Back to Archived Journals » Integrated Blood Pressure Control » Volume 13
Management of Pulmonary Arterial Hypertension in Patients with Systemic Sclerosis
Authors Almaaitah S ![]() , Highland KB, Tonelli AR
, Highland KB, Tonelli AR ![]()
Received 22 September 2019
Accepted for publication 5 March 2020
Published 23 March 2020 Volume 2020:13 Pages 15—29
DOI https://doi.org/10.2147/IBPC.S232038
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Turgay Celik
Saja Almaaitah,1 Kristin B Highland,2 Adriano R Tonelli2
1Medicine Institute, Cleveland Clinic, Cleveland, OH, USA; 2Department of Pulmonary and Critical Care Medicine, Cleveland Clinic, Cleveland, OH, USA
Correspondence: Adriano R Tonelli Email [email protected]
Abstract: Systemic sclerosis (SSc) is a rare and complex immune-mediated connective tissue disease characterized by multi-organ fibrosis and dysfunction. Systemic sclerosis-associated pulmonary arterial hypertension (SSc-PAH) is a leading cause of death in this population. Pulmonary arterial hypertension (PAH) can coexist with other forms of pulmonary hypertension in SSc, including pulmonary hypertension related to left heart disease, interstitial lung disease, chronic thromboembolism and pulmonary venous occlusive disease, which further complicates diagnosis and management. Available pulmonary arterial hypertension therapies target the nitric oxide, endothelin and prostacyclin pathways. These therapies have been studied in SSc-PAH in addition to idiopathic PAH, often with different treatment responses. In this article, we discuss the management as well as the treatment options for patients with SSc-PAH.
Keywords: systemic sclerosis, scleroderma, pulmonary arterial hypertension, pulmonary hypertension
Introduction
Systemic sclerosis (SSc), also called scleroderma, is a complex immune-mediated connective tissue disease characterized by fibrosis and thickening of the skin and internal organs as well as vascular abnormalities that ultimately leads to multi-organ dysfunction.1 These immune, fibrotic and vascular abnormalities, including pulmonary arterial hypertension, are highlighted in the revision of the American College of Rheumatology/European League against Rheumatism (ACR/EULAR) criteria for SSc diagnosis.2 SSc is classified into diffuse and limited cutaneous forms based on the extent of skin involvement. Limited cutaneous systemic sclerosis (lcSSc) has skin involvement distal to the elbows and knees, whereas the skin is involved proximal to the knees and elbows, including the trunk in diffuse cutaneous systemic sclerosis (dcSSc). The term systemic sclerosis sine scleroderma is used when there is no skin involvement, but the patient meets the other criteria for SSc.3
The definition of pulmonary arterial hypertension (PAH) was recently modified by the sixth World Symposium on Pulmonary Hypertension (WSPH) proceedings to include an elevation in the mean pulmonary arterial pressure (mPAP) >20 mmHg, a pulmonary vascular resistance (PVR) ⩾3Wood units (WU) and a pulmonary artery wedge pressure (PAWP) ⩽15 mmHg.4 This change was based on data obtained from healthy individuals, showing that a normal mPAP at rest is 14 ± 3.3 mmHg.5 Two standard deviations from this mean give the current mPAP cut-off for upper limit of normal. Evidence from large databases indicate that patients with a mPAP between 20 and 25 mmHg have worse outcomes than those with a mPAP ≤20 mmHg, further supporting the modification by the 6th WSPH.4 Two studies in patients with systemic sclerosis-associated pulmonary arterial hypertension (SSc-PAH), with mPAP between 21 and 24 mmHg, demonstrated a decrease in functional capacity as shown by an abnormal six-minute walk test (6MWT), even when considering a PVR of ⩾ 2 (instead of ≥3) Wood units.6,7
There are five groups of pulmonary hypertension (PH) that are based on the mechanisms of disease, clinical presentation, hemodynamic characteristics, and therapeutic response.8 Groups 1 to 5 include patients with 1) PAH, 2) PH due to left heart disease, 3) PH due to lung disease and/or hypoxia, 4) PH due to pulmonary artery obstruction and 5) PH due to unclear or multifactorial mechanism, respectively. The prevalence of pulmonary arterial hypertension (PAH) in SSc is estimated to be around 6–12%, a percentage that may increase when using the modified definition for PAH.9,10 It is the second most frequent cause of PAH in both US and European registries following idiopathic pulmonary arterial hypertension (IPAH).11 PAH is more common in lcSSc but can be seen in the other variants. Furthermore, PAH can coexist with other forms of PH in SSc, including PH related to left heart disease, interstitial lung disease/hypoxemia, chronic thromboembolism and pulmonary venous occlusive disease (currently included in group 1PH),8 which further complicates diagnosis and management.
PAH results from an imbalance between vasoconstrictors and proliferative mediators (such as endothelin-1) and vasodilators (such as nitric oxide and prostacyclin). Endothelial injury and intraluminal micro-thrombosis lead to progressive pulmonary arterial remodeling and increase in PVR. The progressive increase in PVR affects the right ventricular function, leading to right ventricular failure and death.9,12 SSc-PAH is a leading cause of mortality, with a mortality rate of 50% within the first 3 years;9,13 which is worse than the one observed in patients with IPAH.14
Types of Pulmonary Hypertension in Systemic Sclerosis
Due to the systemic nature of SSc, an overlap between more than one type of PH is common (Table 1 and Figure 1), making the identification of the predominant type of PH not always straightforward,15 and complicating the management of these patients.
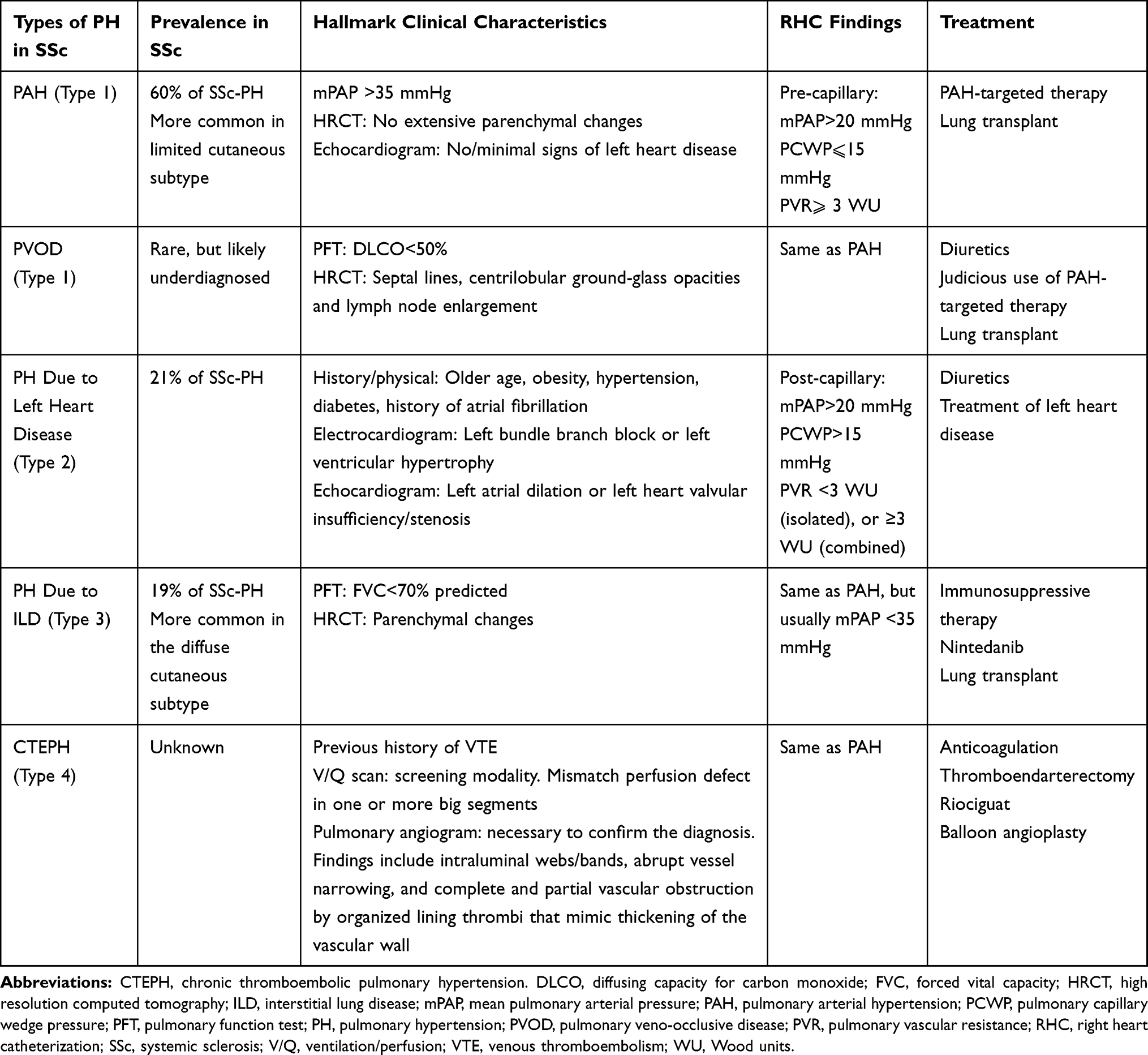 |
Table 1 Summary of Different Phenotypes of Pulmonary Hypertension in SSc |
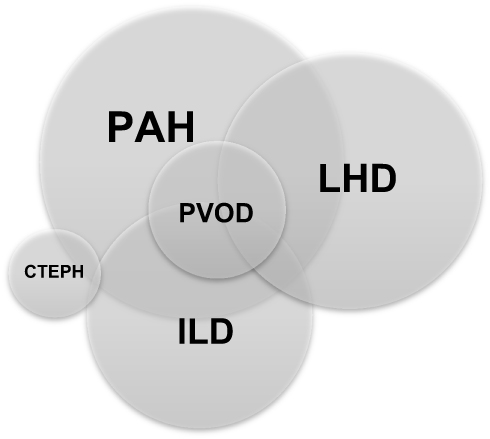 |
