Back to Journals » OncoTargets and Therapy » Volume 13
Malignant Gastrointestinal Neuroectodermal Tumors: Clinicopathological and Prognostic Features of 96 Patients
Authors Li R, Cao J ![]() , Chen L, Cui F, Chen S, Feng Z, Li N
, Chen L, Cui F, Chen S, Feng Z, Li N
Received 10 August 2020
Accepted for publication 10 September 2020
Published 30 September 2020 Volume 2020:13 Pages 9731—9740
DOI https://doi.org/10.2147/OTT.S275633
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Ran Li1 ,* Jintao Cao1 ,* Liucheng Chen2 ,* Fangqin Cui,3 Shaohua Chen,1 Zhenzhong Feng,1 Nan Li1
1Department of Pathology, The First Affiliated Hospital of Bengbu Medical College, Bengbu Medical College, Bengbu, Anhui, People’s Republic of China; 2Department of Radiology, The First Affiliated Hospital of Bengbu Medical College, Department of Imaging Diagnosis, Medical Imaging College, Bengbu Medical College, Bengbu, Anhui, People’s Republic of China; 3Department of Pathophysiology, Bengbu Medical College, Bengbu, Anhui, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Nan Li
Department of Pathology, The First Affiliated Hospital of Bengbu Medical College, 287 Changhuai Road, Bengbu 233000, Anhui, People’s Republic of China
Tel +86-552-3070209
Email [email protected]
Purpose: Gastrointestinal neuroectodermal tumors (GNETs) are uncommon malignant tumors derived from ectodermal primitive neural cells.
Patients and Methods: We retrospectively analyzed 2 GNET cases at our hospital and the remaining 94 cases in the literature to determine clinicopathological prognostic factors.
Results: The patients had a mean age of 36 years and a median tumor size of 4.5 cm. A total of 67.0% of the tumors were located in the small intestine, and 76.4% of the patients presented recurrence or metastasis. There was a significant difference in sex and presence of osteoclast-like cells (P< 0.01). Microscopically, most cells were round or short spindle-like in shape, with weak eosinophilic or clear cytoplasm. Neoplastic cells were always arranged in solid sheets, nests, and pseudoalveoli. Immunohistochemistry showed strong, diffuse S100 and SOX10 expression, with a complete absence of HMB45 and Melan-A expression. A total of 72.9% of the cases revealed genetic EWSR1 recombination, including our 2 cases. The median time to death and first metastasis was 61 months and 12 months, respectively. K-M analysis showed a great difference in survival according to lymph node invasion or distant metastasis (M+N), independent lymph node metastasis (N), lower histological grades (G2), and aggressive chemoradiotherapy (P=0.026, P=0.027, P=0.039 and P=0.037). However, independent T, independent M, and postoperative routine adjuvant therapy showed no statistical influence on overall survival or disease-free survival.
Conclusion: GNET is a new entity distinct in its clinical, morphological, immunochemical, and genetic features. Radical excision, close follow-up and adjuvant therapy may be effective for prolonged survival.
Keywords: gastrointestinal neuroectodermal tumor, clear cell sarcoma-like tumor, GNET, SWI/SNF
Introduction
Gastrointestinal neuroectodermal tumors (GNETs) are rare malignant tumors derived from the neural crest and resembling clear cell sarcoma (CCS) on microscopy. This type of tumor was first distinguished from CCS of the gastrointestinal tract (CCS-GIT) by Zambrano in 2003 due to the lack of melanin expression and the rich presence of osteoclast-like cells.1 No earlier than Zambrano, other researchers also reported the resemblance of gastrointestinal tumors to CCS of soft tissue and gastrointestinal tumors occurring with EWSR1 genetic translocation.2 In 2012, Stockman reclassified this type of tumor as a “gastrointestinal neuroectodermal tumor” considering the neuroectodermal propensity on immunology and the results of the genetic study in their 16 cases.3 Currently, GNET remains a controversial tumor entity and has not been classified as an independent tumor from CCS, as reported by the WHO classification of tumors of soft tissues and bone, 4th edition.4
Currently, only 96 cases have been reported worldwide, and most of the patients have been young and middle-aged adults. GNETs are usually located in the small intestine and normally have EWSR1-ATF1/CREB1 fusions. These tumors are typically negative for melanin expression, such as HBM45 and Melan-A expression, but always positive for S100 and SOX10 expression. On the other hand, these tumors are easily misdiagnosed due to the lack of specificity in terms of morphological features. Due to the lack of specific treatment and the occurrence of early metastasis, most patients have a poor prognosis. Hence, more clinical and experimental studies of this disease should be performed.
In this paper, 2 GNET cases are presented, along with the results of the statistical analysis of clinical and pathological data from 94 cases in the literature. We aimed to improve the understanding and reduce the misdiagnosis of GNETs for clinical therapy.
Patients and Methods
Case Selection
From 2012 to date, 2 patients were diagnosed with GNETs at The First Affiliated Hospital of Bengbu Medical College. Their medical and pathological records were re-examined by 3 experienced pathologists. All medical information was obtained from hospital clinical records, and follow-up investigations were permitted by our patients. Both patients have provided informed consent for their case details to be published. PubMed and CNKI (China National Knowledge Internet) were searched for all cases up to April 2020. Clinical and clinicopathological features, including tumor size, nuclear mitosis, treatment, age, and follow-up information, were carefully analyzed in literature and our 2 cases. This study was approved by the Medical Ethics Committee of The First Affiliated Hospital of Bengbu Medical College and conducted following the ethical guidelines of the Declaration of Helsinki.
Immunohistochemical Staining
After sampling, formalin was used for fixation, followed by dehydration, embedding, sectioning, and HE staining. EnVison’s two-step method was adopted for immunohistochemical staining, and the PBS method was performed as a negative control. The antibodies used in the study are shown in Table 1.
