Back to Journals » Lung Cancer: Targets and Therapy » Volume 12
Managing Acquired Resistance to Third-Generation EGFR Tyrosine Kinase Inhibitors Through Co-Targeting MEK/ERK Signaling
Authors Yu D, Zhao W, Vallega KA ![]() , Sun SY
, Sun SY ![]()
Received 15 December 2020
Accepted for publication 16 January 2021
Published 5 February 2021 Volume 2021:12 Pages 1—10
DOI https://doi.org/10.2147/LCTT.S293902
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Danlei Yu,1,2,* Wen Zhao,2,3,* Karin A Vallega,2 Shi-Yong Sun2
1Department of Oncology, The Second Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 2Department of Hematology and Medical Oncology, Emory University School of Medicine and Winship Cancer Institute, Atlanta, GA, USA; 3Department of Oncology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shi-Yong Sun
Department of Hematology and Medical Oncology, Emory University School of Medicine and Winship Cancer Institute, 1365-C Clifton Road, C3088, Atlanta, GA, 30322, USA
Tel +1 (404) 778-2170
Fax +1 (404) 778-5520
Email [email protected]
Abstract: Although epidermal growth factor receptor (EGFR)-targeted therapy has improved clinical outcomes of patients with advanced non-small-cell lung cancer (NSCLC) carrying activating EGFR mutations, the development of acquired resistance to EGFR tyrosine kinase inhibitors (EGFR-TKIs), including the promising third-generation ones, results in disease progression and has become an unavoidable problem that limits patient long-term benefit. The third-generation EGFR-TKIs, osimertinib and almonertinib, are now approved for the treatment of advanced NSCLC patients harboring activating EGFR mutations (first-line) and/or the resistant T790M mutation (second-line). Clinically, appropriate management of acquired resistance to third-generation EGFR-TKIs will substantially improve their long-term efficacy against EGFR-mutant NSCLC. Recent preclinical and clinical studies suggest that activation of the Ras/Raf/MEK/ERK signaling pathway may be an important resistance mechanism and accordingly co-targeting this pathway effectively overcomes and abrogates acquired resistance to third-generation EGFR-TKIs. This review focuses on discussing the scientific rationale for and potential of co-targeting MEK/ERK signaling in delaying and overcoming acquired resistance to third-generation EGFR-TKIs, particularly osimertinib.
Keywords: acquired resistance, EGFR inhibitors, osimertinib, MEK/ERK, lung cancer
Introduction
Lung cancer is the leading cause of cancer-related death worldwide.1,2 It is generally classified into small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC), which accounts for about 80% of the disease. Oncogenic driver mutations, such as epidermal growth factor receptor (EGFR) and anaplastic factor receptor (ALK), are activated and targetable in patients with NSCLC.3 Currently, patients with activating or sensitive EGFR mutations, most of which are exon 19 deletion (19del) and exon 21 L858R point mutation, have access to targeted therapy with EGFR tyrosine kinase inhibitors (EGFR-TKIs).4
Patients with sensitive EGFR mutations, when treated with first- or second-generation EGFR-TKIs such as gefitinib, erlotinib or afatinib, showed improved progression-free survival, overall response rate and quality of life. Despite this initial good response, most patients eventually relapse after 9–13 months of treatment due to the issue of developing acquired resistance.4 There are multiple underlying resistance mechanisms for the acquired resistance to the first and second-generation EGFR-TKIs, among which acquisition of a secondary resistant EGFR mutation named T790M occurs in about 60% of relapsed patients. The mutation at the “gatekeeper” position 790 of exon 20 results in the substitution of threonine by methionine (ACG to ATG), which restores ATP binding affinity while decreasing the drug-binding affinity due to the increased steric hindrance.4,5
To overcome acquired resistance to EGFR-TKIs owing to the acquisition of the T790M mutation, third-generation EGFR-TKIs that selectively and irreversibly inhibit both EGFR activating (eg, 19del and L858R) and resistant (eg, T790M) mutations with limited activity against wild-type (WT) EGFR have been developed.5–7 The representative one, osimertinib (AZD9291 or TAGRISSO), is the first approved third-generation EGFR-TKI. Compared with first and second-generation EGFR-TKIs, osimertinib significantly improves overall survival (median of 38.6 months compared to 32.8 months) of NSCLC patients harboring mutant EGFR with minimal off-target effects.8,9 Currently, osimertinib is a marketed drug for the treatment of T790M-positive NSCLC patients as a second-line therapy. It is also FDA-approved as a first-line therapy for the treatment of advanced lung cancer with activating EGFR mutations regardless of T790M mutation status.10,11 Almonertinib (Amelie; HS-10296) is another third-generation EGFR-TKI developed by Hansoh Pharma and was recently approved in China for the treatment of NSCLC patients harboring EGFR T790M mutation who have relapsed to other EGFR-TKI therapy. This approval is based on outcomes of the open-label Phase II APOLLO study, in which almonertinib treatment resulted in an objective response rate of 68.9% and a disease control rate of 93.4% in patients with recurrent NSCLC carrying EGFR T790M mutations. The median progression-free survival was 12.3 months. Almonertinib caused an objective response rate of 61.5% in patients with central nervous system metastasis (https://www.onclive.com/view/almonertinib-approved-in-china-for-emegfrem-t79m-nsclc).
Unfortunately, patients still inevitably develop acquired resistance to third-generation EGFR-TKIs, limiting their long-term efficacies. Acquisition of a novel resistant C797S mutation is the most common resistance mechanism when osimertinib is used as a second-line therapy. Beyond the EGFR-dependent resistance mechanism, there are other heterogeneous EGFR-independent mechanisms reported such as MET (c-MET) or HER2 gene amplification, acquired mutations in oncogenes (eg, BRAF), and small-cell or squamous cell transformation (see reviews).7,11–13 However, resistance mechanisms in most cases, particularly when osimertinib is used as a first-line therapy, have not been fully understood. Therefore, it is urgent and clinically desirable to explore the underlying resistance mechanisms in order to develop effective therapies to overcome acquired resistance to osimertinib and other third-generation EGFR-TKIs.