Figure 1 Overlap between different phenotypes of pulmonary hypertension in SSc.Abbreviations: CTEPH, chronic thromboembolic pulmonary hypertension; ILD, interstitial lung disease; LHD, left heart disease; PAH, pulmonary arterial hypertension, PVOD, pulmonary veno-occlusive disease; SSc, systemic sclerosis. |
Systemic sclerosis associated with left heart disease (SSc-LHD, group 2 PH) is defined by an elevated mPAP with left heart disease (characterized by PCWP >15 mmHg). Cardiac dysfunction may be seen in >40% of patients with SSc and includes primary myocardial fibrosis, fibrosis of the conduction system leading to arrhythmias, microvascular and atherosclerotic coronary vessel disease and hypertensive crisis.16–18 Fibrosis of the myocardium may result in either diastolic (heart failure with preserved ejection fraction which is reported in around 18% of SSc patients) or less commonly systolic heart failure (heart failure with reduced ejection fraction which is described in around 2% patients).16 Studies comparing echocardiography findings in patients with SSc-PAH and IPAH with similar hemodynamics showed that patients with SSc-PAH are more likely to have left atrial enlargement and other indications of left ventricular diastolic impairment. Exercise and/or fluid challenge during RHC may be useful in differentiating group 1 and group 2 PH. In the case of elevated PCWP, a markedly elevated transpulmonary (mPAP – PCWP) or diastolic pulmonary gradient (pulmonary artery diastolic pressure – PCWP) suggests the possibility of combined group 1 and group 2 PH.19 The treatment consists of volume status optimization, rate control and heart failure medications.20 Treatment with PAH-specific therapy in group 2 PH or combined group 1 and group 2 is not recommended, as it may result in fluid retention and pulmonary edema (such as the use of macitentan in combined pre and post-capillary pulmonary hypertension, CpcPH).21
Interstitial lung disease (ILD) is common in SSc, with evidence of interstitial changes on imaging in up to 90% and chronic respiratory failure in approximately 10% of patients. PH is seen in up to 31% of patients with clinically significant SSc-ILD and results in higher mortality than in SSc-ILD patients without PH.22 SSc-ILD is more common in the dcSSc type, especially in patients who have positive Scl-70 (anti-topoisomerase)23 but also may occur in all variants. The presence of a positive Scl-70 antibody is somewhat protective against PAH, but these patients may still develop group 3 PH. ILD may be classified as “limited” or “extensive” based on high resolution computed tomography (HRCT) and pulmonary function testing (PFT). It is suggested that PH associated with the extensive form of SSc-ILD (>20% fibrosis on HRCT or forced vital capacity (FVC) <70% in indeterminate HRCT) could be classified as group 3.15 Although the etiology of PH is associated with extent of lung disease, the mPAP does not seem to correlate with extent of fibrosis on imaging or the forced vital capacity.22,24 In patients with SSc-ILD, a diffusion capacity (DLCO) <40% of predicted or a reduction in DLCO out of proportion to FVC (FVC/DLCO ratio >1.6) also suggests the presence of pulmonary vascular disease.11 The PH in group 3 is typically modest with a mPAP <35 mmHg. Likewise, the presence of a mPAP “out of proportion” or >35 mmHg suggests the possibility of concomitant PAH. Treatment of SSc-ILD mainly consists of immune-suppressive therapy, most commonly cyclophosphamide and mycophenolate.25 Most recently, the antifibrotic nintedanib, a tyrosine kinase inhibitor, was shown to decrease progression of SSc-ILD in the SENSCIS trial,26 and murine models have suggested a possible additional benefit of nintedanib on the pulmonary vasculature.27 Treatment of PH due to ILD with PAH-specific therapies may result in worsening oxygenation due to deterioration of the ventilation-perfusion mismatch and is not recommended outside of clinical trials (Figure 2). However, a more recent study demonstrated that the coexistence of type I and type III is common in patients with SSc, and such patients tolerated concomitant targeted therapy and immunosuppressive therapy.28
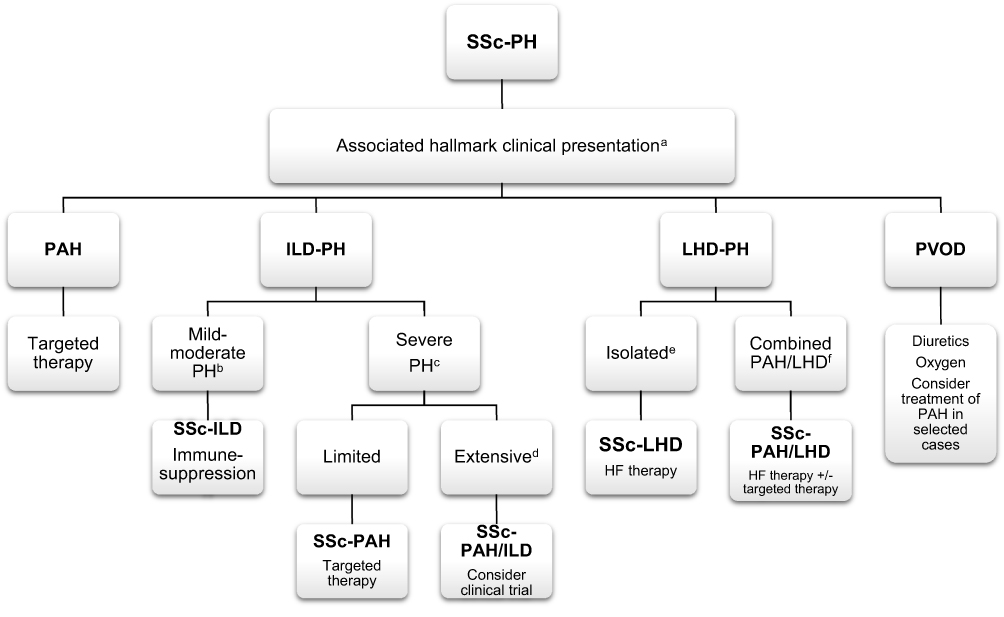 |
Figure 2 Management of different phenotypes of pulmonary hypertension in SSc. aAs suggested by imaging (HRCT, Echocardiogram), PFT and RHC, see Table 1. bMild-moderate PH includes mPAP < 35 mmHg and normal or mildly dysfunctional RV. cSevere PH includes mPAP ≥35 mmHg and/or RV dysfunction more than mild. dExtensive ILD refers to >20% fibrosis on HRCT, or FVC<70% in indeterminate HRCT. eIsolated LHD refers to DPG<7mmHg and/or PVR≤3WU. fCombined PAH/LHD (pre and post capillary PH) refers to DPG≥7 and/or PVR>3 WU.Abbreviations: DPG, diastolic pulmonary gradient; FVC, forced vital capacity; HF, heart failure; HRCT, high resolution computed tomography; ILD, interstitial lung disease; LHD, left heart disease; mPAP, mean pulmonary arterial pressure; PAH, pulmonary arterial hypertension; PFT, pulmonary function test; PH, pulmonary hypertension, PVOD, pulmonary veno-occlusive disease; PVR, pulmonary vascular resistance; RHC, right heart catherization; RV, right ventricle; SSc, systemic sclerosis; WU, wood units. |
Pulmonary veno-occlusive disease (PVOD) is underrecognized in SSc and its presence carries a very poor prognosis.29 Although difficult to distinguish from PAH, it should be suspected in patients who develop pulmonary edema after the initiation of PAH-specific therapies. Other indications are a DLCO <50% on PFTs, severe hypoxemia, and the presence of septal lines, centrilobular ground-glass opacities and lymph node enlargement on HRCT.4 Pathologic findings include intimal fibrosis and obstruction of small pulmonary veins and venules in addition to the arteriopathy seen in PAH.30 Treatment mainly consists of diuretics to optimize fluid status, very careful use of PAH-specific therapies and ultimately lung or heart-lung transplantation.
