Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Magnetically stimulated ciprofloxacin release from polymeric microspheres entrapping iron oxide nanoparticles
Authors Sirivisoot S, Harrison B
Received 13 February 2015
Accepted for publication 16 April 2015
Published 9 July 2015 Volume 2015:10(1) Pages 4447—4458
DOI https://doi.org/10.2147/IJN.S82830
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Sirinrath Sirivisoot,1 Benjamin S Harrison2
1Biological Engineering Program, Faculty of Engineering, King Mongkut’s University of Technology Thonburi, Bangkok, Thailand; 2Wake Forest Institute for Regenerative Medicine, Wake Forest School of Medicine, Wake Forest University Health Sciences, Winston-Salem, NC, USA
Abstract: To extend the external control capability of drug release, iron oxide nanoparticles (NPs) encapsulated into polymeric microspheres were used as magnetic media to stimulate drug release using an alternating magnetic field. Chemically synthesized iron oxide NPs, maghemite or hematite, and the antibiotic ciprofloxacin were encapsulated together within polycaprolactone microspheres. The polycaprolactone microspheres entrapping ciprofloxacin and magnetic NPs could be triggered for immediate drug release by magnetic stimulation at a maximum value of 40%. Moreover, the microspheres were cytocompatible with fibroblasts in vitro with a cell viability percentage of more than 100% relative to a nontreated control after 24 hours of culture. Macrophage cell cultures showed no signs of increased inflammatory responses after in vitro incubation for 56 hours. Treatment of Staphylococcus aureus with the magnetic microspheres under an alternating (isolating) magnetic field increased bacterial inhibition further after 2 days and 5 days in a broth inhibition assay. The findings of the present study indicate that iron oxide NPs, maghemite and hematite, can be used as media for stimulation by an external magnetic energy to activate immediate drug release.
Keywords: antibacterial, maghemite, hematite, Staphylococcus aureus
Introduction
Magnetic nanoparticles (NPs) are potentially powerful biocompatible materials applicable for use in various biomedical applications, ranging from diagnostics (contrast enhancement for magnetic resonance imaging) to therapeutics (potential drug delivery vehicles and hyperthermia agents), as well as cell/protein separation and biosensors.1–3 Among magnetic NPs for biomedical applications, iron oxide NPs are particularly attractive because they exhibit ferromagnetism, low toxicity, and high biocompatibility with cells and tissues.4 Small iron oxide NPs (such as maghemite [γ-Fe2O3], hematite [α-Fe2O3], magnetite [Fe3O4], and other ferrites) exhibit a large magnetic moment that is useful for guiding them to a specific tissue or organ by an external magnetic field.5 Iron oxide magnetic NPs are used as magnetically activated materials to externally control drug release from nanocomposite membranes,6 liposomes,7 microspheres,8,9 microcapsules,10 and nanospheres,11,12 typically by mechanical disruption of the biomaterial matrix-embedded drugs. Iron oxide is also used to magnetically control or target the delivery of anticancer drugs (eg, paclitaxel/rapamycin,13 doxorubicin14) and antibiotics (eg, gentamicin15). Therefore, magnetic targeting using iron oxide NPs containing antibiotics can be localized to pathologic sites by an external magnet to inhibit bacterial growth.
