Back to Journals » Integrated Pharmacy Research and Practice » Volume 12
Management of Patients with Systemic Sclerosis-Associated Interstitial Lung Disease: A Focus on the Role of the Pharmacist
Received 30 November 2022
Accepted for publication 23 March 2023
Published 3 May 2023 Volume 2023:12 Pages 101—112
DOI https://doi.org/10.2147/IPRP.S399518
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jonathan Ling
Jessica Farrell,1 Lawrence Ho2
1Albany College of Pharmacy and Health Sciences and Albany Medical Center Division of Rheumatology, Albany, NY, USA; 2Center for Interstitial Lung Disease, University of Washington, Seattle, WA, USA
Correspondence: Jessica Farrell, Albany College of Pharmacy and Health Sciences and Albany Medical Center Division of Rheumatology, Albany, NY, USA, Email [email protected]
Abstract: Interstitial lung disease (ILD) is a common manifestation of systemic sclerosis (SSc), which becomes fibrosing and progressive in some patients. Regular monitoring of patients with SSc-ILD is important to assess progression and inform treatment decisions. Therapy for SSc-ILD may include immunomodulatory and antifibrotic therapies. Therapeutic decisions should be made on a case-by-case basis, ideally following multidisciplinary discussion. Most patients with SSc-ILD have several organ manifestations of SSc or comorbidities and are taking a complex medication regimen. Patients with SSc are particularly susceptible to gastrointestinal side-effects of medications due to the gastrointestinal manifestations of the disease. Pharmacists play an important role in the management of patients with SSc-ILD by assisting patients with access to medications, optimizing medication regimens, and advising on alternative dosage forms. Pharmacists can also contribute to patient education to help patients better understand their treatment and how to prevent and manage potential side effects.
Keywords: immunosuppressants, lung disease, pulmonary fibrosis, scleroderma, therapeutics
Introduction
Systemic sclerosis (SSc) is a complex and heterogeneous autoimmune disease characterized by thickening of the skin and variable dysfunction in internal organs.1 There are two major subsets of SSc, differentiated by the extent of skin involvement: limited cutaneous SSc (lcSSc) and diffuse cutaneous SSc (dcSSc).2 Interstitial lung disease (ILD) is a common manifestation of SSc and the leading cause of SSc-related death.3 The management of patients with SSc requires a multidisciplinary approach and often involves complex medication regimens to address the main features of the disease: inflammation, autoimmunity, vascular disease, and fibrosis. Pharmacists have the opportunity to be part of the multidisciplinary team treating patients with SSc by enhancing access to treatments and optimizing medication regimens to minimize problems such as adverse events, drug interactions, and issues around administration. In this article, we discuss the management of patients with SSc-ILD, with a focus on the role of pharmacists.
ILD in Patients with SSc
Estimates of the prevalence of ILD in patients with SSc vary depending on the methodology used to identify cases. In a cohort of 1168 patients in the Canadian Scleroderma Research Group registry, 64% of patients with high-resolution computed tomography (HRCT) scans were diagnosed with ILD, compared with 26% of patients based on physical examination and 22% based on chest X-ray.4 At the time of entry into the European Scleroderma Trials and Research (EUSTAR) database, ILD was present on HRCT in 35% of 3099 patients with limited cutaneous SSc and 57% of 1881 patients with diffuse cutaneous SSc.5
Several risk factors for the development of ILD in patients with SSc have been identified, including diffuse cutaneous SSc, older age at onset, African-American race, and the presence of anti-topoisomerase I antibody.6,7 However, it is important to be aware that all patients with SSc are at risk of ILD and should be screened. In a Delphi consensus study, experts agreed that screening for SSc-ILD should involve clinical examination (including chest auscultation), HRCT, pulmonary function tests and assessment of respiratory symptoms (Figure 1).8 Obtaining an HRCT scan is crucial, as lung abnormalities evident on HRCT may not be accompanied by abnormal pulmonary function tests or symptoms.9 It is also important to consider other potential causes of dyspnea, including gastroesophageal reflux, pulmonary hypertension, and anemia, which are common comorbidities in patients with SSc-ILD.10 Diagnosis and assessment of SSc-ILD should be made in the context of multidisciplinary discussion of clinical and radiological data, with input from, at minimum, a pulmonologist, a rheumatologist and a radiologist.