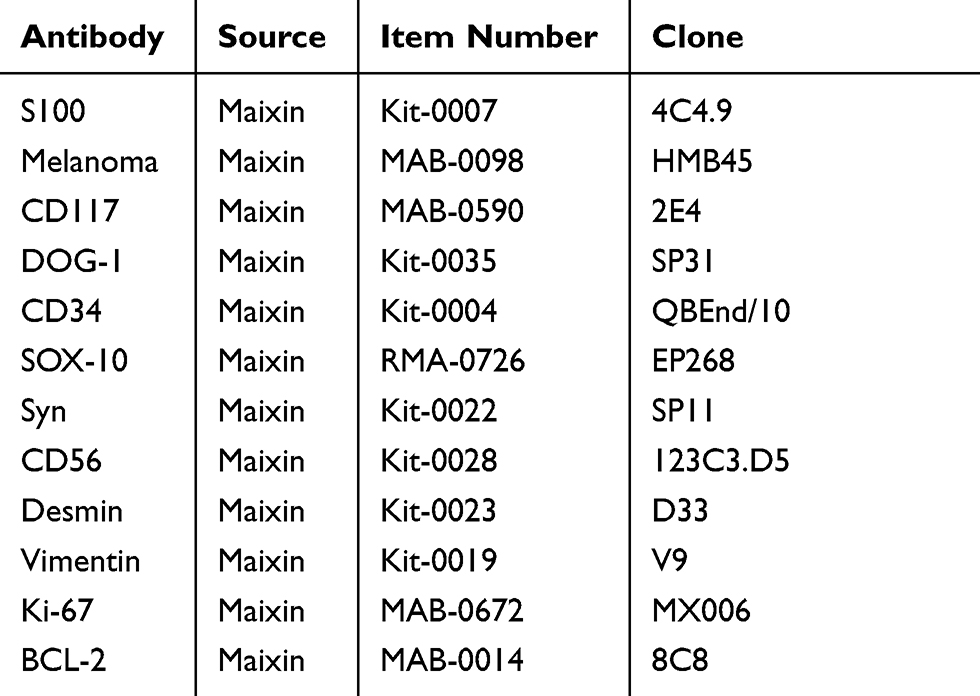 |
Table 1 Antibody Used in the Study |
Fluorescence in situ Hybridization (FISH) Examination
FISH was used to detect EWSR1-related gene fusions. The testing process was strictly controlled. Positive results were critically judged with a critical value of 15%. The probe was obtained from Ambiping Medical Technology Company, Guangzhou, China.
TNM Staging
We performed TNM staging according to the TNM staging criteria for sarcomas of the abdomen and thoracic visceral organs set by the American Joint Committee on Cancer (AJCC, 8th edition). Tumor classifications of T1, T2a, T2b, T3, and T4 indicated tumors that were organically confined, that had invaded to the serosa, that had invaded to beyond the serosa, that invaded to other organs, and that showed multifocal involvement. The histological grade (G) of soft tissue sarcoma is based on the level of tumor differentiation, the mitotic count, and tumor necrosis. At least 3 points are scored for undifferentiated and neuroectodermal tumor cells; hence, the G classification of GNETs should be higher than G2.
Statistical Analysis
Comprehensive analyses were performed for all published GNET cases and our 2 cases. Survival to death and metastasis were carefully defined. Overall survival (OS) was defined as the period from diagnosis to death or the end of follow-up. Disease-free survival (DFS) was defined as the period from diagnosis to first evidence of metastasis or recurrence.
SPSS 17.0 was used for statistical analysis. Differences in proportions and means were analyzed using Student’s t analysis. The Kaplan–Meier (K-M) method was used for the analysis of survival to death and first metastasis. Differences were assessed using the Log rank test. The significance level was set as P< 0.05. The median OS and DFS were determined by K-M analysis.
Results
Clinical Characteristics
One patient was a 17-year-old male. He was admitted to our hospital because of abdominal pain, melena, fatigue, intermittent low fever, and weight loss lasting for more than 2 months. The serological examination showed negative results for tumor markers. Gastroscopy revealed an irregular mass 6.0 cm×5.0 cm×5.0 cm in size located in the stomach, with necrosis and ulceration on the surface (Figure 1A and B). Subsequently, he underwent partial gastrectomy and abdominal lymph node dissection.
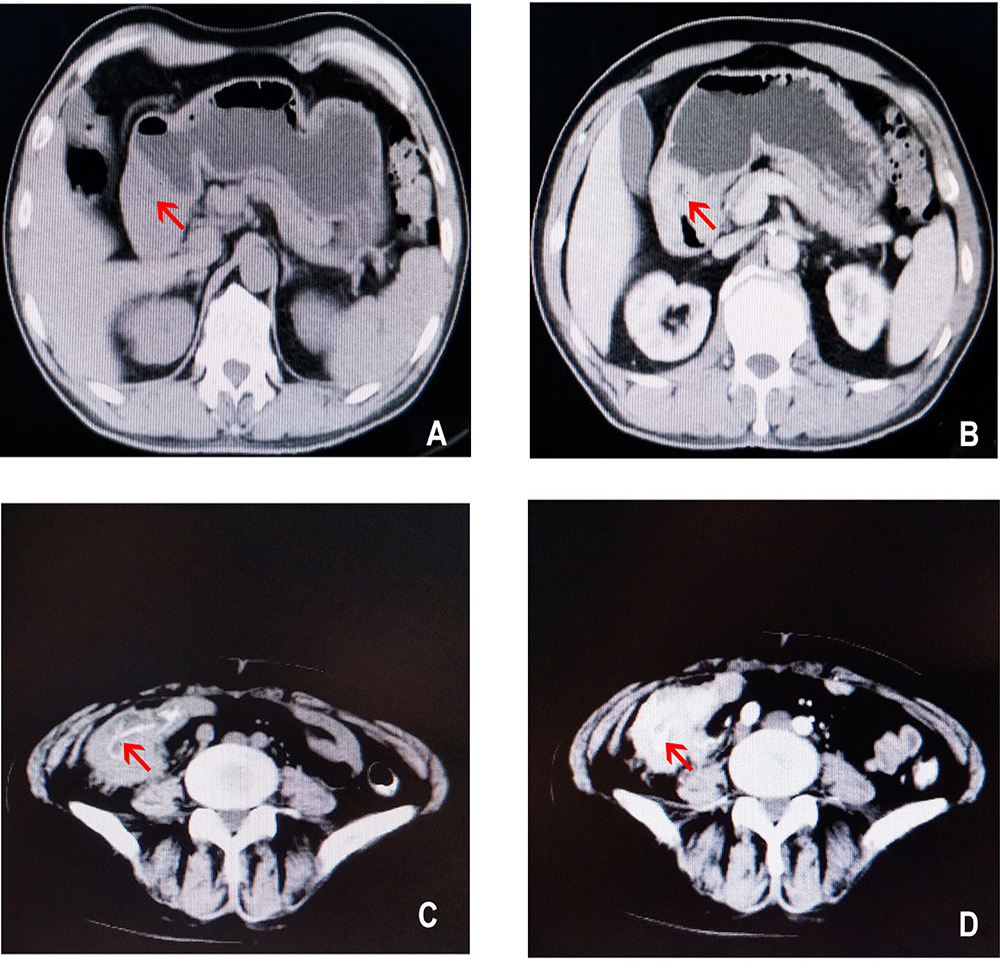 |
Figure 1 CT images from our cases. (A) Precontrast CT image from case 1 revealing an irregular mass 6.0 cm×5.0 cm×5.0 cm in size located in the stomach (red arrow). (B) Contrast-enhanced image from case 1 showing distinct luminal stenosis and heterogeneous enhancement (red arrow). (C) Precontrast CT image from case 2. CT confirmed a mass with a length of 4.0 cm in the right abdomen (red arrow). (D) Contrast-enhanced image from case 2 showing uneven enhancement of the tumor at the ileocecal junction (red arrow). |
The other patient was a 62-year-old female. She had recurrent abdominal pain, diarrhea, and hematochezia lasting for more than 6 years. Due to the aggravation of intermittent abdominal pain and an increase in the size of the abdominal mass, she was admitted to the hospital for further treatment. The serological examination showed no obvious abnormities. The physical examination showed a hard mass in the lower right quadrant with poor mobility. Computed tomography (CT) confirmed a lump with a length of 4.0 cm in the right abdomen (Figure 1C and D). Laparoscopic right hemicolectomy and cholecystectomy were performed. Several hepatic metastases were observed, and enlarged lymph nodes were observed during the operation.
