Back to Journals » OncoTargets and Therapy » Volume 12
Management of recurrent Ewing sarcoma: challenges and approaches
Authors Van Mater D ![]() , Wagner L
, Wagner L
Received 4 December 2018
Accepted for publication 15 February 2019
Published 27 March 2019 Volume 2019:12 Pages 2279—2288
DOI https://doi.org/10.2147/OTT.S170585
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Carlos E Vigil
David Van Mater, Lars Wagner
Department of Pediatrics, Division of Hematology/Oncology, Duke University, Durham, NC, USA
Abstract: Although many patients with newly diagnosed Ewing sarcoma can become long-term survivors, relapse remains an important clinical problem for which there is no standard approach. Several prognostic factors have been identified, and these may help guide patient counseling and therapy decisions. A variety of chemotherapy regimens have produced responses in patients with recurrent Ewing sarcoma, but no comparative studies have been completed to show superiority of any one particular approach. In addition, the optimum length of therapy for salvage regimens and use of local control measures remains unknown. The likelihood of cure remains low and the gaps in our knowledge are great, and so enrollment on clinical trials should be strongly encouraged for these patients when feasible. Because Ewing sarcoma is relatively rare, some pediatric and adult oncologists may be less familiar with the management of relapsed patients. In this review, we address common questions facing the clinician and patient, and provide an update on new strategies for therapy.
Keywords: relapsed Ewing sarcoma, adolescent and young adult oncology, AYA, irinotecan, topotecan
Introduction
Ewing sarcoma is a high-grade sarcoma arising in bone or soft tissue and occurs most commonly in adolescent and young adult patients. The primary tumor is treated with surgery and/or radiation, depending on the feasibility of resection. Because of the high rate of dissemination even in patients with apparently localized disease, patients are also treated with chemotherapy in an effort to eradicate microscopic disease that could lead to distant metastases and death. Three strategies for initial chemotherapy are most commonly used, depending in part on institutional preferences and patient age. For example, younger patients in North America typically receive the combination of vincristine, doxorubicin, and cyclophosphamide (VDC) alternating with ifosfamide and etoposide (IE), using an intensively timed schedule with planned chemotherapy administration every 2 weeks.1 In Europe, a common approach is to use vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) as the initial chemotherapy regimen.2 For adult patients, some centers have simplified the regimen down to vincristine, ifosfamide, and doxorubicin (VID).3 Therapy is administered for up to 14 total cycles, and the vast majority of patients achieve remission by the completion of treatment.
Despite this extensive therapy, at least one-fourth of patients with initially localized disease will relapse after completing all planned therapy. The recurrence rate is even higher for those with initially metastatic disease, with treatment failure seen in 50%–80% of patients depending on the site of metastases.4 For patients in whom relapse is detected, or for those few who are unable to achieve an initial remission, the chance of long-term survival is low. In addition, there is no standard management for this group of patients, raising many questions about how best to proceed. In this review, we address common management issues and discuss current and upcoming strategies for the treatment of this complex disease.
When and where is Ewing sarcoma most likely to recur?
Over 70% of relapses occur within 2 years of initial diagnosis,5–10 and these patients are designated as having “early relapse”. The median time of recurrence for patients with initially localized disease treated on Children’s Oncology Group protocols was 1.4 years from diagnosis, with a median of 1.0 year for those who presented initially with metastases.5 For patients with late relapses, most occur within 2–3 years from initial diagnosis, although very late relapses even 5 or more years from diagnosis have occasionally been reported.11
At least two-thirds of first relapses occur at distant sites, usually the lungs and/or bones. This pattern of recurrence is particularly common in patients who initially presented with metastatic disease. In contrast, isolated local recurrence occurs in about one-fifth of patients, develops later than systemic relapse, and is more typical in patients who initially had localized tumors.6
Of interest, about one-half of relapsed patients in one large retrospective study were identified by scheduled surveillance imaging, while one-half were symptomatic at the time recurrence was noted, with new pain or swelling being the most common complaints.12
What prognostic factors can facilitate risk stratification and patient counseling?
As a group, only one in five patients with recurrent Ewing sarcoma is expected to achieve long-term survival. The disease-free interval (DFI) between diagnosis and first relapse is the single most important prognostic factor, as patients with a DFI >2 years have an estimated 5-year overall survival of approximately 30%. In contrast, 5-year survival is only 7% for the more common group of patients with DFI <2 years.5,6 In terms of median survival, Shankar et al reported that for a group of 61 relapsed patients, the median survival for DFI <1 year was 3 months, compared with 8 months for those with DFI of 12–24 months, and 24 months for those with DFI >2 years.9
In addition to DFI, the site of recurrence is also prognostic. Patients with combined local and distant relapse have the worst outcomes, while those with isolated local recurrences appear to fare better.5,6 This is understandable given that these patients may have less total burden of disease at recurrence and may be amenable to further local therapies in addition to systemic treatment. More debatable is the prognostic impact of isolated pulmonary recurrence, as some series have reported that these patients did better than those with other distant metastases,6,7,10 while others did not find such an association.5
Additional factors that have been associated with improved outcomes in retrospective cases series include normal levels of lactate dehydrogenase and favorable performance status.13 Younger age has been shown to be associated with improved outcome in some case series,10 but results have been variable.13 Some of this variability may relate to the intensity of the initial treatment regimens in younger versus older patients. Understanding the relevance of key prognostic factors such as DFI and relapse site provides important guidance for clinical trial design and interpretation of results, given the substantial differences in survival between patients with favorable vs unfavorable features. Another factor to be considered is the extent of prior therapies, as relapsed patients will typically experience progressively shorter progression-free periods with subsequent lines of therapy.
The prognostic factors may also help guide patient decisions regarding further therapy. As detailed in the sections below, there are several regimens utilizing commercially available drugs that can result in response and/or disease stabilization for some period of time. However, given the very low likelihood of cure with these regimens, and the desperate need for innovative therapies, strong consideration should be given to enrollment on clinical trials testing new strategies when possible.
