Back to Journals » Journal of Blood Medicine » Volume 12
Management of Sickle Cell Disease Complications Beyond Acute Chest Syndrome
Authors Ogu UO ![]() , Badamosi NU, Camacho PE, Freire AX, Adams-Graves P
, Badamosi NU, Camacho PE, Freire AX, Adams-Graves P
Received 10 November 2020
Accepted for publication 7 February 2021
Published 25 February 2021 Volume 2021:12 Pages 101—114
DOI https://doi.org/10.2147/JBM.S291394
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Ugochi O Ogu,1 Nnenna U Badamosi,2 Pamela E Camacho,3 Amado X Freire,4 Patricia Adams-Graves1
1Center for Sickle Cell Disease, University of Tennessee Health Science Center, Memphis, TN, USA; 2Division of Pediatric Hematology and Oncology, Medical College of Georgia, Augusta, GA, USA; 3Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA; 4Division of Pulmonary, Critical Care, and Sleep Medicine, University of Tennessee Health Science Center, Memphis, TN, USA
Correspondence: Patricia Adams-Graves Tel +1 901-545-8538
Fax +1 901-545-6454
Email [email protected]
Abstract: Sickle cell disease results in numerous complications that can lead to significant morbidity and mortality. Amongst them, acute chest syndrome is the leading cause of mortality. As a result, most providers are in tune with this complication and well versed with management. As sickle cell patients now live longer, they face a multitude of other complications that if left unattended, can lead to significant morbidity and mortality as well. It is critical to look beyond acute chest syndrome and adopt a more comprehensive approach to the management of the sickle cell patient.
Keywords: sickle cell disease, acute chest syndrome, complications
Introduction
Sickle cell disease (SCD), an inheritable blood disorder due to a point mutation in the beta-globin gene resulting in the substitution of valine for glutamic acid at the 6th amino acid, was first described over 100 years ago.1–3 Since then, the complex pathophysiology has been elucidated from simply the red blood cell to a multicellular event to include the blood vessel itself.3–5 This hemoglobin (Hb) gene defect is responsible for serious and life-threatening complications of hemolytic anemia, inflammation, an impaired immunity to encapsulated organisms and vascular occlusion (Figure 1).6 Secondary complications to these include stroke, skin ulceration, priapism, acute and chronic organ damage, and a shortened lifespan.7 SCD complications are elusive due to the underestimation of the impact of social barriers, the inability to measure pain, and the failure to look beyond the underlying pathology during painful events. In fact, 22% of deaths associated with SCD complications are preceded by a painful crisis.8 More specifically, acute chest syndrome (ACS) has been one of the most devastating SCD complications with a high mortality rate if there is a delay in diagnosis or mismanagement across all age groups. As such, there is a heightened awareness of ACS among healthcare providers, specifically those in the acute care/emergency setting. We want to help providers shift beyond ACS to a more comprehensive management approach by highlighting the social barriers to quality of care and the other complex, yet elusive, severe and potentially life-threatening complications of SCD.
 |
Figure 1 Pathophysiology of sickle cell disease. Abbreviations: HbS, sickle hemoglobin; NO, nitric oxide; VCAM, vascular cell-adhesion molecule. Notes: Reprinted from Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031, Copyright © 2010 Elsevier Ltd. Licensed under CC BY-NC-ND.6 |
The management of SCD is finally transforming into a more comprehensive and diverse approach. Blood therapy and pain treatment were the mainstay of support therapy until 1998 when Hydroxyurea (HU) became the only Food and Drug Administration (FDA) approved drug that targeted the molecular pathogenesis.
Elucidation of new pathophysiology has resulted in the evolution of new targeted drugs for SCD management.1,2 There is now a transition from having one FDA approved drug over the past 110 years to a robust new arsenal of targeted drug development. Currently, there are over 30 novel pharmaceutical agents being studied and 3 have recently been FDA approved.1,3,9–11 New targets for drug development in SCD include adhesion, antioxidation, inflammation, fetal Hb induction beyond HU, anti-sickling, anticoagulation, and opiate sparing drugs. In addition, we have expanded our approach from supportive care to curative procedures such as bone marrow transplant, gene therapy, and gene editing.
Until recently, the management of SCD has focused on the pathophysiology of the disease with less emphasis on addressing the complex social hardships which ultimately exists as barriers to quality care and management. Individuals with sickle cell disease navigate their lives through the hardships of associated complications such as acute chest syndrome and painful vaso-occlusive events while also facing a barrage of social barriers to quality care such as the transition from pediatric to adult care and health disparities. Now is the time to recognize that there is a need for a national movement to eliminate these barriers in order to truly ensure that the unmet social and medical needs are being addressed in the sickle cell population. Our goal is to review the management of the systemic complications of SCD beyond acute chest syndrome, pipeline novel agents, and health disparity gaps in order to help the health care system have a more heightened awareness of the underlying and elusive complications that if remain overlooked in the shadows of ACS may cause harm to individuals living with sickle cell disease.
Health Disparities
This is a time in which America is becoming more socially and consciously aware of the apparent unresolved consequences of systemic racism. A recent article in the New England Journal of Medicine (NEJM) highlighted racism and solutions in the US and its effect on SCD.12 In the United States (US), the majority of people with sickle cell disease have a 30-year gap in life expectancy compared to the general population and are of African heritage, while on the other hand, there is a lower incidence in people of Hispanic, South Asian, South European, and Middle Eastern descent who are also affected.7 These statistics play an important factor in the way those living with SCD experience real-life consequences of the health disparities that exist in our healthcare system.
The lack of national awareness, funding for research, therapeutic development, and social services for SCD in comparison to other conditions that are prevalent to a more universal racial demographic such as asthma, heart disease, breast cancer, colon cancer, and diabetes supports systemic racism as an underlying issue.13 In fact, even among rare diseases this is true. The genetically acquired, life-threatening impact that SCD has on African Americans is similar to the impact that Cystic Fibrosis (CF) has on Caucasian Americans. Although SCD is more common than CF as it affects three times more Americans, it has been in its shadows with approximately ten times less funding, and has only one quarter of the number of FDA approved therapeutic agents.14–16 Those living with SCD suffer barriers to quality care as there are profound healthcare gaps of socioeconomic status, racial discrimination, lack of sufficient federal funding, lack of consistent disease-specific access to quality care, and provider discord especially for pain management.17–19 The long-standing history of stereotyping as opioid drug seeking fakers of true pain, mistreatment, neglect, and lack of quality care of individuals living with SCD, without a doubt illuminates how racial biases fundamentally obstruct justice in the form of human rights, the rights to proper healthcare, and quality of life.12
We must work together to put in place realistic policies and training to ensure that racial discrimination within the healthcare system, especially as it relates to the barriers to adequate healthcare for those living with SCD, is considered a reportable adverse event and given as much priority to prevent, as medication or surgical errors are given.20–22 Our hope is to see all healthcare settings transform from a place of injustice, fear, and suffering into a place of holistic disease-specific, patient-centered care that provides safety, hope, and healing for those living with SCD.
