal. Contrast enema often reveals a small and unused microcolon and may or may not demonstrate additional foci of atresia (Figure 6A).
 | Figure 6 Intestinal atresias in newborns with distal intestinal obstruction. (A) Image during contrast enema shows blind ending terminal ileum (arrowhead) in ileal atresia. (B) Image during contrast enema shows blind ending descending colon which is dilated with “cobra head” appearance (arrowhead) in colonic atresia. |
Treatment for jejunal and ileal atresia will include nasogastric tube decompression, fluid resuscitation, and prophylactic antibiotics before surgical repair.1 Definitive surgery is resection of the atretic lesions and end-to-end anastomosis.
Colonic atresia is rare by comparison, occurring in approximately 1 in 20,000 live births.1 Patients present with abdominal distention, vomiting, and failure to pass meconium. The initial abdominal radiograph shows multiple dilated loops of bowel, with some air–fluid levels compatible with a distal obstruction. The loops of bowel just proximal to the obstruction are often massively dilated.1 If colonic atresia is suspected, a contrast enema is the imaging investigation of choice and, when conducted, will show a small and unused microcolon. It is difficult to fill the entire colon due to the atresia and the contrast column abruptly ends, sometimes with a rounded “cobra head” appearance (Figure 6B).
Treatment for colonic atresia includes nasogastric tube decompression, fluid resuscitation, and prophylactic antibiotics before surgical repair. Surgical repair should be urgent due to increased risk of perforation or volvulus compared to other atresias, with initial management consisting of decompression of the intestine by an ostomy procedure.33
Meconium ileus
Meconium ileus is an intestinal obstruction at the level of the terminal ileum that occurs when inspissated meconium obstructs the intestinal lumen.34 Approximately 20% of all neonatal intestinal obstruction results from meconium ileus.28 This pathologic entity is usually associated with cystic fibrosis (CF), where abnormally thick meconium creates a functional ileal obstruction; however, only 15% of patients with CF have meconium ileus.25,28 In patients without CF, the pathogenesis of meconium ileus is unclear; however, a spectrum of genetic and pathologic abnormalities are suspected.34
Patients typically present with abdominal distention and failure to pass meconium.34 An abdominal radiograph demonstrates a low intestinal obstruction, as evidenced by four or more dilated air-filled bowel loops. Air–fluid levels may not be present due to the thick consistency of meconium.28 Less commonly, there may be a bubbly gas pattern in the right abdomen.25 Abdominal calcifications may be seen postnatally on radiograph when in utero perforation and meconium peritonitis occur, sometimes, with pseudocyst formation.25,28 Other meconium ileus complications include small bowel or pseudocyst volvulus, perforation, and peritonitis.28
A water-soluble contrast enema should be undertaken for further evaluation of lower intestinal obstruction and is diagnostic, showing filling defects in an extreme microcolon and the distal ileum, with proximal small bowel dilatation (Figure 7A).25,28 Water-soluble contrast enema utilizing hyperosmolar contrast is also therapeutic and may be repeated if necessary to completely cleanse the bowel of meconium.25 Surgery is reserved for the unusual obstruction refractory to therapeutic enemas, and in complicated cases where associated abnormalities are present including bowel atresia, necrosis, and perforation.34
 | Figure 7 Meconium plugs in neonates with meconium ileus and functional immaturity of the colon. (A) Image during contrast enema shows innumerable meconium filling defects (arrowheads) in an extreme microcolon and the terminal ileum, with dilated air-filled bowel proximally, in meconium ileus. (B) Image during contrast enema shows meconium filling defect (arrowhead) in a small left colon (arrows) in functional immaturity of the colon. |
Functional immaturity of the colon
Functional immaturity of the colon is also known as meconium plug syndrome or small left colon syndrome, and is a benign, usually self-limiting, transient, functional colonic obstruction due to immature neuronal (ganglion) cells in the myenteric plexus.28 This is the most common diagnosis in neonates who fail to pass meconium in the first 48 hours, with an incidence of 1 in 500 newborns.28,35 Half of patients with functional immaturity of the colon have a maternal history of diabetes mellitus, and there is a less common association with maternal magnesium sulfate administration for pre-eclampsia.25