The enormous advantages and wide array of functions that can be designed into polymeric systems have encouraged biomedical scientists to use polymers for drug delivery,16,17 tissue engineering,18 and biologic imaging.19 Natural polymers that can degrade into biologically compatible components under physiologic conditions present an attractive alternative for the development of drug delivery systems. Entrapping a drug within a suitable carrier can enhance the aqueous solubility of the drug, increase its circulation half-life, target the drug to certain tissues, and improve tissue penetration of the drug. These features can increase drug bioavailability to diseased tissues, while at the same time decrease drug toxicity to the healthy tissue, thus improving therapeutic efficacy. Polycaprolactone (PCL) is a biocompatible and biodegradable polymer (Food and Drug Administration approved) that undergoes chemical degradation through random hydrolysis of ester bonds, constituting the backbone of polymer chains. PCL creates an acid-free environment during degradation, unlike polylactide or polyglycolide.20 PCL possesses hydrophobicity and high permeability to low molecular-weight drugs21 and has been used to deliver various drugs, such as anticancer drugs (eg, taxol22), anti-inflammatory agents (eg, sulfasalazine/betamethasone23), and proteins (eg, bovine serum albumin24), as well as long-term implants and adhesive barriers. In the present study, we investigated the potential of using a magnetically stimulated antibiotic delivery system of PCL microspheres containing iron oxide NPs and ciprofloxacin (CIP) – to treat Staphylococcus aureus, which commonly contaminates bone implants.
Infection of biomaterial implants is a major cause of implant failure. Biomaterial-associated infection can develop from perioperative bacterial contamination of implant surfaces during implantation, immediately post surgery during hospitalization, or by hematogenous spreading of bacteria from infections elsewhere in the body.25 S. aureus is the most frequently isolated pathogen from biomaterial implant surfaces, detected in ∼23% of infections associated with prosthetic joints.26
CIP is used to treat bacterial infections in human beings (such as urinary tract, gastrointestinal, and skin/bone infections).27 It is the most widely used fluoroquinolone for bacterial bone infections and has potent bactericidal activity against a broad range of clinically relevant Gram-negative and Gram-positive pathogens.28 The minimum inhibitory concentration (MIC) of CIP is as low as 0.25–1 μg/mL for Staphylococcus (frequently found in osteomyelitis).29 Although the usual dosage of CIP is 750 mg twice daily for periods ranging from 6 weeks to several months, the cure rate is only ∼56%.30 Therefore, there is still a need to develop a local and externally controllable release system for immediate antibiotic treatment of infected bone implants.
Our work provides a method to embed CIP and iron oxide NPs (either maghemite [γ-Fe2O3] or hematite [α-Fe2O3]) in PCL microspheres. The CIP was released from the microspheres using an alternating (oscillating) magnetic field (AMF) to suppress the growth of S. aureus. Our hypothesis was that the application of an AMF toward the PCL microspheres entrapping magnetic NPs and antibiotics would trigger faster drug release to achieve a higher local concentration of the drug and eliminate bacteria at the bone implant surface. In addition, we evaluated the fibroblast compatibility and macrophage inflammatory response under standard cell culture conditions in the present study. This study provides a proof of concept of stimulated antibiotic release using a magnetic field, which could be useful for further studies of externally controlled drug delivery devices for bone implants.
Materials and methods
Synthesis of iron oxide NPs
Iron oxide NPs were synthesized by coprecipitation of iron salts. Briefly, ferrous chloride (FeCl2·4H2O; Thermo Fisher Scientific, Waltham, MA, USA) and ferric chloride (FeCl3·6H2O; Thermo Fisher Scientific) in a molar ratio of 1:2 were dissolved in 50 mL of distilled water in the presence of 0.4 N hydrochloric acid (HCl; Thermo Fisher Scientific) and degassed with nitrogen gas. The solution was dropped into 40 mL of ammonium hydroxide (NH4OH; Sigma-Aldrich Co., St Louis, MO, USA) under a nitrogen blanket. The solution of iron oxide NPs was heated to 90°C for 30 minutes under magnetic stirring and aeration. The solution was centrifuged at 4,750 rpm for 10 minutes. The iron oxide NPs were decanted, diluted with distilled water, and dispersed by vortexing. The cleaning process with water was repeated three times. Before the spray-drying process, 0.5 M citric acid (Sigma-Aldrich Co.) was added to the solution of iron oxide NPs to increase NP dispersion. The iron oxide NPs were then spray-dried using a Mini Spray Dryer B-290 (BÜCHI, Flawil, Switzerland) at an inlet temperature of 170°C.