Approach to Children and Transition to Adult Care
Central Nervous System (CNS) Complications
In children, cerebral vasculopathy is the most common CNS complication resulting from progressive inflammation and oxidative endothelial damage within intracranial vessels, leading to increased risk of transient ischemic events and infarctive strokes.23,24 Long-term sequelae can range from mild cognitive or behavioral impairment to devastating neurologic effects or death. Screening for vasculopathy beginning at age two is recommended,25 as well as early initiation of disease modifying therapy to mitigate anemia and endothelial damage.
Another less-common complication that can present with acute neurologic symptoms is acute cranial bony infarction associated with intracranial hemorrhage and/or thrombosis. Cranial bony infarcts are believed to result from diploic vascular disruption due to extramedullary hemopoiesis.26,27 In addition, periosteal elevation following an infarctive episode may result in bleeding and hematoma formation. This constellation of complications is uncommon but should remain on the differential even when an acute stroke is suspected, as acute neurosurgical intervention may be required to prevent significant morbidity or mortality.
Cardiovascular Complications
Many children may present with clinical murmurs due to hyperdynamic blood flow and mild to moderate left ventricular hypertrophy. Pulmonary hypertension and cardiomyopathy due to iron overload may not be clinically apparent until adulthood. Screening echocardiograms are not routinely recommended in children unless new symptoms or new clinical findings are noted on exam.28
Aplastic Crisis
Parvovirus –B19 is an erythrotropic virus that selectively targets human red blood cells and their precursors.29 In patients with SCD, this can lead to life-threatening anemia due to abrupt cessation of erythropoiesis. Features are new or worsening signs of anemia, with an acute drop in hemoglobin and reticulocytopenia in the absence of blood loss or sequestration. Urgent recognition and transfusion are necessary to prevent circulatory failure and death.
Splenic Complications
Splenic complications in children can include impaired immunity to encapsulated organisms such as Streptococcus pneumoniae, Neisseria meningitides and Salmonella species, leaving patients prone to life-threatening infections such as pneumonia, meningitis, osteomyelitis and sepsis. As a result, all febrile illnesses are considered medical emergencies until proven otherwise in infants and under-immunized children. Routine childhood vaccines are monitored and usually administered through primary care providers, with the addition of extended pneumococcal and meningococcal vaccines as standard of care. The initiation of prophylactic antibiotics in early infancy has significantly decreased the morbidity and mortality historically associated with these infections.
Acute splenic sequestration of red cells and/or platelets is another potentially fatal complication in children. Auto-splenectomy is less common in children who are initiated on disease-modifying therapy early in life, and therefore this complication may be seen in older children and teenagers. Reviewing splenic palpation and signs of anemia are an important component of anticipatory guidance for parents, with the need for urgent red cell transfusion if signs of circulatory failure.
Gastrointestinal Complications
Vaso-occlusion can occur in intra-abdominal vessels leading to acute abdominal pain as a presenting sign. A thorough evaluation for other causes of acute abdomen may be unrevealing in these patients. These vaso-occlusive episodes are managed appropriately with pain control and hydration.
Sickle hepatopathy is a broad term for a range of hepatic complications from mild liver dysfunction to chronic liver failure and cirrhosis. Acute vaso-occlusive events within hepatic parenchyma may present with fever, jaundice, transaminitis and intense abdominal pain similar to the acute abdominal presentation above.30 Management is supportive with hydration and pain control. Intrahepatic sequestration, or trapping of red cells within the liver, can also lead to intense pain and transaminitis. A large, tender liver may be felt on examination or noted on ultrasound, in addition to an acute drop in hemoglobin. Management is also supportive with transfusion as needed for symptomatic anemia.
Secondary hemochromatosis may be seen in transfused patients with poorly managed transfusional iron overload with occasional transaminitis noted. However, chronic hepatic or cirrhotic changes are an uncommon presentation in childhood.
Other differentials for acute abdominal pain and jaundice with or without fever in SCD are cholelithiasis and/or cholecystitis resulting from pigmented gall stones due to chronic hemolysis leading to obstruction of hepatobiliary bile flow.30 Obstructive gall stones may also lead to intrahepatic cholestasis or extra-hepatic obstruction and acute pancreatitis. Elevated inflammatory markers, transaminases and bilirubin beyond baseline are suggestive of this diagnosis. While a focused abdominal ultrasound may identify inflammation and/or stones, magnetic resonance or endoscopic cholangiopancreatography are more sensitive in making a diagnosis. Urgent evaluations by gastrointestinal and surgical teams will help determine the need for urgent cholecystectomy vs antibiotics and conservative management with endoscopic stone removal.
Peptic ulcers are not uncommon in children with SCD, especially given frequency of non-steroidal anti-inflammatory drug use. Chronic central to upper abdominal pain that may be relieved by antacids or food, may require evaluation and treatment by a gastroenterologist.
Constipation is another frequent complaint in children, usually as a side effect of narcotic pain medication use. An age-appropriate bowel regimen is recommended for use at home and inpatient, although an aggressive bowel cleanout may occasionally be required.
Genitourinary Complications
Sickle nephropathy results from hypoxia and ischemia within renal medullary vasculature, resulting in micro-infarction and papillary necrosis. In addition, intravascular hemolysis produces free hemoglobin which can lead to oxidative damage in renal tubular cells. Nephropathy can vary in presentation from microalbuminuria and proteinuria to significant renal dysfunction and renal failure. Patients at higher risk are those with significant anemia and hemolysis. Annual screening starting at age 10 can identify microalbuminuria as an early sign of nephropathy. The use of angiotensin-converting enzyme (ACE) inhibitors, in addition to SCD-modifying therapy has been shown to minimize progression of nephropathy.28
In male patients, priapism represents an unwanted persistent painful erection that can present at any age. Conservative measures to redirect penile blood flow such as exercise, distraction, or warm baths are recommended as first line. Pseudoephedrine is an α-adrenergic agonist that may be used as needed at home for intermittent episodes. Painful episodes lasting more than 4 hours are considered an urologic emergency.31
Musculoskeletal
Osteonecrosis can occur in anywhere including cranial bones as described above but are frequently found at the ends of long bones in patients with SCD. These may present as persistent localized pain beyond a typical vaso-occlusive episode with or without fever. Magnetic resonance imaging can aid in the diagnosis, although necrotic bone may be difficult to distinguish from osteomyelitis. Conversely, necrotic bone can serve as a nidus for bacterial overgrowth and osteomyelitis. Management is usually conservative with pain medications, unless associated with osteomyelitis or bone abscess requiring debridement.
