")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Management of bipolar I depression: clinical utility of lurasidone
Authors Findlay LJ, El-Mallakh PL, El-Mallakh R
Received 10 July 2014
Accepted for publication 20 August 2014
Published 8 January 2015 Volume 2015:11 Pages 75—81
DOI https://doi.org/10.2147/TCRM.S57695
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Lillian Jan Findlay,1 Peggy El-Mallakh,1 Rif S El-Mallakh2
1College of Nursing, University of Kentucky, Lexington, KY, USA; 2Department of Psychiatry and Behavioral Sciences, University of Louisville School of Medicine, Louisville, KY, USA
Abstract: Lurasidone is a benzisothiazol derivative second-generation antipsychotic. It has been approved in the United States and Europe for treatment of acute schizophrenia and bipolar depression. In type I bipolar subjects, treatment with lurasidone monotherapy of adjunctive therapy to lithium or valproic acid with doses of 20 to 120 mg once daily with food, results in statistically and clinically significant reduction of depressive symptoms. Patients experience relatively few side effects, which include somnolence, akathisia, nausea, and other gastrointestinal upset. Dopamine related side effects, such as Parkinsonism and elevated prolactin, are rare and mild. Longer term safety data obtained in 6 months long, open continuation observation periods, suggest that metabolic related elevations in weight, glucose, and lipids are absent or minimal. The mechanism of action of lurasidone is not known, but the data are compatible with antagonism of the serotonin 7 receptor. Lurasidone is a new option for the treatment of bipolar depression with relatively few side effects.
Keywords: lurasidone, bipolar disorder, bipolar depression, adjunctive therapy
Introduction
The treatment of depression in patients diagnosed with bipolar disorder presents considerable challenges to the clinician. Depression is a key feature of bipolar disorder, and the total number of depressive episodes may be present in up to a third or half of a patient’s life.1–4 These depressive episodes have a variety of clinical presentations; for example, the symptoms may meet full diagnostic criteria for a major depressive episode, or may be subsyndromal. Alternatively, the symptoms may be a manifestation of the commonly comorbid dysthymic disorder.2,3,5,6 Additionally, depressive symptoms frequently occur due to comorbid medical conditions that are common in patients with bipolar illness. These include sleep apnea, thyroid disease, diabetes, cardiac disease, and other chronic medical conditions.7–12
Pharmacologic treatment of depression in bipolar illness is complicated by a dearth of efficacy and safety data. While the current use of antidepressants in bipolar disorder has declined to as low as 34% of patients,13 their use in this population remains controversial.14 Some reviews report that antidepressants are safe and effective;15 however, others report that antidepressants are ineffective and potentially harmful.16 Current reports on efficacy suggest that antidepressants appear to be effective in bipolar depression when used alone17,18 or when added to an antipsychotic,19 but are ineffective in placebo-controlled trials in which they are added to a mood stabilizer.20–22 Furthermore, while much research has focused on the problem of induction of a manic state due to the use of antidepressants, research suggests that antidepressants can induce at least three distinct problems in bipolar patients. Antidepressants can indeed increase the likelihood of a manic switch,21 or any switch by accelerating cycling,4,23 and increase the likelihood of both acute and chronic depression in rapid cycling patients.24,25
Thus, clinicians are often presented with a quandary in clinical practice when their patients with bipolar disorder present with depression since the evidence-based options available for treating such a patient are limited.26 However, clinical research increasingly supports the use of antipsychotic medications as an effective treatment option for depression in bipolar disorder.27,28 For example, lurasidone, a second-generation antipsychotic that is already approved for the treatment of schizophrenia,29,30 has been approved by the United States Food and Drug Administration (FDA) for the treatment of bipolar depression.31,32 The agent has demonstrated efficacy and safety in this population. Its presumed mechanism of action appears to be unique. This paper will review the available information about lurasidone for the management of bipolar depression.
Pharmacokinetics, pharmacodynamics, and pharmacology
Lurasidone is a benzisothiazol derivative second-generation antipsychotic. It has a complex multi-ring structure that is minimally soluble in water. It is supplied as lurasidone hydrochloride in unscored 20, 40, 60, 80, and 120 mg tablets.