Is a tissue diagnosis required to diagnose recurrent Ewing sarcoma?
The most common scenario for relapse is the development of distant metastases in the lungs and/or bones within 2 years of initial diagnosis. In this context, tissue confirmation is not routinely needed. In patients with a solitary site of metastasis, or when the differential diagnosis includes infectious causes of pulmonary nodules, biopsy may be considered. While biopsy/resection may provide definitive diagnosis of relapse as well as tissue for genetic testing for targetable mutations, one drawback is the possible elimination of measurable or evaluable disease that may be required for clinical trial enrollment or assessment of response to therapy. In general, the decision about obtaining a biopsy to confirm relapse should be customized to the individual patient situation.
What is the role of local control measures for relapsed Ewing sarcoma?
For the approximately 15%–20% of patients with isolated local recurrence, further local control measures with either surgery or radiotherapy may improve outcomes and should strongly be considered when feasible.8 In general, chemotherapy is also used for local recurrences, given the systemic nature of relapsed Ewing sarcoma and the expected high likelihood of developing additional sites of recurrence if therapy is directed only to local sites.
The role of radiotherapy for recurrent pulmonary metastases is less clear. Most cooperative group studies prescribe 15–18 Gy of whole lung irradiation (WLI) as planned treatment for newly diagnosed patients who have pulmonary metastases that can be cleared with either chemotherapy and/or surgery. Therefore, many physicians would provide similar treatment in the relapse setting for those patients who can achieve a second remission and who had not yet received WLI. According to a retrospective review of 136 Ewing sarcoma patients with pulmonary relapse, 88 (65%) were able to achieve second remission after chemotherapy and/or surgery.14 The nonrandomized use of WLI (n=44) showed a trend toward improved 3-year progression-free survival (PFS) of 37% vs 21% in this population, but this was not statistically significant (P=0.18).14 Nevertheless, WLI is generally well tolerated and is often utilized for those patients who can achieve second remission. Larger and more focal radiotherapy doses may be used to treat symptomatic and/or unresectable lesions.
Although local control for patients with localized or oligometastatic disease is often pursued, this approach is not usually feasible for those with widespread metastases. For these unfortunate patients, choosing select lesions for local treatment after administration of salvage chemotherapy may be reasonable. Radiotherapy, including stereotactic body radiotherapy, may be very helpful in palliating painful bone lesions that do not improve with salvage chemotherapy. The timing and extent of local treatment is variable between patients and often is coordinated with chemotherapy decisions. In fact, many patients end up with a customized regimen that incorporates their individual risk factors, tumor burden and sites of disease, and preferences.
What conventional salvage therapies are available outside of a clinical trial?
A variety of commercially available chemotherapy regimens have produced responses in patients with recurrent Ewing sarcoma; however, the superiority of one regimen over another has not yet been established. The rEECur study currently being performed through the Euro Ewing Consortium is designed to address this issue through a multiarm randomized study in which patients aged 2–50 years with recurrent Ewing sarcoma may receive one of the four best-established salvage regimens (ISRCTN 36453794). In this study, patients will receive 1) cyclophosphamide/topotecan (Cy/Topo), 2) gemcitabine/docetaxel (Gem/Doc), 3) high-dose ifosfamide (IFOS), or 4) temozolomide/irinotecan (TEM/IRN). Patients on the IFOS arm will receive four planned cycles, while patients on the other three arms will receive a total of six cycles. Patients with a favorable response may receive additional cycles at the discretion of their oncologist. The design of the study includes sequential elimination of the regimens that produce the lowest objective response rate, which is the primary endpoint of the Phase II portion of the study. Once two regimens have emerged, they will be compared in the Phase III portion of the study, which uses PFS as the primary endpoint. This data will be immensely helpful in defining a more standard approach to recurrent Ewing sarcoma.
Each of the regimens in the rEECur study has been described previously, usually in single-institution retrospective analyses of patients with extensive and often very heavily pretreated disease. Table 1 provides additional data on these four regimens and highlights some differences in expected toxicities. Given that for many patients this therapy will not be curative, understanding the toxicity spectrum and details of treatment administration can become important factors in helping adolescent and young adults make decisions regarding therapy. While all four regimens produce some degree of myelosuppression, this is most pronounced in patients receiving IFOS15 or Cy/Topo,16–18 and so prophylactic myeloid growth factor is routinely used in these patients. Nausea is usually manageable for all regimens, although it may be more significant for those receiving IFOS. Patients often ask about alopecia, which is common with all of these regimens but perhaps less with TEM/IRN.13,19–24 Neuropathy has been a problem for some patients receiving GEM/DOC,25–27 while diarrhea and abdominal pain can be limiting for those treated with TEM/IRN.13,19–24 Of the four regimens, all are routinely given as outpatients except for IFOS.