Avascular necrosis (AVN) is most often located within the femoral head and can be a source of chronic debilitating pain in teenagers and young adults. Management is usually conservative in growing patients, although this complication continues to be a reason for frequent emergency room visits, impaired mobility and diminished quality of life. Long-term pain management is individualized, and often involves a combination of narcotics, non-steroidal anti-inflammatories and other adjunct medications. Unfortunately, optimal pain control may be difficult to achieve in some patients. Conservative surgical techniques such as core decompression and stem cell injection into the joints have had variable results, and many of these patients inevitably require hip-replacement surgery.32
Transition to Adult Care
There are many significant challenges that are unique to the teenage and young adult population as they transition from pediatric to adult care models. Transition in itself is a complication of SCD that requires specific attention and should not be overlooked. It represents yet another example of an important social component that is not only in the shadows of ACS but also exists as a barrier to quality care. Individuals living with SCD are vulnerable to the concept of “falling through the cracks” within the healthcare system, as they transition from pediatric to adult care. Young adults (aged 18–30 years) account for the highest healthcare utilization compared to other age groups (greater than twice the amount of emergency room visits per year, higher inpatient stays and highest frequency of acute care visits).33
Many recurrent childhood complications as described above will persist through the transition years. In addition, early presentations of significant organ damage may start to manifest such as cardiomegaly, pulmonary hypertension, sickle nephropathy and proteinuria, avascular necrosis and chronic pain syndromes. In addition, young adults with a history of strokes in childhood, may present with cognitive deficits that become more notable as the need for health independence and self-efficacy increase. Young patients on chronic transfusions may have difficulty with continuation of transfusion as they transition, if significant iron overload or red cell alloimmunization have developed. Other undesirable outcomes of an unsuccessful transition to adult care include loss of health insurance coverage, limited availability of adult hematologists experienced in SCD, and inability to attend appointments due to transportation issues. The management of SCD beyond ACS must include transition to adult care in order to achieve a truly comprehensive approach to caring for those living with SCD.
Approach to Adults
Sickle cell disease is no longer a disease of childhood, as patients now live well into adulthood. As they progress into adulthood, they are faced with more challenges – increasing rate of comorbidities in the setting of a paucity of skilled and willing adult providers to care for them.34 Acute complications in adulthood include vaso-occlusive crisis (VOC), acute chest syndrome, acute splenic sequestration, aplastic crisis, acute osteomyelitis, cerebrovascular disorders, hepatobiliary issues, acute kidney injury (AKI), priapism, venous thromboembolism (VTE) and multi-system organ failure. Chronic complications include avascular necrosis (AVN), chronic pain, leg ulcers, ocular issues, renal complications and cardiac complications (pulmonary hypertension, diastolic dysfunction). It is important to note that the aging sickle cell population may also experience the typical complications of adulthood and aging (mental health issues, diabetes, gout, hypertension, degenerative joint disease, autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus). These are managed similar to the general population.
Acute chest syndrome is a life-threatening complication in patients suffering from SCD, and the leading cause of mortality. It is usually present at a higher incidence in patients with homozygous SCD, triggers hospital admissions and can have catastrophic consequences.35 Defined by an expert panel as an acute illness characterized by fever and/or respiratory symptoms, accompanied by a new pulmonary infiltrate on chest x-ray, it could also present with multiple infiltrates, hypoxemia conducive to respiratory failure with or without pain syndrome and associated organ failures due to hyper hemolysis, sequestration and/or microvascular occlusions.36 Pneumonia or systemic infections, fat embolism and pulmonary infarction represent the most common mechanisms, with atypical bacterial or viral infections accounting for most cases.37
The standard management for ACS is the use of broad-spectrum antibiotics as well as supportive care such as adequate oxygenation, incentive spirometry, pain control and reduction of abnormal hemoglobin concentration. Decreased concentration can be achieved with transfusions (simple) or with emergent red cell exchange for severe cases. Oxygenation should satisfy patient’s needs and provide symptoms relief with adequate oxygen saturation. Progressive delivery should be started with nasal canula, venturi mask or High Flow Oxygen vs noninvasive ventilation systems. If these options do not achieve desirable effects medical care should proceed to mechanical ventilation with endotracheal tube intubation for support. Patients requiring mechanical ventilation have usually bilateral alveolar infiltrates consistent with pulmonary edema which can be cardiogenic; related to fluid overload consequence of the aggressive hydration to control hemoglobin S concentration; or non-cardiogenic with Acute Respiratory Distress Syndrome (ARDS) physiology secondary to sepsis or Transfusion Associated Lung Injury (TRALI).
Acute pain/VOC is the hallmark of SCD and the most common reason that the sickle cell patient seeks medical attention. Triggers include hypoxia, dehydration, extremes of temperature, acidosis and infection. It is largely managed with pain medications and supportive care.
Pain can also be chronic which is usually multifactorial, ranging from actual tissue/organ damage (due to AVN, leg ulcers) to neuropathic and idiopathic pain. Chronic pain is defined as ongoing pain that is present on most days for over 6 months.38 Opioids are most commonly used. However, use of non-opioid and non-pharmacological modalities such as meditation, massage, acupuncture, etc are garnering more attention.39,40
VTE defined as a deep vein thrombosis or pulmonary embolism (PE), has a high incidence in SCD patients. Up to 12% of patients have a VTE by age 40 years.41 Diagnosis and treatment are as in the general population.
Multisystem organ failure is an acute, fatal complication of SCD, defined as acute decompensation of 2 or more of the following organs: lungs, kidney, liver. It is usually associated with VOC, fever, and an acute drop in Hb and/or platelets. An urgent exchange transfusion is indicated, as well as supportive care such as supplemental oxygen/mechanical ventilation and renal replacement therapy if warranted.
Leg ulcers are an uncommon complication in children and rare under 10 years of age.42,43 However, they pose a significant source of pain and distress in the adult with SCD. Risk factors include hemoglobin SS genotype, severe anemia and increased hemolysis. Leg ulcers usually present initially in the second decade of life and may be present for several years.44 They may arise following a traumatic event to the skin, or spontaneously.45 The most common site is around the medial and lateral malleoli of the ankle. Management is via a multidisciplinary approach which includes wound debridement and dressings, pain management and infection control.44
Ocular manifestations in SCD include proliferative and non-proliferative sickle retinopathy, hyphema, vitreous hemorrhage and central retinal artery occlusion (CRAO). Retinopathy screening is conducted by annual dilated eye exams from age 10 years. Management of proliferative retinopathy involves laser photocoagulation. CRAO is a medical emergency and requires an urgent exchange transfusion.
Splenic complications, aplastic crisis, acute osteomyelitis, acute cerebrovascular events, hepatobiliary issues, renal complications, priapism, AVN, and cardiac complications are covered in detail in a previous section.
Reproductive Health and Pregnancy in Sickle Cell Disease
In men with SCD, reproductive issues include sperm abnormalities, hypogonadism and erectile dysfunction (ED). Up to 91% of males with SCD experience sperm abnormalities manifesting as low sperm count and density, poor motility, and increased abnormal morphology.46 Puberty in general is delayed in SCD. Hypogonadism is present in up to 24% of males with SCD and may manifest as poor testosterone production, infertility, ED and poor libido.47 There remains a debate as to whether it is of primary or secondary etiology. Current therapies include testosterone injections and clomiphene.
ED may result from repeated episodes of priapism. One study demonstrated a 21.4% prevalence of priapism, and 22.2% prevalence of ED within that cohort, 92.3% of the cohort experiencing repeated episodes of priapism.48 Management options include penile implants/prostheses.
In women with SCD, reproductive issues include delayed puberty, choice of contraception and pregnancy-related complications. Puberty in SCD females is delayed by up to 2.4 years compared to the normal population.49 The menstrual cycle is also associated with increase in pain. Choice of contraception remains a source of debate. Due to the theoretical risk of clot predisposition with the estrogen-containing products, providers usually opt for progestin only pills/injectable and IUD. Depo-Provera which also decreases the number of cycles has been helpful for those patients who experience a crisis with their cycles.