It is likely that lurasidone is absorbed in the stomach. For this reason, it is more effectively absorbed when taken with food.33 This effect is similar to that observed with ziprasidone.34,35 In both cases a meal of approximately 350 calories for lurasidone and 500 calories for ziprasidone, maximizes and stabilizes absorption of the drug.34,35 Fat content of the meal does not alter the absorption. Absorption is increased approximately two to three times, and maximal serum concentration is increased about three fold.33 Once absorbed, lurasidone is highly plasma protein bound (99.8% is bound to albumin and α-1-glycoprotein).36
It is metabolized predominantly by the cytochrome P450 isozyme 3A4. It is broken down by oxidative N-dealkylation, hydroxylation of norbornane ring, and S-oxidation. There are two active metabolites (ID-14283 and ID-14326) and three inactive metabolites (ID-20219, ID-20220, and ID-11614).33 The active metabolites comprise only about 28% of the breakdown products of lurasidone, and have a shorter half-life, thus most of the action is believed to be due to the parent drug.36 The half-life of lurasidone is 18 hours for a 40 mg oral dose, so it can be taken once daily. Steady state is achieved within 5 to 7 days.33,36 Because lurasidone is not a substrate for CYP1A2, smoking does not alter metabolism. The majority of the metabolic remnants of lurasidone end up in the feces (80%), with a small fraction in the urine (9%).
Lurasidone interacts with many receptors, and is antagonistic at most of them. The principal ones are dopamine D2 (Ki =1.68 nM), serotonin 5HT7 (Ki =0.5 nM), 5HT2A (Ki =0.5 nM in human and 2.0 nM in rat), and 5HT1A (Ki =6.4 nM in human and 6.8 nM in rat).36 For the 5HT1A receptor, lurasidone is classified as a partial agonist; but the partial agonist effect is only 33% that of serotonin.37 At higher doses it may interact with the adrenergic α2A (Ki =40.7 nM), and α2C (Ki =10.8 nM). It does not interact with the histamine H1 or muscarinic M1 (Ki >1,000 nM for both).36 Receptor binding in humans as measured by positron emission tomography (PET) reveals that doses below 40 mg do not achieve adequate D2 binding for an antipsychotic effect (41%–43% for 10 mg, and 51%–55% for 20 mg).38,39 With a 40 mg dose the D2 binding is 63%–67%, and increases to 77%–79% with 60 mg. Interestingly, higher doses do not increase receptor occupancy (73%–79% with 80 mg dose).39 This observation may explain why Parkinsonism and elevated prolactin are uncommon with lurasidone in these studies, particularly with lower doses.31 However, D2 receptor occupancy is more closely related to blood levels than to dosage of lurasidone, because of the significant variability in drug concentrations across individuals.40
The interaction with 5HT7 is believed to be particularly important for the antidepressant effect in bipolar illness. Blockade of this receptor with selective 5HT7 antagonists or knocking out the 5HT7 gene, improve symptoms of “depression” in animal models of depression.41–43 A pure 5HT7 antagonist reduced depressive symptoms in a preclinical rodent study, but did not separate from placebo due to a high placebo response rate.44 Testing in animal models of depression and anxiety with lurasidone demonstrates that it is effective in reducing depressive symptoms in animals.45 Part of this effect may be due to an increase in dopamine efflux in the frontal cortex that is induced by lurasidone blockade of 5HT7.46,47 This effect is also believed to mediate the pro-cognition effect of lurasidone.48–51 Additionally, lurasidone appears to modulate the reductions in brain-derived neurotrophic factor and promote neuroplasticity in animal models of stress and depression.52,53 A positive effect on brain-derived neurotrophic factor appears to be a marker for effective antidepressant agents.54 None of these potential mechanisms of action have been demonstrated as being central in human studies.
Efficacy of lurasidone in bipolar depression
Two randomized, double-blinded controlled clinical trials have examined the efficacy of lurasidone in the treatment of bipolar I depression (Figures 1 and 2).31,32 Lurasidone was tested as monotherapy at 20–60 mg/day (N=166) and 80–120 mg/day (N=169), with a placebo comparison (N=170)(Figure 1).Findings indicated that after 6 weeks, scores on the Montgomery‐Ȧsberg Depression Scale (MADRS) were equally and significantly decreased in both the 20–60 mg/day group (−15.4) and in the 80–120 mg/day group (−15.4), compared to placebo (−10.7) (Figure 1). Response, defined as >50% improvement on the MADRS, was achieved by 53% of those receiving the low dose, and 51% of those receiving the high dose, compared to 30% of those receiving placebo. The number needed to treat (NNT) was 5 for both groups. Similarly, remission, defined as a MADRS score of ≤12, was achieved by 42% in the low dose group and 40% in the high dose group, compared to 25% on placebo. The NNT for remission was 6 in the low dose group and 7 in the high dose group. In addition, both treatment groups showed significantly greater endpoint decreases in depression severity scores on the Clinical Global Impressions Scale for Bipolar Illness compared to placebo. Endpoint decreases were −1.8 for the 20–60 mg/day group, −1.7 for the 80–120 mg/day group, and −1.1 for the placebo group.