 |
Figure 1 Proposed algorithm for the screening, monitoring and management of patients with SSc-ILD. Reprinted from The Lancet Rheumatology, Vol. 2, Hoffmann-Vold A-M et al, The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements, Pages E71-E83, Copyright 2020, with permission from Elsevier.8 Abbreviations: HRCT, high-resolution computed tomography; ILD, interstitial lung disease; SSc, systemic sclerosis. |
Monitoring and Progression of SSc-ILD
The pre-symptomatic phase of SSc-ILD may be when the most rapid progression occurs. However, patients with early diffuse SSc may be so limited by musculoskeletal manifestations (arthropathy, tendinopathy, myopathy) that dyspnea is not the limiting factor in their activity even when lung disease is present. The clinical course of SSc-ILD is variable and difficult to predict. Patients should be monitored based on pulmonary function tests, assessment of symptoms and repeat HRCT, when required (Figure 1).8 Some patients have relatively stable disease, while in others, SSc-ILD is progressive and life-limiting.11,12 Guidelines issued by international respiratory associations define progressive pulmonary fibrosis (PPF) in a patient with an ILD other than idiopathic pulmonary fibrosis (IPF) as ≥2 of the following occurring within the past year with no alternative explanation: (1) worsening respiratory symptoms; (2) radiological evidence of progression; and (3) physiological evidence of progression based on decline in the percent predicted value for forced vital capacity (FVC) and/or diffusing capacity of the lungs for carbon monoxide (DLco).13 It is important to remember that an isolated decline in DLco in a patient with SSc may reflect the development or progression of pulmonary hypertension rather than ILD.14 Guidelines issued by the European Society of Cardiology and European Respiratory Society recommend that patients with SSc undergo annual screening for pulmonary arterial hypertension based on echocardiography and DLco.15
A greater extent of fibrotic ILD on HRCT is associated with mortality. In a nationwide cohort of Norwegian patients with SSc, the standardized mortality rate over a mean observation period of 10.1 years increased from 2.2 in patients with no lung fibrosis on HRCT at baseline to 8.0 in patients with >25% lung fibrosis.11 Among patients with an FVC of at least 80% predicted at baseline, 5-year survival was 83% in patients with no lung fibrosis on HRCT versus 69% in patients with any lung fibrosis.11 In a UK study of 162 patients with SSc-ILD, a relative decline in FVC (mL) of ≥10% at 1 year, or a relative decline in FVC (mL) of 5–9% with a relative decline in DLco of >15% at 1 year, was associated with an almost two-fold increase in mortality over 15 years (HR 1.96 [95% CI: 1.25, 3.08]).16 Worsening of respiratory symptoms may also suggest progression of ILD, but these need to be carefully evaluated as worsening of cough may reflect an increase in esophageal dysfunction, while worsening of dyspnea may reflect onset or worsening of comorbidities such as pulmonary hypertension or cardiac disease.
Drug Therapies for SSc-ILD
Most patients with SSc-ILD are taking immunomodulatory medication to treat one or more manifestation of their disease.17 These may include corticosteroids, mycophenolate mofetil (MMF), cyclophosphamide, rituximab, or tocilizumab. Corticosteroids are used in varying doses, including pulse therapy, but there is no evidence that they slow the progression of SSc-ILD. Cyclophosphamide and MMF have shown efficacy in slowing the rate of progression of SSc-ILD in randomized, double-blind, controlled clinical trials.18,19 Tocilizumab has been approved by the Food and Drug Administration (FDA) for slowing lung function decline in patients with SSc-ILD based on results observed in a subgroup of patients with SSc-ILD from a randomized, placebo-controlled trial in patients with dcSSc and markers of inflammation.20 Antifibrotic therapy may also be considered. The tyrosine kinase inhibitor nintedanib has been FDA-approved for slowing lung function decline in patients with SSc-ILD based on the results of a randomized placebo-controlled trial in patients with SSc-ILD.21
There are no established treatment guidelines for SSc-ILD, but algorithms for when drug treatment for SSc-ILD should be initiated and escalated, and for which therapeutic options should be considered, have been proposed by expert groups.8,22,23 Treatment decisions should be made on a case-by-case basis, ideally following multidisciplinary discussion, based on considerations including the extent of fibrosis on HRCT, lung function tests, symptoms, risk factors for progression, and evidence of progression, as well as discussion with the patient.8,22,24 Decisions on when to cease or escalate therapy are complicated by the unpredictable course of SSc-ILD. Progression of SSc-ILD while on therapy does not necessarily reflect “treatment failure”, as the patient may have progressed at a faster rate if the therapy had not been initiated.
Mycophenolate Mofetil
MMF is an inhibitor of T- and B-lymphocyte proliferation. In Scleroderma Lung Study II (SLS II), conducted in 142 patients with SSc-ILD, treatment with MMF (at a target dose of 3g/day) for 2 years resulted in similar improvements in FVC % predicted compared with oral cyclophosphamide for 1 year followed by placebo for 1 year (2.2% vs 2.9% predicted, respectively) (Table 1), with better tolerability.25 However, a randomized placebo-controlled trial of MMF (at a target dose of 2g/day) in 41 patients with SSc-ILD and FVC ≥70% predicted at baseline showed no benefit on decline in FVC over 6 months.26 These results suggest that optimizing the dose of MMF to 3g/day in patients with SSc-ILD may be beneficial.
 |
Table 1 Randomized, Double-Blind, Controlled Trials of Drugs in Patients with SSc-ILDa |
Cyclophosphamide
Cyclophosphamide is an alkylating agent used in the treatment of cancers and autoimmune diseases, which prevents cell division by cross-linking DNA strands. Dosing regimens vary but include oral dosing (up to 2 mg/kg/day) for 12 months and monthly intravenous (IV) infusions for 6 months. IV therapy is generally preferred due to a lower cumulative dose and fewer side-effects. In a randomized, double-blind trial conducted in 45 patients with SSc-ILD, treatment with low-dose corticosteroids and IV cyclophosphamide (600 mg/m2) for 6 months followed by oral azathioprine for 6 months resulted in a trend towards an improvement in FVC % predicted compared with treatment with placebo for 1 year (2.4% vs −3.0% predicted).18 In Scleroderma Lung Study I, conducted in 158 patients with SSc-ILD, oral cyclophosphamide (≤2 mg/kg/day) for 1 year slowed the decline in FVC % predicted compared with placebo (−1.0% vs −2.6% predicted).19 The long-term use of cyclophosphamide is limited by its toxicity.27
Rituximab
Rituximab is a monoclonal antibody targeting CD20 positive B-lymphocytes. No randomized, double-blind, placebo-controlled trials of rituximab specifically in patients with SSc-ILD have been published, but a number of open-label and observational trials suggest that rituximab may have beneficial effects on lung function in these patients.28,29 For example, in an open-label randomized trial of 6 months’ treatment with rituximab versus IV cyclophosphamide in 60 patients with SSc-ILD, FVC improved in the rituximab group (from 61.3 to 67.5% predicted) and declined slightly in the cyclophosphamide group (from 59.3 to 58.1% predicted).29 The randomized controlled RECITAL trial evaluated 101 patients with autoimmune-disease related ILDs, including 39 patients with SSc-ILD, who were randomized to receive rituximab or IV cyclophosphamide.30 The primary endpoint of change in FVC (mL) at 24 weeks was not met (mean 97 versus 99; difference based on linear mixed effects model of 40 [95% CI 153, −74] in favor of cyclophosphamide; p=0.49). A trial of rituximab given in combination with MMF versus MMF alone (EvER-ILD; NCT02990286) in patients with various ILDs including SSc-ILD is yet to report results.