One important acquired resistance mechanism to third-generation EGFR-TKIs is the upregulation of the Ras/Raf/MEK/ERK signaling pathway. Targeting this pathway achieved promising therapeutic efficacies against osimertinib-resistant EGFR-mutant NSCLC cell lines and tumors in preclinical studies and even in clinical case studies. This review will focus on discussing the scientific rationale and potential of targeting the MEK/ERK signaling pathway as a promising strategy to overcome and even prevent acquired resistance to third-generation EGFR-TKIs, particularly osimertinib.
Ras/Raf/MEK/ERK Cascade Functions as a Critical Downstream Signaling Pathway of EGFR
The Ras/Raf/MEK/ERK signaling represents a well-established oncogenic pathway that regulates many biological outputs like proliferation, survival, differentiation and apoptosis.14,15 It is also one of the well-known downstream signaling pathways of cell membrane tyrosine kinase growth factor receptors including EGFR.16 Activated cell membrane EGFR can recruit adaptor proteins such as Grb2 to engage guanine nucleotide exchange factors (GEFs) like SOS to the cell membrane. The activated GEFs are capable of interacting with Ras proteins including K-Ras, N-Ras and H-Ras to activate Ras by catalyzing the conversion of GDP (Ras-GDP) to GTP (Ras-GTP), whereas GTPase-activating proteins (GAPs) turn off GTP activity through stimulating GTP hydrolysis to GDP, leading to inactivation of Ras.16 Active Ras has three major downstream effectors, including Raf, PI3K, and Ras-like guanine nucleotide-dissociation stimulator (RalGDS), which all have a Ras binding domain that combines with GTP-Ras. Once Ras is activated, Raf (A-Raf, B-Raf or C-Raf) serine/threonine kinases are activated and recruited to the cell membrane through binding RAS. The activated Raf proteins can then directly activate MEK1 and MEK2 tyrosine and threonine/serine kinases via serine phosphorylation, which subsequently activate ERK1/2 (Figure 1A). The activated ERK1/2 then both activate cytosolic substrates and translocate to the nucleus to stimulate the expression of diverse genes, finally resulting in multiple nuclear and cytoplasmic targets being activated.16,17 Therefore, MEK/ERK are commonly used as the targets to inhibit the Ras/Raf/MEK/ERK signaling pathway.
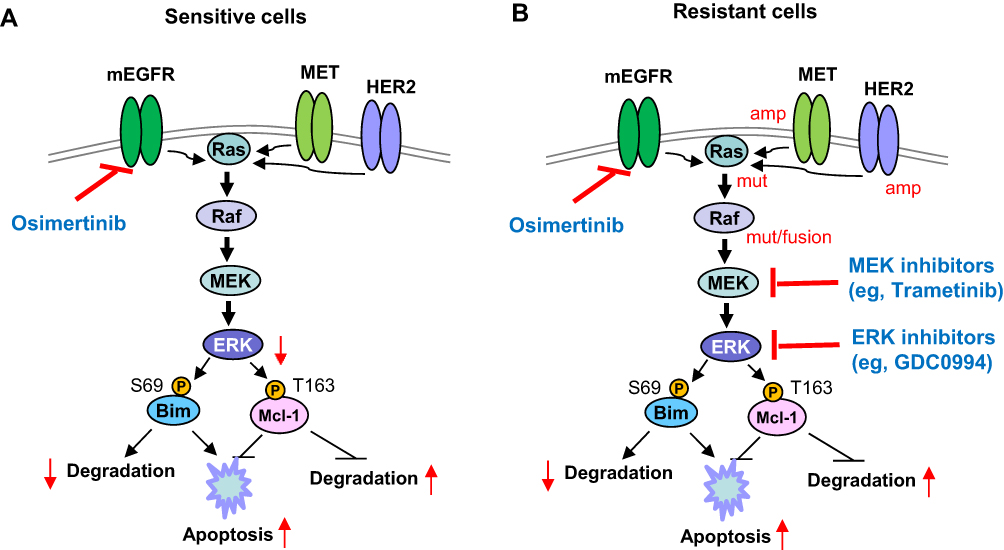 |
Figure 1 Schematic illustration of Ras/Raf/MEK/ERK signaling downstream of cell membranes tyrosine kinase receptors including EGFR that negatively regulate apoptosis through modulating degradation of Bim and Mcl-1 (A), mechanism by which osimertinib induces apoptosis in the sensitive EGFR-mutant NSCLC cells though targeting this pathway (A), and rationale for co-targeting mutant EGFR (mEGFR) and MEK/ERK to enhance apoptosis in the resistant EGFR-mutant NSCLC cells (B). The red arrows indicate the changes caused by osimertinib (A) or the combination of osimertinib with a MEK (eg, trametinib) or ERK (eg, GDC0994) inhibitor (B). amp, amplification; mut, mutation. |
Effective Suppression of the MEK/ERK Signaling Pathway is Critical for Third-Generation TKIs to Exert Their Therapeutic Efficacies in EGFR-Mutant NSCLC Cells
Induction of apoptosis has been considered an important mechanism accounting for the anticancer efficacy of many cancer therapeutic agents including EGFR-TKIs.18–20 Our early work has shown that osimertinib effectively induces apoptosis in EGFR-mutant NSCLC cell lines primarily through concurrent upregulation of Bim and downregulation of Mcl-1.20 A recent study shows that hypoxia-induced resistance to osimertinib is driven by overexpression of fibroblast growth factor receptor 1 (FGFR1) to sustain ERK signaling and subsequent attenuation of Bim induction, which can be overcome by combining with an FGFR1 or MEK inhibitor.21 In agreement, other studies, albeit with different rationales, also demonstrate a critical role of functional Bim in mediating apoptosis induced by osimertinib, other third-generation EGFR-TKIs or the combination of osimertinib with other agents in EGFR-mutant NSCLC cells.22–26
We found that osimertinib quickly inhibited MEK/ERK signaling, evidenced by decreasing levels of p-ERK1/2 primarily in EGFR-mutant NSCLC cell lines accompanied by Bim elevation and Mcl-1 reduction, which are likely due to enhanced Mcl-1 degradation and increased Bim stability. Both Bim and Mcl-1 are phosphorylated by ERK1/2 at S69 and T163, respectively, leading to protein destabilization (Bim) or stabilization (Mcl-1) (Figure 1A). Osimertinib effectively inhibited phosphorylation of both Bim (S69) and Mcl-1 (T163), accompanied with enhanced Mcl-1 degradation and compromised Bim degradation, thus leading to Bim elevation and Mcl-1 decrease (Figure 1A). Gene knockdown-mediated blockade of Bim elevation or Mcl-1 overexpression in the sensitive EGFR-mutant NSCLC cells attenuated or abolished the induction of apoptosis by osimertinib.20 Therefore, inhibition of ERK-dependent modulation of Bim and Mcl-1 is a key mechanism accounting for osimertinib-induced apoptosis in EGFR-mutant NSCLC cells (Figure 1A). This initial preclinical work clearly connects MEK/ERK inhibition to osimertinib-induced apoptosis in EGFR-mutant NSCLC cells.