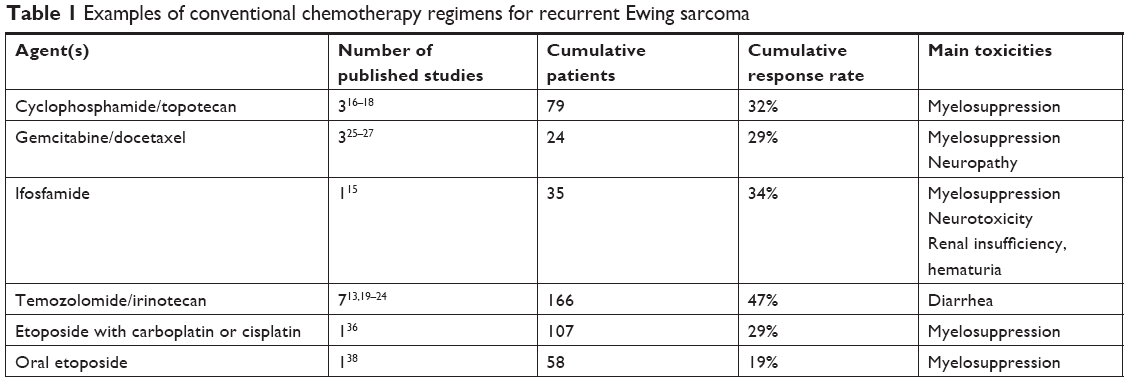 | Table 1 Examples of conventional chemotherapy regimens for recurrent Ewing sarcoma |
TEM/IRN has been the best studied of these four regimens, with a cumulative response rate of 47% reported in seven studies of 166 total patients.13,19–24 Although both IRN and TOPO act on topoisomerase I, the mechanisms of resistance and spectrum of activity appear different, such that progression with one drug does not preclude response to the other.28 The addition of vincristine (VCR) to TEM/IRN has been reported by some authors,22,24,29 in an attempt to exploit the synergy seen between VCR and IRN in rhabdomyosarcoma patients.30 However, that synergy has not been formally assessed in patients with Ewing sarcoma, and the benefit of adding VCR to TEM/IRN for relapsed patients is unclear. VCR is not included in the rEECur study, and this drug may be associated with neuropathy, particularly in older adolescents and young adults. As seen in Table 2, a variety of different schedules and doses have been reported. More recent trials have used a 5-day schedule of irinotecan, based on a rhabdomyosarcoma study which showed that efficacy was similar compared to the more prolonged 2-week schedule.31
 | Table 2 Comparison of studies using temozolomide/irinotecan for relapsed Ewing sarcoma |
There are two other features of the TEM/IRN combination that deserve mention. The first is the possibility for oral administration, in which the intravenous preparation is mixed with cran-grape juice and given orally at least 1 hour after oral administration of temozolomide. Oral administration of irinotecan in this fashion has been studied in multiple trials to date (reviewed in reference32). Importantly, the standard intravenous dose of irinotecan 50 mg/m2/day must be adjusted for poor oral bioavailability. Oral administration of irinotecan 90 mg/m2/day results in exposures of the active metabolite SN-38 similar to those achieved with standard intravenous dosing. Although there have been no direct comparisons between oral and intravenous irinotecan, the similar pharmacokinetics, incidence of grade 3–4 toxicities, and response rate of several tumor types suggest that these dosing strategies are roughly equivalent. Oral administration may reduce the cost of treatment by fivefold, as well as substantially improve patient convenience.32 For these reasons, several ongoing trials are now utilizing orally administered irinotecan, with the addition of prophylactic cephalosporins to reduce irinotecan-associated diarrhea.33
Second, because TEM/IRN generally is the least myelosuppressive of these four regimens, it has been more commonly used as a backbone on which to add investigational agents. Given that single-agent targeted therapy is unlikely to be curative, coupling novel drugs with a standard well-tolerated backbone is attractive and would likely reflect how newer agents would eventually be used for upfront therapy. TEM/IRN has been used in this fashion in recently completed studies combining this regimen with bevacizumab34 and temsirolimus,35 as well as ongoing clinical trials adding in metformin [NCT01528046], palbociclib [NCT03709680], PARP inhibitors [NCT01858168, NCT02044120], and immunotherapy [NCT03495921].
Finally, other regimens have also been reported to have activity in relapsed Ewing sarcoma. For example, the combination of intravenous etoposide with either cisplatin or carboplatin showed an encouraging response rate and PFS in a retrospective review of patients mostly in first relapse from five European sarcoma centers,36 consistent with an earlier report about the use of ifosfamide, carboplatin, and etoposide (ICE).37 Less intensive options include the use of oral etoposide given on a protracted metronomic schedule.38
The optimal number of chemotherapy cycles for relapsed patients is not established and depends in part on response as well as tolerance of therapy. Many physicians try to administer at least six to eight cycles of therapy in responding patients, as suggested by the rEECur study design in which most patients receive six planned cycles of therapy. However, the length of treatment often needs to be customized to the patient. Cumulative toxicity of therapy, both physical and psychological, can limit continued therapy in some adolescent and young adult patients, especially given that salvage therapy is often not curative and treatment decisions must factor in quality of life considerations.
Which clinical trials may be the most well suited for recurrent Ewing sarcoma patients?
Clinical trials of new strategies for treating patients with recurrent Ewing sarcoma will be essential for improving outcomes. Available clinical trials can be divided into three general categories, including 1) general cytotoxic chemotherapy, 2) agents targeting pathways specific to tumor cells, and 3) immunotherapy. Examples of each group are discussed below; however, this list is not exhaustive.
General cytotoxic chemotherapy
The microtubule inhibitor eribulin has received regulatory approval for adult soft tissue sarcoma and has shown preclinical activity against mouse models of Ewing sarcoma,39 as well as a response in the recently completed pediatric Phase I trial.40 Ewing sarcoma is one of the target tumors included in an ongoing single-agent Phase II trial of eribulin [NCT03441360], as well as in combination with irinotecan [NCT03245450]. New formulations of older cytotoxic agents are also being studied for Ewing sarcoma, including the nanoparticle albumin-bound formulation of paclitaxel (nab-paclitaxel) either alone41 or in combination with gemcitabine [NCT02945800], based on preclinical reports of additive activity of these agents42 as well as past responses seen with gemcitabine/taxane combinations.26 Finally, a nanoliposomal preparation of irinotecan (MM-398; Onivyde®, Ipsen Biopharmaceuticals, Inc., Cambridge, MA, USA) may yield higher plasma and tissue concentrations of SN-38 and has shown in vivo activity against Ewing sarcoma xenografts.43 This drug is now being studied in combination with cyclophosphamide [NCT02013336].