Tocilizumab
Tocilizumab, an interleukin-6 (IL-6) receptor antagonist, is used in the treatment of autoimmune and inflammatory conditions. IL-6 levels are elevated in patients with SSc and are associated with the development of SSc-ILD.31 Tocilizumab was investigated as a treatment for SSc in a randomized placebo-controlled Phase 2 trial in patients with early SSc (≤60 months) (faSScinate), which failed to meet the primary endpoint of change in skin fibrosis measured using the modified Rodnan skin score (mRSS), but suggested that tocilizumab may have a benefit on FVC.32,33 The Phase 3 focuSSced trial evaluated 210 SSc patients with early diffuse SSc (≤60 months) who were randomized to receive tocilizumab 162 mg or placebo subcutaneously once weekly for 48 weeks. The primary endpoint of change in mRSS was not statistically significant; however, the secondary outcomes of change in FVC % predicted at week 48 favored the tocilizumab group with a mean difference of 4.2 (95% CI 2.0–6.4; nominal p=0.0002).34 Among the subgroup of 136 patients with SSc-ILD, lung function was preserved in the tocilizumab group, with a mean change in FVC % predicted at week 48 of −0.1 compared with −6.4 in the placebo group (difference: 6.5]; p<0.0001).34
Anti-Fibrotic Agents
Nintedanib
Nintedanib is an inhibitor of multiple tyrosine kinases that inhibits processes fundamental to the progression of pulmonary fibrosis, such as the proliferation and migration of fibroblasts and the deposition of extracellular matrix.35 Nintedanib has been FDA-approved for slowing decline in lung function in patients with SSc-ILD, as well as for the treatment of IPF and other chronic fibrosing ILDs with a progressive phenotype. It received a conditional recommendation for use in patients with PPF in the latest international guidelines issued by respiratory associations.13 Nintedanib is given orally 150 mg every 12 hours. The SENSCIS trial was a randomized, double-blind, placebo-controlled trial that evaluated the efficacy and safety of nintedanib in 576 patients with SSc with first non-Raynaud’s symptom within ≤7 years and an extent of fibrotic ILD on HRCT ≥10%.21 A broad population of patients was enrolled, including patients with limited cutaneous and diffuse cutaneous disease. Inclusion criteria allowed patients who were receiving prednisone (up to 10 mg per day), mycophenolate or methotrexate at a stable dose for at least 6 months before randomization. Nintedanib significantly reduced the rate of decline in FVC in mL/year over 52 weeks compared with placebo (−52.4 versus −93.3; difference 41.0 [95% CI 2.9, 79.0]; p=0.04) (Figure 2). It is important to note that 48% of the patients in the trial had been receiving a stable dose of mycophenolate for ≥6 months at baseline.36 Nintedanib can be an appropriate add-on therapy for patients who progress, especially within the first few years of treatment with MMF or cyclophosphamide. It may also be an alternative treatment for patients who experience toxicities or are unable to take MMF or cyclophosphamide. Limitations to the use of nintedanib include side-effects such as gastrointestinal (GI) side-effects, mainly diarrhea.
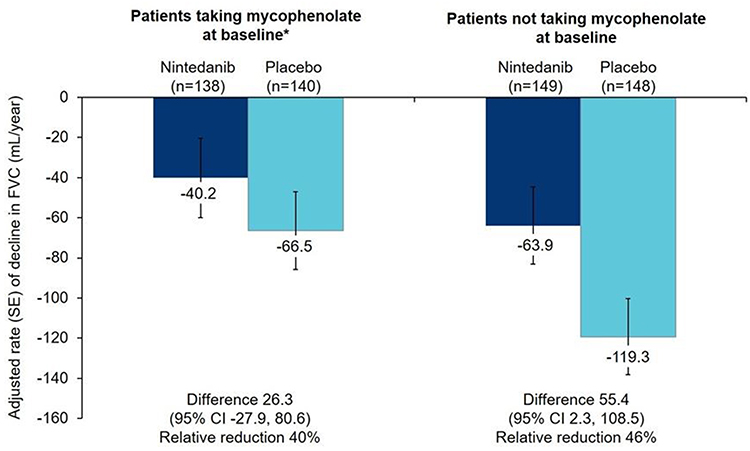 |
Figure 2 Rate of decline in FVC (mL/year) over 52 weeks in the SENSCIS trial in patients randomized to nintedanib or placebo. *Patients taking mycophenolate had been taking a stable dose of mycophenolate for at least 6 months before randomization. Reprinted from Lancet Respir Med, Vol (issue) 9(1), Highland KB, Distler O, Kuwana M, et al, Efficacy and safety of nintedanib in patients with systemic sclerosis-associated interstitial lung disease treated with mycophenolate: a subgroup analysis of the SENSCIS trial, Pages 96–106, Copyright 2021, with permission from Elsevier.36 Abbreviation: FVC, forced vital capacity. |
Pirfenidone
Pirfenidone is an approved treatment for IPF. Its mechanism of action remains poorly understood. An open-label study suggested that the tolerability of pirfenidone in patients with SSc-ILD is similar to that in patients with IPF.37 In a randomized placebo-controlled study in 34 patients with SSc-ILD, pirfenidone showed no benefit on FVC over 6 months.38 The efficacy and safety of pirfenidone in combination with mycophenolate versus mycophenolate alone in approximately 150 patients with SSc-ILD are being investigated in Scleroderma Lung Study III (NCT03221257).