Drug encapsulation (oil-in-water emulsion)
PCL (Cat# 19561, Mn ∼43–50 k; Polysciences, Inc., Warrington, PA, USA) was dissolved at 5% w/v in chloroform (Sigma-Aldrich Co.). Then, 2 mL (140 mg/mL) of maghemite (nonspray-dried NPs) fluid or 380 mg of the hematite (spray-dried NPs) was added to the organic solution and vortexed for 10 minutes. Next, 4.5 mM CIP (Sigma-Aldrich Co.) was added to 10 mL of the organic solution and vortexed for 10 minutes. The organic mixture was poured into a mixture of 0.5% w/v polyvinyl alcohol (Sigma-Aldrich Co.) and distilled water. The mixture was sonicated using a Model 100 Sonic Dismembrator (Thermo Fisher Scientific) for 10 minutes at the room temperature. The PCL–CIP–NP microspheres were collected from the emulsified solution by centrifugation at 4,750 rpm. The microspheres were then decanted, diluted with distilled water, and centrifuged at 4,750 rpm three times. The microspheres were then dispersed in phosphate-buffered saline (PBS; 1 M, pH 7.4) at a concentration of 40 μg/mL and gamma irradiated at 1 mrad before the cell culture and bacterial inhibition studies.
Nano- and microparticle characterizations
The PCL–CIP–NP microspheres were characterized using a Fourier transform infrared spectrometer (Spectrum 400; PerkinElmer Inc., Waltham, MA, USA) equipped with a universal attenuated total reflectance sampling accessory (PerkinElmer Inc.). The samples were also measured by dynamic light scattering (DLS) measurements. The DLS measurements were performed on a Zetasizer Nano S (Malvern Instruments, Malvern, UK) equipped with an HeNe laser at 632.8 nm and a scattering detector at 173°C using water as a dispersant (refraction index =1.33 and viscosity =0.8872) at the room temperature with cumulants analysis. Raman spectra of the synthesized NPs were obtained using a Raman spectrometer (RamanStation 400; PerkinElmer Inc.) equipped with an excitation source of 350 mW near-infrared 785 nm laser. The spectra were obtained from 100 cm−1 to 1,800 cm−1 with a peak resolution of 4 cm−1. The solutions of NPs and PCL–CIP–NPs were dropped and dried overnight on carbon-coated Formvar 200 mesh copper grids (Ted Pella, Inc., Redding, CA, USA). The grids were visualized using a Philips 400 120 keV transmission electron microscope (TEM; Philips, Hillsboro, OR, USA) with a high-tilt goniometer stage.
Cell culture and assay
Mouse fibroblasts NIH/3T3 (CRL-1658, passage number =4–7; American Type Culture Collection (ATCC), Manassas, VA, USA) or mouse macrophage Raw 264.7 (TIB-71, passage number =7–12; ATCC) was seeded at a density of 40,000 cells/cm2 in Dulbecco’s Modified Eagle’s Medium (HyClone, Logan, UT, USA) with 10% bovine calf serum (HyClone) and 1% penicillin/streptomycin (HyClone) and incubated under standard cell culture conditions (37°C, 5% CO2 in humidified atmosphere). After 4 hours of incubation, the solutions of samples (maghemite, hematite, PCL–CIP–maghemite, and PCL–CIP–hematite) in deionized water were added to the cell culture media at concentrations of 4 μg/mL, 2 μg/mL, 1 μg/mL, and 0.5 μg/mL. The samples were further cultured with fibroblasts for 24 hours and with macrophages for 56 hours. The CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS; Promega Corporation, Fitchburg, WI, USA) was used to determine the cell viability of the fibroblasts and macrophages. The cell suspensions treated with the samples in triplicate were incubated with 500 μL of 10% v/v MTS assay at 37°C for 3 hours. The formazan product, which was bioreduced from MTS by living cells, was read at an absorbance of 490 nm using a spectrophotometer (SpectraMax® M5; Molecular Devices LLC, Sunnyvale, CA, USA). Untreated cells were used as controls to calculate the percentages of cell viability. Experiments using both cell types were repeated three times. The media were changed every other day.