 | Figure 1 The efficacy outcome at the end of 6 weeks of treatment of acutely depressed type I bipolar patients treated with placebo, or monotherapy with lurasidone 20–60 mg/day, or lurasidone 80–120 mg/day. |
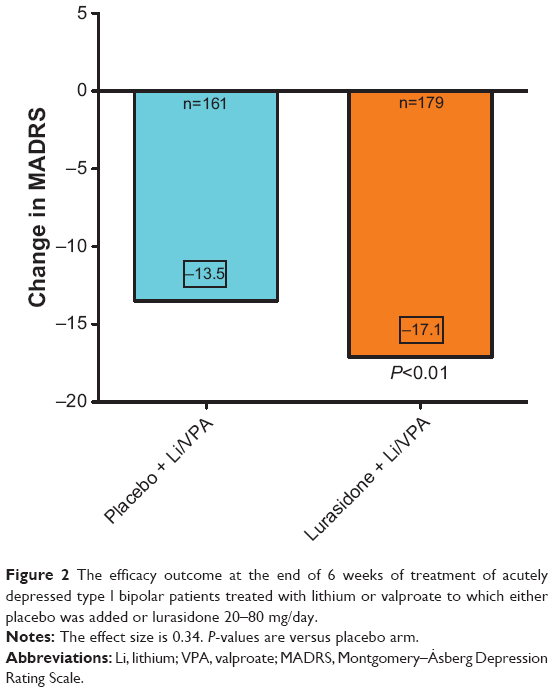 | Figure 2 The efficacy outcome at the end of 6 weeks of treatment of acutely depressed type I bipolar patients treated with lithium or valproate to which either placebo was added or lurasidone 20–80 mg/day. |
For both treatment groups, significant improvements were seen on several domains of the MADRS, including in reported and apparent sadness, inner tension, “inability to feel”, lassitude, and pessimistic thoughts. The lurasidone 20–60 mg/day group showed significant improvements in sleep, and the 80–120 mg/day group showed significant improvements in concentration. No significant changes were seen in appetite or suicidal ideation in either treatment group compared to placebo. Both treatment groups also reported significant reductions in anxiety compared to placebo, as measured by the Hamilton Anxiety Rating Scale. Findings from this study further indicate that participants in both treatment groups reported significantly improved quality of life and life satisfaction, along with significantly reduced disability.32
A second randomized, controlled trial examined the effectiveness of lurasidone as an adjunct to lithium or valproate in the treatment of bipolar I depression.32 All patients met criteria for a moderate major depression despite documented therapeutic levels of lithium or valproate. The treatment group (N=183) was started on lurasidone at 20 mg, which was increased to 60 mg by the end of the first week of the study. After the first week, dosages could be adjusted within a range of 20–120 mg/day at weekly intervals, with increases or decreases of 20 mg, based on clinical judgment.32 The control group (N=165) received placebo added to the lithium or valproate.
Depression, anxiety, quality of life, life satisfaction, and disability were measured at baseline and at 6 weeks. Findings indicate that the group treated with lurasidone + lithium/valproate had significantly improved depression, as measured by the MADRS (Figure 2). The least squares mean change of the MADRS score for the lurasidone + lithium/valproate group was −17.1, compared to −13.5 for the placebo group. In addition, significantly shorter time to remission was seen in the lurasidone + lithium/valproate group (35 days) compared to the placebo group (43 days). Response was achieved by 57% of patients receiving lurasidone + lithium/valproate compared to 42% of those on placebo + lithium/valproate. The NNT was 7. Remission rates were 50% in the lurasidone + mood stabilizer group, versus 35% in the placebo + mood stabilizer group; with an NNT of 7.