Managing Medication Safety and Tolerability
Management of SSc, a multisystem disorder, often involves complex medication regimens. Pharmacists can help minimize medication-related problems, assist with alternative dosage forms, and provide patient education to help improve the tolerability of treatments.
Corticosteroids
Adverse effects associated with chronic corticosteroid use are well-known, including risk of infection, weight gain, hypertension, hyperglycemia, myopathy, cardiovascular disease, osteoporosis, and neuropsychiatric effects. Specific to patients with SSc, the use of medium to high-dose corticosteroids has been associated with an increased risk of scleroderma renal crisis.39 This risk may be further elevated in patients with anti-RNA polymerase autoantibodies.40 While low-dose (<15 mg/day) corticosteroids can be used to treat skin and musculoskeletal manifestations of SSc, it is important to use the lowest effective dose for the shortest duration. Pharmacists can educate patients to recognize the early signs of steroid-induced adverse effects and ensure that screening such as bone mineral density testing is performed regularly. Tools such as the “renal crisis prevention card”, which aims to improve outcomes in patients at high risk of SSc renal crisis,41 may be of value in educating patients and HCPs.
Mycophenolate Mofetil
Side-effects of MMF include gastrointestinal events, cytopenia, and malignancy.42 The most commonly seen issues with MMF in patients with SSc are diarrhea, nausea, and difficulty swallowing the large tablet. MMF should be started at a low dose and titrated up to a target of 1.5–3 g daily. Generally dosing that starts at 500 mg orally twice daily and is increased by 500 mg weekly or biweekly is tolerated well. In patients who do not develop tolerance to gastrointestinal effects, switching to mycophenolic acid may be beneficial, although this has not been studied in patients with SSc. It is important to note that MMF and mycophenolic acid are not interchangeable. Based on data from renal transplant patients, mycophenolate acid (as mycophenolate sodium) 720 mg bid is considered equivalent to MMF 1000 mg bid.43 MMF is available in three forms for oral dosage: 250 mg capsules, 500 mg tablets, 200 mg/mL suspension for reconstitution. Patients with difficulty swallowing may find it easier to take the capsule than the tablet and may benefit from using saliva substitute products. Some patients may find the suspension product more tolerable; however, this should not be given with any other medications or liquids and a lack of insurance coverage may be a barrier.
All patients taking immunosuppressants should take steps to minimize their risk of infections. Pharmacists can remind patients about techniques such as hand washing, cleaning hard surfaces, and wearing masks in public places. Patients should be counseled to hold MMF if they develop a serious infection. Based on guidance from the American College of Rheumatology (ACR), if disease is stable, MMF should be held for 1 week after each COVID-19 vaccination to allow for a more robust immune response.44 Pharmacists can assist with monitoring for neutropenia; if a patient becomes neutropenic, therapy should be held or the dose reduced. Antacids with magnesium or aluminum hydroxide, proton pump inhibitors (PPIs), drugs that interfere with enterohepatic recirculation and calcium-free phosphate binders may decrease exposure to mycophenolate and possibly reduce its efficacy.45 In a small study in patients with autoimmune diseases, plasma concentrations of mycophenolic acid were lower in patients who received a PPI 30 minutes after MMF intake, but not in those who received a PPI 90 minutes after MMF intake.46 Patients should at least avoid taking a PPI at the same time as MMF. Alternatively, the drug-drug interaction can be avoided by switching to mycophenolic acid.