Patients with SSc have a 3-fold increased risk of pulmonary thromboembolic disease, especially if antiphospholipid antibodies are present.31 Furthermore, SSc is a potential risk factor for developing chronic thromboembolic pulmonary hypertension (CTEPH, group 4 PH),32 that could be related, at least in part, to higher levels of vWF in patients with SSc.33,34 As such, patients with SSc-PH should be screened for CTEPH as a potential etiology of PH. Screening involves obtaining a ventilation/perfusion (V/Q) scan in all patients newly diagnosed with SSc-PH, even in patients without prior history of pulmonary embolism, as approximately 25% of patients diagnosed with CTEPH have no known history of pulmonary embolism.35 In the case of abnormal V/Q scan, a pulmonary angiogram should follow to confirm the diagnosis.36
Screening for Pulmonary Hypertension in Systemic Sclerosis
All SSc patients should have pulmonary function testing (PFT), consisting of spirometry, lung volumes and DLCO, to screen for both ILD and PH. A decrease in DLCO <60% or >20% in one year in the absence of significant lung volume abnormalities, or a FVC/DLCO percent >1.6 suggests PH.11 An elevated NT-proBNP showed a sensitivity and specificity of 90% for the presence of SSc-PAH in one study, and may suggest PAH if elevated > two-fold the upper limit of normal.37 However, the role of pro-BNP in screening for SSc-PAH is yet to be determined. Transthoracic echocardiography is the best screening tool for PH. It assesses right- and left-sided morphology and function, detects valvular abnormalities and is useful for the estimation of right ventricular pressures.11,38 When PH is suggested by echocardiography (RVSP > 40 mmHg and/or any degree of RV dysfunction) a RHC is warranted.39
HRCT is performed mainly to screen for interstitial lung disease and/or PVOD. However, a HRCT might show an enlarged pulmonary artery and right ventricle in advanced PAH.38,40 A cardiopulmonary exercise test (CPET) can suggest pulmonary vascular disease in cases of low end-tidal partial pressure of carbon dioxide (EtPCO2), high ventilator equivalents for carbon dioxide (VE/VCO2), low oxygen pulse (VO2/HR) and low peak oxygen uptake (VO2).20,41 The six-minute walk test (6MWT) is a non-invasive submaximal functional test that correlates with maximum exercise capacity as measured by CPET.42 In addition, heart rate recovery (HRR, as measured by the difference of heart rate at the end of 6MWT and after 1 min of resting) may predict clinical worsening in patients with connective tissue disease associated PAH (CTD-PAH), including SSc.43 However, the performance characteristics of the 6MWT in SSc are less robust than in IPAH secondary to non-cardiopulmonary limitations of exercise, such as joint pain, skin contractures and muscle weakness. Recently, a combination of features obtained during the 6MWT, ie, distance walked, degree of oxygenation and Borg dyspnea index was found useful in identifying patients who need further cardiopulmonary evaluation.44
Compared with echocardiography, cardiac MRI (CMR) may provide information on the underpinnings of cardiac involvement in patients with SSc, including inflammatory, microvascular and fibrotic mechanisms.45 Furthermore, CMR provides a more comprehensive assessment of the right ventricular function.46
Due to the high morbidity and mortality of PAH, patients newly diagnosed with SSc should be screened for PAH at presentation and annually thereafter for life.41 Data suggest that SSc-PAH patients who are diagnosed and treated early as a result of screening have improved mortality as compared to patients diagnosed as a result of clinical suspicion.47–49 The European Society of Cardiology/European Respiratory Society recommends annual echocardiography.20 If features suggestive of PH are noted, ie, elevated tricuspid regurgitant jet velocity or abnormal right ventricular morphology and function, a right heart catheterization is recommended. The Australian Scleroderma Interest Group (ASIG) recommends screening with NT-proBNP and PFT. An elevated NT-proBNP and/or an elevated FVC/DLCO ratio is an indication to consider RHC.50 The 2-step DETECT algorithm combines clinical, physiologic and laboratory data to decide who should get an echocardiogram and based on its result who should undergo a RHC to confirm the presence of PH.13 All three screening strategies13,20,50 have similar sensitivity, specificity, positive and negative predictive values (Table 2).
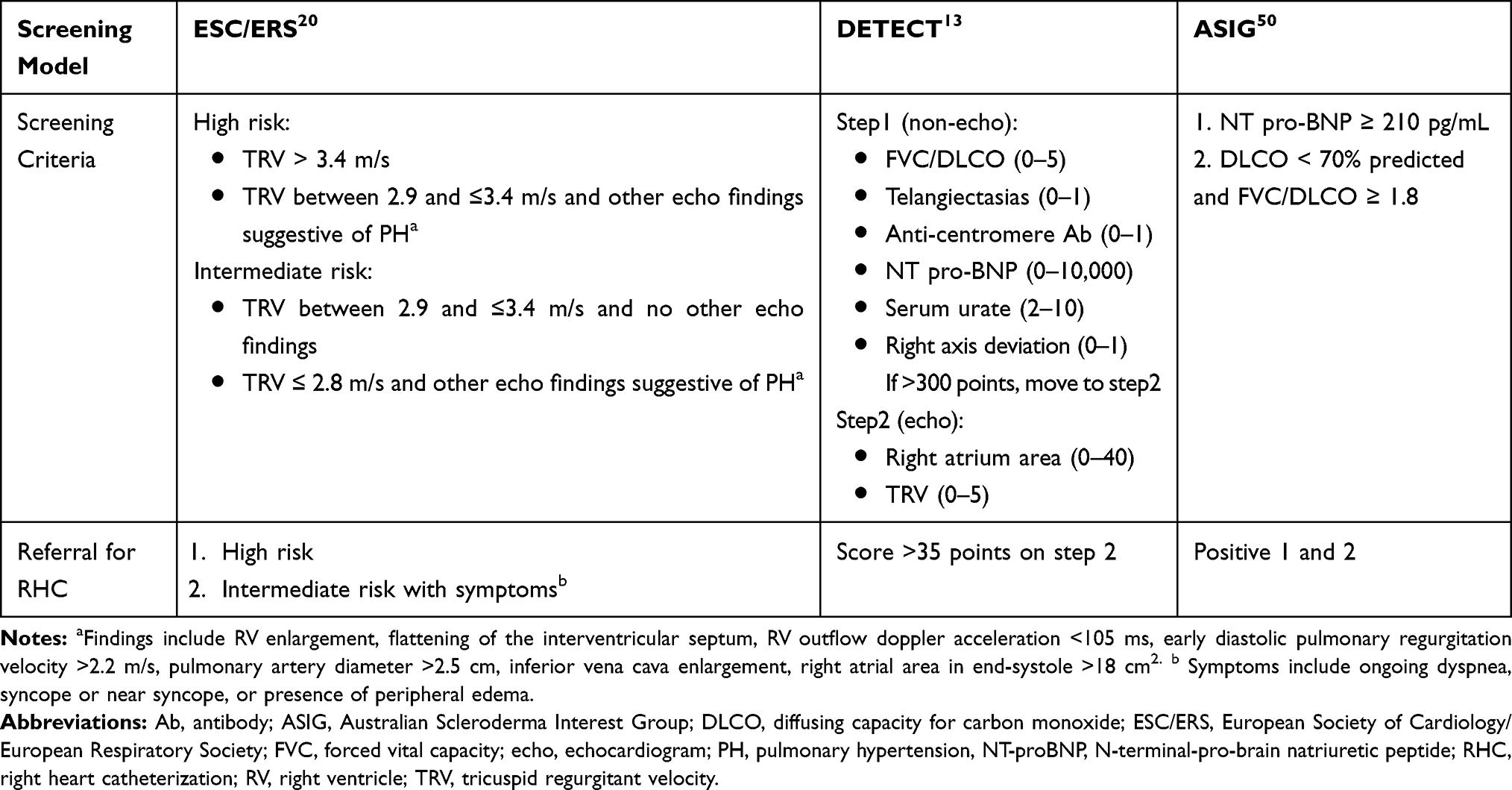 |
Table 2 Summary of Screening Models |
Treatment
As SSc is yet to be a treatable disease, the treatment of SSc-PAH is directed at controlling the progression of PAH.51 General measures and supportive therapy should be offered to all patients. PAH-specific treatment is generally offered for patients with WHO functional class (WHO FC) II, III or IV.9 Currently available treatments target the nitric oxide, endothelin or prostanoid pathways (Table 3). The 6th World Symposium Proceedings in PH recommend a treatment strategy based on a multi-parametric risk stratification approach,52 with the main objective of achieving a low-risk status that is associated with a reduced mortality (annual mortality of <5%).20 Several risk stratification strategies have been used, including the REVEAL 2.0,53 French Pulmonary Hypertension Network (FPHN),54 COMPERA55 and the Swedish Pulmonary Arterial Hypertension Registrar (SPAHR),56 (Table 4). The two methodologies most commonly used to assess risk are the FPHN and REVEAL 2.0. The FPHN risk assessment totals the number of low-risk criteria (WHO functional class I or II, 6MW distance >440 m, RA pressure <8 mmHg, and cardiac index ≥2.5 L/min/m2).57 The REVEAL 2.0 risk score includes a larger number of variables but provides a greater risk discrimination than the FPHN.53
 |
Table 3 Pivotal Trials for the Medications Used in SSc-PAH with Comparison of the Treatment Response Between SSc-PAH and IPAH |
 |
Table 4 Summary of PAH Risk Scoring System |
Vasoreactivity Testing
Vasoreactivity testing is recommended predominantly for patients with idiopathic PAH to identify individuals that can benefit from calcium channel blockers. However, vasoreactivity testing in SSc-PAH is not mandated in recent guidelines as most patients are non-reactive.58 Therefore, calcium channel blockers are not recommended as they may worsen right heart failure; nevertheless, and under careful vigilance, some SSc individuals receive this medication for their Raynaud phenomenon.59
Treatment Approach
After confirmation of the diagnosis of SSc-PAH, the 6th World Symposium proceedings recommend initiating PAH targeted therapy based on risk stratification (Figure 3).60 Patients with low-intermediate risk are started generally on combination therapy,52 with a few exceptions in which monotherapy is an adequate alternative. Choice of medication is usually based on a number of factors, including comorbidities, side effects, route of administration and patient preference.52 High-risk patients should be treated with combination therapy that includes a parenteral prostacyclin analogue.52