Newborns typically present with abdominal distention and failure to pass meconium in the first 48 hours of life.28 Abdominal radiograph shows signs of a low intestinal obstruction as evidenced by four or more dilated air-filled loops of bowel, with absence of gas in the rectum.28,35 A water-soluble contrast enema should be conducted, which demonstrates a normal rectal size and meconium plugs in the colon.28 Frequently, a small left colon extending from the sigmoid colon to the level of the splenic flexure is seen with proximal colonic distention – hence, the name small left colon syndrome (Figure 7B).28 While meconium intraluminal filling defects, also known as meconium “plugs” are seen in this abnormality, they are nonspecific and are seen in other etiologies of low intestinal obstruction.25
Water-soluble contrast enema is usually curative. If the patient has persistent stooling symptoms, Hirschsprung disease should be excluded by rectal biopsy, as up to 13% of patients initially diagnosed with functional immaturity of the colon go on to have biopsy-proven Hirschsprung disease.28,36
Hirschsprung disease
Hirschsprung disease occurs when there is absence of normal neuronal (ganglion) cells in the submucosal and myenteric plexus of the large bowel.1 The embryologic process of neuronal cell migration in the gastrointestinal tract occurs in a proximal-to-distal fashion from the mouth to the anus and, thus, large bowel involvement in Hirschsprung disease is always contiguous, extending proximally from the level of the anus.28 While 75% of cases affect a short segment involving the rectum and a portion of the sigmoid colon, involvement may range from an ultrashort segment to the entire colon. Rarely, the distal small bowel could also be affected.28
Hirschsprung disease occurs in 1 in 4,000 live births with a 4:1 male to female ratio.25 While 70% of cases are isolated, Hirschsprung disease is associated with other congenital anomalies.37 Trisomy 21 is the most frequently associated chromosomal abnormality, and is seen in 2%–15% of cases of Hirschsprung disease.37,38
Abnormal neuronal cells in the large bowel result in the inability of the affected bowel to relax normally, which creates a functional bowel obstruction with proximal bowel dilation and problems with defecation.28 The most common clinical presentation in the neonatal period is progressive abdominal distention, failure to pass meconium by 24–48 hours of life, and bilious emesis.25,38 Hirschsprung enterocolitis occurs in 25%–35% of patients with Hirschsprung disease, and is the presenting symptom in 25% of patients.39 This complication is the most common cause of mortality in Hirschsprung disease and classically presents with abdominal distention, fever, and diarrhea, with other nonspecific symptoms including vomiting, rectal bleeding, lethargy, loose stools, and obstipation.39,40 In Hirschsprung enterocolitis, mural inflammation and ischemia can result in sepsis, bowel perforation, and death.25,39 It is most frequently seen in long-segment disease, patients with Trisomy 21 and those with a positive family history of Hirschsprung enterocolitis.40
While the diagnosis of Hirschsprung disease requires tissue sampling and histologic confirmation of abnormal neuronal cells, radiologic workup may suggest or confirm the diagnosis.25 Abdominal radiograph may demonstrate nonspecific findings of low intestinal obstruction, as evidenced by four or more loops of dilated air-filled bowel in the abdomen (Figure 8A).25,41 A water-soluble contrast enema should be done for further evaluation of low intestinal obstruction.25,41 Possible findings on contrast enema include abnormal rectosigmoid ratio of less than 1, a transition point at the junction of normal and abnormal bowel, irregular rectal contractions with a saw-tooth appearance, and retained contrast material on delayed radiographs (Figure 8B).28 When imaging suggests a transition point, there is often discordance with the histopathological transition point.40 Contrast enema may also be normal in cases of ultrashort segment or total colonic Hirschsprung disease, and false negative rates for transition point and abnormal rectosigmoid ratio are as high as 35%. Rectal suction biopsy is indicated if there is persistent clinical concern for Hirschsprung disease, with a reported sensitivity of 97% and specificity of 97%.38,40 Imaging findings in Hirschsprung enterocolitis include dilated bowel, pneumatosis, portal venous gas, or free intraperitoneal air.25
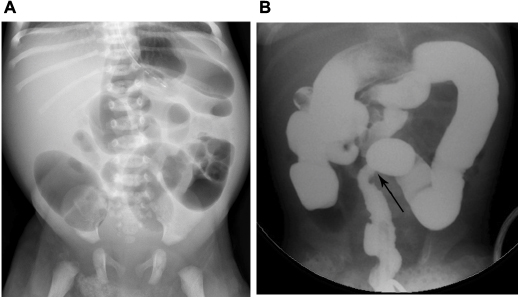 | Figure 8 Hirschsprung disease in neonates with distal intestinal obstruction. (A) Supine abdominal radiograph shows more than three dilated air-filled loops of bowel and absence of air in the rectum. (B) Image during contrast enema in a different patient shows abnormal rectosigmoid ratio and transition point (arrow). |