Pregnancy in SCD is associated with increased risk of VTE, pre-eclampsia/eclampsia, increased pain crisis, preterm labor, prematurity, intra-uterine growth retardation (IUGR), small for gestational age (SGA) babies and fetal demise.50 Pregnancy in SCD is considered high risk and a multi-disciplinary approach inclusive of a sickle cell expert and a knowledgeable maternal fetal medicine provider is recommended for close monitoring during this delicate period. The decision to prophylactically transfuse or not is usually provider dependent. Management of acute and chronic pain also poses a challenge with pregnancy in SCD. A retrospective study demonstrated that compared to non-SCD pregnant mothers who are on methadone for opioid dependence, neonatal abstinence syndrome (NAS) occurs at a similar rate in SCD pregnant mothers on daily opioids, and at a significantly lower rate in those treated episodically with opioids.51 However, opioids must be used judiciously in pregnancy to mitigate these adverse effects on the fetus/newborn.
Sickle Cell Disease in the Age of COVID-19
Severe acute respiratory syndrome coronavirus 2 (SARS CoV-2), also known as COVID-19 has now affected over 38 million patients worldwide.52 Our knowledge base is rapidly evolving, but this virus poses a significant concern in our patient population. Sickle cell disease is an immunocompromised condition which puts patients at risk of complications from respiratory infections. Our experience of this particular complication is currently limited and a standard of care has not been established; however, there have been several collaborative groups that have aimed to identify patterns and suggest management options for our patients.53,54
Early publications have suggested increased morbidity in SCD patients. A recent French case series described outcomes of eighty-three inpatient individuals.55 The experience reported an ICU admission rate of 20%, fifty-three percent of which required mechanical ventilation, including two patients which required extracorporeal membrane oxygenation, and two patients who died in the ICU with COVID-19 pneumopathy. This underscores the importance of early identification and intervention in our patients. The challenge is that there is significant overlap in presenting signs and symptoms of patients with ACS and COVID-19 infection. Fever, shortness of breath, cough, and myalgias are all overlapping symptoms of ACS, pulmonary embolus, vaso-occlusive crisis, and SARS-CoV-2.
The Medical College of Wisconsin has developed a voluntary international registry of patients with SCD and COVID-19 infection in the hopes to better understand its pathophysiology.54 Interestingly, the most common presenting sign, in the approximately 350 patients registered, is pain, with less than 30% of patients presenting with pneumonia. This highlights the importance of looking beyond ACS and becoming hypervigilant of possible COVID-19 infection in patients without respiratory complaints. Recent case reports also support VOC and fever as the most common reported symptoms in patients with SCD and COVID-19 infection.56 Other respiratory viral infections often trigger “sickle cell crisis”, and COVID-19 appears to have a similar effect. It is imperative that we obtain SARS-CoV-2 PCR testing in any patient with SCD presenting with ACS and/or VOC symptoms.
The lack of large published studies investigating this disease also poses a challenge in the management of known COVID-19 infected patients. A recent review of the literature of nineteen SCD patients with COVID-19 reported from December 2019 to May 2020 described a varied combination of management all similar to those used in the treatment of ACS.57 Their approach included supportive care with hydration, analgesics, empirical broad-spectrum antibiotics, red blood cell exchange, and simple blood transfusions. Oxygen-support ranged from low flow (2 L/min) to high flow, non-invasive and mechanical ventilation in critically ill patients. A single dose of tocilizumab (8 mg/kg) was reported in 2 cases (adult and pediatric) with success.
The American Society of Hematology has provided community resources as guidance for the management of suspected COVID-19 in patients with SCD.58 A useful checklist has been developed in collaboration with ED physicians. Suggested interventions include supplemental oxygen to raise pO2 >94%, judicious fluid replacement and avoidance of fluid bolus as this may exacerbate pulmonary edema, broad-spectrum antibiotics after blood cultures if febrile, macrolide in addition to a third-generation cephalosporin in the event of pneumonia, vancomycin if there is concern for line or skin infection. Any patient with COVID-19 who develops respiratory symptoms should have a type and cross available. Transfusion recommendations include simple transfusions if hemoglobin drops by more than 2 g/dL from baseline, or for patients with respiratory compromise with a goal hemoglobin of approximately 10 g/dL. If the hemoglobin is greater than 10 g/dl admissions should be transferred to an institution where exchange transfusion is available due to the risk of hyperviscosity with further simple transfusions. If patients are discharged home, close follow up is needed with telemedicine or in person visits within 24 hours. Patients should have a low threshold to return to the ED, especially if dyspnea worsens and should be provided with a pulse oximeter if possible.
Further management to consider during the age of COVID-19 includes the use of bronchodilators. The most common comorbidity reported in patients with SCD and COVID-19 is asthma.54 Many institutions have suspended the use of nebulizers due to the risk of aerosolizing virus particles, however metered-dose inhalers should be used as replacement when appropriate. Additional management should also follow institutional standards of care for managing SCD and fever. These may include evaluation for typical seasonal viral infections and empiric oseltamivir, until influenza is ruled out; adequate pulmonary toileting, which includes ambulation as tolerated as well as incentive spirometry. Good pain control is also especially important to reduce atelectasis.
Lastly, it is of particular importance to address COVID-19-associated coagulopathy. Patients with sickle cell disease exhibit a baseline hypercoagulable state and are at an increased risk for venous thrombosis and pulmonary embolism. Some symptomatic patients may benefit from a PE protocol spiral computed tomography (CT) scan during work up. All children and adults hospitalized with SCD and COVID-19 should receive prophylactic anticoagulation dosing or “intermediate intensity” dosing (enoxaparin 0.5mg/kg twice daily), unless the risk of bleeding outweighs the risk of thrombosis. Inpatients should also have close monitoring of disseminated intravascular coagulation (DIC) markers (PT, aPTT, PTT, hepzyme, thrombin time, fibrinogen, D dimer) and inflammatory markers (CRP, ferritin, fibrinogen, IL-6, factor 8) to address thromboembolic risk throughout hospitalization.58
Sickle cell disease is a chronic medical condition requiring a multidisciplinary approach with continued “maintenance care” and constant education. It is of utmost importance that these interactions and collaboration continue during this global pandemic. Telemedicine has become an excellent tool to continue chronic sickle cell maintenance while aiming to avoid exposure. It is important to provide virtual appointments when possible rather than canceling regular maintenance appointments. It is also imperative to educate our patients on the importance of physical distancing, masking, and proper handwashing for the prevention of further spread.
Old and New Therapies
Blood Transfusion
Blood transfusion was the first therapy used in SCD, even at a time when the pathophysiology was still poorly understood.59 Transfusions (simple and exchange transfusions) have remained a critical therapeutic and prophylactic intervention in sickle cell disease to date. Transfusion exerts its effect by dilution of Hb-S containing red blood cells (thereby decreasing the percentage of the abnormal Hb S), and increasing the oxygen carrying capacity of blood.60 Therapeutic blood transfusions have been used for several indications such as: ACS, stroke, acute symptomatic anemia, aplastic crisis, splenic and hepatic sequestration, sickle hepatopathy, central retinal artery occlusion, and multisystem organ failure. Several randomized clinical trials have demonstrated the efficacy of prophylactic/chronic blood transfusions in primary and secondary stroke prevention, pre-operative management, and pregnancy.61–67
In addition to the above indications, providers have also utilized blood transfusions to alleviate other sickle cell disease complications, though not supported by concrete evidence. Before embarking on a chronic blood transfusion regimen with a patient, the potential benefits must be weighed against risks such as iron overload, red cell alloimmunization, transfusion reactions, and blood-borne viral infections.