Pathological Morphology
Tumor cells in these 2 cases were mainly located in the lamina propria and mucosa, with focal expansion to the serous membrane and the extramural fat. In addition, neoplastic cells in the 2 patients were predominantly arranged in solid sheets and nests, separated by fibers (Figure 2A). In case 2, focal cells were loosely arranged, presenting pseudocapillary and pseudoalveolar growth patterns (Figure 2B and C). Most tumor cells in the 2 cases were round or short spindle-like in shape, with round, oval, or partially vacuolated nuclei. In case 1, rich osteoclast-like cells and mitotic nuclei were easily found, with clear cytoplasm and large atypical nuclei in most of the cells (Figure 2D). The majority of cells in case 2 were comparatively transparent, with weak eosinophilic cytoplasm and vacuolated nuclei (Figure 2B and C). Atypical mitotic nuclei could also be observed. Osteoclast-like cells were absent in this case.
 |
Figure 2 Morphology of neoplastic cells. (A) Neoplastic cells were mainly arranged in solid sheets and nests, separated by fibers (×100). (B and C) Focal cells were present in pseudocapillary and pseudoalveolar architectures (×400, red arrow). Most cells were round or short spindle-like in shape, with weak eosinophilic or clear cytoplasm. (D) Microscopic findings revealed tissue rich in osteoclast-like cells in one case (400×, red arrow). The nuclei were obviously atypical, round, or oval, with vacuolation in many cells. (E and F) Staining for S100 and SOX10 was diffuse and strongly positive (×400). |
Results of Immunohistochemistry
S100 and SOX10 showed diffuse, strongly positive expression (Figure 2E and F). There was a complete lack of expression for melanin markers, such as HMB45 and Melan-A, in the two cases. Vimentin and BCL-2 expression was strongly positive in one patient, while focal vimentin expression was found in the other patient. The results for other immunohistochemistry markers, such as WT-1, CK, CD117, CD34, CD99, EMA, NSE, CD56, Syn, and MyoD1, were all negative. Tissue rich in osteoclast-like cells was positive for CD68.
Genetic Testing
The proportion of neoplastic cells with an abnormal signal was 75% and 80% in our two cases, which far exceeded the critical value of 15%. The results indicated fracture rearrangement of the EWSR1 gene (Figure 3A and B, respectively).
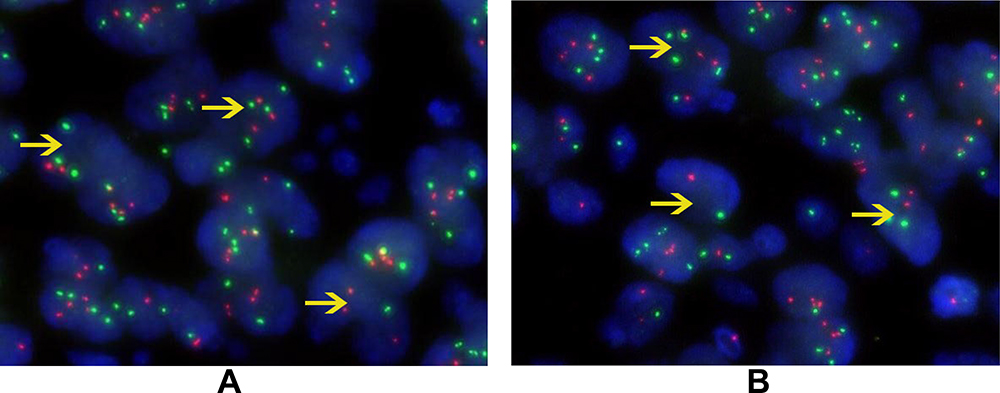 |
Figure 3 FISH genetic testing of EWSR1. The proportion of neoplastic cells with an abnormal signal was 75% (A) and 80% (B) in our two cases (yellow arrow). The threshold value was 15%. The excessive number of abnormal cells indicated rearrangement of the EWSR1 gene. |
Results of Treatment and Follow-Up
The two patients underwent surgical treatment. However, neither patient received adjuvant chemotherapy or radiotherapy after the operation. Patient 1 has survived for 10 months without metastasis or recurrence, while the other patient survived for more than 6 months, with small liver metastases discovered during the operation.