Several domains of the MADRS showed significant improvement in the lurasidone + lithium/valproate group compared to placebo, including reported and apparent sadness, sleep, lassitude, inability to feel, and pessimistic thoughts. However, no significant differences were seen in inner tension, appetite, concentration difficulties, and suicidal ideation. In addition, the lurasidone + lithium/valproate group reported significant reductions in anxiety and disability, along with significant improvements in quality of life, enjoyment, and life satisfaction, compared to the placebo group.
A separate adjunctive, placebo-controlled study failed to separate from placebo. This study had a design similar to the previous study, while improvement was noted in the lurasidone arm, it did not separate from placebo. The safety data (see the “Lurasidone Safety and tolerability” section) includes patients from this unpublished failed study.
A 6 months open observation period was performed for patients in both of these studies who wished to remain in the study. Patients on placebo were switched to active medication. The study has not yet been published and symptom data are not available for this review.
Lurasidone safety and tolerability in bipolar depression
The FDA approved lurasidone for schizophrenia in 2010 and subsequently in 2013 for bipolar depression.55 It is considered as safe, well-tolerated, and efficacious in these conditions.31,32,56,57 In a systematic review of the literature from June 2009 to February 2014, lurasidone, compared to other recently approved second-generation atypical antipsychotics such as paliperidone, iloperidone, and asenapine, was considered by researchers to be less likely to produce the metabolic side effects of weight gain, hyperlipidemia, hypercholesterolemia, and hyperglycemia, and was not linked to significant electrocardiogram (ECG) abnormalities.58–61
Among a total of eleven articles found that described the safety and tolerability of lurasidone in the treatment of schizophrenia or major depressive episodes associated with bipolar I disorder, four were peer-reviewed, double-blind, placebo-controlled studies,31,32,56,57 and another was a meta-analysis.62 Two of the peer-reviewed studies examined the use of lurasidone in the treatment of bipolar I disorder.31,32
In the monotherapy study, 505 adults 18–75 years of age were recruited.31 The three study groups comprised of patients receiving 6 weeks of lurasidone flexible daily oral doses in the ranges of either 20–60 mg or 80–120 mg, or placebo.31 The majority of adverse events were rated as mild to moderate and ≤10% ranked as severe across groups.31 Dropout rates due to adverse events were similar across groups (5.9%–6.6%). There was no statistically or clinically-significant change in weight, lipid, cholesterol, or prolactin levels, glycemic control, waist circumference, or ECG readings.31 The proportion of patients with ≥7% increase in weight from baseline was 4.2% in the 20–60 mg group, 0.7% in both the 80–120 mg and placebo groups.31 The high dose group experienced more adverse effects with a modest occurrence of the incidence of nausea (N=29, 17.4%),akathisia (N=18, 10.8%), somnolence (N=11, 13.8%), sedation (N=12, 7.2%), vomiting (N=10, 6.0%) and extrapyramidal side effects (N=15, 9.0%) (Table 1). Females experienced a higher mean increase in prolactin (7.5 ng/mL) compared to males (2.6 ng/mL).31 Treatment-emergent mania did not differ across the groups. There were low rates of serious adverse events across groups with no deaths, suicidal behaviors or suicides during the study.31
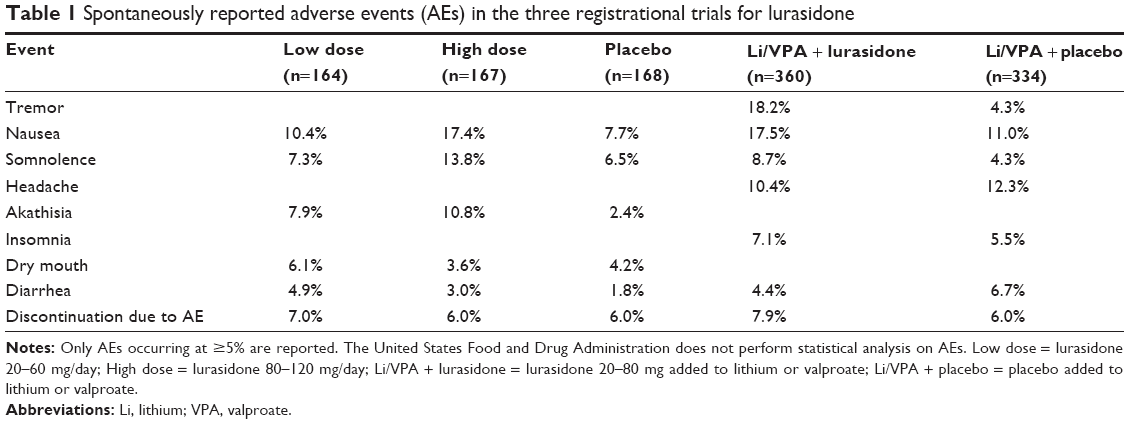 | Table 1 Spontaneously reported adverse events (AEs) in the three registrational trials for lurasidone |