Determination of MIC
Stock solutions and dilutions were prepared according to the Clinical and Laboratory Standards Institute (M100-S16 CLSI).31 The MIC was determined by broth and agar dilution tests.32 The MIC of CIP was defined as the lowest concentration inhibiting visible growth after incubation at 37°C for 24 hours with a shaker (speed at 220 rpm). Briefly, S. aureus (passage number =3–4, ATCC 2913) preserved in 20% glycerol and stored at −80°C was purchased from the Cell and Virus Vector Core Laboratory (Wake Forest Baptist Medical Center, Winston-Salem, NC, USA). At the time of use, a frozen tube was thawed to make a streak plate. A sterilized 10 μm loop was dipped into the bacterial tube and streaked onto a 5% sheep blood agar plate (SBA plate; BD, Franklin Lakes, NJ, USA). The streaked plate was incubated for 24 hours under standard cell culture conditions (37°C, 5% CO2 in humidified air). The next day, the colony count plate was prepared and the MIC was determined. A single colony was lifted from the streak plate using a sterilized cotton-tipped applicator and dissolved with 5 mL sterilized PBS (HyClone) in a 14 mL glass tube with snap cap (BD). The bacterial suspension was adjusted to be equal to 108 cfu/mL by comparison with the McFarland 0.5 turbidity standard (Thermo Fisher Scientific) and the addition of sterilized PBS. The bacterial suspension (108 cfu/mL equivalent) was diluted to 105 cfu/mL with sterilized PBS and 103 cfu/mL with Mueller-Hinton broth (BD), respectively. To prepare the colony count plate, 50 μL of bacterial suspension (103 cfu/mL equivalent) was pipetted onto an SBA plate and spread using an L-shape spreader (Thermo Fisher Scientific) and incubated in 37°C for 24 hours. The bacterial colonies were counted and recorded the next day. Then, the serial dilutions of 500 μg/mL of CIP in 0.1 N HCl (stock solution) were prepared to a range of concentrations (10–0.039 μg/mL). The control group was a standard nutrient broth without CIP. Next, 10 μL bacterial suspension (105 cfu/mL equivalent in broth) was added to 10 mL of serial dilutions of CIP and a control tube. All tubes were incubated at 37°C for 24 hours with a shaker (220 rpm). A 100 μL sample was obtained from the CIP-bacterial suspension and streaked onto SBA plates and incubated for 24 hours. The bacterial colonies were counted and recorded the next day. The study showed that the bacteria were inhibited by CIP at a concentration between 0.156 μg/mL and 0.31 μg/mL. At a CIP concentration of 0.156 μg/mL, bacterial colonies were too many to count (>300) in the plate. At a concentration of 0.31 μg/mL, there was only one colony. Therefore, an inhibited concentration at 50% (MIC50) of CIP for S. aureus used in the present study was 0.23 μg/mL.
Determination of CIP
The percentage of drug encapsulation was investigated by measuring the absorbance of CIP in the PCL–CIP–NPs using a spectrophotometer (SpectraMax® M5) to determine the amount of CIP loaded within the microspheres at 270 nm, a previously validated wavelength for determining CIP concentrations.33 Before the drug release study, the PCL–CIP–NP microspheres were purified by resuspension with PBS (1 M, pH 7.4) and centrifuged three times. A linear relationship between concentration and absorbance at 270 nm of CIP in PBS was detected with a correlation coefficient of ~0.99.