Treatment of Hirschsprung disease includes stabilization of the patient and decompression of the bowel initially, and fluid resuscitation to replace lost fluids. Fluid resuscitation should be done to replace lost fluids.38 Feeding can resume once bowel decompression is achieved.38 Rectal washouts should be carried out routinely until definitive Hirschsprung disease treatment with surgical resection of the aganglionic bowel segment is conducted.38,40 Historically, this has been done in a two-step process with diverting colostomy placement then takedown, however, many surgeons perform the repair in a single stage when possible.40 Treatment for Hirschsprung enterocolitis includes antibiotic therapy, intravenous fluid resuscitation, and saline rectal washouts, with diverting colostomy reserved for severe cases with sepsis refractory to resuscitation.40
Anorectal malformation
The spectrum of anorectal malformations includes imperforate anus and ranges from isolated perineal fistula to complex cloacal malformations where there is a common channel between the genitourinary system and rectum.28,42,43 Approximately 80% of anorectal malformation have associated genitourinary anomalies.38 Other associated congenital anomalies include spinal, cardiac, other gastrointestinal as well as limb abnormalities, with the most common association being VACTERL.1,28,38 While the etiology remains unknown, it is likely multifactorial, with genetic factors suspected as anorectal malformation is associated with many syndromes and in members of the same family.1,42 anorectal malformation occur in 1–4 out of 5,000 newborn babies, with males and females equally affected.25,28
Neonates with anorectal malformation present with abdominal distention, failure to pass meconium, and absence of a normal anal opening on physical examination. A perineal fistula may also be visible.28,42 Traditionally, lesions where the rectum ends above the puborectalis sling were termed “high” lesions, often with fistulous communication to the bladder, urethra, or vagina.28 Lesions were deemed “low” when the rectum terminates below the puborectalis sling.28 More recently, the Krickenbeck classification was created to group anorectal malformation by their type of fistula, including rare and regional types.44
Physical examination shows the absence of a normal anal opening.28 Abdominal radiograph demonstrates low intestinal obstruction, and a lateral abdominal radiograph in the prone position may help determine the distance of the air-filled blind-ending rectal pouch to the expected location of the anus (Figure 9).28 More recently, ultrasound of the perineum has been shown to more accurately differentiate high and low lesions, with an accuracy of 95% and specificity of 86%.25,28 MRI of the pelvis may be used to further characterize anatomy prior to, and following, surgery.28 Workup for associated abnormalities includes echocardiogram, renal/bladder ultrasound, spinal ultrasound, voiding cystourethrogram, and radiographs of the abdomen and spine.1,38
 | Figure 9 Anorectal malformation in neonate with sacral agenesis. (A) Supine abdominal radiograph shows absence of sacrum and air in the rectum. (B) Lateral abdominal radiograph shows absence of sacrum and blind ending air-filled rectum (arrow). |
As associated congenital anomalies may be present, assessment for possible life-threatening anomalies should be undertaken prior to corrective surgery.42 By 24 hours of life, careful examination for evidence of perineal or urinary fistulae is possible, where meconium is seen at the perineum or in the urine, respectively.42
High lesions are treated with colostomy as a temporizing measure, with later definitive surgical repair after better delineation of anatomy.28,42 A posterior sagittal surgical approach is predominantly undertaken, with an adjunct abdominal approach reserved for high lesions where the rectum, prostate, bladder, or vagina are very high.38,42 Low lesions are treated surgically with anoplasty or dilation.28
Conclusion
Neonatal bowel obstruction is common and may involve the upper or lower gastrointestinal tract. Workup usually involves abdominal radiography, which may direct further evaluation with fluoroscopic examination such as an UGI contrast study or contrast enema. Depending on the etiology and the level of obstruction, surgery may be indicated for treatment. If intestinal malrotation is a differential consideration, prompt surgical exploration or evaluation with UGI contrast study is paramount to decrease morbidity and mortality.