Cyclophosphamide
Side-effects of cyclophosphamide, which include hematuria, leukopenia, nausea and malignancy, limit its long-term use.27 Hemorrhagic cystitis, pyelitis, ureteritis, and hematuria have been reported in patients treated with cyclophosphamide.47 Aggressive hydration and frequent micturition can reduce bladder toxicity, and mesna can be used to prevent severe bladder toxicity.48 Similar to MMF, the ACR guidance for COVID-19 vaccination recommends timing cyclophosphamide administration for approximately 1 week after each vaccine dose, when feasible.44
Rituximab
Adverse events associated with rituximab vary based on indication and dosing regimen, but include an increased risk of infection (including reactivation of viral infections such as hepatitis B), risk of infusion-related reactions, late-onset neutropenia, development of human antichimeric antibodies and rare mucocutaneous events. Due to the prolonged depletion of B-cells, risk of serious infections is a significant concern in patients with SSc treated with rituximab. Pharmacists can play an important role in educating patients about infection control techniques and in monitoring patients between provider visits. Patients treated with rituximab may develop hypogammaglobulinemia, increasing the risk for serious or recurrent infections.49 Immunoglobulin levels should be measured prior to repeat treatments. It may be helpful to assess risk factors for an increased risk of infection (eg advanced lung disease, use of corticosteroids, older age). Some patients receiving repeat courses of rituximab experience sustained low IgG.50 Patients with low IgG who experience recurrent or serious infections may require therapy with intravenous immunoglobulin (IVIg). The ACR COVID-19 vaccine guidance recommends initiating the vaccine approximately 4 weeks prior to the next scheduled rituximab cycle and delaying rituximab until 2–4 weeks after the final vaccine, if disease activity allows.44
Tocilizumab
Side effects associated with the use of tocilizumab include risk of infection (including reactivation of herpes zoster), neutropenia, thrombocytopenia, increased lipids and liver enzymes, and intestinal perforations in patients with a history of diverticulitis. Adverse events seen in trials in patients with SSc were similar to those seen in other studies of tocilizumab,32,34 with infections the most common adverse events. Pharmacists can assist providers in laboratory monitoring and adverse event counseling. Due to the lack of an effect of IL-6 inhibition on vaccine response, the ACR COVID-19 vaccine guidance does not recommend adjustment of the dosing or timing of tocilizumab administration around vaccination.44
Nintedanib
The most common side-effect associated with nintedanib is diarrhea.51 It is recommended that nintedanib be taken with food to improve gastrointestinal tolerability. In cases of diarrhea, patients should be treated promptly with adequate hydration and medication such as loperamide. Dose reduction to 100 mg bid and/or treatment interruption may be necessary. Weight loss occurs in some patients, but rarely requires permanent discontinuation of treatment.51 Elevations in liver enzymes may occur in patients treated with nintedanib and periodic monitoring is required.52 Data from the SENSCIS trial suggest that the dose adjustments used to manage adverse events did not impact the benefit of nintedanib on reducing FVC decline.51 However, the most effective dose of nintedanib in reducing the progression of ILD is 150 mg bid53 and it is not possible to predict which patients will develop side-effects. Some clinicians use uptitration in patients sensitive to GI intolerance, but the recommended usage is to start patients on the 150 mg bid dose and then use dose reduction or treatment interruption to manage side-effects if they occur.
Nintedanib reaches its maximum plasma concentration 2 to 4 hours after oral administration and steady state is reached within 1 week.54 As nintedanib is a substrate for p-glycoprotein (P-gp) and, to a minor extent, CYP3A, concomitant treatment with potent inhibitors or inducers of p-gp/CYP3A can affect exposure to nintedanib.55 Patients receiving nintedanib and P-gp/CYP3A4 inhibitors, such as ketoconazole and erythromycin, should be monitored for tolerability; co-administration of nintedanib and P-gp inducers, such as rifampicin and St. John’s wort, should be avoided.52 Exposure to nintedanib is increased in subjects with mild or moderate hepatic impairment.52 Patients with mild hepatic impairment (Child-Pugh A) can be treated with nintedanib 100 mg bid. Nintedanib is not recommended for patients with moderate or severe hepatic impairment (Child-Pugh B or C). As nintedanib is an inhibitor of the vascular endothelial growth factor receptor, bleeding is a potential risk.52 Patients at known risk of bleeding were excluded from trials of nintedanib. Arterial thromboembolic events have been reported and caution is recommended when using nintedanib in patients at higher cardiovascular risk, including known coronary artery disease.52 Nintedanib may increase the risk of gastrointestinal perforation and caution is recommended when treating patients who have had recent abdominal surgery, have a history of diverticular disease, or are receiving corticosteroids or non-steroidal anti-inflammatory drugs.52
Other Medication-Related Issues
Male and female fertility may be impaired in patients treated with cyclophosphamide.47 Cyclophosphamide and nintedanib can cause fetal harm and should not be used by women who are or may become pregnant. Use of mycophenolate during pregnancy is associated with an increased risk of pregnancy loss and congenital malformations and should be avoided if safer treatment options are available.45
It is important that pharmacists involved with counseling patients with SSc-ILD be aware of how SSc may affect organ systems and how those effects can impact medication administration, absorption and tolerability. Management of gastrointestinal problems is particularly important, as gastrointestinal complications, including dysmotility and diarrhea, are common manifestations of SSc.56 Issues with swallowing may increase the risk of aspiration and put patients at risk of aspiration pneumonia. Historically there have been concerns over the toxicity of methotrexate in patients with autoimmune diseases, including concerns over damage to the lungs. The use of methotrexate in patients with SSc remains controversial,17 particularly in patients at risk of kidney dysfunction or pericardial or pleural effusions.
Pharmacists can advise on access to medications and assist with insurance authorization. Nintedanib is usually initiated in the outpatient setting and must be dispensed through a specialty pharmacy. Patients with commercial or government insurance may be eligible for the manufacturer’s copay program or for copay assistance through independent foundations. Obtaining insurance coverage for off-label use of drugs can be challenging and pharmacists can help navigate the complex system by providing the insurance plan with the necessary information and helping patients navigate barriers in communicating with specialty pharmacies.
Stem Cell Transplant and Lung Transplant
In patients with progressive SSc, hematopoietic stem cell transplantation (HSCT) improves long-term survival but is associated with significant short-term mortality.57 Treatment guidelines issued by the European Alliance of Associations for Rheumatology (EULAR) recommend that HSCT be considered for patients with rapidly progressive SSc at risk of organ failure based on careful assessment of a patient’s risk-benefit profile.58 HSCT should only be performed at expert centers.