Antimicrobial susceptibility testing of PCL–CIP–NPs
The schematic for the bacteria study is shown in Figure 1. Briefly, 16 μL of PCL–CIP–NP suspension in PBS (40 μg/mL) containing MIC50 of CIP (determined by fluorescent intensity measurement) was added into each tube containing 1 mL bacterial suspension (105 cfu/mL in broth), which was diluted from 108 cfu/mL equivalent suspension. Then, the bacterial-sample suspension was incubated along with control (only bacteria and broth) for 24 hours at 37°C in a shaker at a speed of 220 rpm. The next day, bacterial dilutions were prepared from the incubated bacterial-sample solutions to make the streak plates. Serial dilutions of the bacterial-sample stocks and sterilized PBS from 1:101 (stock: PBS) to 1:109 were used in the study. Each bacterial dilution (10 μL) was pipetted onto an SBA plate and streaked using an L-shaped spreader. After incubation for 1 day, 2 days, and 5 days without changing broth, the bacterial colonies were counted at each time point. To study the effect of the magnetic field on bacteria growth, the suspensions of the NPs, PCL–CIP–NPs, and bacteria alone (control) were incubated in the presence of an AMF for 1 day, 2 days, and 5 days.
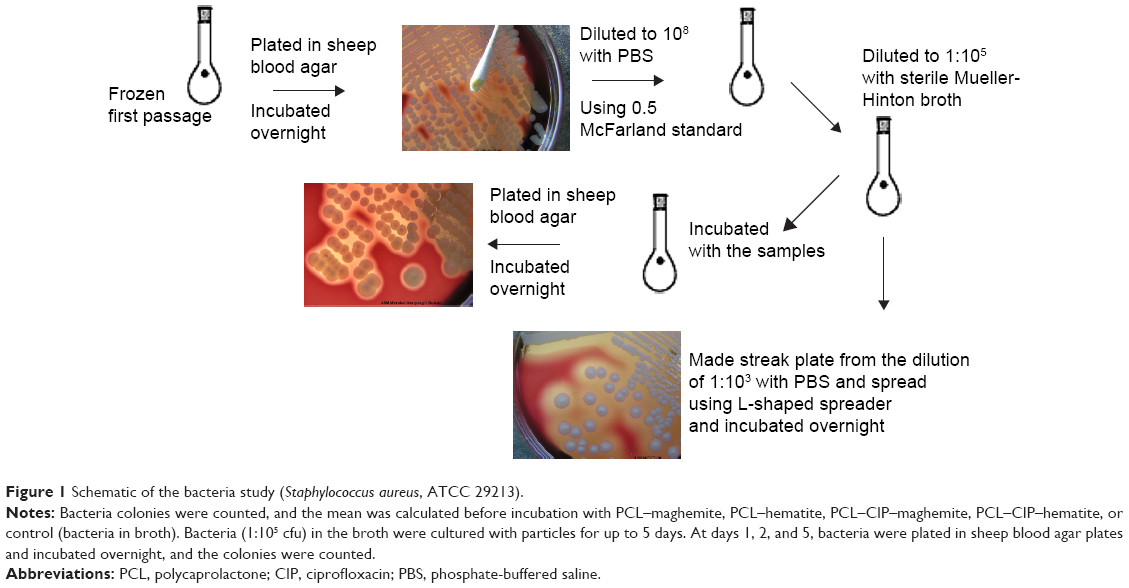 | Figure 1 Schematic of the bacteria study (Staphylococcus aureus, ATCC 29213). |
Application of AMF
An AMF was generated using a bed magnet (USWP Manufacturing, Spokane Valley, WA, USA) placed on a lab rotator model 1314 (Barnstead Intl. Lab-Line, Melrose Park, IL, USA) with an orbital diameter of 0.75 in at 220 rpm. The AMF was measured using a 5180 Gauss/Tesla Meter (FW Bell/Pacific Scientific-OECO, Milwaukie, OR, USA) at a height of 1 cm (approximately the mean height of a bacterial suspension in a 1.5 mL tube) above the bed magnet.