Comparative Analysis of Published Cases
A total of 96 cases, including our 2 cases, were identified (Supplementary file 1). The data of 54 females, 37 males, and 5 patients without sex information were collected. Patients were aged from 5 to 82 years old, with a median age of 36 years (Table 2, Figure 4A). GNETs were mostly located in the small intestine (67.0%).5 The stomach (13.8%) and colon (9.6%) were also frequently influenced (Table 2). The tumor size ranged from 1.2 cm to 15.0 cm, with a median size of 4.5 cm. The lymphatic system and liver were two of the most frequent sites of GNET metastasis (Table 2). Microscopically, most cells were round or short spindle-like in shape, with weak eosinophilic or clear cytoplasm. Neoplastic cells were always arranged in solid sheets, nests, and pseudoalveoli. The prevalence of nuclear mitosis and necrosis was variable. The mitotic number ranged from 3 to 30 per 10 high-power field (HPF), with an average number of 8.0 and a median count of 9.53. Osteoclast-like cells are a specific characteristic of GNETs. Osteoclast-like cells were present in 36 of 71 cases, and Student’s t-test revealed the importance of these bizarre cells (P<0.01). The immunohistochemical results showed strong, diffuse S100 and SOX10 expression, with a complete absence of HMB45 and Melan-A expression. Genetic recombination of EWSR1 was found in 72.9% of the patients.
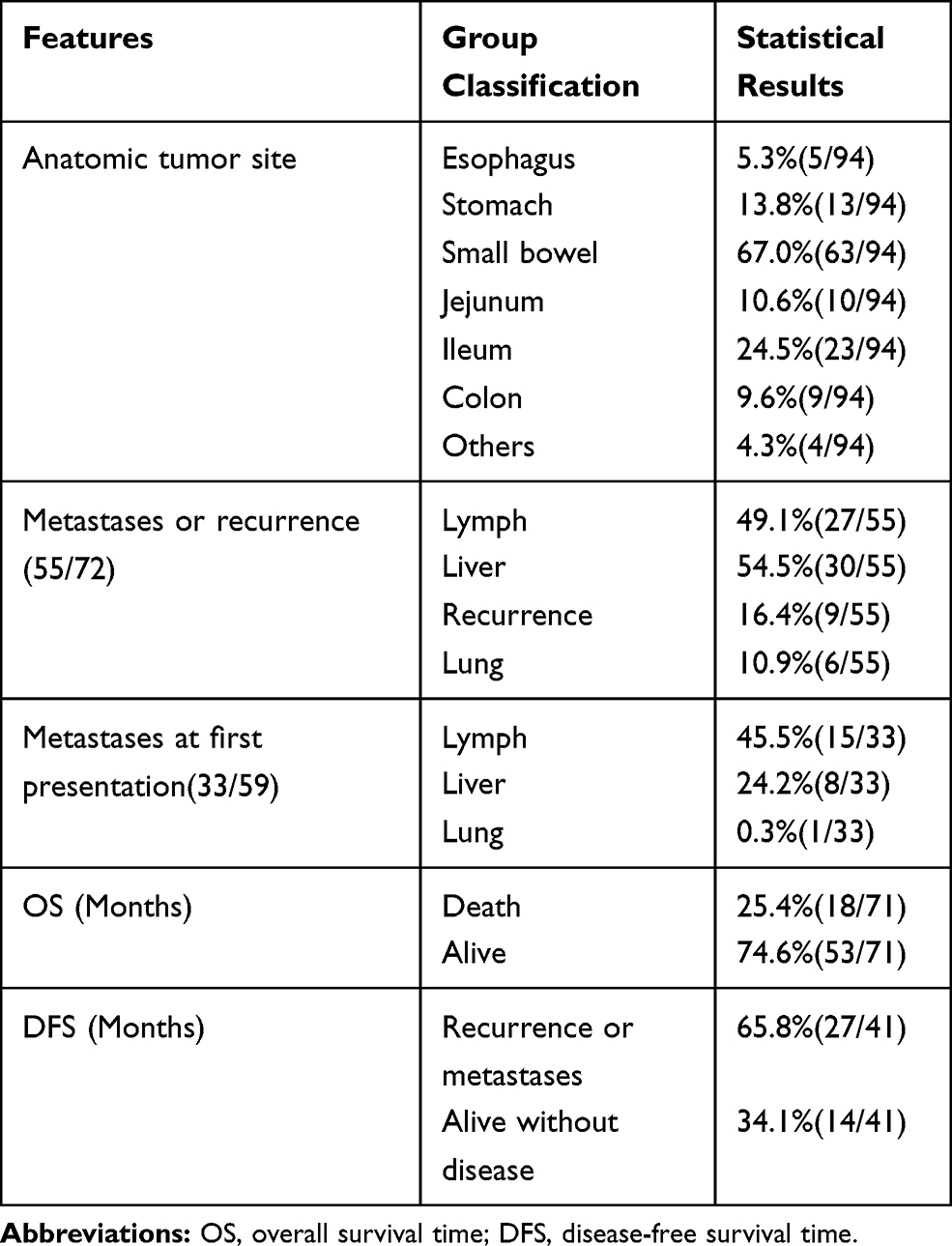 |
Table 2 Clinical Features of GNET Patients |
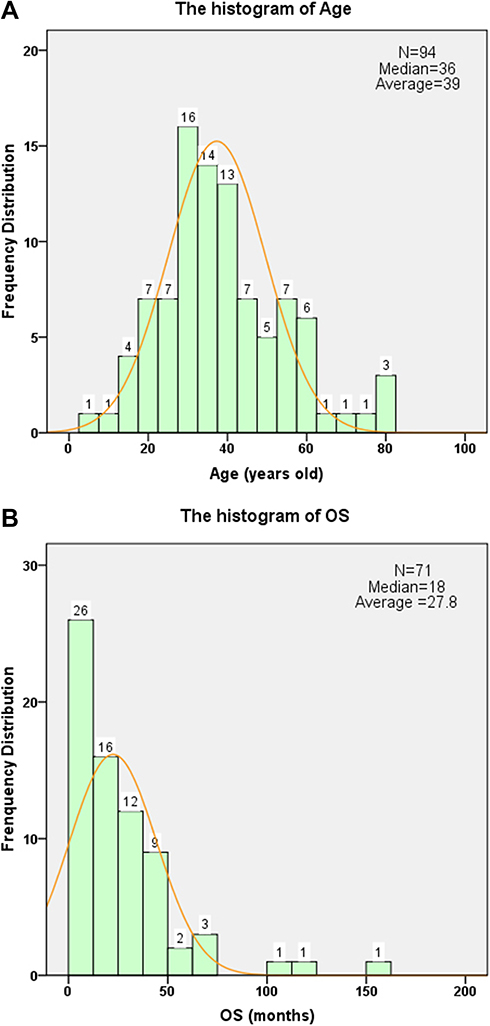 |
Figure 4 General distribution of GNET patients. (A) Distribution of the patients’ age follows a normal curve, with a range of 5 to 82 and a median of 36. The majority of patients were young adults aged 20 to 40 years. (B) Distribution of the follow-up time. The follow-up date was mainly within 48 months, with a median of 18 months and an average of 27.8 months. |
The TNM staging results and follow-up data are shown in Table 3. Sixty-eight percent of the patients presented with transmural tumors (T2a or T2b). The histological grade was G2 (88.1%) in most patients, with G3 (11.9%) accounting for the remaining proportion. In addition, 26 patients had lymph node invasion and 14 had distant metastasis at the time of diagnosis (N and M). The median follow-up time was 18 months, and the majority of follow-up times were within 48 months (89.2%) (Figure 4B). OS ranged from 0.69 to 161 months, with a median of 61 months (Figure 5A). DFS ranged from 1 to 109 months, with a median of 10.0 months and an average of 14.0 months; the median time to first metastasis was 12 months.