In the trial comparing the safety and efficacy of lurasidone added to stable doses of lithium or valproate versus placebo added to lithium or valproate in depressed bipolar I patients, 348 adults 18–65 years of age were studied.32 The lurasidone + lithium/valproate group most commonly experienced the adverse events of nausea (17.5%), somnolence (8.7%), tremor (8.2%), akathisia (7.7%), insomnia (7.1%), and Parkinsonism (15.3%). Discontinuation rates due to adverse events were 6% in this group versus 7.9% in the placebo + lithium/valproate group. Additionally, lurasidone did not significantly increase body weight, lipids, glucose, or prolactin levels.32
In both bipolar I studies, overall adverse events with lurasidone versus placebo included extrapyramidal events 24% versus 13%, akathisia 11% versus 5%, and Parkinsonism 13% versus 8%.56 Similarly, in the two studies examining the safety, tolerability, and efficacy of lurasidone in the treatment of acute schizophrenia, researchers generally found that the most commonly experienced side effects were akathisia, headache, gastrointestinal upset (nausea, vomiting, and dyspepsia), insomnia, somnolence, and sedation ( Table 1).56,57
In the 6 months open observation study of patients receiving lurasidone monotherapy (N=130), there was no additional weight gain (0.0 kg).56 Similarly fasting glucose increased by 1.2 mg/dL, total cholesterol dropped by 0.5 mg/dL, triglycerides dropped by 1.0 mg/dL, and prolactin dropped by 1.2 ng/dL.56 Patients receiving lurasidone as adjunct (N=88) had more changes, but still quite modest. Over the 6 months period, weight increased by 1.3 kg, fasting glucose increased by 1.7 mg/dL, total cholesterol dropped by 0.9 mg/dL, triglycerides increased by 5.3 mg/dL, and prolactin dropped by 2.9 ng/dL.56
Lurasidone is metabolized primarily through the cytochrome P450 3A4, and it should be used with caution in combination with strong CYP3A4 inhibitors such as ketoconazole, clarithromycin, and voriconazole (the latter two no longer in the American market), or inducers such as rifampin, St John’s Wort, phenytoin, and carbamazepine.56 Dose adjustments are generally recommended in patients with renal and hepatic impairment.56 Renal impairment may require initial dosing at 20 mg/day with a maximum dose of 80 mg/day for creatinine clearance <50 mg/min. However, since most of the deactivation of the drug is hepatic, these recommendations are precautionary. For hepatic impairment patients with a Child–Pugh Class B, starting dose is 20 mg/day with a maximum dose of 80 mg/day; Child–Pugh Class C patients should start at 20 mg/day with a maximum dose of 40 mg/day.56
Last, safety and tolerability in pediatric and geriatric populations diagnosed with either bipolar I disorder or schizophrenia has not been established.56 However, in a review of the pharmacologic and clinical profile of lurasidone, according to industry data, no dose adjustment was needed in patients 65–85 years of age since no significant differences were found in concentrations of lurasidone among adult and geriatric patients diagnosed with schizophrenia.62 Furthermore, because of improved cognition experienced in geriatric patients taking lurasidone and its lower potential for anticholinergic and hypotensive effects, further study is warranted in this age group.62 It should be remembered that all antipsychotics carry an FDA class warning that death in severely demented, agitated patients with psychosis is nearly twice as common as in patients not receiving such medications.
Conclusion
Lurasidone is a new second-generation antipsychotic that appears to have the desired property of antagonizing 5HT7 receptors at relatively low doses. It is believed that this 5HT7 blockade mediates the clinical improvement in depression. This effect occurs when lurasidone is administered alone or in combination with a mood stabilizer. The adverse effects seen in patients with bipolar illness receiving lurasidone are uncommon and generally mild. The effect on metabolic parameters is very favorable. Lurasidone is a new safe and effective option for the management of bipolar depression.
Disclosure
RSE has research funding from Merck and AssureRx. He is also a speaker for AstraZeneca, Lundbeck, Otsuka, Takeda, and Sunovion. Neither of the other authors have conflicts of interest to declare. This research was not funded by any extramural agency.