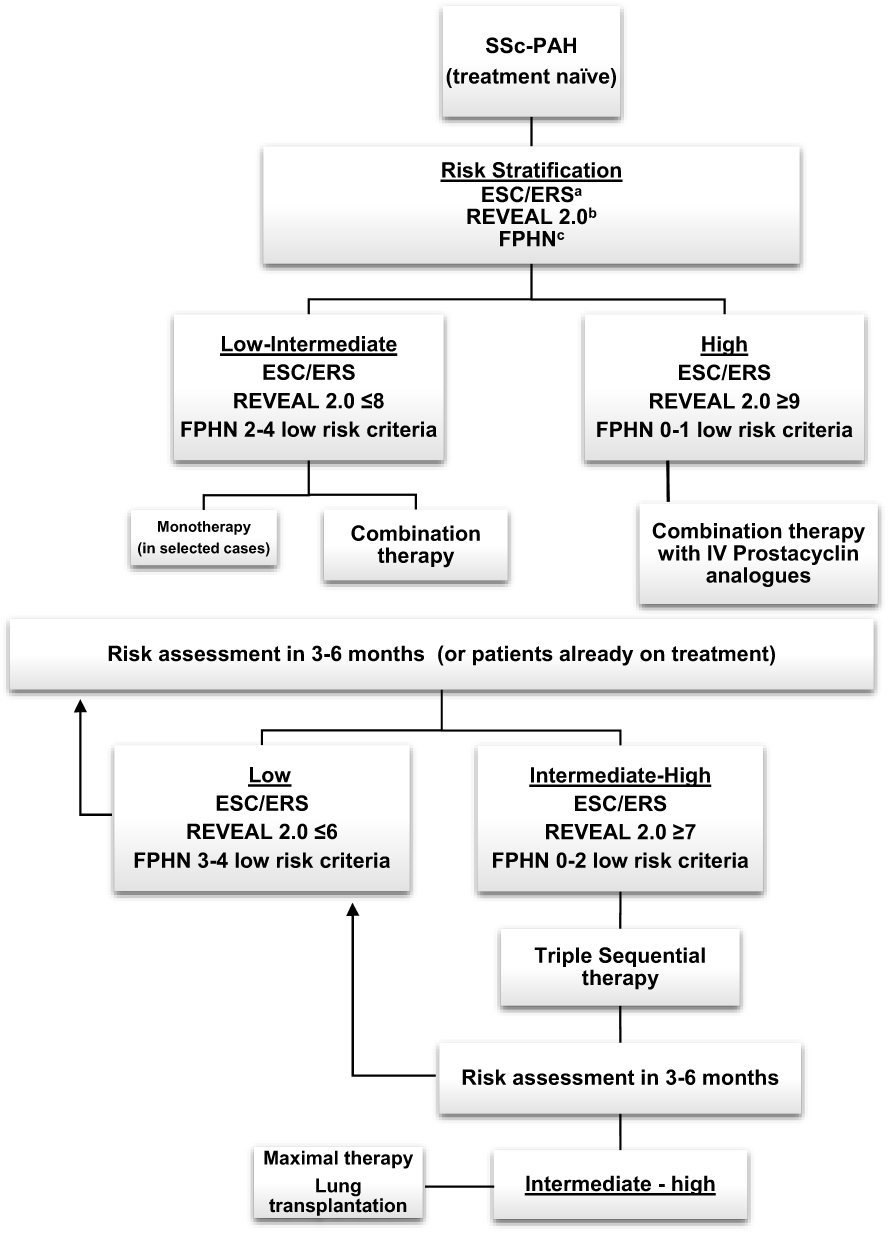 |
Figure 3 Treatment approach to pulmonary arterial hypertension in Systemic Sclerosis. aESC/ERS risk assessment is based on clinical signs of right heart failure, syncope, WHO functional class, 6MWT, CPET, NT-proBNP, Imaging (echocardiography, MRI) and cardiac hemodynamics. bREVEAL criteria: see table 4. cFPHN low risk criteria: WHO class I/II, 6MWT >440m, RAP <8, CI ≥2.5.Abbreviations: 6MWT, six-minute walk test; CPET, cardiopulmonary exercise testing; CI, cardiac index; ESC/ERS, European Society of Cardiology/European Respiratory Society; FPHN, French pulmonary hypertension Network; IV, intravenous; MRI, magnetic resonance imaging; NT-proBNP, N-terminal- pro brain natriuretic peptide; RAP, right atrial pressure; REVEAL, Registry to Evaluate Early and Long-Term PAH Disease Management; SSc-PAH, systemic sclerosis associated pulmonary arterial hypertension; WHO, world health organization. Data from these studies.14,44,45 |
Patients are usually followed up within 1–3 months after initiating therapy to evaluate treatment response, and thereafter, every 3–6 months depending on the patient.9 Tests suggested during follow up include clinical assessment (WHO FC), 6MWT, NT-proBNP and echocardiogram. A RHC should be considered 3–6 months after initiation or change in therapy, and yearly thereafter.20 Treatment should be escalated in patients who fail to achieve a low-risk status within 3–6 months. Those failing triple therapy should be considered for lung transplantation.52
Targeted Therapy
Nitric Oxide Pathway
In healthy individuals, endothelium-derived nitric oxide activates soluble guanylate cyclase (sGC), which increases the production of cyclic guanosine monophosphate (cGMP) that mediates smooth muscle relaxation. cGMP is degraded by phosphodiesterase type 5 (PDE-5). PDE-5 inhibitors (sildenafil and tadalafil) act as selective and potent inhibitors of this enzyme, increasing the levels of cGMP. In contrast, riociguat acts upstream, stimulating sGC.61,62 The concomitant administration of PDE-5 inhibitors and riociguat is contraindicated due to the increase in adverse effects such as headache, hypotension, dizziness, vomiting and even death, as was shown in the PATENT Plus trial.63
Phosphodiesterase-5 Inhibitors
Both sildenafil and tadalafil are approved for the treatment of SSc-PAH.51 SUPER-1/SUPER-2 trials demonstrated the efficacy of sildenafil therapy in improving the 6MW distance, cardiac hemodynamics and WHO FC in patients with PAH, including those with CTD-PAH (45% of whom had SSc). Improvements in the distance walked during the 6MWT were similar for patients with CTD-PAH or IPAH.64,65 Tadalafil was studied in the PHIRST-1/PHIRST-2 trials, which demonstrated its ability to improve 6MW distance, quality of life and reduce clinical worsening, in a PAH population that included CTD-PAH patients. However, treatment with tadalafil in patients with CTD-PAH was less efficacious than in patients with IPAH.66,67 Interestingly, PDE-5 inhibitors were shown to improve healing and prevent the development of digital ulcers in patients with SSc.51
Guanylate Cyclase Stimulator
Riociguat was evaluated in the PATENT-1/PATENT-2 trials and demonstrated considerable efficacy in CTD-PAH including SSc-PAH, improving the 6MW distance, pulmonary hemodynamics (such as PVR and the cardiac index) and WHO FC. These improvements were less pronounced in patients with CTD-PAH than IPAH; however, 2-year survival rates were similar in both PAH types.68,69 In addition, a case series demonstrated that in patients with SSc-PAH with unsatisfactory response to PDE-5 inhibitors, switching to riociguat was associated with improved respiratory and cardiac hemodynamics.70 Long-term treatment with riociguat has been associated with a reduction in right heart size and an improvement in right ventricular function in patients with PAH (14% of patients with CTD-PAH) and CTEPH.71,72
Endothelin Receptor Antagonists
Endothelin-1 binds to endothelin receptors on the pulmonary vasculature and results in vasoconstriction.73 Bosentan, ambrisentan and macitentan are endothelin receptor antagonists (ERA) approved for the treatment of SSc-PAH. In the BREATHE-1 trial, bosentan prevented the deterioration of walking distance in 6MWT, predominantly in patients with IPAH (3-m improvement in SSc-PAH vs 46 m in IPAH).74 In the ARIES-1/ARIES-2 trials, ambrisentan improved the 6MW distance and slowed clinical worsening in CTD-PAH, though survival was better in the IPAH population.75,76 The SERAPHIN trial showed that macitentan reduced morbidity and mortality in PAH patients, after-which a meta-analysis showed similar outcomes between IPAH and CTD-PAH.77,78 ERA side effects include elevated liver function tests (bosentan), peripheral edema and anemia (all).20 In the RAPIDS-2 study,79 bosentan prevented the formation of new digital ulcers but did not improve the rate of ulcer healing.
Prostacyclin Pathway Agonists
Prostacyclin is produced by endothelial cells and causes potent pulmonary artery vasodilation. Dysregulation of the prostacyclin pathway has been shown in patients with PAH.80 Epoprostenol, treprostinil and iloprost are approved for the treatment of SSc-PAH.51 Epoprostenol is given intravenously due to its short half-life. Treprostinil may be administered intravenously, subcutaneously, orally or by inhalation.81 Iloprost is only administered by inhalation in the US, although an intravenous formulation is available in Europe. Intravenous epoprostenol improved exercise capacity and cardiopulmonary hemodynamics (decrease in mPAP and PVR and improvement in WHO FC) in patients with SSc-PAH.82,83 Similar results were demonstrated with intravenous and subcutaneous treprostinil as well as inhaled iloprost.84–88 Intermittent intravenous iloprost infusion (composed of iloprost infusion for 6 hrs per day for 5 days every 6 weeks) decreased the systolic PAP, improved the distance walked during the 6MWT and protected against PAH progression in patients with SSc.89 Prostaglandins are also commonly used to treat ischemic digital ulcers in patients with SSc.51 Selexipag, an oral selective IP-receptor agonist, was evaluated in the GRIPHON trial, where it decreased the risk of death or complications related to PAH patients including SSc, findings that were consistent with those seen in patients with IPAH.90,91
Combination Therapy versus Initial Monotherapy