The Aberrant Activation of Ras/Raf/MEK/ERK Signaling Pathway is Connected to Development of Acquired Resistance to Third-Generation EGFR-TKIs
Clinical Observations
Other than the C797S mutation, MET amplification has been the most characterized and recognized resistance mechanism to third-generation EGFR-TKIs. In different major trials with osimertinib, including both first- and second-line therapy, MET amplification was detected in 7–24% of resistance cases.11,27 In a trial with another early third-generation EGFR-TKI named rociletinib (CO1686), MET amplification was detected in 26% of resistance cases.28 HER2 and RET are other cell membrane tyrosine kinase receptors. HER2 gene amplification was detected in 1–5% of cases resistant to osimertinib.9 RET fusion mutations (eg, RET-RUFY2, RET-CCDC6 and RET-NCOA4) were also detected in 1–3% EGFR-mutant tissues relapsed to osimertinib.29–31 These gene alterations can all lead to EGFR-independent activation of downstream Ras/Raf/MEK/ERK signaling.9,27
Moreover, alterations directly in the Ras/Raf/MEK/ERK axis can also activate this signaling pathway. KRAS G12S mutation was detected in tumor biopsies (1/7) with acquired resistance to second-line osimertinib.32 One patient harboring KRAS Q61K mutation was identified among 28 NSCLC patients that relapsed from osimertinib treatment with T790M loss.33 One KRAS Q61R mutation was found among 21 osimertinib-resistant cases with T790M loss.13 In a cohort of patients with NSCLC harboring positive T790M mutation enrolled in AURA trial, KRAS G12V mutation (1/13) was detected.34
Beyond RAS mutations, an early case report identified a BRAF V600E mutation in a patient at the time of progression while being treated with osimertinib as a second-line therapy.35 Other studies with different cohorts of EGFR-mutant NSCLC patients that relapsed from osimertinib second-line treatment also detected a BRAF mutation33,34 and even BRAF fusions,33 although the frequencies were not high.
Currently, we know little about the resistance mechanisms when osimertinib is used as a first-line treatment. In the AURA clinical trial (NCT01802632) using osimertinib as a first-line treatment of EGFR mutation-positive advanced NSCLC, one case each of MEK1 and RAS mutation was identified among 19 relapsed patients.36 In the Phase 3 FLAURA trial (NCT02296125), an acquired concurrent BRAF V600E and MET amplification was detected in an EGFR-mutant NSCLC patient experiencing resistance to first-line osimertinib treatment.37
Preclinical Evidence
The early study by Eberlein et al38 analyzed EGFR-mutant NSCLC cell lines with acquired resistance to osimertinib or WZ4002 (another early third-generation EGFR-TKI) established from EGFR-mutant PC-9 and H1975 cell lines and found these resistant cell lines were in general sensitive to MEK inhibitors. Moreover, they identified N-RAS mutations including E63K, G12V and G12R mutations and copy-number gains of MAPK1, CRK like proto-oncogene (CRKL), N-RAS, and K-RAS across the resistant populations. These results suggest that the Ras/Raf/MEK/MAPK signaling activation is important for driving resistance to third-generation EGFR-TKIs in the absence of EGFR signaling. Ortiz-Cuaran et al32 tested the impact of an acquired K-RAS G12S mutation on cell response to osimertinib or rociletinib by ectopically expressing K-RAS G12S in sensitive PC-9 and HCC827 cell lines. They found that the cells expressing K-RAS-G12S were less sensitive to osimertinib or rociletinib compared with the corresponding control cells expressing WT K-RAS and maintained sustained ERK phosphorylation under osimertinib treatment, suggesting MAPK signaling has a role in inducing resistance to third-generation EGFR-TKIs. Moreover, these K-RAS G12S cell lines were sensitive to co-treatment with osimertinib and trametinib (a MEK inhibitor). Nakatani et al39 detected K-RAS amplification in both rociletinib- and osimertinib-resistant PC-9 cells established from afatinib-resistant PC-9 cells with EGFR 19del and T790M mutations. These resistant cell lines did not respond to osimertinib-mediated suppression of ERK phosphorylation but were sensitive to the concomitant inhibition of MEK and EGFR.
By analyzing PC-9 cells with acquired resistance to osimertinib, Ku et al40 identified an H-RAS G13R mutation with aberrant ERK signaling activation through an EGFR-independent fashion, which is also resistant to osimertinib modulation. La Monica et al41 established osimertinib-resistant cell lines by culturing PC-9 cells in the presence of increasing concentrations of osimertinib and identified a B-RAF G469A mutation in all three resistant clones that developed acquired resistance, which shows a constitutive ERK signaling activation. Stable expression of B-RAF G469A in both PC-9 and HCC827 cell lines conferred cell resistance to osimertinib.