In vitro drug release study
To study the drug release, 2.4 μg of microspheres were suspended in 1 mL of sterilized PBS (1 M, pH 7.4). The drug release study was performed at 37°C at a stirring rate of 220 rpm in the presence of an AMF. A 100 μL aliquot of the solution was sampled, and the absorbance was measured with a spectrophotometer (SpectraMax® M5) at 270 nm before replacing the aliquot back into the solution. Optical measurements were obtained daily on days 1–9 and then at 19 days after release. The drug release profiles of CIP from PCL–CIP–NPs with and without AMF were plotted as the percentage of drug release (weight/weight) vs time (days) of observation. All experiments were performed in triplicate. The percentage of drug release was calculated using the equation:
|
|
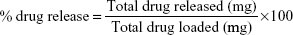
Results and discussion
Chemical synthesis of iron oxide NPs
The synthesized iron oxide NPs were characterized by TEM imaging (Figure 2A). The magnetic NPs were then spray-dried at a temperature of 170°C (Figure 2B). Although the NP diameter was mostly <10 nm, they formed aggregated clusters. The magnetic NPs were synthesized by coprecipitation of Fe2+ and Fe3+ aqueous chloride salt solution. Fe3O4 was obtained according to the following chemical equation: Fe2+ + 2Fe3+ + 8OH− → Fe3O4 + 4H2O. To obtain the colloidal suspension of Fe2O3 (brown-red color), the aqueous Fe3O4 magnetic NPs were heated at 90°C for 30 minutes under aeration (nitrogen gas) to directly oxidize the magnetite (black color).34,35 Crystalline Fe2O3 is commonly obtained as either maghemite (γ-phase) or hematite (α-phase). The Raman spectra results (Figure 3) confirmed that maghemite (γ-Fe2O3) NPs changed to hematite (α-Fe2O3) by oxidation following the spray-drying process at 170°C.
 | Figure 2 TEM micrographs of (A) maghemite (nonspray-dried iron oxide NPs), (B) hematite (spray-dried iron oxide NPs), (C) PCL–CIP–maghemite microsphere, and (D) PCL–CIP–hematite microsphere. |
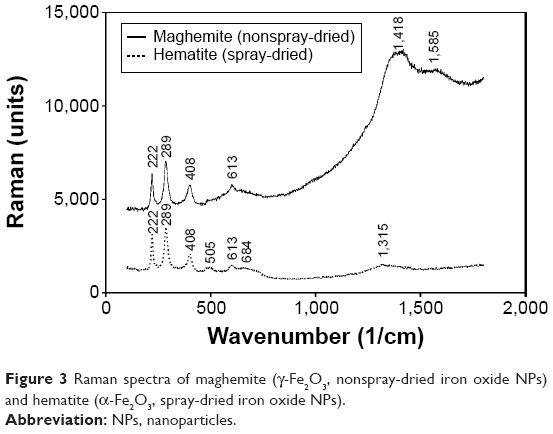 | Figure 3 Raman spectra of maghemite (γ-Fe2O3, nonspray-dried iron oxide NPs) and hematite (α-Fe2O3, spray-dried iron oxide NPs). |
Iron oxide NPs and drug encapsulation
PCL microspheres entrapping the NPs and CIP were observed using TEM (Figure 2C and D). The nonspray-dried iron oxide NPs were clearly observed throughout the PCL microspheres, whereas the spray-dried iron oxide NPs were observed as a group of aggregated NPs. Hematite was loaded near the wall of the PCL–CIP–hematite microspheres (Figure 2D). Furthermore, PCL–CIP–hematite was embedded in resin and sectioned (80–100 nm thick) before TEM imaging to confirm the presence of iron oxide particles in the middle of the microspheres (data not shown). The findings indicated iron oxide NPs in the middle of some PCL–CIP–hematite microspheres that could not be observed by TEM imaging of the whole microspheres alone, without sectioning. Attenuated total reflectance-Fourier transform infrared spectra of maghemite, hematite, PCL, CIP, PCL–CIP–maghemite, and PCL–CIP–hematite are shown in Figure 4. The results confirmed that NPs and CIP were successfully encapsulated inside the PCL microspheres.