Combination therapy is the approach of choice for drug naïve SSc-PAH patients. The ATPAHSS-O and the AMBITION trials showed that upfront combination therapy with ambrisentan and tadalafil in SSc-PAH improved cardiac hemodynamics (PVR and stroke volume, NT-proBNP) and 6MW distance, and lowered the risk of clinical worsening, when compared to monotherapy with either drug alone.92,93 More recently, the SERAPHIN and the long-term GRIPHON trials demonstrated lower morbidity and mortality rates with the addition of macitentan and selexipag on a background therapy, respectively.94,95
General Measures
Oral Anticoagulation
Data on the use of oral anticoagulation remain conflicting for patients with IPAH;96 recent data have discouraged the use of anticoagulation for patients with SSc-PAH, since these patients are at increased risk of bleeding secondary to gastric antral vascular ectasia and arterial vascular malformations in the intestines. In the COMPERA trial, oral anticoagulation was associated with a survival benefit in IPAH, but not in other forms of PAH, such as SSc-PAH.97 In fact, long-term use of warfarin was associated with worse prognosis in patients with SSc-PAH in the REVEAL Registry.98 Furthermore, anticoagulation with warfarin was not associated with an effect on survival in PAH (12% had SSc-PAH) or idiopathic PAH patients treated with SQ treprostinil.99 A prospective cohort study showed that anticoagulation with warfarin was associated with improved survival; however, 46% of SSc-PAH received treatment for indications other than PAH.48 There is an ongoing trial (SPHInX) that aims to evaluate the efficacy and safety of apixaban, a direct oral anticoagulant, in SSc-PAH.100
Immunosuppressive Therapy
While immunosuppressive therapy has been associated with improved clinical outcome and survival in PAH associated with some forms of CTD (such as glucocorticoids and cyclophosphamide in systemic lupus erythematosus and mixed connective tissue disease),101 SSc-PAH is refractory to corticosteroids or immunosuppressive therapy.102
Supportive Therapy
Despite the lack of specific data, experts recommend the use of diuretics to manage volume overload and digoxin for atrial arrhythmias and right heart failure. Long-term oxygen therapy is recommended to maintain arterial blood oxygen pressure above 60 mm Hg or oxygen saturation above 90%.103,104 In terms of non-medical management, exercise training was found to improve work capacity, quality of life and possibly survival in patients with CTD-PAH including SSc.105 Pregnancy is contraindicated in SSc-PAH given substantial mortality rate, estimated to be up to 50%,106 and teratogenic effect of some PAH-specific medications such as riociguat and ERAs. If a patient becomes pregnant, a discussion about pregnancy termination should ensue. Patients who decide to continue with pregnancy are optimized on PAH therapies, followed-up closely in an expert center with experience in managing this high-risk pregnancy.20 Other general measures also include immunization against influenza and pneumococcal infections.20
Refractory Disease
Despite the continuous advances in the management of PAH, a significant proportion of patients with SSc-PAH experience disease progression.107 Atrial septostomy has been investigated as a palliative measure for patients with SSc-PAH who continue to deteriorate despite maximal medical treatment, and was found to improve exercise capacity and possibly survival in selective cases.108 In patients with SSc-PAH refractory or poorly responsive to PAH therapy, lung transplantation is currently the only option. At least one study showed that patients with SSc-PAH experience similar 2-year survival rates after lung transplant when compared to IPAH or idiopathic pulmonary fibrosis.109,110 Unfortunately, patients with SSc are commonly deemed inappropriate candidates to lung transplantation due to risk of post-transplant aspiration in the setting of esophageal dysmotility, severe renal impairment, severe Raynaud phenomenon, non-healing digital ulcerations that pose a risk of infection, and very rarely, severe chest wall skin thickening leading to restriction.111 Available data have mainly focused on lung transplantation for patients with SSc interstitial lung disease with or without PH rather than isolated SSc-PAH.112,113
Prognosis
Despite important advances in the treatment of PAH in the recent years, SSc-PAH still carries a poor prognosis, with survival rates of 81%, 64% and 52% over the first, second and third years, respectively.114 Moreover, the mortality rate of SSc-PAH is worse than that of IPAH and non SSc CTD-PAH, probably due to the multi-organ involvement of SSc (when compared to IPAH) and the poorer response to treatment (when compared to IPAH and CTD-PAH).14,115 Predictors of worse outcome include age >60 years old, male sex, WHO FC IV, higher mPAP, low systolic blood pressure <110 mmHg, 6MW distance <165 m, DLCO <39% predicted, presence of pericardial effusion and anti-U1 ribonucleoprotein (RNP) negative status.114,116,117
Future Studies and Medications
Several medications are currently being studied for the treatment of SSc-PAH. One of the medications is ifetroban, a thromboxane A2/prostaglandin H2 receptor antagonist. Ifetroban works by alleviating blood vessel contraction, increasing vasodilation and thereby decreasing PAH.118 Another potential treatment is rituximab, a monoclonal antibody against a protein called CD20, which is found on the surface of B-cells. It is thought that rituximab may slow the progression of fibrosis in the lungs by lowering antibodies against the platelet-derived growth factor. Rituximab is currently being tested for this indication in a Phase 2 clinical trial in patients with SSc-PAH, though preliminary data did not show statistical significance in improvement of the 6MW distance.119,120 Bardoxolone methyl is currently being studied in patients with PAH including CTD-PAH. Bardoxolone methyl works by inducing nuclear factor erythroid 2-related factor 2 (Nrf2) and suppressing nuclear factor-kB (NF-kB). Bardoxolone methyl is thought to target several cells involved in SSc-PAH, such as smooth muscle cells, endothelial cells and macrophages.121
Conclusion
Systemic sclerosis-associated pulmonary arterial hypertension is a devastating complication carrying a poor prognosis. Current therapies targeting SSc-PAH have resulted in improved quality of life, cardiac hemodynamics and survival. Most recent guidelines focus on routine screening for PAH in patients with SSc, and when PAH-SSc is diagnosed, initial aggressive combination treatment is important to reach a low-risk status. Several promising medications are still being studied in this patient population.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Dr. Highland receives grants/contracts, consults and/or is on the Speaker’s Bureau of Actelion Pharmaceuticals, Bayer Healthcare, Boehringer Ingelheim, Eiger Pharmaceuticals, Gilead Sciences, Reata Pharmaceuticals, and United Therapeutics. The other authors have no significant conflicts of interest with any companies or organization whose products or services may be discussed in this article.