FDA Approved Drugs
There are currently 4 FDA approved drugs for sickle cell disease, most gaining approval since 2017.
Hydroxyurea
Hydroxyurea, approved in 1998, was the first FDA-approved drug for sickle cell disease. It gained approval based on the randomized clinical trial that demonstrated decreased rates of vaso-occlusive crises, increased median time to first and second crises, and decreased rates of acute chest syndrome.68 Initially approved for adults >18 years with sickle cell anemia, in 2017 it gained approval for children >2 years of age, based on an open-label trial conducted with pediatric patients.69
Hydroxyurea is a ribonucleotide reductase inhibitor that inhibits DNA replication. Its mechanism of action is not completely understood, but it is known to induce fetal hemoglobin (Hb F), which in turn inhibits intracellular Hb S polymerization and prevents sickling within the red blood cells.70 In addition to increased Hb F synthesis and decreased Hb S polymerization, effects include increased Hb synthesis, decreased neutrophil count, hemolysis, RBC membrane damage, endothelial cell activation and adhesion, to mention a few.71
Endari (l-Glutamine)
L-glutamine is a conditionally essential amino acid; during periods of stress and severe illness, the body’s production becomes insufficient. It is required for the synthesis of nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP), and indirectly regulates the metabolism of glutathione. NAD and its reduced form, NADH are involved in maintaining redox balance and sickle red cells have a low redox ratio compared to normal red cells.72 The mechanism of action of l-glutamine in SCD is not fully understood but it has been shown to increase NADH in sickle RBCs, which is believed to reduce oxidative stress.72 L-glutamine gained FDA approval in 2017 based on a randomized trial that demonstrated significantly reduced pain crises and hospitalizations in the drug group.72 It is approved for sickle cell patients of all genotypes, age 5 years and above. This was hailed as the first SCD drug approval in almost 20 years since Hydroxyurea. However, concerns about patient accessibility and adherence have been reported.73
Adakveo (Crizanlizumab)
Crizanlizumab is a monoclonal antibody against the adhesion molecule P-selectin. P-selectin, expressed on the surface of endothelial cells mediates abnormal adhesion of sickle RBCs to the endothelium, and this process is implicated in the painful vaso-occlusion in SCD.74 A randomized trial demonstrated significantly decreased rate of vaso-occlusive crises, and prolonged time to first and second crises on Crizanlizumab.75 This led to its FDA approval in 2019 for sickle cell patients of all genotypes, age 16 years and above.
Oxbryta (Voxelotor)
Voxelotor is a small molecule that binds to the alpha chain of hemoglobin, increasing Hb S affinity for oxygen, delaying Hb S polymerization and preventing RBC sickling.76 In essence, it is an HbS polymerization inhibitor.77 A randomized trial demonstrated significantly higher percentage of participants with a Hb response (>1 g/dl from baseline), fewer instances of worsening anemia and significant reduction in baseline markers of hemolysis with the drug.77 This led to its FDA approval in 2019 for sickle cell patients of all genotypes, age 12 years and above.
Curative Modalities
Bone Marrow Transplant
Bone marrow transplant (BMT) works by replacing the faulty “machinery” in the sickle cell patient: the marrow which produces abnormal sickle RBCs is replaced with a functioning marrow that produces normal RBCs that do not sickle. The first successful BMT as a cure for SCD was serendipitous. A young pediatric patient with both leukemia and sickle cell anemia underwent transplant as treatment for her leukemia, with resultant cure of her sickle cell disease as well.78 This was followed by a quick succession of other institutional reports of their experience with BMT as a cure for SCD.79–83 Outcomes have been impressive with overall and event free survival well above 90% with HLA-matched sibling donors.84,85 Expanded donor pools such as matched unrelated donors and haploidentical donors have been explored, albeit at an increased risk of graft rejection and increased mortality.86
Gene Therapy
Gene therapy portends a potential for cure for SCD but is still in its nascent stages. Viral vectors are employed as vehicles to introduce a therapeutic anti-sickling coding gene (gene addition), to induce Hb F via silencing of repressors of the gamma-globin gene (Hb F induction), or to correct the sickle mutation via genomic engineering tools such as Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 (gene correction).87 The first successful gene therapy for SCD was reported in 2017, using a lentiviral vector-mediated addition of an anti-sickling beta-globin gene into the subject’s hematopoietic stem cells.88 Currently, multiple other clinical trials are in progress to investigate the efficacy of gene therapy in SCD. Anecdotal reports of successful gene therapy have been highlighted.88–90 However, caution must be exercised, because as with every thorough clinical research, the ongoing trials need to be concluded to determine the efficacy of the intervention.
Pipeline Agents
In contrast to a few decades ago, there now exists numerous ongoing clinical trials with novel agents for the treatment of sickle cell disease in the pipeline.91 Table 1 reviews this information. Completed or terminated trials, as well as trials in gene therapy are omitted.