Beyond the above mechanisms conferring acquired resistance through genomic alterations that activate ERK1/2 signaling, Tricker et al,25 reported that ERK1/2 reactivation occurred rapidly following exposure to WZ4002, inhibition of which by the MEK inhibitor, trametinib, enhanced WZ4002-induced apoptosis and inhibited the development of acquired resistance to WZ4002 in WZ4002-sensitive tumor models. ERK reactivation also occurred on EGFR-mutant NSCLC cells exposed to osimertinib.42,43 Osimertinib plus a MEK inhibitor such as trametinib-enhanced induction of apoptosis in different EGFR-mutant NSCLC cell lines and significantly delayed the emergence of acquired resistance to osimertinib as we recently demonstrated.43 Another related aspect is that osimertinib lost its capacity to inhibit MEK/ERK signaling in different cell lines resistant to osimertinib evidenced by limited inhibitory effects on ERK phosphorylation as documented in several studies.20,42,44 This mechanism may also contribute to the acquired resistance to osimertinib and other third-generation EGFR-TKIs since co-targeting MEK or ERK can efficiently sensitize osimertinib-resistant cells and tumors to osimertinib as discussed below.
Targeting MEK/ERK Signaling Effectively Overcomes Acquired Resistance to Third-Generation EGFR-TKIs
The above findings on activation of the Ras/Raf/MEK/ERK signaling pathway as an important resistance mechanism to osimertinib and other third-generation EGFR-TKIs provide a strong scientific rationale for targeting this pathway to achieve the goal of overcoming acquired resistance to third-generation EGFR-TKIs.
In a preclinical study by Eberlein et al,38 with a number of cell populations resistant to WZ4002 or osimertinib, which have an N-RAS mutation and a gain of copy number of WT N-RAS or K-RAS, the MEK inhibitor, selumetinib (AZD6244; ARRY-142,886), when combined with osimertinib, was effective in inhibiting the growth of the resistant cell populations. Moreover, concomitant dosing of osimertinib plus selumetinib induced regression of osimertinib-resistant tumors in an EGFR mutant/T790M transgenic model in vivo. Similarly, a combination of osimertinib plus a MEK inhibitor, trametinib or selumetinib, was highly synergistic in inhibiting the growth of osimertinib-resistant cells with a K-RAS G12S mutation with full inhibition of downstream MAPK signaling.32 Moreover, a MEK inhibitor (eg, selumetinib) combined with osimertinib also restored the sensitivity of osimertinib-resistant cells with increased MET expression and an H-RAS G13R mutation both in vitro and in vivo.40 Beyond the RAS-dependent resistance models, MEK inhibition with trametinib synergized with osimertinib to suppress the growth of EGFR-mutant NSCLCs expressing a B-Raf fusion45 or resistant tumors with elevated expression of FGFR1 and activated MEK/ERK signaling.21
In agreement with the preclinical findings, a clinical case study reported that an osimertinib-resistant patient with NSCLC carrying an EGFR 19del mutation and a B-RAF fusion (AGK-BRAF) partially responded to the combination of osimertinib and trametinib during a 5-month treatment period.46 An NSCLC patient harboring EGFR L858R, T790M and B-RAF V600E mutations that received treatment of osimertinib and dabrafenib (a B-Raf inhibitor) plus trametinib showed significantly improved clinical symptoms after 3 weeks and could stay in the outpatient department (recovery from ECOG PS of 3 to 1). All treatment-related adverse events, including a grade II rash and decreased appetite, were tolerable. Obvious tumor mass shrinkage in the right lower lobe was observed with chest CT scans and the efficacy was confirmed 6 weeks later.47 Another patient with advanced lung adenocarcinoma carrying EGFR-19del, T790M and B-RAF V600E mutations that received a similar three-drug combination treatment achieved a striking progression-free survival of 9 months and did not show any serious adverse reaction.48 The same treatment regimen was also used to treat two NSCLC patients carrying EGFR 19del, T790M, and B-RAF V600E mutations in another case report, in which clinical response in one patient lasted for 13.4 months, but not in the other patient.49 Similarly, the treatment of two cases of advanced NSCLC patients carrying EGFR 19del or L858R, T790M and B-RAF V600E mutations with osimertinib and the B-Raf V600E inhibitor, vemurafenib, combination achieved a noticeable improvement in clinical symptoms including physical mobility within 4 to 6 weeks from the initiation of the combination treatment. This combination treatment was well tolerated by patient 2 with well-controlled disease by the treatment; patient 1 responded to the treatment, but developed grade IV vomiting, leading to discontinuation of the treatment.50
Our preclinical studies have also suggested that targeting MEK/ERK signaling is an efficacious and promising strategy for overcoming acquired resistance to osimertinib and possibly other third-generation EGFR-TKIs, albeit having a different scientific rationale.20,51 Toward understanding the mechanisms by which osimertinib and other third-generation EGFR-TKIs exert their therapeutic efficacies against NSCLC cells with activating EGFR mutations, we have shown that modulation of ERK-dependent Bim and Mcl-1 proteasomal degradation plays an important role in mediating osimertinib-induced apoptosis in EGFR-mutant NSCLC cells. Osimertinib lost its ability to suppress ERK phosphorylation and modulate Bim and Mcl-1 levels in different cell lines with acquired osimertinib resistance owing to MET amplification, EGFR C797S mutation or unknown mechanisms. Thus, the inability of osimertinib to alter Bim and Mcl-1 levels by modulating ERK-dependent Bim and Mcl-1 degradation in osimertinib-resistant cells is an important mechanism accounting for how sensitive EGFR-mutant NSCLC cells could become resistant to osimertinib-induced apoptosis. Our initial hypothesis was that enforced elevation of Bim levels and decrease in Mcl-1 levels via abrogation of ERK-dependent Bim and Mcl-1 phosphorylation with a MEK inhibitor could restore the sensitivity of osimertinib-resistant EGFR-mutant NSCLC cells to die of apoptosis (Figure 1B), achieving promising treatment against the growth of osimertinib-resistant NSCLC cells or tumors. With three different MEK inhibitors, trametinib, selumetinib and mirdametinib (PD0325901), we generated identical results, ie, osimertinib synergized with either MEK inhibitor to decrease the survival and induce apoptosis of osimertinib-resistant cell lines regardless of the underlying mechanism. This therapeutic strategy also generated impressive in vivo activity against osimertinib-resistant tumors either with the concurrent or intermittent schedule.20 In agreement with our findings, another study using EGFR-mutant NSCLC xenograft models to evaluate the efficacy of selumetinib and osimertinib combination as a second-line treatment in reverting osimertinib resistance found that the combination was effective to revert the osimertinib in the majority of tested mice, achieving a response rate of 50% to 80%.52