References
1. Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390(10103):1685–1699. doi:10.1016/S0140-6736(17)30933-9
2. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737–2747. doi:10.1002/art.38098
3. LeRoy EC, Medsger TA
4. Simonneau G, Montani D. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913.
5. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34(4):888–894. doi:10.1183/09031936.00145608
6. Xanthouli P, Jordan S, Milde N, et al. Haemodynamic phenotypes and survival in patients with systemic sclerosis: the impact of the new definition of pulmonary arterial hypertension. Ann Rheumatic Dis. 2019;79(3):370–378.
7. Nagel C, Marra AM, Benjamin N, et al. Reduced right ventricular output reserve in patients with systemic sclerosis and mildly elevated pulmonary artery pressure. Arthritis Rheumatol. 2019;71(5):805–816. doi:10.1002/art.2019.71.issue-5
8. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. doi:10.1183/13993003.01913-2018
9. Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2013;144(4):1346–1356. doi:10.1378/chest.12-2396
10. Avouac J, Airo P, Meune C, et al. Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J Rheumatol. 2010;37(11):2290–2298. doi:10.3899/jrheum.100245
11. Khanna D, Gladue H, Channick R, et al. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum. 2013;65(12):3194–3201. doi:10.1002/art.38172
12. Johnson SR, Granton JT. Pulmonary hypertension in systemic sclerosis and systemic lupus erythematosus. Eur Respir Rev. 2011;20(122):277–286. doi:10.1183/09059180.00003811
13. Coghlan JG, Denton CP, Grunig E, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis. 2014;73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
14. Fisher MR, Mathai SC, Champion HC, et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006;54(9):3043–3050. doi:10.1002/(ISSN)1529-0131
15. Launay D, Sobanski V, Hachulla E, Humbert M. Pulmonary hypertension in systemic sclerosis: different phenotypes. Eur Respir Rev. 2017;26(145):170056. doi:10.1183/16000617.0056-2017
16. Lambova S. Cardiac manifestations in systemic sclerosis. World J Cardiol. 2014;6(9):993–1005. doi:10.4330/wjc.v6.i9.993
17. de Groote P, Gressin V, Hachulla E, et al. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis. 2008;67(1):31–36. doi:10.1136/ard.2006.057760
18. Fernandez-Codina A, Simeon-aznar CP, Pinal-Fernandez I, et al. Cardiac involvement in systemic sclerosis: differences between clinical subsets and influence on survival. Rheumatol Int. 2017;37(1):75–84. doi:10.1007/s00296-015-3382-2
19. Vachiery JL, Adir Y, Barbera JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100–D108. doi:10.1016/j.jacc.2013.10.033
20. Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67–119. doi:10.1093/eurheartj/ehv317
21. Vachiery JL, Delcroix M. Macitentan in pulmonary hypertension due to left ventricular dysfunction. Eur Respir J. 2018;51(2):1701886.
22. Hinchcliff M, Fischer A, Schiopu E, Steen VD. Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma (PHAROS): baseline characteristics and description of study population. J Rheumatol. 2011;38(10):2172–2179. doi:10.3899/jrheum.101243
23. Schoenfeld SR, Castelino FV. Interstitial lung disease in scleroderma. Rheum Dis Clin North Am. 2015;41(2):237–248. doi:10.1016/j.rdc.2014.12.005
24. Hurdman J, Condliffe R, Elliot CA, et al. ASPIRE registry: assessing the spectrum of pulmonary hypertension identified at a REferral centre. Eur Respir J. 2012;39(4):945–955. doi:10.1183/09031936.00078411
25. Cappelli S, Bellando Randone S, Camiciottoli G, De Paulis A, Guiducci S, Matucci-Cerinic M. Interstitial lung disease in systemic sclerosis: where do we stand? Eur Respir Rev. 2015;24(137):411–419. doi:10.1183/16000617.00002915
26. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis–associated interstitial lung disease. N Engl J Med. 2019;380(26):2518–2528.
27. Huang J, Maier C, Zhang Y, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis. 2017;76(11):1941–1948. doi:10.1136/annrheumdis-2016-210823
28. Young A, Vummidi D, Visovatti S, et al. Prevalence, treatment, and outcomes of coexistent pulmonary hypertension and interstitial lung disease in systemic sclerosis. Arthritis Rheumatol. 2019;71(8):1339–1349. doi:10.1002/art.v71.8
29. Gupta S, Gupta A, Rehman S, et al. Pulmonary veno-occlusive disease is highly prevalent in scleroderma patients undergoing lung transplantation. ERJ Open Res. 2019;5(1):00168–2018. doi:10.1183/23120541.00168-2018
30. Humbert M, Guignabert C. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53(1):1801887.
31. Schoenfeld SR, Choi HK, Sayre EC, Avina-zubieta JA. Risk of pulmonary embolism and deep venous thrombosis in systemic sclerosis: a general population-based study. Arthritis Care Res. 2016;68(2):246–253. doi:10.1002/acr.22673
32. Martinez C, Wallenhorst C, Teal S, Cohen AT, Peacock AJ. Incidence and risk factors of chronic thromboembolic pulmonary hypertension following venous thromboembolism, a population-based cohort study in England. Pulm Circ. 2018;8(3):2045894018791358. doi:10.1177/2045894018791358
33. Bonderman D, Turecek PL, Jakowitsch J, et al. High prevalence of elevated clotting factor VIII in chronic thromboembolic pulmonary hypertension. Thromb Haemost. 2003;90(3):372–376. doi:10.1160/TH03-02-0067
34. Pendergrass SA, Hayes E, Farina G, et al. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS One. 2010;5(8):e12106. doi:10.1371/journal.pone.0012106
35. Pepke-zaba J, Delcroix M, Lang I, et al. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation. 2011;124(18):1973–1981. doi:10.1161/CIRCULATIONAHA.110.015008