Targeted therapy
A variety of targeted therapies have been developed in the past decade, and they represent a rational and specific approach to treating relapsed Ewing sarcoma. The insulin growth factor receptor 1 (IGF-1R) is highly expressed on Ewing sarcoma tumor cells and appears to drive tumor growth.44 Targeting IGF-1R with monoclonal antibodies produced convincing and durable responses in early trials of Ewing sarcoma patients.45,46 However, larger studies failed to show consistently high response rates,47,48 even when combined with an mTOR inhibitor.49–51 Disappointingly, the inability to identify biomarkers predictive of the small subset of responding patients has severely limited the use of these agents, as IGF-1R expression alone is not correlated with clinical benefit. The IGF-1R antibody ganitumab is currently being studied in combination with conventional chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma, and it is hoped that improved activity and predictive biomarkers may be identified [NCT02306161]. A second target for antibody-directed therapy is placenta growth factor (PGF), which has been implicated in the invasiveness and metastatic potential of Ewing sarcoma.52 A clinical trial is underway which targets PGF with the monoclonal antibody TB-403 [NCT02748135].53
Targeting important tumor pathways with receptor tyrosine kinase inhibitors is another attractive strategy. Many such agents are now commercially available, and the oral route of administration is convenient for patients. Some modest success has been seen with the VEGFR inhibitor regorafenib, with a response rate of 10% in heavily pretreated adult patients (median five prior regimens, median age 32 years) with recurrent Ewing sarcoma.54 The primary endpoint was 8-week PFS, with the observed rate of 73% exceeding the defined bar for activity of 25%. Similarly, a response rate of 28% and 6-month PFS of 24% have been described in relapsed Ewing sarcoma patients treated with cabozantinib,55 an inhibitor of the MET protein which is expressed in 62% of Ewing tumors.56 These drugs generally have more toxicity than monoclonal antibodies, with many patients requiring dose reductions when multiple cycles are given. As seen with IGF-1R antibodies, the identification of predictive biomarkers will be essential for more successful use of these agents.
Another focus for targeted therapy has been the DNA repair protein PARP1. Expression of this enzyme is elevated in Ewing tumors, and there is a positive feedback loop with the EWS-FLI1 fusion transcript.57 Although mechanistically attractive, the clinical experience of PARP inhibitors as single agents has been disappointing.58 Current trials now combine PARP inhibitors with DNA damaging agents such as temozolomide and/or irinotecan, based on preclinical demonstration of synergy.59 Results from these studies are eagerly awaited, although the doses of conventional chemotherapy may need to be substantially reduced in order to make combination regimens feasible.
Other strategies include specifically focusing on the EWS-FLI1 translocation that characterizes this disease and drives tumor growth. Although transcription factors such as the fusion product produced by this translocation have been notoriously difficult to target, Zollner et al have identified an inhibitor to the RNA helicase A which binds to EWS-FLI1 and disrupts its protein interactions, leading to activity against Ewing xenografts.60 TK216 is now in a Phase I clinical trial, although administration requires a 7-day continuous infusion because of its short half-life [NCT02657005]. BRD4 is another factor required for the EWS-FLI1 fusion protein to function.61 BRD4 is a member of the bromodomain and extraterminal domain (BET) family of proteins that act as “readers” of chromatin to regulate gene expression. Recently, bromodomain inhibitors have been shown to negatively impact gene expression mediated by the EWS fusion protein, and the BET family of proteins represents a potential vulnerability that can be exploited by BET inhibitors as monotherapy or (more likely) in combination with other agents.61–65 Clinical trials of bromodomain inhibitors are now open [NCT02419417, NCT03220347].
Another rational strategy is inhibition of lysine-specific demethylase 1 (LSD-1), which is highly expressed in Ewing sarcoma and represses the transcriptional activation of downstream targets of EWS/FLI1 that lead to tumor growth.66 Two LSD-1 inhibitors are now entering clinical trials [NCT03514407, NCT03600649]. The above studies are particularly exciting because they represent efforts to directly target the specific underlying molecular biology of Ewing sarcoma. A summary of several targeted therapies currently in clinical trials is provided in Table 3.
 | Table 3 Selected studies of targeted therapies for Ewing sarcoma |
Immunotherapy
Previous studies in adult malignancies have suggested that widespread expression of programed death ligand-1 (PD-L1) in malignant cells, high mutational tumor burden, and extensive infiltration of tumors with CD8+ T cells were all associated with responses to immune checkpoint blockade.67 Although results have varied between studies, Machado et al identified PD-L1 expression in tumor cells in 19% of Ewing sarcoma samples in the largest series to date.68 However, Ewing sarcoma has a low mutational tumor burden when compared to carcinomas or melanoma.69–72 Further, Ewing sarcomas typically have only a low level of infiltrating T cells, identified in only 15% of tumor samples in the Machado series.68 These laboratory findings suggest that Ewing sarcoma would be a relatively “cold” tumor in terms of responding to immunotherapy, and indeed two cooperative group clinical trials reported to date are consistent with this impression. Specifically, no responses have been reported in 13 patients treated with the anti-PD-1 antibody pembrolizumab73 or in ten patients treated with the anti-PD-1 antibody nivolumab.74 While it is hoped that combination immunotherapy regimens now under investigation will make sarcomas more “hot” and therefore responsive,75 at present there is little evidence that checkpoint inhibitors should routinely be used as monotherapy for relapsed Ewing sarcoma.
An important caveat is the use of an innovative immunotherapy approach for Ewing sarcoma called Vigil.76,77 This novel strategy involves administration of a vaccine that comprised autologous tumor cells to provide patient-specific tumor antigens to provoke an anti-tumor response. These cells are transfected with the rhGMCSF transgene and the RNAibi-shRNAfurin in order to recruit and activate dendritic cells while reducing local immune tolerance through the blockade of furin-mediated activation of endogenous TGF-beta1 and 2. An early trial reported a 1-year survival of 73% for patients with relapsed Ewing sarcoma treated with Vigil compared to 23% of historical controls treated with conventional chemotherapy.78 Given the low toxicity of Vigil, a randomized Phase III trial is now underway that combines Vigil with TEM/IRN and compares this group with patients treated with TEM/IRN alone [NCT03495921]. This trial represents one of the few Phase III studies for relapsed Ewing sarcoma, and hopefully accrual of patients from both pediatric and adult sarcoma centers will allow for meaningful conclusions.
Should relapsed patients be treated with high-dose chemotherapy and autologous stem cell transplantation?