With the same rationale (Figure 1B), our recent study has further shown that osimertinib combined with an ERK inhibitor such as GDC0994 (ravoxertinib) or VRT752,271 (ulixertinib or BVD-523) also synergistically decreased the survival and enhanced apoptosis of osimertinib-resistant cell lines with an effective and significant effect on suppressing the growth of osimertinib-resistant tumors in vivo. While our osimertinib-resistant cell lines were cross-resistant to first or second-generation EGFR-TKIs (eg, erlotinib and afatinib), the combination of a MEK or ERK inhibitor even with a first-generation (eg, erlotinib) or second-generation (eg, afatinib) EGFR-TKI remarkably suppressed the growth of osimertinib-resistant cells in vitro and of tumors in vivo.51
These findings together strongly suggest the necessity of targeting MEK/ERK signaling in maintaining the long-term efficacy of osimertinib by overcoming osimertinib acquired resistance and thus warrant further clinical investigation of this promising therapeutic strategy to improve the long-term benefit of osimertinib. An attempt has been made in the clinic in the recent phase IB TATTON study (NCT02143466). The strategy of osimertinib 80 mg orally (p.o.) once a day with selumetinib (25–75 mg p.o. twice a day; continuous or intermittent) was tested in 36 patients with advanced EGFR-mutant NSCLC and disease progression on a prior EGFR-TKI and found feasible in terms of antitumor activity and manageable toxicity.53
Targeting MEK/ERK Signaling Delays or Abrogates Emergence of Acquired Resistance to Third-Generation EGFR-TKIs
In addition to the above discussion on overcoming acquired resistance to third-generation EGFR-TKIs through co-targeting MEK/ERK signaling, co-inhibition of MEK or ERK has a high potential to delay or prevent the emergence of acquired resistance to third-generation EGFR-TKIs. Based on the finding that WZ4002 treatment induces quick ERK1/2 reactivation, Tricker et al,25 reported that WZ4002 and trametinib combination effectively prevented ERK1/2 reactivation, enhanced induction of apoptosis and retarded the development of resistance in WZ4002-sensitive models that can develop acquired resistance through both T790M-dependent and -independent mechanisms. Our recent study has also shown that osimertinib combined with a MEK or ERK inhibitor exerts similar activity.43 Osimertinib synergized with a MEK (eg, trametinib) or ERK (eg, VRT-752271) inhibitor in decreasing cell survival and enhancing induction of apoptosis in EGFR-mutant NSCLC cells, but not in NSCLC cells with WT EGFR. These combinations very effectively killed several clones that were intrinsically resistant to osimertinib via enhanced induction of apoptosis. Concurrent and intermittent (two-weeks osimertinib followed by two-weeks combination with trametinib or four-weeks osimertinib followed by two-weeks combination with trametinib) application of trametinib with osimertinib substantially retarded the development of osimertinib resistance both in vitro and in vivo.43 It deserves mentioning that we observed some mice without detectable tumors in each combination treatment group; this collectively accounted for a cure rate of 27.8% (5 of 18 mice in total).43 This suggests that some patients may achieve long-term remission with these combination treatments and thus is clinically meaningful.
Although we believe delaying the development of acquired resistance is a promising strategy, the application of combination approaches, particularly those vertically targeting the same pathway (eg, MEK/ERK signaling here), in this process for prolonged treatment periods may also have the risk of increased toxicity for patients. Hence, the potential concern is that the osimertinib and trametinib combination, particularly for prolonged applications, may accordingly enhance toxicity while augmenting therapeutic efficacy. In our previous therapeutic studies for overcoming osimertinib resistance, no increased toxicity was observed in nude mice carrying osimertinib-resistant tumors treated with the combination of osimertinib with a MEK or ERK inhibitor for 2 or 3 weeks.20,51 Similarly, mice receiving the combination of osimertinib with trametinib in a concurrent way for more than 3 months in the study for delaying the emergence of acquired resistance did not exhibit apparent toxicity either.43 These findings together indicate its safety.
Interestingly, the combination of osimertinib with either a MEK or ERK inhibitor did not further enhance the killing of NSCLC cell lines harboring WT EGFR although they synergistically decreased the growth of EGFR-mutant NSCLC cell lines,43 suggesting that this combinatorial strategy may not be toxic to cells or tissues (eg, normal tissues) harboring normal or WT EGFR. Whether this can explain the safety of osimertinib combined with either MEK or ERK inhibition in abrogating and overcoming acquired resistance to osimertinib warrants further study.
In another study by Della Corte et al,52 which evaluated the efficacy and safety of osimertinib and selumetinib combination as a first-line therapy in preclinical xenografts harboring EGFR activating mutations, they demonstrated that the combination is able to increase the response rate (up to 90%): taking all groups of mice treated with combinations together, only 7 of 60 mice showed progression disease (3 of H1975-, 2 HCC827-, and 2 PC-9/T790M-xenografts, respectively). Moreover, median progression-free survival was not reached in all combination groups, indicating that the combination treatments are better than single-agent osimertinib regarding median progression-free survival (17–18 weeks). Importantly, all mice with clinical complete response or partial response maintained these responses for the period of the experiment (fixed to 52 weeks) without relevant toxicities.52
In addition to directly targeting the MEK/ERK signaling with MEK or ERK inhibitors, another study by Romaniello et al54 identified a triple combination of osimertinib at a low dose with clinically approved drugs, cetuximab and trastuzumab (an anti-HER2 mAb), as an effective and long-lasting treatment to prevent the onset of resistance to osimertinib by blocking bypass pathways, particularly ERK activation. Both continuous and intermittent treatments using this triple combination robustly prevented resistance to osimertinib. Mechanistically, this triple combination induced EGFR and HER2 degradation, inhibited activation of ERK, reduced abundance of several bypass proteins, namely MET, AXL, and HER3, and cooperatively enhanced apoptosis. Moreover, this triple combination strategy also effectively eradicated tumors after they acquired resistance to osimertinib. Through similar mechanisms, the combination of a neutralizing anti-HER3 antibody that can clear HER3 from the cell surface, with osimertinib and cetuximab generated similar results.55 These studies thus further underline the importance of targeting MEK/ERK pathway in preventing and overcoming acquired resistance to osimertinib.