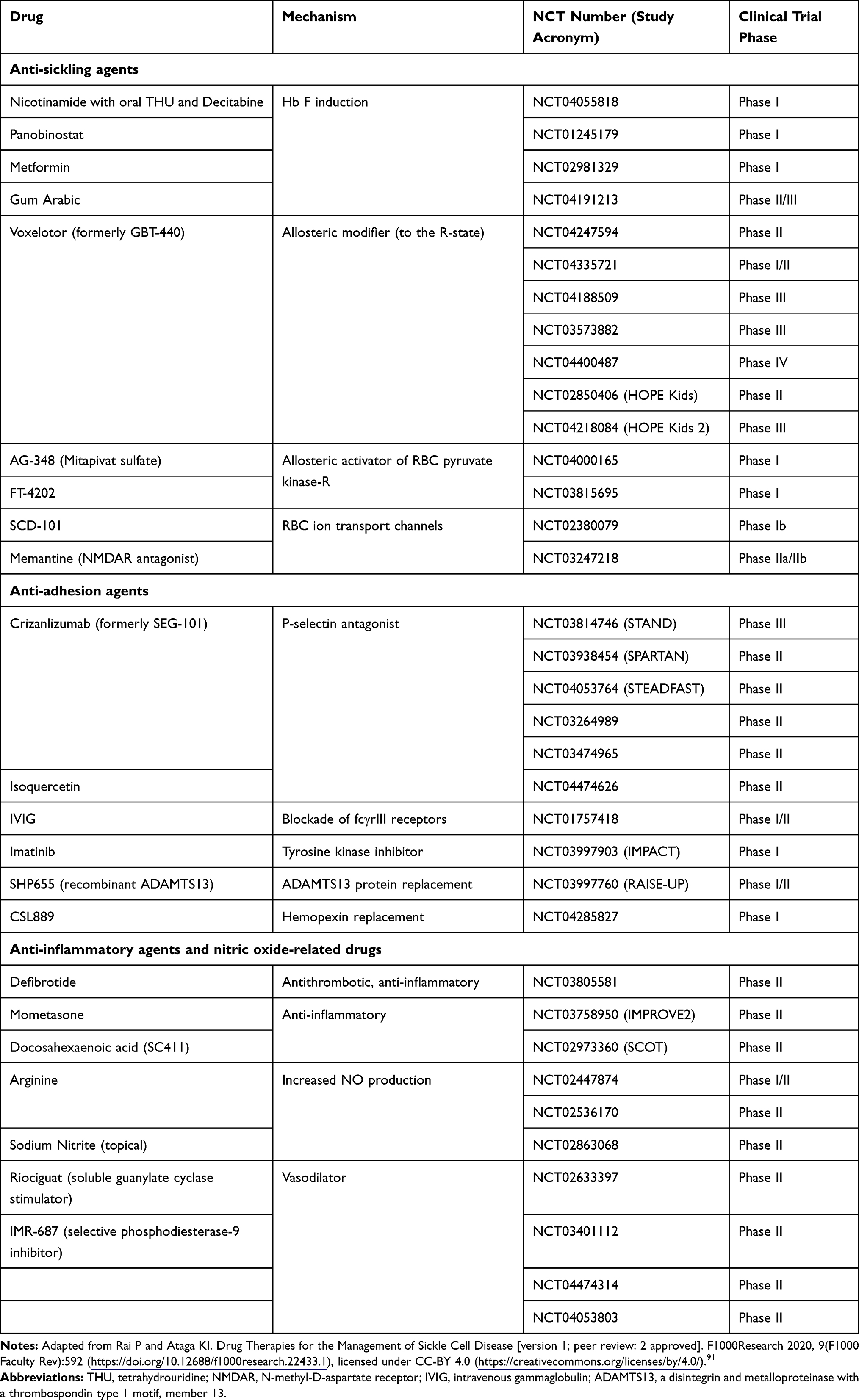 |
Table 1 Ongoing Clinical Trials of Novel Agents in Sickle Cell Disease |
Conclusion
The complications and comorbidities of sickle cell disease are numerous. Acute chest syndrome remains the leading cause of mortality and must be taken keenly. However, various other sickle cell complications deserve attention, in order to ensure the wellbeing of the patient as a whole and improve their quality of life. Other complications that may lead to long-term comorbidities should not be overlooked. Whether it is acute chest syndrome, pain, or a social challenge like reduced quality of care due to racial discrimination, there is a need to look beyond the usual, and shift focus to a more comprehensive approach to the patient.
Multidisciplinary management as with management of leg ulcers or the pregnant sickle cell patient should be emphasized, so as to attain more favorable outcomes. Reproductive counseling can generate a profound impact on the burden of sickle cell disease. Interventions as simple as a routine annual ophthalmologic exam to rule out sickle cell retinopathy can have grave consequences if neglected in a subgroup of patients. Cerebrovascular events should be promptly followed up with intense rehabilitation to preserve residual gross and fine motor functioning. Updated vaccinations, to provide added protection against encapsulated organisms should be ensured. Routine health maintenance as in the general population, such as yearly flu vaccines, colonoscopies, and mammograms should be highly encouraged as well.
It is refreshing that several novel agents are in the pipeline, to deal with the underlying cause of sickle cell disease. Experimental gene therapy also promises a potential cure, if successful. However, true success in the clinical trials realm will entail successful implementation of their significant findings beyond the developed world – in low- and middle-income countries, where most of the world’s sickle cell population reside. As discussed with the complications of sickle cell disease, the race to the cure must be attained on a global level.
Finally, sickle cell disease is a clear example of the fact that systemic racism is an unfortunate truth in American society. Therefore, we as healthcare providers should serve as role models to the rest of the country by dedicating ourselves to eradicating racially motivated healthcare disparities such as inadequate access to healthcare, suboptimal patient care, limited funding for research and therapy development, and poor quality of life in order to truly go beyond to new heights in the management of SCD.
Disclosure
Dr Ugochi O Ogu received Consultancy fees from Vertex Pharmaceuticals, outside the submitted work. Dr Patricia Adams-Graves is Consultant and speaker for Novartis and GBT, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Fernandes Q. Therapeutic strategies in sickle cell anemia: the past present and future. Life Sci. 2017;178:100–108. doi:10.1016/j.lfs.2017.03.025
2. Vekilov PG. Sickle-cell haemoglobin polymerization: is it the primary pathogenic event of sickle-cell anaemia? Br J Haematol. 2007;139(2):173–184. doi:10.1111/j.1365-2141.2007.06794.x
3. Moerdler S, Manwani D. New insights into the pathophysiology and development of novel therapies for sickle cell disease. Hematology Am Soc Hematol Educ Program. 2018;2018(1):493–506. doi:10.1182/asheducation-2018.1.493
4. Piccin A, Murphy C, Eakins E, et al. Insight into the complex pathophysiology of sickle cell anaemia and possible treatment. Eur J Haematol. 2019;102(4):319–330. doi:10.1111/ejh.13212
5. Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016;127(7):801–809. doi:10.1182/blood-2015-09-618538
6. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi:10.1016/S0140-6736(10)61029-X
7. Thein MS, Igbineweka NE, Thein SL. Sickle cell disease in the older adult. Pathology. 2017;49(1):1–9. doi:10.1016/j.pathol.2016.10.002
8. Payne R. Sickle cell anemia and pain: will data prevail over beliefs? Ann Emerg Med. 2009;53(5):596–597. doi:10.1016/j.annemergmed.2008.10.022
9. Shah F, Dwivedi M. Pathophysiology and recent therapeutic insights of sickle cell disease. Ann Hematol. 2020;99(5):925–935. doi:10.1007/s00277-020-03977-9
10. Adebiyi MG, Manalo JM, Xia Y. Metabolomic and molecular insights into sickle cell disease and innovative therapies. Blood Adv. 2019;3(8):1347–1355. doi:10.1182/bloodadvances.2018030619
11. Singh PC, Ballas SK. Emerging drugs for sickle cell anemia. Expert Opin Emerg Drugs. 2015;20(1):47–61. doi:10.1517/14728214.2015.985587
12. Power-Hays A, McGann PT. When actions speak louder than words - racism and sickle cell disease. N Engl J Med. 2020;383:1902–1903. doi:10.1056/NEJMp2022125
13. Bundy DG, Strouse JJ, Casella JF, Miller MR. Urgency of emergency department visits by children with sickle cell disease: a comparison of 3 chronic conditions. Acad Pediatr. 2011;11(4):333–341. doi:10.1016/j.acap.2011.04.006
14. Centers for Disease Control and Prevention. Data and statistics: sickle cell disease; 2019. Available from: https://www.cdc.gov/ncbddd/sicklecell/data.html.
15. National Organization for Rare Disorders. Cystic Fibrosis; 2017. Available from: https://rarediseases.org/rare-diseases/cystic-fibrosis/.