Several retrospective studies have suggested that treatment with myeloablative doses of alkylators followed by autologous stem cell transplantation may improve outcomes in patients with recurrent Ewing sarcoma.79–81 Most patients in these studies first received conventional-dose chemotherapy, which was then followed by high-dose busulfan and melphalan. To date, no prospective randomized studies have been performed, and the difficulty in identifying a suitable control population has made interpretation of results more complicated. For example, patients who receive high-dose chemotherapy generally have recurrent tumor that is responsive to typical salvage therapy. At best, this describes only about half of relapsed patients,80 with the majority being those with initially localized disease who often have longer survival than relapsed patients who had metastases at diagnosis. In addition, patients must remain progression-free until high-dose chemotherapy is administered, must have adequate stem cells collected, and must have no comorbidities or organ dysfunction that would preclude such intensive treatment. Finally, patients must be willing to undergo this intensive treatment, which is not always the case given that their prognosis still remains unfavorable, despite the prolonged therapy they have already received.
Taken together, the data would suggest that certain patients with favorable features at relapse may possibly have prolonged PFS with high-dose therapy. In a different clinical context, newly diagnosed patients with localized higher-risk tumors were randomized to receive either myeloablative busulfan/melpahalan with autologous stem cells or continuation of standard chemotherapy.82 Although patients receiving high-dose therapy had superior outcomes, it should be emphasized that the treatment backbone used in European studies of newly diagnosed patients differs from that used in North America,83 and the implications of this study for relapsed patients are unclear. Moreover, a study incorporating high-dose chemotherapy in patients with responsive recurrent disease was not able to show it to be an independent variable influencing post-relapse survival.10 The analysis was complicated by the fact that high-dose chemotherapy was reserved for patients achieving a complete or partial response to IFOS, no prior history of myeloablative therapy, and willingness to undergo high-intensity treatment.10 As such, only 20/107 evaluable patients underwent treatment.10 This study illustrates some of the complexities of designing a clinical trial to rigorously demonstrate a role for high-dose therapy and autologous stem cell transplantation in the relapse setting. It is clear that this strategy is not well suited for all relapsed patients, and it remains an intensive therapy for which the unequivocal benefits have not yet been established.
What is the role of genetic testing of tumor tissue for actionable mutations in patients with recurrent Ewing sarcoma?
Apart from the characteristic EWSR1 translocations that characterize this tumor, recurring genetic changes in Ewing sarcoma are relatively infrequent. The most common mutations occur in genes such as STAG2, CDKN2A, and TP53, which have not been easily targetable.69–71 Although potentially actionable mutations have been reported with Ewing sarcoma, these are uncommon.84 Therefore, the likelihood of identifying a molecular change that will drive effective therapy is low but not zero. Ongoing translational studies that prospectively collect data on patients with relapsed Ewing sarcoma will be helpful to further characterize the genetic changes that may occur in these patients, especially given the potential of new targeted agents being developed. However, extensive molecular testing outside the context of a clinical trial is expensive, and decisions regarding genetic analysis should be individualized given that the cost/benefit relationship of such testing has not been clarified.
Conclusion
The outcome for patients with recurrent Ewing sarcoma remains poor, and the standard approach to their management has not yet been established. Many patients may initially benefit from salvage chemotherapy in terms of reducing symptoms and prolonging time to further progression, but consistent cures remain elusive. Knowledge of prognostic factors that affect survival of these patients may help guide therapy decisions. Enrollment on clinical trials should strongly be considered when feasible, as a variety of mechanistically novel Phase I to Phase III studies are currently underway and represent the best way to better understand which treatments may be beneficial in the future. In this regard, close cooperation between pediatric and adult oncology centers, as well as between continents, will help drive advances for this rare disease of adolescence and young adulthood.
Disclosure
The authors report no conflicts of interest in this work.
References
Womer RB, West DC, Krailo MD, et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(33):4148–4154. doi:10.1200/JCO.2011.41.5703 | ||
Juergens C, Weston C, Lewis I, et al. Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer. 2006;47(1):22–29. doi:10.1002/pbc.20820 | ||
Wagner MJ, Gopalakrishnan V, Ravi V, et al. Vincristine, ifosfamide, and doxorubicin for initial treatment of Ewing sarcoma in adults. Oncologist. 2017;22(10):1271–1277. doi:10.1634/theoncologist.2016-0464 | ||