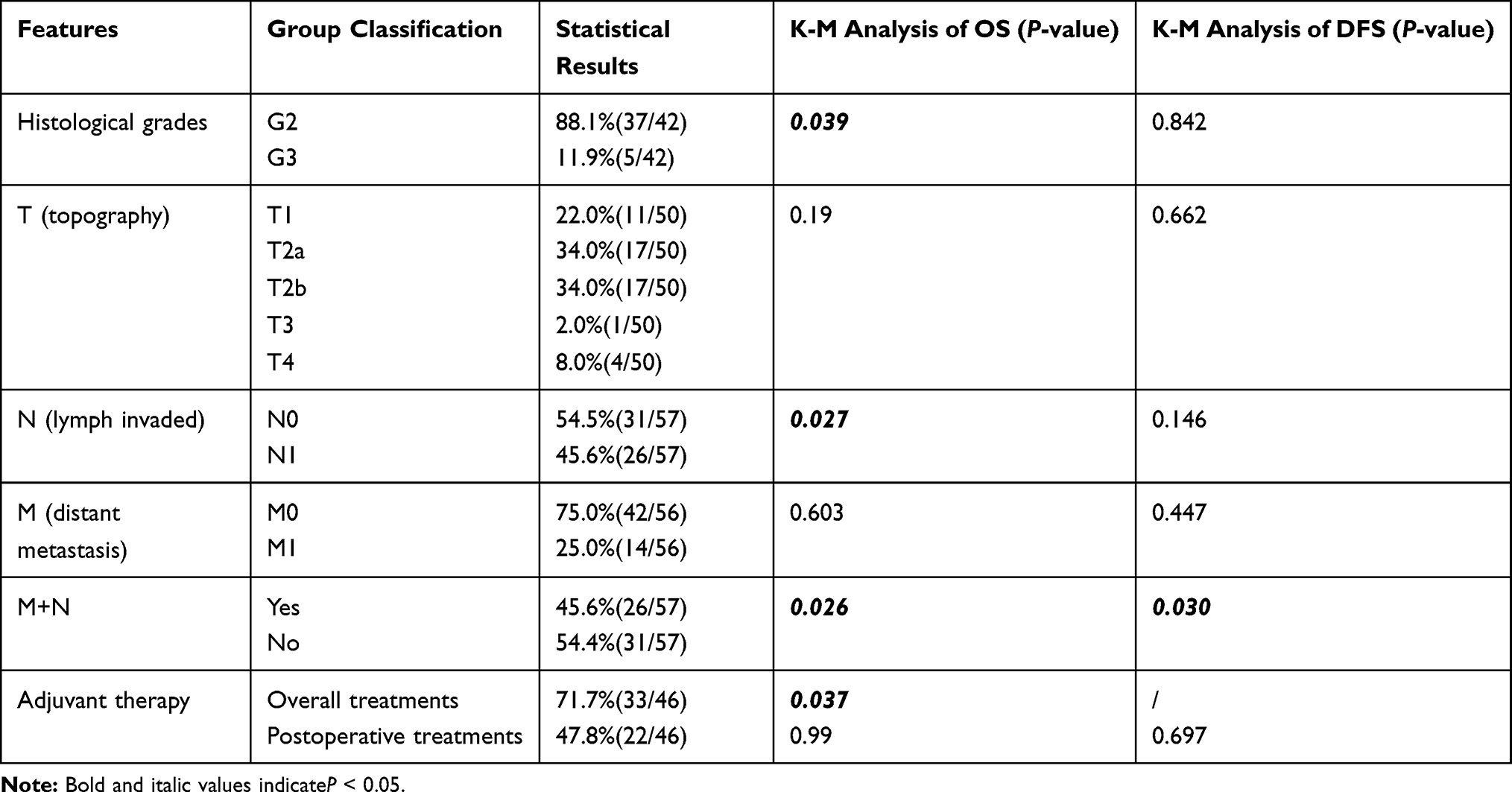 |
Table 3 TNM Staging and K-M Analyses of GNET Patients |
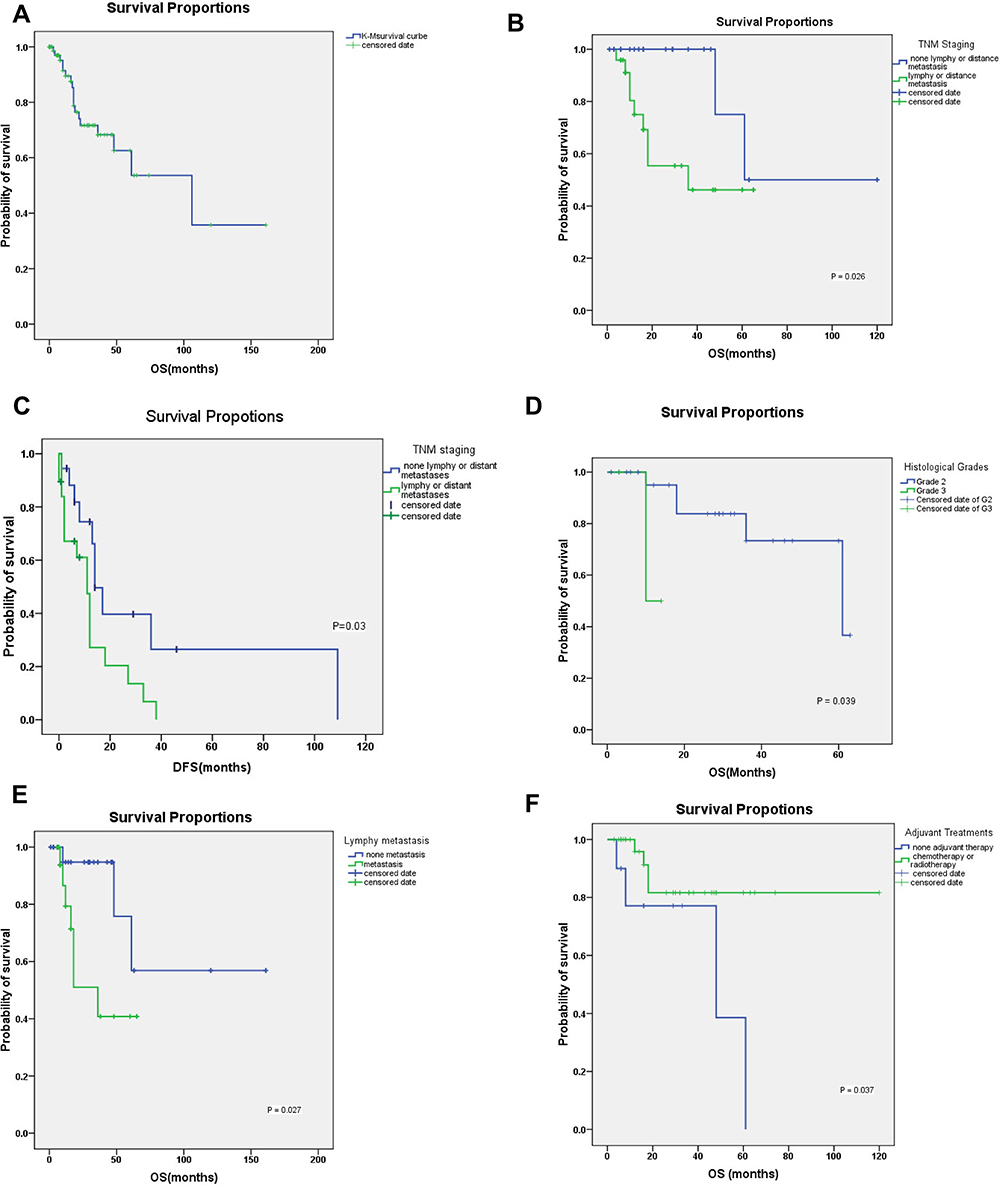 |
Figure 5 The Kaplan–Meier survival curves of GNET patients. (A) The Kaplan–Meier’s survival curve of all patients. The two-year disease-specific survival rate was approximately 71%. The median time to death and first metastasis was 61 and 12 months, respectively. The median follow-up time was 18 months. (B) The Kaplan–Meier’s survival curve of TNM staging. People without lymph invaded or distant metastasis at first diagnosed also present of a better prognosis. (C) Staging of metastasis has a significant influence on disease-free survival time. People without metastases at diagnosis have an advantage to decrease postoperative metastases or recurrence. (D) K-M analysis shows a great difference in the overall survival time between populations of Grade 2 and Grade 3. It means that lower nuclear mitosis and lacking necrosis may link to a better prognosis. (E) Staging of independent lymphy metastasis (N) has a significant influence on overall survival time. People without lymphy metastasis when first diagnosed live a better predictive life. (F) The Kaplan–Meier’s survival curve of adjuvant therapy. Aggressive chemoradiotherapy of GNET treatments are effective in patients’ survival time. |
There are currently no specific GNET staging scores according to the AJCC. Hence, topography (T), lymph node invasion (N), distant metastasis (M), and histological grade (G) were analyzed using the K-M method. Lymph node invasion or distant metastasis (M+N) had a significant influence on OS and DFS (Table 3) (Figure 5B and C). Independent lymph node metastasis (N), lower histological grades (G2), and aggressive chemoradiotherapy affected OS (P=0.027, P=0.039 and P=0.037) (Table 3) (Figure 5D–F). However, independent T, independent M, and postoperative routine adjuvant therapy showed no statistical influence on OS or DFS (Table 3).
Discussion