References
Altshuler LL, Kupka RW, Hellemann G, et al. Gender and depressive symptoms in 711 patients with bipolar disorder evaluated prospectively in the Stanley Foundation bipolar treatment outcome network. Am J Psychiatry. 2010;167(6):708–715. | ||
Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59(6):530–537. | ||
Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry. 2003;60(3):261–269. | ||
Post RM, Leverich GS, Nolen WA, et al. A re-evaluation of the role of antidepressants in the treatment of bipolar depression: data from the Stanley Foundation Bipolar Network. Bipolar Disord. 2003;5(6): 396–406. | ||
Altshuler LL, Post RM, Black DO, et al. Subsyndromal depressive symptoms are associated with functional impairment in patients with bipolar disorder: results of a large, multisite study. J Clin Psychiatry. 2006;67(10):1551–1560. | ||
Goldstein BI, Herrmann N, Shulman KI. Comorbidity in bipolar disorder among the elderly: results from an epidemiological community sample. Am J Psychiatry. 2006;163(2):319–321. | ||
McElroy SL, Keck PE Jr. Metabolic syndrome in bipolar disorder: a review with a focus on bipolar depression. J Clin Psychiatry. 2014;75(1): 46–61. | ||
Rezaeitalab F, Moharrari F, Saberi S, Asadpour H, Rezaeetalab F. The correlation of anxiety and depression with obstructive sleep apnea syndrome. J Res Med Sci. 2014;19(3):205–210. | ||
Saunamäki T, Jehkonen M. Depression and anxiety in obstructive sleep apnea syndrome: a review. Acta Neurol Scand. 2007;116(5):277–288. | ||
Post RM, Altshuler LL, Leverich GS, et al. More medical comorbidities in patients with bipolar disorder from the United States than from the Netherlands and Germany. J Nerv Ment Dis. 2014;202(4):265–270. | ||
Sierra P, Cámara R, Tobella H, Livianos L. What is the real significance and management of major thyroid disorders in bipolar patients? Rev Psiquiatr Salud Ment. 2014;7(2):88–95. | ||
Williams MD, Shah ND, Wagie AE, Wood DL, Frye MA. Direct costs of bipolar disorder versus other chronic conditions: an employer-based health plan analysis. Psychiatr Serv. 2011;62(9):1073–1078. | ||
Cascade EF, Reites J, Kalali AH, Ghaemi N. Antidepressants in bipolar disorder. Psychiatry (Edgmont). 2007;4(3):56–58. | ||
Pacchiarotti I, Bond DJ, Baldessarini RJ, et al. The International Society of Bipolar Disorders (ISBD) task force report on antidepressant use in bipolar disorders. Am J Psychiatry. 2013;70(11):1249–1262. | ||
Gijsman HJ, Geddes JR, Rendell JM, Nolen WA, Goodwin GM. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry. 2004;161(9):1537–1547. | ||
El-Mallakh RS, Karippot A. Chronic depression in bipolar disorder. Am J Psychiatry. 2006;163(8):1137–1341. | ||
Amsterdam J. Efficacy and safety of venlafaxine in the treatment of bipolar II major depressive episode. J Clin Psychopharmacol. 1998;18(5): 414–417. | ||
Amsterdam JD, Garcia-España F, Fawcett J, et al. Efficacy and safety of fluoxetine in treating bipolar II major depressive episode. J Clin Psychopharmacol. 1998;18(6):435–440. | ||
Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60(11):1079–1088. | ||
Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158(6):906–912. | ||
El-Mallakh RS. Adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;357(6):615. | ||
Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356(17):1711–1722. | ||
Wehr TA, Goodwin FK. Can antidepressants cause mania and worsen the course of affective illness? Am J Psychiatry. 1987;144(11):1403–1411. | ||
El-Mallakh RS, Ghaemi SN, Sagduyu K, et al. Antidepressant-Associated Chronic Irritable Dysphoria (ACID) in STEP-BD Patients. J Affect Disord. 2008;111(2–3):372–377. | ||
Ghaemi SN, Ostacher MM, El-Mallakh RS, et al. Antidepressant discontinuation in bipolar depression: a Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD) randomized clinical trial of long-term effectiveness and safety. J Clin Psychiatry. 2010;71(4):372–380. | ||