Gaspar N, Hawkins DS, Dirksen U, et al. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol. 2015;33(27):3036–3046. doi:10.1200/JCO.2014.59.5256 | ||
Leavey PJ, Mascarenhas L, Marina N, et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2008;51(3):334–338. doi:10.1002/pbc.21618 | ||
Stahl M, Ranft A, Paulussen M, et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer. 2011;57(4):549–553. doi:10.1002/pbc.23040 | ||
Bacci G, Longhi A, Ferrari S, et al. Pattern of relapse in 290 patients with nonmetastatic Ewing’s sarcoma family tumors treated at a single institution with adjuvant and neoadjuvant chemotherapy between 1972 and 1999. Eur J Surg Oncol. 2006;32(9):974–979. doi:10.1016/j.ejso.2006.01.023 | ||
Rodriguez-Galindo C, Billups CA, Kun LE, et al. Survival after recurrence of Ewing tumors: the St Jude Children’s Research Hospital experience, 1979–1999. Cancer. 2002;94(2):561–569. doi:10.1002/cncr.10192 | ||
Shankar AG, Ashley S, Craft AW, Pinkerton CR. Outcome after relapse in an unselected cohort of children and adolescents with Ewing sarcoma. Med Pediatr Oncol. 2003;40(3):141–147. doi:10.1002/mpo.10248 | ||
Ferrari S, Luksch R, Hall KS, et al. Post-relapse survival in patients with Ewing sarcoma. Pediatr Blood Cancer. 2015;62(6):994–999. doi:10.1002/pbc.25388 | ||
Wasilewski-Masker K, Liu Q, Yasui Y, et al. Late recurrence in pediatric cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2009;101(24):1709–1720. doi:10.1093/jnci/djp417 | ||
Heinemann M, Ranft A, Langer T, et al. Recurrence of Ewing sarcoma: is detection by imaging follow-up protocol associated with survival advantage? Pediatr Blood Cancer. 2018;65(7):e27011. doi:10.1002/pbc.27011 | ||
Palmerini E, Jones RL, Setola E, et al. Irinotecan and temozolomide in recurrent Ewing sarcoma: an analysis in 51 adult and pediatric patients. Acta Oncol. 2018;57(7):958–964. doi: 10.1080/0284186X.2018.1449250. | ||
Scobioala S, Ranft A, Wolters H, et al. Impact of whole lung irradiation on survival outcome in patients with lung relapsed Ewing sarcoma. Int J Radiat Oncol Biol Phys. 2018;102(3):584–592. doi:10.1016/j.ijrobp.2018.06.032 | ||
Ferrari S, Del Prever AB, Palmerini E, et al. Response to high-dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer. 2009;52(5):581–584. doi:10.1002/pbc.21917 | ||
Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jurgens H. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer. 2006;47(6):795–800. doi:10.1002/pbc.20719 | ||
Saylors RL 3rd, Stine KC, Sullivan J, et al. Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol. 2001;19(15):3463–3469. doi:10.1200/JCO.2001.19.15.3463 | ||
Farhat R, Raad R, Khoury NJ, et al. Cyclophosphamide and topotecan as first-line salvage therapy in patients with relapsed ewing sarcoma at a single institution. J Pediatr Hematol Oncol. 2013;35(5):356–360. doi:10.1097/MPH.0b013e318270a343 | ||
Wagner LM, McAllister N, Goldsby RE, et al. Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer. 2007;48(2):132–139. doi:10.1002/pbc.20697 | ||
Casey DA, Wexler LH, Merchant MS, et al. Irinotecan and temozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatr Blood Cancer. 2009;53(6):1029–1034. doi:10.1002/pbc.22206 | ||
Anderson P, Kopp L, Anderson N, et al. Novel bone cancer drugs: investigational agents and control paradigms for primary bone sarcomas (Ewing’s sarcoma and osteosarcoma). Expert Opin Investig Drugs. 2008;17(11):1703–1715. doi:10.1517/13543784.17.11.1703 | ||
Raciborska A, Bilska K, Drabko K, et al. Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Pediatr Blood Cancer. 2013;60(10):1621–1625. doi:10.1002/pbc.24621 | ||
Kurucu N, Sari N, Ilhan IE. Irinotecan and temozolamide treatment for relapsed Ewing sarcoma: a single-center experience and review of the literature. Pediatr Hematol Oncol. 2015;32(1):50–59. doi:10.3109/08880018.2014.954070 | ||
Buyukkapu Bay S, Kebudi R, Gorgun O, Zulfikar B, Darendeliler E, Cakir FB. Vincristine, irinotecan, and temozolomide treatment for refractory/relapsed pediatric solid tumors: a single center experience. J Oncol Pharm Pract. 2018:1078155218790798. Epub 2018 Aug 6. | ||
Fox E, Patel S, Wathen JK, et al. Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or locally recurrent chondrosarcoma: results of Sarcoma Alliance for Research Through Collaboration Study 003. Oncologist. 2012;17(3):321. doi:10.1634/theoncologist.2010-0265 | ||
Mora J, Cruz CO, Parareda A, de Torres C. Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. J Pediatr Hematol Oncol. 2009;31(10):723–729. doi:10.1097/MPH.0b013e3181b2598c | ||
Tanaka K, Joyama S, Chuman H, et al. Feasibility and efficacy of gemcitabine and docetaxel combination chemotherapy for bone and soft tissue sarcomas: multi-institutional retrospective analysis of 134 patients. World J Surg Oncol. 2016;14(1):306. doi:10.1186/s12957-016-1059-2 | ||
Wagner L. Camptothecin-based regimens for treatment of ewing sarcoma: past studies and future directions. Sarcoma. 2011;2011:957957. doi:10.1155/2011/957957 | ||
Wagner LM, Perentesis JP, Reid JM, et al. Phase I trial of two schedules of vincristine, oral irinotecan, and temozolomide (VOIT) for children with relapsed or refractory solid tumors: a Children’s Oncology Group phase I consortium study. Pediatr Blood Cancer. 2010;54(4):538–545. doi:10.1002/pbc.22407 | ||
Pappo AS, Lyden E, Breitfeld P, et al. Two consecutive phase II window trials of irinotecan alone or in combination with vincristine for the treatment of metastatic rhabdomyosarcoma: the Children’s Oncology Group. J Clin Oncol. 2007;25(4):362–369. doi:10.1200/JCO.2006.07.1720 | ||