Lung transplant is a therapeutic option for a select minority of patients with SSc-ILD who progress despite therapy and do not have contraindications.59 Post-transplant mortality in patients with SSc-ILD is similar to that in patients with other forms of ILD.60 Early referral is important to allow timely assessment of any contraindications for lung transplantation.
Supportive Care
Patients with SSc-ILD should be offered supportive care as needed throughout the course of their disease. This may include symptom relief, pulmonary rehabilitation, or supplemental oxygen.61,62 Many patients with SSc find support groups such as those run by the Scleroderma Foundation (https://www.scleroderma.org) highly valuable for providing emotional support and a sense of community, as well as information to help them understand their disease and be involved in decisions about their care.63 Patients with SSc often have high health literacy and would like to receive as much information as they can. Pharmacists have the opportunity to provide medication-focused support to these patients.
Conclusions
Pharmacists have an important role to play within the care team for patients with SSc-ILD by advising on the appropriate use of drugs and on how to avoid, monitor, and manage medication-related problems. Patients with SSc often have multiple systemic disease manifestations and comorbidities, which complicate their care, increase the risk of adverse effects of medications, and make the expertise of a pharmacist particularly valuable. Patients with SSc-ILD are often taking several medications, which may include immunomodulatory and antifibrotic therapies. They require regular monitoring to assess the progression of their ILD, inform treatment decisions, and ensure that side effects can be managed promptly. Pharmacists can contribute to patient education, helping patients to understand the aims of therapy, and how to avoid or manage medication-related problems.
Acknowledgments
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors did not receive payment for development of this article. The authors acknowledge the contribution of Lee Shapiro to an earlier draft of this article. Writing support was provided by Julie Fleming and Wendy Morris of Fleishman-Hillard, London, UK, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc. Boehringer Ingelheim was given the opportunity to review the article for medical and scientific accuracy as well as intellectual property considerations.
Disclosure
Jessica Farrell serves on the Rheumatology Advance Practice Provider board of directors and the Scleroderma Foundation medical and scientific advisory board and is an Associate Medical Officer at the Steffens Scleroderma Foundation. Lawrence Ho reports no conflicts of interest in this work.
References
1. van den Hoogen F, Khanna D, Fransen J, et al. classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;2013(65):2737–2747. doi:10.1002/art.38098
2. LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–205.
3. Rubio-Rivas M, Royo C, Simeón CP, Corbella X, Fonollosa V. Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Semin Arthritis Rheum. 2014;44:208–219.
4. Steele R, Hudson M, Lo E, Baron M. Canadian Scleroderma Research Group. Clinical decision rule to predict the presence of interstitial lung disease in systemic sclerosis. Arthritis Care Res. 2012;64:519–524. doi:10.1002/acr.21583
5. Frantz C, Huscher D, Avouac J, et al. Outcomes of limited cutaneous systemic sclerosis patients: results on more than 12,000 patients from the EUSTAR database. Autoimmun Rev. 2020;19:102452. doi:10.1016/j.autrev.2019.102452
6. Steen V, Domsic RT, Lucas M, et al. A clinical and serologic comparison of African American and Caucasian patients with systemic sclerosis. Arthritis Rheum. 2012;64:2986–2994. doi:10.1002/art.34482
7. Nihtyanova SI, Schreiber BE, Ong VH, et al. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheumatol. 2014;66:1625–1635. doi:10.1002/art.38390
8. Hoffmann-Vold AM, Maher TM, Philpot EE, et al. The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. Lancet Rheumatol. 2020;2:E71–E83. doi:10.1016/S2665-9913(19)30144-4
9. Tashkin DP, Volkmann ER, Tseng CH, et al. Relationship between quantitative radiographic assessments of interstitial lung disease and physiological and clinical features of systemic sclerosis. Ann Rheum Dis. 2016;75:374–381. doi:10.1136/annrheumdis-2014-206076
10. Gayle A, Schoof N, Alves M, et al. Healthcare resource utilization among patients in England with systemic sclerosis-associated interstitial lung disease: a retrospective database analysis. Adv Ther. 2020;37:2460–2476. doi:10.1007/s12325-020-01330-0
11. Hoffmann-Vold AM, Fretheim H, Halse AK, et al. Tracking impact of interstitial lung disease in systemic sclerosis in a complete nationwide cohort. Am J Respir Crit Care Med. 2019;200:1258–1266. doi:10.1164/rccm.201903-0486OC
12. Volkmann ER, Tashkin DP, Sim M, et al. Short-term progression of interstitial lung disease in systemic sclerosis predicts long-term survival in two independent clinical trial cohorts. Ann Rheum Dis. 2019;78:122–130. doi:10.1136/annrheumdis-2018-213708
13. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205:e18–e47. doi:10.1164/rccm.202202-0399ST