16. Farooq F, Mogayzel PJ, Lanzkron S, Haywood C, Strouse JJ. Comparison of US federal and foundation funding of research for sickle cell disease and cystic fibrosis and factors associated with research productivity. JAMA Netw Open. 2020;3(3):e201737. doi:10.1001/jamanetworkopen.2020.1737
17. Haywood C, Lanzkron S, Bediako S, et al. Perceived discrimination, patient trust, and adherence to medical recommendations among persons with sickle cell disease. J Gen Intern Med. 2014;29(12):1657–1662. doi:10.1007/s11606-014-2986-7
18. Lee L, Smith-Whitley K, Banks S, Puckrein G. Reducing health care disparities in sickle cell disease: a review. Public Health Rep. 2019;134(6):599–607. doi:10.1177/0033354919881438
19. Pulte D, Lovett PB, Axelrod D, Crawford A, McAna J, Powell R. Comparison of emergency department wait times in adults with sickle cell disease versus other painful etiologies. Hemoglobin. 2016;40(5):330–334. doi:10.1080/03630269.2016.1232272
20. Hsu LL, Green NS, Donnell Ivy E, et al. Community health workers as support for sickle cell care. Am J Prev Med. 2016;51(1 Suppl 1):S87–S98. doi:10.1016/j.amepre.2016.01.016
21. Adams-Graves P, Bronte-Jordan L. Recent treatment guidelines for managing adult patients with sickle cell disease: challenges in access to care, social issues, and adherence. Expert Rev Hematol. 2016;9(6):541–552. doi:10.1080/17474086.2016.1180242
22. Whiteman LN, Lanzkron S, Stewart RW, Haywood C, Strouse JJ, Feldman L. Quality improvement process in a sickle cell infusion center. Am J Med. 2015;128(5):541–544. doi:10.1016/j.amjmed.2014.11.020
23. Corvest V, Blais S, Dahmani B, et al. [Cerebral vasculopathy in children with sickle cell disease: key issues and the latest data]. Arch Pediatr. 2018;25(1):63–71. French. doi:10.1016/j.arcped.2017.11.015
24. Kassim AA, Galadanci NA, Pruthi S, DeBaun MR. How I treat and manage strokes in sickle cell disease. Blood. 2015;125(22):3401–3410. doi:10.1182/blood-2014-09-551564
25. Adams RJ. TCD in sickle cell disease: an important and useful test. Pediatr Radiol. 2005;35(3):229–234. doi:10.1007/s00247-005-1409-7
26. Medepalli V, Bajaj S, Bajaj M, Badamosi N Recurrent multifocal calvarial bony infarcts and intracranial hematomas in a patient with sickle cell anemia; 2020. Available from: https://www.eurorad.org/case/16842.
27. Hamm J, Rathore N, Lee P, et al. Cranial epidural hematomas: a case series and literature review of this rare complication associated with sickle cell disease. Pediatr Blood Cancer. 2017;64(3):e26237. doi:10.1002/pbc.26237
28. Liem RI, Lanzkron S, Coates T, et al. American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease. Blood Adv. 2019;3(23):3867–3897. doi:10.1182/bloodadvances.2019000916
29. Wyrick-Glatzel J, Conway-Klaassen J. Clinical utility of the IRF: assessment of erythroid regeneration following parvo B19 infection. Clin Lab Sci. 2002;15(4):208–212.
30. Shah R, Taborda C, Chawla S. Acute and chronic hepatobiliary manifestations of sickle cell disease: a review. World J Gastrointest Pathophysiol. 2017;8(3):108–116. doi:10.4291/wjgp.v8.i3.108
31. Anele UA, Le BV, Resar LMS, Burnett AL. How I treat priapism. Blood. 2015;125(23):3551–3558. doi:10.1182/blood-2014-09-551887
32. Martí‐Carvajal AJ, Solà I, Agreda‐Pérez LH. Treatment for avascular necrosis of bone in people with sickle cell disease. Cochrane Database Syst Rev. 2016;8.
33. Lebensburger JD, Bemrich-Stolz CJ, Howard TH. Barriers in transition from pediatrics to adult medicine in sickle cell anemia. J Blood Med. 2012;3:105–112. doi:10.2147/JBM.S32588
34. Ogu UO, Billett HH. Comorbidities in sickle cell disease: adult providers needed! Indian J Med Res. 2018;147(6):527–529. doi:10.4103/ijmr.IJMR_1019_18
35. Jain S, Bakshi N, Krishnamurti L. Acute chest syndrome in children with sickle cell disease. Pediatr Allergy Immunol Pulmonol. 2017;30(4):191–201. doi:10.1089/ped.2017.0814
36. Ballas SK, Lieff S, Benjamin LJ, et al. Definitions of the phenotypic manifestations of sickle cell disease. Am J Hematol. 2010;85(1):6–13. doi:10.1002/ajh.21550
37. Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med. 2000;342(25):1855–1865. doi:10.1056/NEJM200006223422502
38. Dampier C, Palermo TM, Darbari DS, Hassell K, Smith W, Zempsky W. AAPT diagnostic criteria for chronic sickle cell disease pain. J Pain. 2017;18(5):490–498. doi:10.1016/j.jpain.2016.12.016
39. Edwards LY, Edwards CL. Psychosocial treatments in pain management of sickle cell disease. J Natl Med Assoc. 2010;102(11):1084–1094. doi:10.1016/S0027-9684(15)30737-9
40. Williams H, Tanabe P. Sickle cell disease: a review of nonpharmacological approaches for pain. J Pain Symptom Manage. 2016;51(2):163–177. doi:10.1016/j.jpainsymman.2015.10.017
41. Shet AS, Wun T. How I diagnose and treat venous thromboembolism in sickle cell disease. Blood. 2018;132(17):1761–1769. doi:10.1182/blood-2018-03-822593
42. Olatunya OS, Albuquerque DM, Adekile AD, Costa FF. Evaluation of sociodemographic, clinical, and laboratory markers of sickle leg ulcers among young Nigerians at a tertiary health institution. Niger J Clin Pract. 2018;21(7):882–887. doi:10.4103/njcp.njcp_4_18
43. Serjeant GR, Serjeant BE, Mohan JS, Clare A. Leg ulceration in sickle cell disease: medieval medicine in a modern world. Hematol Oncol Clin North Am. 2005;19(5):943–ix. doi:10.1016/j.hoc.2005.08.005
44. Minniti CP, Kato GJ. Critical reviews: how we treat sickle cell patients with leg ulcers. Am J Hematol. 2016;91(1):22–30. doi:10.1002/ajh.24134
45. Vasconcelos A, Prior AR, Ferrão A, Morais A. An adolescent with sickle cell anaemia experiencing disease-related complications: priapism and leg ulcer–a management challenge. BMJ Case Rep. 2012;2012:bcr1120115146–bcr1120115146. doi:10.1136/bcr.11.2011.5146
46. Smith-Whitley K. Reproductive issues in sickle cell disease. Blood. 2014;124(24):3538–3543. doi:10.1182/blood-2014-07-577619
47. Taddesse A, Woldie IL, Khana P, et al. Hypogonadism in patients with sickle cell disease: central or peripheral? Acta Haematol. 2012;128(2):65–68. doi:10.1159/000337344
48. Madu AJ, Ubesie A, Ocheni S, et al. Priapism in homozygous sickle cell patients: important clinical and laboratory associations. Med Princ Pract. 2014;23(3):259–263. doi:10.1159/000360608
49. Serjeant GR, Singhal A, Hambleton IR. Sickle cell disease and age at menarche in Jamaican girls: observations from a cohort study. Arch Dis Child. 2001;85(5):375–378. doi:10.1136/adc.85.5.375
50. Smith-Whitley K. Complications in pregnant women with sickle cell disease. Hematology Am Soc Hematol Educ Program. 2019;2019(1):359–366. doi:10.1182/hematology.2019000039
51. Nnoli A, Seligman NS, Dysart K, Baxter JK, Ballas SK. Opioid utilization by pregnant women with sickle cell disease and the risk of neonatal abstinence syndrome. J Natl Med Assoc. 2018;110(2):163–168. doi:10.1016/j.jnma.2017.04.002
52. Johns Hopkins University & Medicine. COVID-19 map; 2020. Available from: https://coronavirus.jhu.edu/map.html.
53. Sickle Cell Society. Coronavirus (COVID-19) & sickle cell disorder; 2020. Available from: https://www.sicklecellsociety.org/coronavirus-and-scd/.