Mascarenhas L, Lyden ER, Breitfeld PP, et al. Randomized phase II window trial of two schedules of irinotecan with vincristine in patients with first relapse or progression of rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2010;28(30):4658–4663. doi:10.1200/JCO.2010.29.7390 | ||
Wagner LM. Oral irinotecan for treatment of pediatric solid tumors: ready for prime time? Pediatr Blood Cancer. 2010;54(5):661–662. doi:10.1002/pbc.22410 | ||
Wagner LM, Crews KR, Stewart CF, et al. Reducing irinotecan-associated diarrhea in children. Pediatr Blood Cancer. 2008;50(2):201–207. doi:10.1002/pbc.21280 | ||
Wagner L, Turpin B, Nagarajan R, Weiss B, Cripe T, Geller J. Pilot study of vincristine, oral irinotecan, and temozolomide (VOIT regimen) combined with bevacizumab in pediatric patients with recurrent solid tumors or brain tumors. Pediatr Blood Cancer. 2013;60(9):1447–1451. doi:10.1002/pbc.24547 | ||
Bagatell R, Norris R, Ingle AM, et al. Phase 1 trial of temsirolimus in combination with irinotecan and temozolomide in children, adolescents and young adults with relapsed or refractory solid tumors: a Children’s Oncology Group Study. Pediatr Blood Cancer. 2014;61(5):833–839. doi:10.1002/pbc.24874 | ||
van Maldegem AM, Benson C, Rutkowski P, et al. Etoposide and carbo-or cisplatin combination therapy in refractory or relapsed Ewing sarcoma: a large retrospective study. Pediatr Blood Cancer. 2015;62(1):40–44. doi:10.1002/pbc.25230 | ||
Van Winkle P, Angiolillo A, Krailo M, et al. Ifosfamide, carboplatin, and etoposide (ICE) reinduction chemotherapy in a large cohort of children and adolescents with recurrent/refractory sarcoma: the Children’s Cancer Group (CCG) experience. Pediatr Blood Cancer. 2005;44(4):338–347. doi:10.1002/pbc.20227 | ||
Podda MG, Luksch R, Puma N, et al. Oral etoposide in relapsed or refractory Ewing sarcoma: a monoinstitutional experience in children and adolescents. Tumori. 2016;102(1):84–88. doi:10.5301/tj.5000419 | ||
Kolb EA, Gorlick R, Reynolds CP, et al. Initial testing (stage 1) of eribulin, a novel tubulin binding agent, by the pediatric preclinical testing program. Pediatr Blood Cancer. 2013;60(8):1325–1332. doi:10.1002/pbc.24517 | ||
Schafer ES, Rau RE, Berg S, et al. A phase 1 study of eribulin mesylate (E7389), a novel microtubule-targeting chemotherapeutic agent, in children with refractory or recurrent solid tumors: a Children’s Oncology Group Phase 1 Consortium study (ADVL1314). Pediatr Blood Cancer. 2018;65(8):e27066. doi:10.1002/pbc.27066 | ||
Moreno L, Casanova M, Chisholm JC, et al. Phase I results of a phase I/II study of weekly nab-paclitaxel in paediatric patients with recurrent/refractory solid tumours: a collaboration with innovative therapies for children with cancer. Eur J Cancer. 2018;100:27–34. doi:10.1016/j.ejca.2018.05.002 | ||
Wagner LM, Yin H, Eaves D, Currier M, Cripe TP. Preclinical evaluation of nanoparticle albumin-bound paclitaxel for treatment of pediatric bone sarcoma. Pediatr Blood Cancer. 2014;61(11):2096–2098. doi:10.1002/pbc.25062 | ||
Kang MH, Wang J, Makena MR, et al. Activity of MM-398, nanoliposomal irinotecan (nal-IRI), in Ewing’s family tumor xenografts is associated with high exposure of tumor to drug and high SLFN11 expression. Clin Cancer Res. 2015;21(5):1139–1150. doi:10.1158/1078-0432.CCR-14-1882 | ||
Kolb EA, Gorlick R. Development of IGF-IR inhibitors in pediatric sarcomas. Curr Oncol Rep. 2009;11(4):307–313. | ||
Pappo AS, Patel SR, Crowley J, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research Through Collaboration Study. J Clin Oncol. 2011;29(34):4541–4547. doi:10.1200/JCO.2010.34.0000 | ||
Juergens H, Daw NC, Geoerger B, et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol. 2011;29(34):4534–4540. doi:10.1200/JCO.2010.33.0670 | ||
Malempati S, Weigel B, Ingle AM, et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(3):256–262. doi:10.1200/JCO.2011.37.4355 | ||
Tap WD, Demetri G, Barnette P, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol. 2012;30(15):1849–1856. doi:10.1200/JCO.2011.37.2359 | ||
Naing A, LoRusso P, Fu S, et al. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumors. Clin Cancer Res. 2012;18(9):2625–2631. doi:10.1158/1078-0432.CCR-12-0061 | ||
Schwartz GK, Tap WD, Qin LX, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013;14(4):371–382. doi:10.1016/S1470-2045(13)70049-4 | ||
Wagner LM, Fouladi M, Ahmed A, et al. Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2015;62(3):440–444. doi:10.1002/pbc.25334 | ||
Richter GH, Fasan A, Hauer K, et al. G-Protein coupled receptor 64 promotes invasiveness and metastasis in Ewing sarcomas through PGF and MMP1. J Pathol. 2013;230(1):70–81. doi:10.1002/path.4170 | ||
Nielsen DL, Sengelov L. Inhibition of placenta growth factor with TB-403: a novel antiangiogenic cancer therapy. Expert Opin Biol Ther. 2012;12(6):795–804. doi:10.1517/14712598.2012.679655 | ||
Attia S, Bolejack V, Ganjoo KN, et al. A phase II trial of regorafenib (REGO) in patients (pts) with advanced Ewing sarcoma and related tumors (EWS) of soft tissue and bone: SARC024 trial results [abstract]. J Clin Oncol. 2017;35:11005. doi:10.1200/JCO.2017.35.15_suppl.11005 | ||
Italiano A, Penel N, Toulmonde M, et al. Cabozantinib in patients with advanced osteosarcomas and Ewing sarcomas: a French Sarcoma Group (FSG)/US National Cancer Institute phase II collaborative study. Poster presented at: European Society for Medical Oncology; October 19–23; 2018; Munich, Germany. | ||