Conclusions and Perspectives
Despite the promising clinical outcomes of osimertinib as both a second-line and first-line therapy, the development of acquired resistance impedes its long-term success in curing cancer. The aberrant activation of the Ras/Raf/MEK/ERK signaling pathway has been recognized as one of the important resistant mechanisms. Co-targeting the MEK/ERK signaling pathway proves to be a promising therapeutic strategy not only for overcoming acquired resistance to third-generation EGFR-TKIs but also for delaying or preventing the emergence of acquired resistance to third-generation EGFR-TKIs as demonstrated in some preclinical studies. Several clinical case reports and a phase IB trial have also demonstrated its feasibility based on the preliminary activities and manageable toxicity. Several ongoing trials testing these combinations (Table 1) may eventually provide an answer.
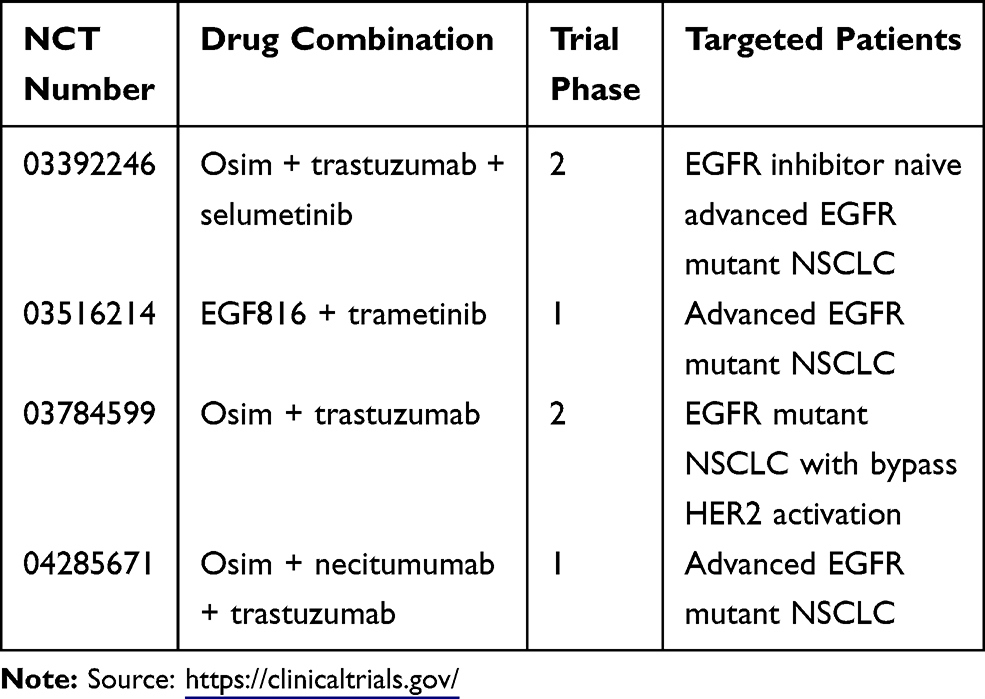 |
Table 1 Ongoing Clinical Trials That Test Osimertinib (Osim) or EGF816 Combined with a MEK Inhibitor or Other Inhibitor That Blocks MEK/ERK Activation |
Biomarkers to predict the potential efficacy of the combination strategy are generally needed to guide the therapy. Detection of acquired gene alterations such as K-RAS and B-RAF mutations including gene rearrangements is definitely important in this regard. However, there is no study that detects phosphorylation of MEK1/2 and ERK1/2 in post-relapse tumor samples, which represents the activated state of the Ras/Raf/MEK/ERK signaling given that ERK is reactivated during treatment with a third-generation EGFR-TKI and resistant to modulation by third-generation EGFR-TKIs. This direction can be explored. In fact, our findings show that co-targeting either MEK or ERK works to overcome osimertinib acquired resistance in different resistant cell models owing to MET amplification, EGFR C797S mutation or unknown mechanisms, strongly suggesting that this strategy works to overcome acquired resistance regardless of the underlying mechanisms. From this point of view, patient selection may not be a critical issue. Future clinical trials are definitely needed to validate this potential strategy in effectively overcoming acquired resistance to osimertinib and other third-generation EGFR-TKIs.
It is important to take effective action to treat patients who relapsed to osimertinib or other third-generation EGFR-TKIs. Compared to this passive practice, it is more important to take action early before disease progression to manage the unavoidable development of acquired resistance. We believe that the early intervention using effective and tolerable combinational regimens may substantially improve the outcomes of EGFR-mutant NSCLC treatment with third-generation EGFR-TKIs. The combination of osimertinib and a MEK inhibitor has shown impressive efficacy as first-line therapy in the treatment of mice with EGFR-mutant NSCLC tumors, which is superior to single-agent osimertinib, with no apparent toxicity during a long period of treatment in preclinical studies.43,52 With further optimized combination schedules, particularly intermittent ones as we recently demonstrated,43 safe and effective combination regimens including those based on the inhibition of MEK/ERK signaling as discussed in this review are likely to be developed for delaying and even preventing the emergence of acquired resistance, maximizing the therapeutic efficacies of third-generation EGFR-TKIs.
Sustained treatment of certain types of cancer such as melanoma with MEK inhibitors may cause ERK reactivation, leading to resistance or treatment failure,56,57 although this has not been demonstrated in lung cancer. Whether there is an advantage of targeting ERK1/2 over MEK in delaying and overcoming acquired resistance to osimertinib or other third-generation EGFR-TKIs should be considered and explored.
Acknowledgment
SYS is a Georgia Research Alliance Distinguished Cancer Scientist and supported by NIH/NCI R01 CA223220 (to SYS), R01 CA245386 (to SYS), UG1 CA233259 and Lung Cancer SPORE P50 CA217691 DRP award (to S-YS) and Emory University Winship Cancer Institute lung cancer pilot fund (to SYS).