GNETs are rare malignant tumors with a poor prognosis. In 2003, Zambrano initially described a new entity distinct from CCS.1 In 2012, Stockman analyzed 16 cases and first identified the tumors as GNETs using immunohistochemical and ultrastructural evidence. In his study, it was hypothesized that neoplastic cells may originate from autonomic primitive neural cells with a complete lack of melanocytic differentiation.3,6 Yuan Li provided further proof of the derivation of GNETs by reporting the first case of a GNET occurring in the bronchia.7 In this paper, we conducted a statistical analysis of numerous GNET cases reported worldwide for the first time and aimed to identify the clinicopathological and prognostic features of GNETs.
Many GNETs might be misdiagnosed, most commonly as CCS-GIT lesions. Generally, we concluded that GNETs are a new entity distinct from CCS-GIT lesions. In contrast to the absence of melanocytic differentiation in GNETs, CSS lesions are always positive for the melanin markers HMB45/Melan-A, and melanin granules are commonly visible on electron microscopy. Compared to the wider distribution of CCS lesions, 90.4% of GNETs were present in the GIT, 5.3% in the esophagus, one in the anal canal, and one in the bronchia.7–10 In addition, GNETs affect younger patients than CSS, with a median of 36 years and 57 years, respectively.6 The majority of GNET patients are young adults aged 20 to 40 years.11 Only 5 patients were over 60 years old, and 1 was under 10 years.8,12 Distinct from the male predominance of CCS-GIT lesions, women prevailed in the GNET group, with a ratio of 1:1.5 between males and females (P < 0.01). This result is different from that in previous reports that noted sexual equality in their analyses.13 Osteoclast-like cells are also a specific characteristic of GNETs and were present in 36 of 71 cases (P<0.01).14 FISH genetic testing could also improve the confidence in the diagnosis, as 72.9% of GNETs presented EWSR1-ATF1/CREB1 restructuring. Genetic restructuring could also be found in many other tumors, such as CCS, Ewing sarcoma, and myxolipoma.1,3,15 Thus, it is essential to take full consideration of the histopathological characteristics, immunochemical features, and FISH results for diagnosing GNETs.