Fleuren ED, Roeffen MH, Leenders WP, et al. Expression and clinical relevance of MET and ALK in Ewing sarcomas. Int J Cancer. 2013;133(2):427–436. doi:10.1002/ijc.28047 | ||
van Maldegem AM, Bovee JV, Peterse EF, Hogendoorn PC, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171–180. doi:10.1016/j.ejca.2015.09.009 | ||
Choy E, Butrynski JE, Harmon DC, et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer. 2014;14:813. doi:10.1186/1471-2407-14-813 | ||
Stewart E, Goshorn R, Bradley C, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014;9(3):829–841. doi:10.1016/j.celrep.2014.09.028 | ||
Zollner SK, Selvanathan SP, Graham GT, et al. Inhibition of the oncogenic fusion protein EWS-FLI1 causes G2-M cell cycle arrest and enhanced vincristine sensitivity in Ewing’s sarcoma. Sci Signal. 2017;10:499. doi:10.1126/scisignal.aam8429 | ||
Gollavilli PN, Pawar A, Wilder-Romans K, et al. EWS/ETS-driven Ewing sarcoma requires BET bromodomain proteins. Cancer Res. 2018;78(16):4760–4773. doi:10.1158/0008-5472.CAN-18-0484 | ||
Mancarella C, Pasello M, Ventura S, et al. Insulin-like growth factor 2 mRNA-binding protein 3 is a novel post-transcriptional regulator of Ewing sarcoma malignancy. Clin Cancer Res. 2018;24(15):3704–3716. doi:10.1158/1078-0432.CCR-17-2602 | ||
Jacques C, Lamoureux F, Baud’huin M, et al. Targeting the epigenetic readers in Ewing sarcoma inhibits the oncogenic transcription factor EWS/Fli1. Oncotarget. 2016;7(17):24125–24140. doi:10.18632/oncotarget.8214 | ||
Loganathan SN, Tang N, Fleming JT, et al. BET bromodomain inhibitors suppress EWS-FLI1-dependent transcription and the IGF1 autocrine mechanism in Ewing sarcoma. Oncotarget. 2016;7(28):43504–43517. doi:10.18632/oncotarget.9762 | ||
Hensel T, Giorgi C, Schmidt O, et al. Targeting the EWS-ETS transcriptional program by BET bromodomain inhibition in Ewing sarcoma. Oncotarget. 2016;7(2):1451–1463. doi:10.18632/oncotarget.6385 | ||
Theisen ER, Pishas KI, Saund RS, Lessnick SL. Therapeutic opportunities in Ewing sarcoma: EWS-FLI inhibition via LSD1 targeting. Oncotarget. 2016;7(14):17616–17630. doi:10.18632/oncotarget.7124 | ||
Tong M, Wang J, He W, et al. Predictive biomarkers for tumor immune checkpoint blockade. Cancer Manag Res. 2018;10:4501–4507. doi:10.2147/CMAR.S179680 | ||
Machado I, Lopez-Guerrero JA, Scotlandi K, Picci P, Llombart-Bosch A. Immunohistochemical analysis and prognostic significance of PD-L1, PD-1, and CD8+tumor-infiltrating lymphocytes in Ewing’s sarcoma family of tumors (ESFT). Virchows Arch. 2018;472(5):815–824. doi:10.1007/s00428-018-2316-2 | ||
Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326–1341. doi:10.1158/2159-8290.CD-13-1037 | ||
Tirode F, Surdez D, Ma X, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4(11):1342–1353. doi:10.1158/2159-8290.CD-14-0622 | ||
Brohl AS, Solomon DA, Chang W, et al. The genomic landscape of the Ewing sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10(7):e1004475. doi:10.1371/journal.pgen.1004541 | ||
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi:10.1126/science.1235122 | ||
Tawbi HA, Burgess M, Bolejack V, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18(11):1493–1501. doi:10.1016/S1470-2045(17)30624-1 | ||
Davis KL, Fox E, Reid JM, et al. ADVL1412: initial results of a phase I/II study of nivolumab and ipilimumab in pediatric patients with relapsed/refractory solid tumors – A COG study [abstract]. J Clin Oncol. 2017;35:10526. doi:10.1200/JCO.2017.35.15_suppl.10526 | ||
D’Angelo SP, Mahoney MR, Van Tine BA, et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018;19(3):416–426. doi:10.1016/S1470-2045(18)30006-8 | ||
Ghisoli M, Barve M, Schneider R, et al. Pilot trial of FANG immunotherapy in Ewing’s sarcoma. Mol Ther. 2015;23(6):1103–1109. doi:10.1038/mt.2015.43 | ||
Rao DD, Jay C, Wang Z, et al. Preclinical Justification of pbi-shRNA EWS/FLI1 lipoplex (LPX) treatment for Ewing’s sarcoma. Mol Ther. 2016;24(8):1412–1422. doi:10.1038/mt.2016.93 | ||
Ghisoli M, Barve M, Mennel R, et al. Three-year follow up of GMCSF/bi-shRNA(furin) DNA-transfected autologous tumor immunotherapy (Vigil) in metastatic advanced Ewing’s sarcoma. Mol Ther. 2016;24(8):1478–1483. doi:10.1038/mt.2016.86 | ||
Rasper M, Jabar S, Ranft A, Jurgens H, Amler S, Dirksen U. The value of high-dose chemotherapy in patients with first relapsed Ewing sarcoma. Pediatr Blood Cancer. 2014;61(8):1382–1386. doi:10.1002/pbc.25042 | ||
Barker LM, Pendergrass TW, Sanders JE, Hawkins DS. Survival after recurrence of Ewing’s sarcoma family of tumors. J Clin Oncol. 2005;23(19):4354–4362. doi:10.1200/JCO.2005.05.105 | ||
McTiernan A, Driver D, Michelagnoli MP, Kilby AM, Whelan JS. High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing’s sarcoma family of tumours. Ann Oncol. 2006;17(8):1301–1305. doi:10.1093/annonc/mdl108 | ||
Whelan J, Le Deley MC, Dirksen U, et al. High-dose chemotherapy and blood autologous stem-cell rescue compared with standard chemotherapy in localized high-risk Ewing sarcoma: results of Euro-E.W.I.N.G.99 and Ewing-2008. J Clin Oncol. 2018;36(31):3110–3119. doi:10.1200/JCO.2018.78.251. | ||
Gorlick R, Janeway KA, Adamson PC. Dose intensification improves the outcome of Ewing sarcoma. J Clin Oncol. 2018;38(31):3072–3073. doi:10.1200/JCO.2018.79.3489. | ||
Jiang Y, Subbiah V, Janku F, et al. Novel secondary somatic mutations in Ewing’s sarcoma and desmoplastic small round cell tumors. PLoS One. 2014;9(8):e93676. doi:10.1371/journal.pone.0093676 |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.