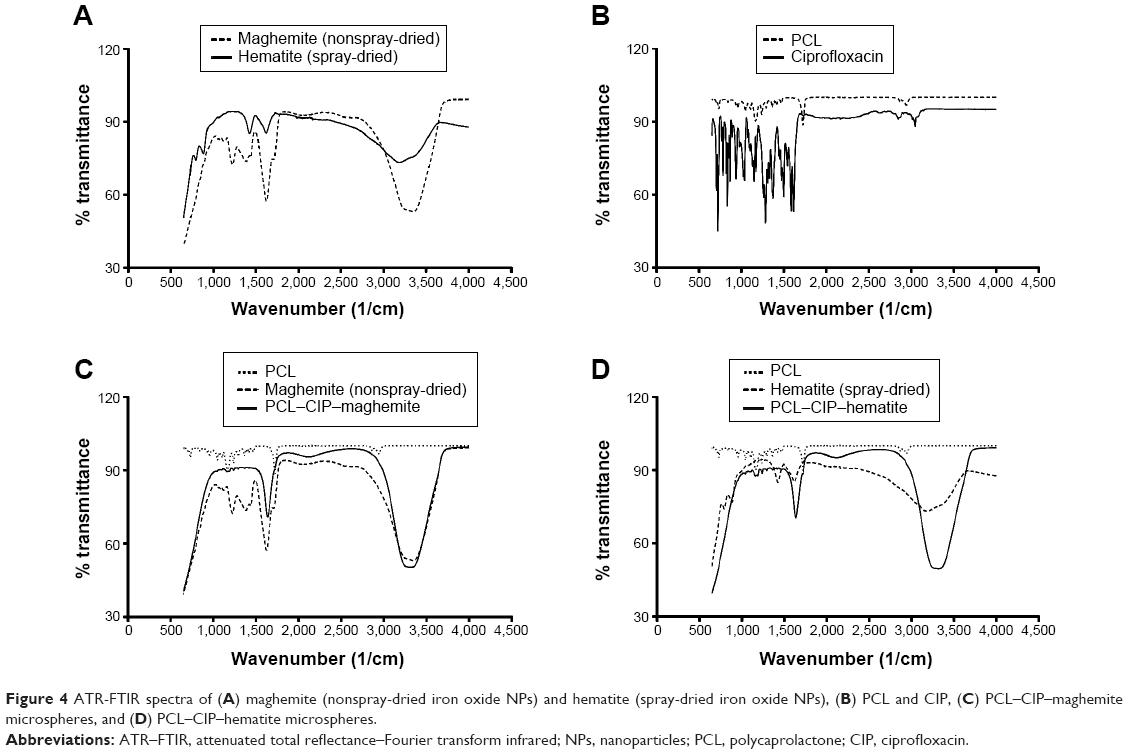 | Figure 4 ATR-FTIR spectra of (A) maghemite (nonspray-dried iron oxide NPs) and hematite (spray-dried iron oxide NPs), (B) PCL and CIP, (C) PCL–CIP–maghemite microspheres, and (D) PCL–CIP–hematite microspheres. |
The hydrodynamic size distribution of the samples measured using DLS and the mean diameter are shown in Figure 5A–D. The hydrodynamic diameters of the maghemite in deionized water ranged from 4.19 nm to 24.36 nm, whereas the diameters of the hematite in deionized water ranged from 531.20 nm to 1,480 nm because the NPs aggregated during measurement. The hydrodynamic diameters of the PCL–CIP–maghemite and PCL–CIP–hematite ranged from 255 nm to 530 nm and from 190 nm to 1,110 nm, respectively. The aggregation of hematite (Figure 2B) might contribute to the larger hydrodynamic diameters of the PCL–CIP–hematite microspheres, compared with the PCL–CIP–maghemite microspheres, after the encapsulation process.