GNETs have been reported as an aggressive malignancy with rapid progression.5 The results of our statistical analyses are comparable to those of previous reports. Among the 16 cases reported by Stockman, approximately 50% of the patients died, and 43.6% had metastasis at presentation.3 According to the review of 58 cases reported by Green, the rates of death and first metastasis were 20.7% and 46.6%, respectively.6 The median time to death and metastasis was 9.5 months and 1 month, respectively, in Green’s study.6 Meanwhile, in our review of the follow-up data, approximately 76 cases were included; among them, 18 patients (25.4%) died of the tumor, 55 patients had metastasis or recurrence (76.4%), and 33 GNET patients had metastasis at diagnosis (55.9%) (Table 2). Approximately 40% of the patients experienced metastasis or recurrence within 0.5 years after surgical treatment, and 81.8% did within 2 years. OS was within 48 months for most patients (89.2%), and only 8 patients survived for 5 years or more. The two-year disease-specific survival rate was approximately 71%. The median time to death and first metastasis was 61 and 12 months, respectively (Figure 5A). In our understanding, the long survival time may be attributed to the improvement of adjuvant therapies and lack of survival data. This may indicate that GNETs might not be as progressively malignant as in previous reports. Further retrospective analyses should be conducted in the future.
As a new entity originating from gastrointestinal neuroectodermal precursor cells, GNETs cannot be appropriately staged by diameter. Thus, instead of using guidelines for gastrointestinal carcinoma, we staged GNETs according to the TNM staging criteria for sarcomas of the abdomen and thoracic visceral organs (AJCC, 8th edition). On TNM staging and K-M analysis, many features showed a significant influence on survival, such as the histological grade, metastasis at diagnosis, and independent lymph node metastasis. These results suggest that patients with metastasis are more likely to experience recurrence and further metastasis and have a poor prognosis. Lymph metastasis may play a more important role in GNET patient survival. In addition, a lower prevalence of nuclear mitosis and a lack of necrosis may be linked to a better prognosis. Hence, metastasis and mitotic activity should be taken into consideration when evaluating clinical risk.
Further, for the limitation of the retrospective study, adequate follow-up data, clinical trials, and more molecular studies were not available for the rare malignancies. Generally, radical surgical excision, close follow-up and various adjuvant therapies are common treatments performed in many patients.16 Approximately 33 patients underwent chemotherapy or radiotherapy, and among them, 22 underwent postoperative routine adjuvant therapy (Table 3). Clinical management varied among the 8 patients who survived for more than 60 months.16,17 Treatment with common antitumor drugs, such as ifosfamide, doxorubicin, crizotinib, and pazopanib, was noted.18,19 Radiotherapy after lung and liver metastasis was also performed in some cases.16,20 Two scholars mentioned the possible influence of focal radiotherapy on the neoplastic origin, but no experimental evidence was provided to confirm this influence.21 Drug resistance and the lack of specific targeted treatments are the main problems of clinical treatment.11 According to the K-M analysis, aggressive chemotherapy or radiotherapy was effective in increasing the time to death but not to first metastasis. Postoperative routine adjuvant therapy showed no effect on survival. Hence, in our opinion, GNET patients need to be diagnosed and treated early, before tumor metastasis. Chemotherapy or focal radiotherapy may be useful for prolonging survival. Annual endoscopic examination is another effective and sensible preventive method.
The mechanism of GNETs is still unclear. Some scholars have suggested abdominal radiotherapy.21 Luigi narrowed the mechanism to that of Ewing sarcoma because of the expression of FLI-1 and a history of Ewing sarcoma.22 Gao preferred immunological factors given the macrophages in the abdominal nodules in their cases.23 Controversies regarding these isolated cases aside, the SWI/SNF-related hypothesis described by Stockman may be more reliable.3 Current findings have revealed possible oncogenic mechanisms, including the potential relation of GNET neoplastic oncogenes to the SWI/SNF complex.24 Binding of the SWI/SNF complex could induce nuclear localization, change the balance of tumor inhibitors and lead to similar changes in terms of SWI/SNF gene mutations.25,26
GNETs could also be easily misdiagnosed as many other tumors. Neoplastic cells similar to those in monophasic synovial sarcoma (MSS) have been reported. CD99 and SOX10 are not expressed in MSS lesions, and genetic SYT fusion could be used to distinguish these lesions from GNETs. Additionally, gastrointestinal stromal tumors (GISTs) should be considered. The positive markers of GISTs, such as CD117, DOG1, and CD34, are negative in GNETs. Furthermore, osteoclast-like cells are rare in GIST cases. Regarding oncogenic performance, c-kit fusion is common in GISTs.
Conclusion
In summary, with a young onset age, poor prognosis, undefined pathogenesis, and unspecific treatments, GNETs are malignant tumors that merit further study. GNET should be suspected in cases of a tumor of gastrointestinal tract, which comprising epithelioid or spindle cells arranged in various patterns. Osteoclast–like cells and absence of melanin markers will be helpful. ESWSR1-ATF1/CREB1 genetic rearrangement is necessary to confirm the diagnosis of GNET.
Acknowledgments
We would like to appreciate the parents and clinicians for help in our report and providing a detailed history. Thanks to AJE for the polishing of the article. This study was supported by the Natural Science Foundation of Anhui Province (No.1608085QH207, No. 1908085MH275), the Nature Science Key Program of College and University of Anhui Province (No.KJ2018A0986) and the Natural Science Foundation of Bengbu Medical College (BYKF1711).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Zambrano E, Reyes-Mugica M, Franchi A, Rosai J. An osteoclast-rich tumor of the gastrointestinal tract with features resembling clear cell sarcoma of soft parts: reports of 6 cases of a GIST simulator. Int J Surg Pathol. 2003;11(2):75–81. doi:10.1177/106689690301100202
2. Antonescu CR, Nafa K, Segal NH, Dal Cin P, Ladanyi M. EWS-CREB1: a recurrent variant fusion in clear cell sarcoma–association with gastrointestinal location and absence of melanocytic differentiation. Clin Cancer Res. 2006;12(18):5356–5362. doi:10.1158/1078-0432.CCR-05-2811
3. Stockman DL, Miettinen M, Suster S, et al. Malignant gastrointestinal neuroectodermal tumor: clinicopathologic, immunohistochemical, ultrastructural, and molecular analysis of 16 cases with a reappraisal of clear cell sarcoma-like tumors of the gastrointestinal tract. Am J Surg Pathol. 2012;36(6):857–868. doi:10.1097/PAS.0b013e31824644ac
4. Zambo I, Vesely K. WHO classification of tumours of soft tissue and bone 2013: the main changes compared to the 3rd edition]. Cesk Patol. 2014;50(2):64–70.