Disclosure
The authors report no conflicts of interest in this work.
References
Juang D, Snyder CL. Neonatal bowel obstruction. Surg Clin North Am. 2012;92(3):685–711. | ||
Kuhn J, Slovis T, Haller J, editors. Caffey’s Pediatric Diagnostic Imaging. 10th ed; volume 1. Philadelphia: Elsevier’s Mosby; 2004. | ||
Clark DC. Esophageal atresia and tracheoesophageal fistula. Am Fam Physician. 1999;59(4):910–916, 919–920. | ||
Feldman M, Friedman LS, Brandt LJ. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia : Saunders; 2016. | ||
Patiño Mayer J, Bettolli M. Alimentary tract duplications in newborns and children: diagnostic aspects and the role of laparoscopic treatment. World J Gastroenterol. 2014;20(39):14263–14271. | ||
Stern LE, Warner BW. Gastrointestinal duplications. Semin Pediatr Surg. 2000;9(3):135–140. | ||
Richards DS, Langham MR, Anderson CD. The prenatal sonographic appearance of enteric duplication cysts. Ultrasound Obstet Gynecol. 1996;7(1):17–20. | ||
Hur J, Yoon CS, Kim MJ, Kim OH. Imaging features of gastrointestinal tract duplications in infants and children: from oesophagus to rectum. Pediatr Radiol. 2007;37(7):691–699. | ||
Cribbs RK, Gow KW, Wulkan ML. Gastric volvulus in infants and children. Pediatrics. 2008;122(3):e752–e762. | ||
Kunisaki SM, Dakhoub A, Jarboe MD, Geiger JD. Gastric dissociation for the treatment of congenital microgastria with paraesophageal hiatal hernia. J Pediatr Surg. 2011;46(6):e1–e4. | ||
Dicken BJ, Novotny NM, Breckler FD, Yim JP, Rescorla FJ. Use of the Hunt-Lawrence pouch in congenital microgastria—a report of 2 cases. J Pediatr Surg. 2010;45(11):2238–2240. | ||
Heinz-Erian P, Gassner I, Klein-Franke A, et al. Gastric lactobezoar – a rare disorder? Orphanet J Rare Dis. 2012;7:3. | ||
Bajorek S, Basaldua R, McGoogan K, Miller C, Sussman CB. Neonatal gastric lactobezoar: management with N-acetylcysteine. Case Rep Pediatr. 2012;2012:412412. | ||
Ilce Z, Erdogan E, Kara C, et al. Pyloric atresia: 15-year review from a single institution. J Pediatr Surg. 2003;38(11):1581–1584. | ||
Knouff S, Klein A, Kaminski M. Pyloric atresia in the neonate. Neonatal Netw. 2014;33(6):329–335. | ||
Kuhn J, Slovis T, Haller J, editors. Caffey’s Pediatric Diagnostic Imaging. 10th ed; volume 2. Philadelphia: Elsevier’s Mosby; 2004. | ||
Pandya S, Heiss K. Pyloric stenosis in pediatric surgery: an evidence-based review. Surg Clin North Am. 2012;92(3):527–539. | ||
Peters B, Oomen MW, Bakx R, Benninga MA. Advances in infantile hypertrophic pyloric stenosis. Expert Rev Gastroenterol Hepatol. 2014;8(5):533–541. | ||
Sathya C, Wayne C, Gotsch A, Vincent J, Sullivan KJ, Nasr A. Laparoscopic versus open pyloromyotomy in infants: a systematic review and meta-analysis. Pediatr Surg Int. 2017;33(3):325–333. | ||
Adams SD, Stanton MP. Malrotation and intestinal atresias. Early Hum Dev. 2014;90(12):921–925. | ||
Chung PH, Wong CW, Ip DK, Tam PK, Wong KK. Is laparoscopic surgery better than open surgery for the repair of congenital duodenal obstruction? A review of the current evidences. J Pediatr Surg. 2017;52(3):498–503. | ||
Carroll AG, Kavanagh RG, Ni Leidhin C, Cullinan NM, Lavelle LP, Malone DE. Comparative effectiveness of imaging modalities for the diagnosis of intestinal obstruction in neonates and infants: a critically appraised topic. Acad Radiol. 2016;23(5):559–568. | ||