36. Kim NH, Delcroix M, Jais x. Chronic thromboembolic pulmonary hypertension. Eur Respir J. 2019;53(1):1801915.
37. Allanore Y, Borderie D, Meune C, et al. N-terminal pro-brain natriuretic peptide as a diagnostic marker of early pulmonary artery hypertension in patients with systemic sclerosis and effects of calcium-channel blockers. Arthritis Rheum. 2003;48(12):3503–3508. doi:10.1002/(ISSN)1529-0131
38. Ahmed M, Dweik RA, Tonelli AR. What is the best approach to a high systolic pulmonary artery pressure on echocardiography? Cleve Clin J Med. 2016;83(4):256–260. doi:10.3949/ccjm.83a.14186
39. Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev. 2013;22(130):515–525. doi:10.1183/09059180.00006013
40. Zompatori M, Leone MB, Giannotta M, et al. Pulmonary hypertension and systemic sclerosis: the role of high-resolution computed tomography. Radiol Med. 2013;118(8):1360–1372. doi:10.1007/s11547-013-0934-1
41. Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J. 2019;53(1):1801904. doi:10.1183/13993003.01904-2018
42. Camargo VM, Martins Bdo C, Jardim C, Fernandes CJ, Hovnanian A, Souza R. Validation of a treadmill six-minute walk test protocol for the evaluation of patients with pulmonary arterial hypertension. J Bras De Pneumol. 2009;35(5):423–430. doi:10.1590/S1806-37132009000500006
43. Minai OA, Nguyen Q, Mummadi S, Walker E, McCarthy K, Dweik RA. Heart rate recovery is an important predictor of outcomes in patients with connective tissue disease-associated pulmonary hypertension. Pulm Circ. 2015;5(3):565–576. doi:10.1086/682432
44. Gadre A, Ghattas C, Han X, Wang X, Minai O, Highland KB. Six-minute walk test as a predictor of diagnosis, disease severity, and clinical outcomes in scleroderma-associated pulmonary hypertension: the DIBOSA study. Lung. 2017;195(5):529–536. doi:10.1007/s00408-017-0034-1
45. Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis. 2009;68(12):1878–1884. doi:10.1136/ard.2008.095836
46. Bezante GP, Rollando D, Sessarego M, et al. Cardiac magnetic resonance imaging detects subclinical right ventricular impairment in systemic sclerosis. J Rheumatol. 2007;34(12):2431–2437.
47. Humbert M, Gerry Coghlan J, Khanna D. Early detection and management of pulmonary arterial hypertension. Eur Respir Rev. 2012;21(126):306–312. doi:10.1183/09059180.00005112
48. Morrisroe K, Stevens W, Huq M, et al. Survival and quality of life in incident systemic sclerosis-related pulmonary arterial hypertension. Arthritis Res Ther. 2017;19(1):122. doi:10.1186/s13075-017-1341-x
49. Vachiery JL, Coghlan G. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev. 2009;18(113):162–169. doi:10.1183/09059180.00003209
50. Thakkar V, Stevens WM, Prior D, et al. N-terminal pro-brain natriuretic peptide in a novel screening algorithm for pulmonary arterial hypertension in systemic sclerosis: a case-control study. Arthritis Res Ther. 2012;14(3):R143. doi:10.1186/ar3876
51. Kowal-bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017;76(8):1327–1339. doi:10.1136/annrheumdis-2016-209909
52. Galie N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889. doi:10.1183/13993003.01889-2018
53. Benza RL, Gomberg-Maitland M, Elliott CG, et al. Predicting survival in patients with pulmonary arterial hypertension: the REVEAL risk score calculator 2.0 and comparison with ESC/ERS-based risk assessment strategies. Chest. 2019;156(2):323–337. doi:10.1016/j.chest.2019.02.004
54. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023–1030. doi:10.1164/rccm.200510-1668OC
55. Hoeper MM, Kramer T, Pan Z, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J. 2017;50(2):1700740.
56. Kylhammar D, Kjellstrom B, Hjalmarsson C, et al. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur Heart J. 2018;39(47):4175–4181. doi:10.1093/eurheartj/ehx257
57. Boucly A, Weatherald J, Savale L, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J. 2017;50(2):1700889. doi:10.1183/13993003.00889-2017
58. Frech T, Walker AE, Barrett-o’keefe Z, et al. Systemic sclerosis induces pronounced peripheral vascular dysfunction characterized by blunted peripheral vasoreactivity and endothelial dysfunction. Clin Rheumatol. 2015;34(5):905–913. doi:10.1007/s10067-014-2834-5
59. Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum. 2011;41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
60. Galie N, McLaughlin VV, Rubin LJ, Simonneau G. An overview of the 6th world symposium on pulmonary hypertension. Eur Respir J. 2019;53(1):1802148. doi:10.1183/13993003.02148-2018
61. Barnett CF, Machado RF. Sildenafil in the treatment of pulmonary hypertension. Vasc Health Risk Manag. 2006;2(4):411–422. doi:10.2147/vhrm.2006.2.issue-4
62. Khaybullina D, Patel A, Zerilli T. Riociguat (adempas): a novel agent for the treatment of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. P TPeer-Rev J Formulary Manage. 2014;39(11):749–758.
63. Galie N, Muller K, Scalise AV, Grunig E. PATENT PLUS: a blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. Eur Respir J. 2015;45(5):1314–1322. doi:10.1183/09031936.00105914
64. Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–2157. doi:10.1056/NEJMoa050010
65. Badesch DB, Hill NS, Burgess G, et al. Sildenafil for pulmonary arterial hypertension associated with connective tissue disease. J Rheumatol. 2007;34(12):2417–2422.
66. Galie N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894–2903. doi:10.1161/CIRCULATIONAHA.108.839274
67. Galie N, Denton CP, Dardi F, et al. Tadalafil in idiopathic or heritable pulmonary arterial hypertension (PAH) compared to PAH associated with connective tissue disease. Int J Cardiol. 2017;235:67–72. doi:10.1016/j.ijcard.2017.02.094
68. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–340. doi:10.1056/NEJMoa1209655
69. Humbert M, Coghlan JG, Ghofrani HA, et al. Riociguat for the treatment of pulmonary arterial hypertension associated with connective tissue disease: results from PATENT-1 and PATENT-2. Ann Rheum Dis. 2017;76(2):422–426. doi:10.1136/annrheumdis-2015-209087
70. Raina A, Benza RL, Farber HW. Replacing a phosphodiesterase-5 inhibitor with riociguat in patients with connective tissue disease-associated pulmonary arterial hypertension: a case series. Pulm Circ. 2017;7(3):741–746. doi:10.1177/2045893217721694
71. Marra AM, Halank M, Benjamin N, et al. Right ventricular size and function under riociguat in pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension (the RIVER study). Respir Res. 2018;19(1):258. doi:10.1186/s12931-018-0957-y
72. Marra AM, Egenlauf B, Ehlken N, et al. Change of right heart size and function by long-term therapy with riociguat in patients with pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Int J Cardiol. 2015;195:19–26. doi:10.1016/j.ijcard.2015.05.105
73. Chester AH, Yacoub MH. The role of endothelin-1 in pulmonary arterial hypertension. Global Cardiology Sci Pract. 2014;2014(2):62–78. doi:10.5339/gcsp.2014.29
74. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903. doi:10.1056/NEJMoa012212
75. Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010–3019. doi:10.1161/CIRCULATIONAHA.107.742510
76. Fischer A, Denton CP, Matucci-Cerinic M, et al. Ambrisentan response in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH) - A subgroup analysis of the ARIES-E clinical trial. Respir Med. 2016;117:254–263. doi:10.1016/j.rmed.2016.06.018
77. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. doi:10.1056/NEJMoa1213917
78. Rhee RL, Gabler NB, Sangani S, Praestgaard A, Merkel PA, Kawut SM. Comparison of treatment response in idiopathic and connective tissue disease-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(9):1111–1117. doi:10.1164/rccm.201507-1456OC
79. Matucci-Cerinic M, Denton CP, Furst DE, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2011;70(1):32–38. doi:10.1136/ard.2010.130658
80. Del Pozo R, Hernandez Gonzalez I, Escribano-subias P. The prostacyclin pathway in pulmonary arterial hypertension: a clinical review. Expert Rev Respir Med. 2017;11(6):491–503. doi:10.1080/17476348.2017.1317599
81. Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142(6):1383–1390. doi:10.1378/chest.11-2212
82. Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132(6):425–434. doi:10.7326/0003-4819-132-6-200003210-00002
83. Klings ES, Hill NS, Ieong MH, Simms RW, Korn JH, Farber HW. Systemic sclerosis-associated pulmonary hypertension: short- and long-term effects of epoprostenol (prostacyclin). Arthritis Rheum. 1999;42(12):2638–2645. doi:10.1002/(ISSN)1529-0131
84. Launay D, Hachulla E, Hatron PY, et al. Aerosolized iloprost in CREST syndrome related pulmonary hypertension. J Rheumatol. 2001;28(10):2252–2256.