5. Kong J, Li N, Wu S, Guo X, Gu C, Feng Z. Malignant gastrointestinal neuroectodermal tumor: a case report and review of the literature. Oncol Lett. 2014;8(6):2687–2690. doi:10.3892/ol.2014.2524
6. Green C, Spagnolo DV, Robbins PD, Fermoyle S, Wong DD. Clear cell sarcoma of the gastrointestinal tract and malignant gastrointestinal neuroectodermal tumour: distinct or related entities? A review. Pathology. 2018;50(5):490–498. doi:10.1016/j.pathol.2018.05.001
7. Zheng Q, Chen H, Li Y. Primary gastrointestinal-type clear cell sarcoma-like tumor of the bronchus: a hitherto unreported bronchial tumor. J Thorac Oncol. 2019;14(9):e202–e205. doi:10.1016/j.jtho.2019.04.030
8. Allanson BM, Weber MA, Jackett LA, et al. Oral malignant gastrointestinal neuroectodermal tumour with junctional component mimicking mucosal melanoma. Pathology. 2018;50(6):648–653. doi:10.1016/j.pathol.2018.07.002
9. Kim SB, Lee SH, Gu MJ. Esophageal subepithelial lesion diagnosed as malignant gastrointestinal neuroectodermal tumor. World J Gastroenterol. 2015;21(18):5739–5743. doi:10.3748/wjg.v21.i18.5739
10. Breton S, Dubois M, Geay J-F, et al. Clear cell sarcoma or gastrointestinal neuroectodermal tumor (GNET) of the tongue? Case report and review of the literature of an extremely rare tumor localization. Ann Pathol. 2019;39(2):167–171. doi:10.1016/j.annpat.2018.10.004
11. Chang B, Yu L, Guo WW, et al. Malignant gastrointestinal neuroectodermal tumor: clinicopathologic, immunohistochemical, and molecular analysis of 19 cases. Am J Surg Pathol. 2020;44(4):456–466. doi:10.1097/PAS.0000000000001396
12. D’Amico FE, Ruffolo C, Romeo S, Massani M, Dei Tos AP, Bassi N. Clear cell sarcoma of the ileum: report of a case and review of the literature. Int J Surg Pathol. 2012;20(4):401–406. doi:10.1177/1066896911428073
13. Alyousef MJ, Alratroot JA, ElSharkawy T, et al. Malignant gastrointestinal neuroectodermal tumor: a case report and review of the literature. Diagn Pathol. 2017;12(1):29.
14. Kato T, Ichihara S, Gotoda H, et al. Imprint cytology of clear cell sarcoma-like tumor of the gastrointestinal tract in the small intestine: a case report. Diagn Cytopathol. 2017;45(12):1137–1141. doi:10.1002/dc.23786
15. Thway K, Fisher C. Tumors with EWSR1-CREB1 and EWSR1-ATF1 fusions: the current status. Am J Surg Pathol. 2012;36(7):e1–e11. doi:10.1097/PAS.0b013e31825485c5
16. Shah AA, Grosh WW, Frierson HF
17. Subbiah V, Holmes O, Gowen K, et al. Activity of c-Met/ALK inhibitor crizotinib and multi-kinase VEGF inhibitor pazopanib in metastatic gastrointestinal neuroectodermal tumor harboring EWSR1-CREB1 fusion. Oncology. 2016;91(6):348–353. doi:10.1159/000449204
18. Washimi K, Takagi M, Hisaoka M, et al. Clear cell sarcoma-like tumor of the gastrointestinal tract: a clinicopathological review. Pathol Int. 2017;67(10):534–536. doi:10.1111/pin.12573
19. Song SH, Shin JH, Ryu HJ, Kim DJ, Park SY. Successful surgical treatment of a recurrent esophageal malignant gastrointestinal neuroectodermal tumor. Korean J Thorac Cardiovasc Surg. 2018;51(2):142–145. doi:10.5090/kjtcs.2018.51.2.142
20. Libertini M, Thway K, Noujaim J, et al. Clear cell sarcoma-like tumor of the gastrointestinal tract: clinical outcome and pathologic features of a molecularly characterized tertiary center case series. Anticancer Res. 2018;38(3):1479–1483.
21. Thway K, Judson I, Fisher C. Clear cell sarcoma-like tumor of the gastrointestinal tract, presenting as a second malignancy after childhood hepatoblastoma. Case Rep Med. 2014;2014:984369. doi:10.1155/2014/984369
22. Insabato L, Guadagno E, Natella V, et al. An unusual association of malignant gastrointestinal neuroectodermal tumor (clear cell sarcoma-like) and ewing sarcoma. Pathol Res Pract. 2015;211(9):688–692. doi:10.1016/j.prp.2015.06.001
23. Huang GX, Chen QY, Zhong LL, et al. Primary malignant gastrointestinal neuroectodermal tumor occurring in the ileum with intra-abdominal granulomatous nodules: a case report and review of the literature. Oncol Lett. 2019;17(4):3899–3909.
24. Linden M, Thomsen C, Grundevik P, et al. FET family fusion oncoproteins target the SWI/SNF chromatin remodeling complex. EMBO Rep. 2019;20(5). doi:10.15252/embr.201845766
25. Boulay G, Sandoval GJ, Riggi N, et al. Cancer-specific retargeting of BAF complexes by a prion-like domain. Cell. 2017;171(1):163–178 e119. doi:10.1016/j.cell.2017.07.036
26. Fisher C. The diversity of soft tissue tumours with EWSR1 gene rearrangements: a review. Histopathology. 2014;64(1):134–150. doi:10.1111/his.12269
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.