Schroeder E, Gao Y, Roberts RJ, El-Mallakh RS. Recognition and management of bipolar depression. J Clin Outcomes Manag. 2011;18(11):515–525. | ||
Cruz N, Sanchez-Moreno J, Torres F, Goikolea JM, Valenti M, Vieta E.Efficacy of modern antipsychotics in placebo-controlled trials in bipolar depression: a meta-analysis. Int J Neuropsychopharmacol. 2010;13(1):5–14. | ||
DeFruyt J, Deshepper E, Audenaert K, et al. Second generation antipsychotic in the treatment of bipolar depression: a systematic review and meta-analysis. J Psychopharmacology. 2012;26(5):603–617. | ||
Nakamura M, Ogasa MS, Guarino J, et al. Lurasidone in the treatment of acute schizophrenia: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(6):829–836. | ||
Nasrallah HA, Silva R, Phillips D, et al. Lurasidone for the treatment of acutely psychotic patients with schizophrenia: a 6-week, randomized, placebo-controlled study. J Psychiatr Res. 2013;47(5):670–677. | ||
Loebel A, Cucchiaro J, Silva R, et al. Lurasidone monotherapy in the treatment of bipolar I depression: a randomized, double-blind, placebo-controlled study. Am J Psychiatry. 2014;171(2):160–168. | ||
Loebel A, Cucchiaro J, Silva R, et al. Lurasidone as adjunctive therapy with lithium or valproate for the treatment of bipolar I depression: A randomized, double-blind, placebo-controlled study. Am J Psychiatry. 2014;171(2):169–177. | ||
Preskorn S, Ereshefsky L, Chiu Y-Y, Poola N, Loebel A. Effect of food on the pharmacokinetics of lurasidone: results of two randomized, open-label, crossover studies. Hum Psychopharmacol. 2013; 28(5):495–505. | ||
.Hamelin BA, Allard S, Laplante L, et al. The effect of timing of a standard meal on the pharmacokinetics and pharmacodynamics of the novel atypical antipsychotic agent ziprasidone. Pharmacotherapy. 1998;18(1):9–15. | ||
Gandelman K, Alderman JA, Glue P, et al. The impact of calories and fat content of meals on oral ziprasidone absorption: a randomized, open-label, crossover trial. J Clin Psychiatry. 2009;70(1):58–62. | ||
Meyer JM, Loebel AD, Schweizer E. Lurasidone: a new drug in development for schizophrenia.Expert Opin Investig Drugs. 2009;18(11): 1715–1726. | ||
Ishibashi T, Horisawa T, Tokuda K, et al. Pharmacological profile of lurasidone, a novel antipsychotic agent with potent 5-hydroxytryptamine 7 (5-HT7) and 5-HT1A receptor activity. J Pharmacol Exp Ther. 2010;334(1):171–181. | ||
Uhida H, Takeuchi H, Graff-Guerrero A, Suzuki T, Watanabe K, Mamo DC. Dopamine D2 receptor occupancy and clinical effects: a systematic review and pooled analysis. J Clin Psychopharmacol. 2011; 31(4):497–502. | ||
Wong DF, Kuwabara H, Brašić JR, et al. Determination of dopamine D2 receptor occupancy by lurasidone using positron emission tomography in healthy male subjects. Psychopharmacology (Berl). 2013;229(2):245–252. | ||
Potkin SG, Keator DB, Kesler-West ML, et al. D2 receptor occupancy following lurasidone treatment in patients with schizophrenia and schizoaffective disorder. CNS Spectr. 2014;19(2):176–181. | ||
Hedlund PB, Huitron-Resendiz S, Henriksen SJ, Sutcliffe JG. 5-HT7 receptor inhibition and inactivation induce antidepressant like behavior and sleep pattern. Biol Psychiatry. 2005;58(10):831–837. | ||
Wesolowska A, Nikiforuk A, Stachowicz K, Tatarczynska E. Effect of the selective-5HT7 receptor antagonist SB 269970 in animal models of anxiety and depression. Neuropharmacology. 2006;51(3):578–586. | ||
Bonaventure P, Dugovic C, Kramer M, et al. Translational evaluation of JNJ-18038683, a 5-HT7 receptor antagonist, on REM sleep and in major depressive disorder. J Pharmacol Exp Ther. 2012;342(2):429–440. | ||
Bonaventure P, Kelly L, Aluisio L, et al. Selective blockade of 5-hydroxytryptamine 5HT7 receptors enhances 5-HT transmission, antidepressant-like behavior, and rapid eye movement sleep suppression induced by citalopram in rodents. J Pharmacol Exp Ther. 2007;321(2): 690–698. | ||