14. Steen V, Medsger TA. Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum. 2003;48:516–522. doi:10.1002/art.10775
15. Galiè N, Humbert M, Vachiery JL, et al. ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;2015(46):903–975.
16. Goh NS, Hoyles RK, Denton CP, et al. Short-term pulmonary function trends are predictive of mortality in interstitial lung disease associated with systemic sclerosis. Arthritis Rheumatol. 2017;69:1670–1678. doi:10.1002/art.40130
17. Adler S, Huscher D, Siegert E, et al. Systemic sclerosis associated interstitial lung disease - individualized immunosuppressive therapy and course of lung function: results of the EUSTAR group. Arthritis Res Ther. 2018;20:17. doi:10.1186/s13075-018-1517-z
18. Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54:3962–3970. doi:10.1002/art.22204
19. Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354:2655–2666. doi:10.1056/NEJMoa055120
20. Roofeh D, Lin CJF, Goldin J, et al. Tocilizumab prevents progression of early systemic sclerosis associated interstitial lung disease. Arthritis Rheumatol. 2021;73:1301–1310. doi:10.1002/art.41668
21. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. 2019;380:2518–2528. doi:10.1056/NEJMoa1903076
22. Roofeh D, Lescoat A, Khanna D. Treatment for systemic sclerosis-associated interstitial lung disease. Curr Opin Rheumatol. 2021;33:240–248.
23. Khanna D, Lescoat A, Roofeh D, et al. Systemic sclerosis-associated interstitial lung disease: how to incorporate two Food and Drug Administration-approved therapies in clinical practice. Arthritis Rheumatol. 2022;74:13–27. doi:10.1002/art.41933
24. Cheema TJ, Young M, Rabold E, et al. Patient and physician perspectives on systemic sclerosis-associated interstitial lung disease. Clin Med Insights Circ Respir Pulm Med. 2020;14:1179548420913281.
25. Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4:708–719. doi:10.1016/S2213-2600(16)30152-7
26. Naidu GSR, Sharma SK, Adarsh MB, et al. Effect of mycophenolate mofetil (MMF) on systemic sclerosis-related interstitial lung disease with mildly impaired lung function: a double-blind, placebo-controlled, randomized trial. Rheumatol Int. 2020;40:207–216.
27. Barnes H, Holland AE, Westall GP, et al. Cyclophosphamide for connective tissue disease-associated interstitial lung disease. Cochrane Database Syst Rev. 2018;1:CD010908.
28. Daoussis D, Melissaropoulos K, Sakellaropoulos G, et al. A multicenter, open-label, comparative study of B-cell depletion therapy with rituximab for systemic sclerosis-associated interstitial lung disease. Semin Arthritis Rheum. 2017;46:625–631. doi:10.1016/j.semarthrit.2016.10.003
29. Sircar G, Goswami RP, Sircar D, et al. Intravenous cyclophosphamide vs rituximab for the treatment of early diffuse scleroderma lung disease: open label, randomized, controlled trial. Rheumatology. 2018;57:2106–2113.
30. Maher TM, Tudor V, Saunders P, et al. Rituximab versus intravenous cyclophosphamide in patients with connective tissue disease-associated interstitial lung disease in the UK (RECITAL): a double-blind, double-dummy, randomised, controlled, phase 2b trial. Lancet Respir Med. 2022;11S2213–S2600.
31. Gourh P, Arnett FC, Assassi S, et al. Plasma cytokine profiles in systemic sclerosis: associations with autoantibody subsets and clinical manifestations. Arthritis Res Ther. 2009;11:R147.
32. Khanna D, Denton CP, Jahreis A, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet. 2016;387:2630–2640.
33. Khanna D, Denton CP, Lin CJF, et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: results from the open-label period of a Phase II randomised controlled trial (faSScinate). Ann Rheum Dis. 2018;77:212–220. doi:10.1136/annrheumdis-2017-211682
34. Khanna D, Lin CJF, Furst DE, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2020;8:963–974.
35. Wollin L, Distler JH, Denton CP, et al. Rationale for the evaluation of nintedanib as a treatment for systemic sclerosis–associated interstitial lung disease. J Scleroderma Relat Disord. 2019;4:212–218. doi:10.1177/2397198319841842
36. Highland KB, Distler O, Kuwana M, et al. Efficacy and safety of nintedanib in patients with systemic sclerosis-associated interstitial lung disease treated with mycophenolate: subgroup analysis of the SENSCIS trial. Lancet Respir Med. 2021;9:96–106.
37. Khanna D, Albera C, Fischer A, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS trial. J Rheumatol. 2016;43:1672–1679. doi:10.3899/jrheum.151322
38. Acharya N, Sharma SK, Mishra D, Dhooria S, Dhir V, Jain S. Efficacy and safety of pirfenidone in systemic sclerosis-related interstitial lung disease-a randomised controlled trial. Rheumatol Int. 2020;40:703–710.
39. Trang G, Steele R, Baron M, Hudson M. Corticosteroids and the risk of scleroderma renal crisis: a systematic review. Rheumatol Int. 2012;32:645–653.
40. Penn H, Howie J, Kingdon EJ, et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. QJM. 2007;100:485–494. doi:10.1093/qjmed/hcm052
41. Shapiro L, Saketkoo LA, Farrell J, Fligelstone K. Development of a “Renal crisis prevention card” as an education tool to improve outcomes in high risk patients with systemic sclerosis (SSc). Ann Rheum Dis. 2015;74:1136. doi:10.1136/annrheumdis-2015-eular.3605
42. Omair MA, Alahmadi A, Johnson SR. Safety and effectiveness of mycophenolate in systemic sclerosis. A systematic review. PLoS One. 2015;10:e0124205. doi:10.1371/journal.pone.0124205
43. Budde K, Glander P, Diekmann F, et al. Enteric-coated mycophenolate sodium: safe conversion from mycophenolate mofetil in maintenance renal transplant recipients. Transplant Proc. 2004;36:524S–27S. doi:10.1016/j.transproceed.2003.12.042
44. Curtis JR, Johnson SR, Anthony DD, et al. American College of Rheumatology guidance for COVID-19 vaccination in patients with rheumatic and musculoskeletal diseases: version 3. Arthritis Rheumatol. 2021;73:e60–e75. doi:10.1002/art.41928
45. Genentech. CellCept® (mycophenolate mofetil) prescribing information; 2018. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/050722s035,050723s035,050758s033,050759s041lbl.pdf.