Both authors share the first authorship: Danlei Yu and Wen Zhao.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Barta JA, Powell CA, Wisnivesky JP. Global epidemiology of lung cancer. Ann Glob Health. 2019;85:1. doi:10.5334/aogh.2419
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
3. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17(11):637–658.
4. Shah R, Lester JF. Tyrosine kinase inhibitors for the treatment of EGFR mutation-positive non-small-cell lung cancer: a clash of the generations. Clin Lung Cancer. 2020;21(3):e216–e228. doi:10.1016/j.cllc.2019.12.003
5. Skoulidis F, Papadimitrakopoulou VA. Targeting the gatekeeper: osimertinib in EGFR T790M mutation-positive non-small cell lung cancer. Clin Cancer Res. 2017;23(3):618–622. doi:10.1158/1078-0432.CCR-15-2815
6. Russo A, Franchina T, Ricciardi GRR, et al. Third generation EGFR TKIs in EGFR-mutated NSCLC: where are we now and where are we going. Crit Rev Oncol Hematol. 2017;117:38–47. doi:10.1016/j.critrevonc.2017.07.003
7. Murtuza A, Bulbul A, Shen JP, et al. Novel third-generation EGFR tyrosine kinase inhibitors and strategies to overcome therapeutic resistance in lung cancer. Cancer Res. 2019;79(4):689–698. doi:10.1158/0008-5472.CAN-18-1281
8. Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382(1):41–50. doi:10.1056/NEJMoa1913662
9. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725–737. doi:10.1038/s41416-019-0573-8
10. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–125. doi:10.1056/NEJMoa1713137
11. Piper-Vallillo AJ, Sequist LV, Piotrowska Z. Emerging treatment paradigms for EGFR-mutant lung cancers progressing on osimertinib: a review. J Clin Oncol. 2020;JCO1903123. doi:10.1200/JCO.19.03123
12. Tang ZH, Lu JJ. Osimertinib resistance in non-small cell lung cancer: mechanisms and therapeutic strategies. Cancer Lett. 2018;420:242–246. doi:10.1016/j.canlet.2018.02.004
13. Le X, Puri S, Negrao MV, et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin Cancer Res. 2018;24(24):6195–6203. doi:10.1158/1078-0432.CCR-18-1542
14. Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14(2):342–346. doi:10.1158/1078-0432.CCR-07-4790
15. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–3310. doi:10.1038/sj.onc.1210422
16. Martinelli E, Morgillo F, Troiani T, Ciardiello F. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: the role of MEK. Cancer Treat Rev. 2017;53:61–69. doi:10.1016/j.ctrv.2016.12.001
17. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. 2017;9:5. doi:10.3390/cancers9050052
18. Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9(7):501–507. doi:10.1038/nrc2663
19. Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5(11):876–885. doi:10.1038/nrc1736
20. Shi P, Oh YT, Deng L, et al. Overcoming acquired resistance to AZD9291, a third-generation EGFR inhibitor, through modulation of MEK/ERK-dependent Bim and Mcl-1 degradation. Clin Cancer Res. 2017;23(21):6567–6579. doi:10.1158/1078-0432.CCR-17-1574
21. Lu Y, Liu Y, Oeck S, Zhang GJ, Schramm A, Glazer PM. Hypoxia induces resistance to EGFR inhibitors in lung cancer cells via upregulation of FGFR1 and the MAPK pathway. Cancer Res. 2020;80(21):4655–4667. doi:10.1158/0008-5472.CAN-20-1192
22. Tanimoto A, Takeuchi S, Arai S, et al. Histone deacetylase 3 inhibition overcomes BIM deletion polymorphism-mediated osimertinib resistance in EGFR-mutant lung cancer. Clin Cancer Res. 2017;23(12):3139–3149. doi:10.1158/1078-0432.CCR-16-2271
23. Yochum ZA, Cades J, Wang H, et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene. 2019;38(5):656–670. doi:10.1038/s41388-018-0482-y
24. Song KA, Niederst MJ, Lochmann TL, et al. Epithelial-to-mesenchymal transition antagonizes response to targeted therapies in lung cancer by suppressing BIM. Clin Cancer Res. 2018;24(1):197–208. doi:10.1158/1078-0432.CCR-17-1577
25. Tricker EM, Xu C, Uddin S, et al. Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR-mutant lung cancer. Cancer Discov. 2015;5:960–971. doi:10.1158/2159-8290.CD-15-0063