Gilbertson-Dahdal DL, Dutta S, Varich LJ, Barth RA. Neonatal malrotation with midgut volvulus mimicking duodenal atresia. Am J Roentgenol. 2009;192(5):1269–1271. | ||
Applegate KE, Anderson JM, Klatte EC. Intestinal malrotation in children: a problem-solving approach to the upper gastrointestinal series. Radiographics. 2006;26(5):1485–1500. | ||
Reid JR. Practical imaging approach to bowel obstruction in neonates: a review and update. Semin Roentgenol. 2012;47(1):21–31. | ||
Sizemore AW, Rabbani KZ, Ladd A, Applegate KE. Diagnostic performance of the upper gastrointestinal series in the evaluation of children with clinically suspected malrotation. Pediatr Radiol. 2008;38(5):518–528. | ||
Marine MB, Karmazyn B. Imaging of malrotation in the neonate. Semin Ultrasound CT MR. 2014;35(6):555–570. | ||
Vinocur DN, Lee EY, Eisenberg RL. Neonatal intestinal obstruction. AJR Am J Roentgenol. 2012;198(1):W1–W10. | ||
Raske ME, Dempsey ME, Dillman JR, et al. ACR appropriateness criteria vomiting in infants up to 3 months of age. J Am Coll Radiol. 2015;12(9):915–922. | ||
Slam KD, Teitelbaum DH. Multiple sequential intussusceptions causing bowel obstruction in a preterm neonate. J Pediatr Surg. 2007;42(7):1279–1281. | ||
Avansino JR, Bjerke S, Hendrickson M, Stelzner M, Sawin R. Clinical features and treatment outcome of intussusception in premature neonates. J Pediatr Surg. 2003;38(12):1818–1821. | ||
Wang NL, Yeh ML, Chang PY, et al. Prenatal and neonatal intussusception. Pediatr Surg Int. 1998;13(4):232–236. | ||
Karnak I, Ciftci AO, Senocak ME, Tanyel FC, Buyukpamukcu N. Colonic atresia: surgical management and outcome. Pediatr Surg Int. 2001;17(8):631–635. | ||
Karimi A, Gorter RR, Sleeboom C, Kneepkens CM, Heij HA. Issues in the management of simple and complex meconium ileus. Pediatr Surg Int. 2011;27(9):963–968. | ||
Al-Salem AH. Meconium plug syndrome. In: Al-Salem AH, editor. An Illustrated Guide to Pediatric Surgery. Cham: Springer International Publishing; 2014:195–197. | ||
Keckler SJ, St Peter SD, Spilde TL, et al. Current significance of meconium plug syndrome. J Pediatr Surg. 2008;43(5):896–898. | ||
Duess JW, Hofmann AD, Puri P. Prevalence of Hirschsprung’s disease in premature infants: a systematic review. Pediatr Surg Int. 2014;30(8):791–795. | ||
Sloan K, Wagener S. Updates in: Hirschsprung’s disease and anorectal malformations. J Pediatr Surg Specialties. 2016;10(3):43–51. | ||
Gosain A. Established and emerging concepts in Hirschsprung’s-associated enterocolitis. Pediatr Surg Int. 2016;32(4):313–320. | ||
Burkardt DD, Graham JM Jr, Short SS, Frykman PK. Advances in Hirschsprung disease genetics and treatment strategies: an update for the primary care pediatrician. Clin Pediatr (Phila). 2014;53(1):71–81. | ||
Das K, Mohanty S. Hirschsprung disease – current diagnosis and management. Indian J Pediatr. 2017;84(8):618–623. | ||
Levitt MA, Peña A. Anorectal malformations. Orphanet J Rare Dis. 2007;2:33. | ||
Peña A, Hong A. Advances in the management of anorectal malformations. Am J Surg. 2000;180(5):370–376. | ||
Holschneider A, Hutson J, Peña A, et al. Preliminary report on the International Conference for the Development of Standards for the Treatment of Anorectal Malformations. J Pediatr Surg. 2005;40(10):1521–1526. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.