85. Li JH, Zhang HD, Wang ZZ, et al. Acute iloprost inhalation improves right ventricle function in pulmonary artery hypertension: a cardiac magnetic resonance study. Front Pharmacol. 2018;9:1550. doi:10.3389/fphar.2018.01550
86. Tapson VF, Gomberg-maitland M, McLaughlin VV, et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: a prospective, multicenter, open-label, 12-week trial. Chest. 2006;129(3):683–688. doi:10.1378/chest.129.3.683
87. Oudiz RJ, Schilz RJ, Barst RJ, et al. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest. 2004;126(2):420–427. doi:10.1378/chest.126.2.420
88. Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800–804. doi:10.1164/ajrccm.165.6.2106079
89. Caravita S, Wu SC, Secchi MB, Dadone V, Bencini C, Pierini S. Long-term effects of intermittent Iloprost infusion on pulmonary arterial pressure in connective tissue disease. Eur J Intern Med. 2011;22(5):518–521. doi:10.1016/j.ejim.2011.02.005
90. Gaine S, Chin K, Coghlan G, et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur Respir J. 2017;50(2):1602493. doi:10.1183/13993003.02493-2016
91. Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522–2533. doi:10.1056/NEJMoa1503184
92. Hassoun PM, Zamanian RT, Damico R, et al. Ambrisentan and tadalafil up-front combination therapy in scleroderma-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(9):1102–1110. doi:10.1164/rccm.201507-1398OC
93. Coghlan JG, Galie N, Barbera JA, et al. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis. 2017;76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
94. Jansa P, Pulido T. Macitentan in pulmonary arterial hypertension: a focus on combination therapy in the SERAPHIN trial. Am J Cardiovasc Drugs. 2018;18(1):1–11. doi:10.1007/s40256-017-0260-1
95. Coghlan JG, Channick R, Chin K, et al. Targeting the prostacyclin pathway with selexipag in patients with pulmonary arterial hypertension receiving double combination therapy: insights from the randomized controlled GRIPHON study. Am J Cardiovasc Drugs. 2018;18(1):37–47. doi:10.1007/s40256-017-0262-z
96. Barst RJ, Gibbs JS, Ghofrani HA, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S78–S84. doi:10.1016/j.jacc.2009.04.017
97. Olsson KM, Delcroix M, Ghofrani HA, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation. 2014;129(1):57–65. doi:10.1161/CIRCULATIONAHA.113.004526
98. Preston IR, Roberts KE, Miller DP, et al. Effect of warfarin treatment on survival of patients with Pulmonary Arterial Hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation. 2015;132(25):2403–2411. doi:10.1161/CIRCULATIONAHA.115.018435
99. Ascha M, Zhou X, Rao Y, Minai OA, Tonelli AR. Impact on survival of warfarin in patients with pulmonary arterial hypertension receiving subcutaneous treprostinil. Cardiovasc Ther. 2017;35(5):e12281. doi:10.1111/cdr.2017.35.issue-5
100. Calderone A, Stevens W, Prior D, et al. Multicentre randomised placebo-controlled trial of oral anticoagulation with apixaban in systemic sclerosis-related pulmonary arterial hypertension: the SPHInX study protocol. BMJ Open. 2016;6(12):e011028. doi:10.1136/bmjopen-2016-011028
101. Aithala R, Alex AG, Danda D. Pulmonary hypertension in connective tissue diseases: an update. Int J Rheum Dis. 2017;20(1):5–24. doi:10.1111/1756-185X.13001
102. Sanchez O, Sitbon O, Jais X, Simonneau G, Humbert M. Immunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertension. Chest. 2006;130(1):182–189. doi:10.1378/chest.130.1.182
103. Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep. 2010;12(1):8–18. doi:10.1007/s11926-009-0078-1
104. Galie N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60–D72. doi:10.1016/j.jacc.2013.10.031
105. Grunig E, Maier F, Ehlken N, et al. Exercise training in pulmonary arterial hypertension associated with connective tissue diseases. Arthritis Res Ther. 2012;14(3):R148. doi:10.1186/ar3883
106. Miniati I, Guiducci S, Mecacci F, Mello G, Matucci-Cerinic M. Pregnancy in systemic sclerosis. Rheumatol. 2008;47(Suppl 3):iii16–iii18. doi:10.1093/rheumatology/ken174
107. Kawut SM, Taichman DB, Archer-chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest. 2003;123(2):344–350. doi:10.1378/chest.123.2.344
108. Allcock RJ, O’sullivan JJ, Corris PA. Palliation of systemic sclerosis-associated pulmonary hypertension by atrial septostomy. Arthritis Rheum. 2001;44(7):1660–1662. doi:10.1002/(ISSN)1529-0131
109. Schachna L, Medsger TA
110. Gadre SK, Minai OA, Wang XF, Zhang Q, Budev M, Tonelli AR. Lung or heart-lung transplant in pulmonary arterial hypertension: what is the impact of systemic sclerosis? Exp Clin Transplant. 2017;15(6):676–684. doi:10.6002/ect.2016.0209
111. Shah RJ, Boin F. Lung transplantation in patients with systemic sclerosis. Curr Rheumatol Rep. 2017;19(5):23. doi:10.1007/s11926-017-0650-z
112. Fernandez-codina A, Berastegui C, Pinal-fernandez I, et al. Lung transplantation in systemic sclerosis: a single center cohort study. Joint Bone Spine. 2018;85(1):79–84. doi:10.1016/j.jbspin.2017.03.012
113. Sottile PD, Iturbe D, Katsumoto TR, et al. Outcomes in systemic sclerosis-related lung disease after lung transplantation. Transplantation. 2013;95(7):975–980. doi:10.1097/TP.0b013e3182845f23
114. Lefevre G, Dauchet L, Hachulla E, et al. Survival and prognostic factors in systemic sclerosis-associated pulmonary hypertension: a systematic review and meta-analysis. Arthritis Rheum. 2013;65(9):2412–2423. doi:10.1002/art.38029
115. Chung L, Farber HW, Benza R, et al. Unique predictors of mortality in patients with pulmonary arterial hypertension associated with systemic sclerosis in the REVEAL registry. Chest. 2014;146(6):1494–1504. doi:10.1378/chest.13-3014
116. Chung L, Domsic RT, Lingala B, et al. Survival and predictors of mortality in systemic sclerosis-associated pulmonary arterial hypertension: outcomes from the pulmonary hypertension assessment and recognition of outcomes in scleroderma registry. Arthritis Care Res (Hoboken). 2014;66(3):489–495. doi:10.1002/acr.v66.3
117. Sobanski V, Giovannelli J, Lynch BM, et al. Characteristics and Survival of anti-U1 RNP antibody-positive patients with connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheumatol. 2016;68(2):484–493. doi:10.1002/art.39432
118. ClinicalTrials.gov. A phase 2 multicenter, randomized, double-blind, placebo-controlled study to assess the safety and efficacy of ifetroban in patients with diffuse cutaneous Systemic Sclerosis (SSc) or SSc-associated Pulmonary Arterial Hypertension (SSc-PAH). NCT02682511. Available from: https://clinicaltrials.gov/ct2/show/NCT02682511.
119. Center NIoHC. A randomized, double-blind, placebo-controlled, phASE II Multicenter trial of a monoclonal antibody to CD20 (Rituximab) for the Treatment of Systemic Sclerosis-Associated Pulmonary Arterial Hypertension (SSc-PAH). NCT01086540. Available from: https://clinicaltrials.gov/ct2/show/NCT01086540.
120. Zamanian R, Badesch D, Chung L, et al. Late Breaking Abstract - Safety and efficacy of B-cell depletion with rituximab for the treatment of systemic sclerosis-associated pulmonary arterial hypertension. Eur Respir J. 2019;54(suppl63):RCT1884.
121. ClinicalTrials.gov. A dose-ranging study of the efficacy and safety of bardoxolone methyl in patients with pulmonary hypertension. NCT02036970 [May 16, 2018]. Available from: https://clinicaltrials.gov/ct2/show/NCT02036970.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.