Cates LN, Roberts AJ, Huitron-Resendiz S, Hedlund PB. Effects of lurasidone in behavioral models of depression. Role of the 5-HT7 receptor subtype. Neuropharmacology. 2013;70:211–217. | ||
Huang M, Horiguchi M, Felix AR, Meltzer HY. 5-HT1A and 5-HT7 receptors contribute to lurasidone-induced dopamine efflux. Neuroreport. 2012;23(7):436–440. | ||
Huang M, Panos JJ, Kwon S, Oyamada Y, Rajagopal L, Meltzer HY. Comparative effect of lurasidone and blonanserin on cortical glutamate, dopamine, and acetylcholine efflux: role of relative serotonin 5HT2A and DA D2 antagonism and 5-HT1A partial agonism. J Neurochem. 2014; 128(6):938–949. | ||
Meltzer HY, Massey BW. The role of serotonin receptors in the action of atypical antipsychotic drugs. Curr Opin Pharmacol. 2011;11(1): 59–67. | ||
Horisawa T, Ishibashi T, Nishikawa H, et al. The effects of selective antagonists of serotonin 5-HT7 and 5-HT1A receptors on MK-801-induced impairment of learning and memory in the passive avoidance and Morris water maze tests in rats: mechanistic implications for the beneficial effects of the novel atypical antipsychotic lurasidone. Behav Brain Res. 2011;220(1):83–90. | ||
Horisawa T, Nishikawa H, Toma S, et al. The role of 5-HT7 receptor antagonism in the amelioration of MK-801-induced learning and memory deficits by the novel atypical antipsychotic drug lurasidone. Behav Brain Res. 2013;244:66–69. | ||
Yuen EY, Li X, Wei J, Horiguchi M, Meltzer HY, Yan Z. The novel antipsychotic drug lurasidone enhances N-methyl-D-aspartate receptor-mediated synaptic responses. Mol Pharmacol. 2012;81(2):113–119. | ||
Fumagalli F, Calabrese F, Luoni A, Bolis F, Racagni G, Riva MA. Modulation of BDNF expression by repeated treatment with the novel antipsychotic lurasidone under basal condition and in response to acute stress. Int J Neuropscyhophramcol. 2012;15(2):235–246. | ||
Luoni A, Berry A, Calabrese F, et al. Delayed BDNF alterations in the prefrontal cortex of rats exposed to prenatal stress: preventive effect of lurasidone treatment during adolescence. Eur Neuropsychopharmacol. 2014;24(6):986–995. | ||
Castrén E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev Neurobiol. 2010;70(5):289–297. | ||
Sunovion Pharmaceuticals Inc. The US Food and Drug Administration approved Latuda® (lurasidone HCl) for the treatment of adult patients with depressive episodes associated with bipolar I disorder (bipolar depression), both as monotherapy and as an adjunct to lithium or valproate; June 28, 2013. Available from:http://www.sunovion.com/news/latuda-press-room.html. Accessed July 4, 2014. | ||
Latuda® (lurasidone HCI tablets) [prescribing information]. Sunovion Pharmaceuticals Inc; 2013. | ||
Meltzer HY, Cucchiaro J, Silva R, et al. Lurasidone in the treatment of schizophrenia: a randomized, double-blind, placebo-and-olanzapine-controlled study. Am J Psychiatry. 2011;168(9): 957–967. | ||
Citrome L. Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract. 2011;65(2):189–210. | ||
Citrome L, Ketter TA, Cucchiaro J, Loebel A. Clinical assessment of lurasidone benefit and risk in the treatment of bipolar I depression using number needed to treat, number needed to harm, and likelihood to be helped or harmed. J Affect Disord. 2014;155:20–27. | ||
De Hert M, Yu W, Detraux J, Sweers K, van Winkel R, Correll CU. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder. CNS Drugs. 2012;26(9):733–759. | ||
Howland RH. Update on newer antipsychotic drugs. J Psychosoc Nurs Ment Health Serv. 2011;49(4):13–15. | ||
Rado J, Janicak PG. Pharmacological and clinical profile of recently approved second-generation antipsychotics. Drugs Aging. 2012; 29(10):783–791. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.