46. Schaier M, Scholl C, Scharpf D, et al. Proton pump inhibitors interfere with the immunosuppressive potency of mycophenolate mofetil. Rheumatology. 2010;49:2061–2067. doi:10.1093/rheumatology/keq238
47. Baxter Healthcare. Cyclophosphamide prescribing information; 2013. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/012141s090,012142s112lbl.pdf.
48. Dan D, Fischer R, Adler S, Förger F, Villiger PM. Cyclophosphamide: as bad as its reputation? Long-term single centre experience of cyclophosphamide side effects in the treatment of systemic autoimmune diseases. Swiss Med Wkly. 2014;144:w14030. doi:10.4414/smw.2014.14030
49. Barmettler S, Ong MS, Farmer JR, et al. Association of immunoglobulin levels, infectious risk, and mortality with rituximab and hypogammaglobulinemia. JAMA Netw Open. 2018;1:e184169. doi:10.1001/jamanetworkopen.2018.4169
50. van Vollenhoven RF, Emery P, Bingham CO, et al. Long-term safety of patients receiving rituximab in rheumatoid arthritis clinical trials. J Rheumatol. 2010;37:558–567. doi:10.3899/jrheum.090856
51. Seibold JR, Maher TM, Highland KB, et al. Safety and tolerability of nintedanib in patients with systemic sclerosis-associated interstitial lung disease: data from the SENSCIS trial. Ann Rheum Dis. 2020;79:1478–1484. doi:10.1136/annrheumdis-2020-217331
52. Boehringer Ingelheim Pharmaceuticals, Inc. OFEV® (nintedanib) prescribing information; 2020. Available from: https://docs.boehringer-ingelheim.com/Prescribing%20Information/PIs/Ofev/ofev.pdf.
53. Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–1087. doi:10.1056/NEJMoa1103690
54. Wind S, Schmid U, Freiwald M, et al. Clinical pharmacokinetics and pharmacodynamics of nintedanib. Clin Pharmacokinet. 2019;58:1131–1147. doi:10.1007/s40262-019-00766-0
55. Luedtke D, Marzin K, Jungnik A, et al. Effects of ketoconazole and rifampicin on the pharmacokinetics of nintedanib in healthy subjects. Eur J Drug Metab Pharmacokinet. 2018;43:533–541. doi:10.1007/s13318-018-0467-9
56. McMahan ZH, Hummers LK. Gastrointestinal involvement in systemic sclerosis: diagnosis and management. Curr Opin Rheumatol. 2018;30:533–540. doi:10.1097/BOR.0000000000000545
57. Sullivan KM, Goldmuntz EA, Keyes-Elstein L, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med. 2018;378:35–47. doi:10.1056/NEJMoa1703327
58. Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017;76:1327–1339. doi:10.1136/annrheumdis-2016-209909
59. Leard LE, Holm AM, Valapour M, et al. Consensus document for the selection of lung transplant candidates: an update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40:1349–1379. doi:10.1016/j.healun.2021.07.005
60. Crespo MM, Bermudez CA, Dew MA, et al. Lung transplant in patients with scleroderma compared with pulmonary fibrosis. Short- and long-term outcomes. Ann Am Thorac Soc. 2016;13:784–792. doi:10.1513/AnnalsATS.201503-177OC
61. Wijsenbeek MS, Holland AE, Swigris JJ, Renzoni EA. Comprehensive supportive care for patients with fibrosing interstitial lung disease. Am J Respir Crit Care Med. 2019;200:152–159. doi:10.1164/rccm.201903-0614PP
62. Jacobs SS, Krishnan JA, Lederer DJ, et al. Home oxygen therapy for adults with chronic lung disease. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2020;202:e121–e141. doi:10.1164/rccm.202009-3608ST
63. Pépin M, Kwakkenbos L, Carrier ME, et al. Reasons for attending support groups and organizational preferences: a replication study using the North American Scleroderma Support Group Survey. J Scleroderma Relat Disord. 2019;4:173–186. doi:10.1177/2397198319849806
64. Seibold JR, Denton CP, Furst DE, et al. Randomized, prospective, placebo-controlled trial of bosentan in interstitial lung disease secondary to systemic sclerosis. Arthritis Rheum. 2010;62:2101–2108. doi:10.1002/art.27466
65. Khanna D, Tashkin DP, Wells AU, et al. STRATUS: a phase II study of abituzumab in patients with systemic sclerosis-associated interstitial lung disease. J Rheumatol. 2021;48:1295–1298. doi:10.3899/jrheum.191365
66. Hsu VM, Denton CP, Domsic RT, et al. Pomalidomide in patients with interstitial lung disease due to systemic sclerosis: a Phase II, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. J Rheumatol. 2018;45:405–410. doi:10.3899/jrheum.161040
67. Ebata S, Yoshizaki A, Oba K, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatol. 2021;3:e489–e497. doi:10.1016/S2665-9913(21)00107-7
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.