54. Medical College of Wisconsin. SECURE-SCD registry; 2020. Available from: https://covidsicklecell.org/updates-data/.
55. Arlet JB, de Luna G, Khimoud D, et al. Prognosis of patients with sickle cell disease and COVID-19: a French experience. Lancet Haematol. 2020;7(9):e632–e634. doi:10.1016/S2352-3026(20)30204-0
56. Hussain FA, Njoku FU, Saraf SL, Molokie RE, Gordeuk VR, Han J. COVID-19 infection in patients with sickle cell disease. Br J Haematol. 2020;189(5):851–852. doi:10.1111/bjh.16734
57. Sahu KK, Siddiqui AD, Cerny J. Managing sickle cell patients with COVID-19 infection: the need to pool our collective experience. Br J Haematol. 2020;190(2):e86–e89. doi:10.1111/bjh.16880
58. American Society of Hematology. COVID-19; 2020. Available from: https://www.hematology.org/covid-19.
59. Baker JP. Sickle Cell Anemia. Trans Am Clin Climatol Assoc. 1941;57:203–212.
60. Howard J. Sickle cell disease: when and how to transfuse. Hematology Am Soc Hematol Educ Program. 2016;2016(1):625–631. doi:10.1182/asheducation-2016.1.625
61. Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339(1):5–11. doi:10.1056/NEJM199807023390102
62. Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005;353(26):2769–2778.
63. Ware RE, Helms RW. Stroke with transfusions changing to hydroxyurea (SWiTCH). Blood. 2012;119(17):3925–3932. doi:10.1182/blood-2011-11-392340
64. DeBaun MR, Gordon M, McKinstry RC, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med. 2014;371(8):699–710. doi:10.1056/NEJMoa1401731
65. Ware RE, Davis BR, Schultz WH, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, Phase 3, non-inferiority trial. Lancet. 2016;387(10019):661–670. doi:10.1016/S0140-6736(15)01041-7
66. Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet. 2013;381(9870):930–938. doi:10.1016/S0140-6736(12)61726-7
67. Koshy M, Burd L, Wallace D, Moawad A, Baron J. Prophylactic red-cell transfusions in pregnant patients with sickle cell disease. A randomized cooperative study. N Engl J Med. 1988;319(22):1447–1452. doi:10.1056/NEJM198812013192204
68. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. 1995;332(20):1317–1322. doi:10.1056/NEJM199505183322001
69. U.S. Food and Drug Administration. FDA approves hydroxyurea for treatment of pediatric patients with sickle cell anemia; 2020. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-hydroxyurea-treatment-pediatric-patients-sickle-cell-anemia.
70. McGann PT, Ware RE. Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf. 2015;14(11):1749–1758. doi:10.1517/14740338.2015.1088827
71. Verma HK, Lakkakula S, Lakkakula BVKS. Retrospection of the effect of hydroxyurea treatment in patients with sickle cell disease. Acta Haematol Pol. 2018;49(1):1. doi:10.2478/ahp-2018-0001
72. Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226–235. doi:10.1056/NEJMoa1715971
73. Ogu UO, Thomas M, Chan F, et al. L-glutamine use in adults with sickle cell disease: clinical trials where success meets reality. Am J Hematol. 2020.
74. Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A, Embury SH. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood. 2001;98(6):1955–1962. doi:10.1182/blood.V98.6.1955
75. Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429–439. doi:10.1056/NEJMoa1611770
76. Oksenberg D, Dufu K, Patel MP, et al. GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br J Haematol. 2016;175(1):141–153. doi:10.1111/bjh.14214
77. Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509–519. doi:10.1056/NEJMoa1903212
78. Johnson FL, Look AT, Gockerman J, Ruggiero MR, Dalla-Pozza L, Billings FT. Bone-marrow transplantation in a patient with sickle-cell anemia. N Engl J Med. 1984;311(12):780–783. doi:10.1056/NEJM198409203111207
79. Vermylen C, Fernandez Robles E, Ninane J, Cornu G. Bone marrow transplantation in five children with sickle cell anaemia. Lancet. 1988;331(8600):1427–1428. doi:10.1016/S0140-6736(88)92239-8
80. Ferster A, De Valck C, Azzi N, Fondu P, Toppet M, Sariban E. Bone marrow transplantation for severe sickle cell anaemia. Br J Haematol. 1992;80(1):102–105. doi:10.1111/j.1365-2141.1992.tb06407.x
81. Vermylen C, Cornu G. Bone marrow transplantation for sickle cell disease. The European experience. Am J Pediatr Hematol Oncol. 1994;16(1):18–21.
82. Johnson FL, Mentzer WC, Kalinyak KA, Sullivan KM, Abboud MR. Bone marrow transplantation for sickle cell disease. The United States experience. Am J Pediatr Hematol Oncol. 1994;16(1):22–26.
83. Walters MC, Patience M, Leisenring W, et al. Bone marrow transplantation for sickle cell disease. N Engl J Med. 1996;335(6):369–376. doi:10.1056/NEJM199608083350601
84. Walters MC, De Castro LM, Sullivan KM, et al. Indications and results of HLA-identical sibling hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2016;22(2):207–211. doi:10.1016/j.bbmt.2015.10.017
85. Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017;129(11):1548–1556. doi:10.1182/blood-2016-10-745711
86. Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010.
87. Demirci S, Uchida N, Tisdale JF. Gene therapy for sickle cell disease: an update. Cytotherapy. 2018;20(7):899–910. doi:10.1016/j.jcyt.2018.04.003
88. Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376(9):848–855. doi:10.1056/NEJMoa1609677
89. Magrin E, Semeraro M, Magnani A, et al. Results from the Completed Hgb-205 Trial of Lentiglobin for Β-Thalassemia and Lentiglobin for Sickle Cell Disease Gene Therapy. Washington, DC: American Society of Hematology; 2019.
90. Kanter J, Tisdale JF, Mapara MY, et al. Resolution of Sickle Cell Disease Manifestations in Patients Treated with Lentiglobin Gene Therapy: Updated Results from the Phase 1/2 Hgb-206 Group C Study. Washington, DC: American Society of Hematology; 2019.
91. Rai P, Ataga KI. Drug Therapies for the Management of Sickle Cell Disease. F1000Res. 2020; 9.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.