26. Kurppa KJ, Liu Y, To C, et al. Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell. 2020;37(1):104–122 e112.
27. Wang Q, Yang S, Wang K, Sun SY. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J Hematol Oncol. 2019;12(1):63. doi:10.1186/s13045-019-0759-9
28. Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi:10.1038/ncomms11815
29. Schoenfeld AJ, Chan JM, Kubota D, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin Cancer Res. 2020;26(11):2654–2663. doi:10.1158/1078-0432.CCR-19-3563
30. Piotrowska Z, Isozaki H, Lennerz JK, et al. Landscape of acquired resistance to osimertinib in EGFR-Mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 2018;8(12):1529–1539. doi:10.1158/2159-8290.CD-18-1022
31. Offin M, Somwar R, Rekhtman N. et al. Acquired ALK and RET gene fusions as mechanisms of resistance to osimertinib in EGFR-mutant lung cancers. JCO Precis Oncol;2018. 2. 10.1200/PO.18.00126
32. Ortiz-Cuaran S, Scheffler M, Plenker D, et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res. 2016;22:4837–4847. doi:10.1158/1078-0432.CCR-15-1915
33. Oxnard GR, Hu Y, Mileham KF, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA oncol. 2018;4(11):1527–1534. doi:10.1001/jamaoncol.2018.2969
34. Lin CC, Shih JY, Yu CJ, et al. Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: a genomic study. Lancet Respir Med. 2018;6(2):107–116. doi:10.1016/S2213-2600(17)30480-0
35. Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Yang JC. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thoracic Oncol. 2017;12(3):567–572. doi:10.1016/j.jtho.2016.11.2231
36. Ramalingam SS, Yang JC, Lee CK, et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J Clin Oncol. 2018;36(9):841–849. doi:10.1200/JCO.2017.74.7576
37. Minari R, Bordi P, La Monica S, et al. Concurrent acquired BRAF V600E mutation and MET amplification as resistance mechanism of first-line osimertinib treatment in a patient with EGFR-Mutated NSCLC. J Thoracic Oncol. 2018;13(6):e89–e91. doi:10.1016/j.jtho.2018.03.013
38. Eberlein CA, Stetson D, Markovets AA, et al. Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. 2015;75(12):2489–2500. doi:10.1158/0008-5472.CAN-14-3167
39. Nakatani K, Yamaoka T, Ohba M, et al. KRAS and EGFR amplifications mediate resistance to rociletinib and osimertinib in acquired afatinib-resistant nsclc harboring exon 19 deletion/t790m in EGFR. Mol Cancer Ther. 2019;18(1):112–126. doi:10.1158/1535-7163.MCT-18-0591
40. Ku BM, Choi MK, Sun JM, et al. Acquired resistance to AZD9291 as an upfront treatment is dependent on ERK signaling in a preclinical model. PLoS One. 2018;13(4):e0194730. doi:10.1371/journal.pone.0194730
41. La Monica S, Minari R, Cretella D, et al. Acquired BRAF G469A mutation as a resistance mechanism to first-line osimertinib treatment in NSCLC cell lines harboring an EGFR exon 19 deletion. Target Oncol. 2019;14(5):619–626. doi:10.1007/s11523-019-00669-x
42. Ichihara E, Westover D, Meador CB, et al. SFK/FAK signaling attenuates osimertinib efficacy in both drug-sensitive and drug-resistant models of EGFR-mutant lung cancer. Cancer Res. 2017;77(11):2990–3000. doi:10.1158/0008-5472.CAN-16-2300
43. Gu J, Yang W, Shi P, et al. MEK or ERK inhibition effectively abrogates emergence of acquired osimertinib resistance in the treatment of EGFR-mutant lung cancers. Cancer. 2020;126:3788–3799. doi:10.1002/cncr.32996
44. Tang ZH, Jiang XM, Guo X, Fong CM, Chen X, Lu JJ. Characterization of osimertinib (AZD9291)-resistant non-small cell lung cancer NCI-H1975/OSIR cell line. Oncotarget. 2016;7(49):81598–81610. doi:10.18632/oncotarget.13150
45. Vojnic M, Kubota D, Kurzatkowski C, et al. Acquired BRAF rearrangements induce secondary resistance to EGFR therapy in EGFR-mutated lung cancers. J Thoracic Oncol. 2019;14(5):802–815. doi:10.1016/j.jtho.2018.12.038
46. Dagogo-Jack I, Piotrowska Z, Cobb R, et al. Response to the combination of osimertinib and trametinib in a patient with EGFR-mutant NSCLC harboring an acquired BRAF fusion. J Thoracic Oncol. 2019;14(10):e226–e228. doi:10.1016/j.jtho.2019.05.046
47. Zhou F, Zhao W, Chen X, Zhang J, Zhou C. Response to the combination of dabrafenib, trametinib and osimertinib in a patient with EGFR-mutant NSCLC harboring an acquired BRAF (V600E) mutation. Lung Cancer. 2020;139:219–220. doi:10.1016/j.lungcan.2019.10.014
48. Ding H, Zhuang Z, Xie J, Huang H, Tao Z, Liu Z. Durable clinical response of advanced lung adenocarcinoma harboring EGFR-19del/t790m/BRAF (v600e) mutations after treating with osimertinib and dabrafenib plus trametinib: a case report. Onco Targets Ther. 2020;13:7933–7939. doi:10.2147/OTT.S240775
49. Meng P, Koopman B, Kok K, et al. Combined osimertinib, dabrafenib and trametinib treatment for advanced non-small-cell lung cancer patients with an osimertinib-induced BRAF V600E mutation. Lung Cancer. 2020;146:358–361. doi:10.1016/j.lungcan.2020.05.036
50. Xie Z, Gu Y, Xie X, et al. Lung adenocarcinoma harboring concomitant EGFR mutations and BRAF v600e responds to a combination of osimertinib and vemurafenib to overcome osimertinib resistance. Clin Lung Cancer. 2020. doi:10.1016/j.cllc.2020.06.008
51. Li Y, Zang H, Qian G, Owonikoko TK, Ramalingam SR, Sun SY. ERK inhibition effectively overcomes acquired resistance of epidermal growth factor receptor-mutant non-small cell lung cancer cells to osimertinib. Cancer. 2020;126(6):1339–1350. doi:10.1002/cncr.32655
52. Della Corte CM, Ciaramella V, Cardone C, et al. Antitumor efficacy of dual blockade of EGFR signaling by osimertinib in combination with selumetinib or cetuximab in activated EGFR human NCLC tumor models. J Thoracic Oncol. 2018;13(6):810–820. doi:10.1016/j.jtho.2018.02.025
53. Oxnard GR, Yang JC, Yu H, et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol. 2020;31(4):507–516. doi:10.1016/j.annonc.2020.01.013
54. Romaniello D, Mazzeo L, Mancini M, et al. A combination of approved antibodies overcomes resistance of lung cancer to osimertinib by blocking bypass pathways. Clin Cancer Res. 2018;24(22):5610–5621. doi:10.1158/1078-0432.CCR-18-0450
55. Romaniello D, Marrocco I, Belugali Nataraj N, et al. Targeting HER3, a catalytically defective receptor tyrosine kinase, prevents resistance of lung cancer to a third-generation EGFR kinase inhibitor. Cancers. 2020;12:9. doi:10.3390/cancers12092394
56. Kun E, Tsang YTM, Ng CW, Gershenson DM, Wong KK. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev. 2020;92:102137. doi:10.1016/j.ctrv.2020.102137
57. Dumaz N, Lebbe C. New perspectives on targeting RAF, MEK and ERK in melanoma. Curr Opin Oncol. 2020. doi:10.1097/CCO.0000000000000708
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.