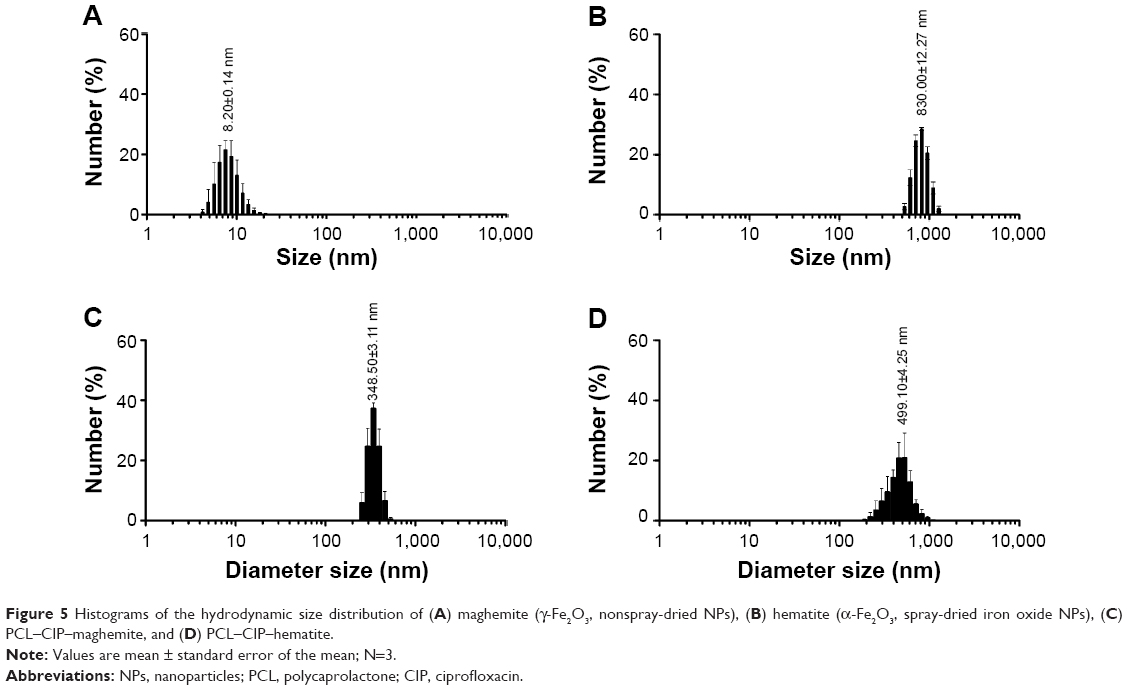 | Figure 5 Histograms of the hydrodynamic size distribution of (A) maghemite (γ-Fe2O3, nonspray-dried NPs), (B) hematite (α-Fe2O3, spray-dried iron oxide NPs), (C) PCL–CIP–maghemite, and (D) PCL–CIP–hematite. |
Bacterial inhibition study
In this study, a bed magnet generated a weak AMF. The bacterial populations decreased in the presence of PCL–CIP–maghemite, PCL–CIP–maghemite (AMF), PCL–CIP–hematite, and PCL–CIP–hematite (AMF) in comparison with the two control groups (bacteria alone with and without AMF) on days 2 and 5 of the cultures (Figure 6). On day 5, the bacterial population was significantly decreased in the presence of all samples when compared with the control groups. The PCL–CIP–maghemite and PCL–CIP–hematite inhibited bacterial growth after 2 days and 5 days of culture, but the efficiency improved in the presence of a magnetic field. For the control group, the application of an AMF did not inhibit bacterial growth from days 1 to 5. The findings from the broth inhibition assay suggest that the growth inhibition effects of PCL–CIP–NPs on S. aureus growth were enhanced in the presence of an AMF after 2 days and 5 days of incubation compared to the effects in the absence of AMF. The results from the bacterial inhibition study confirmed that CIP, antibiotic against S. aureus, was released from the PCL–CIP–NP microspheres.
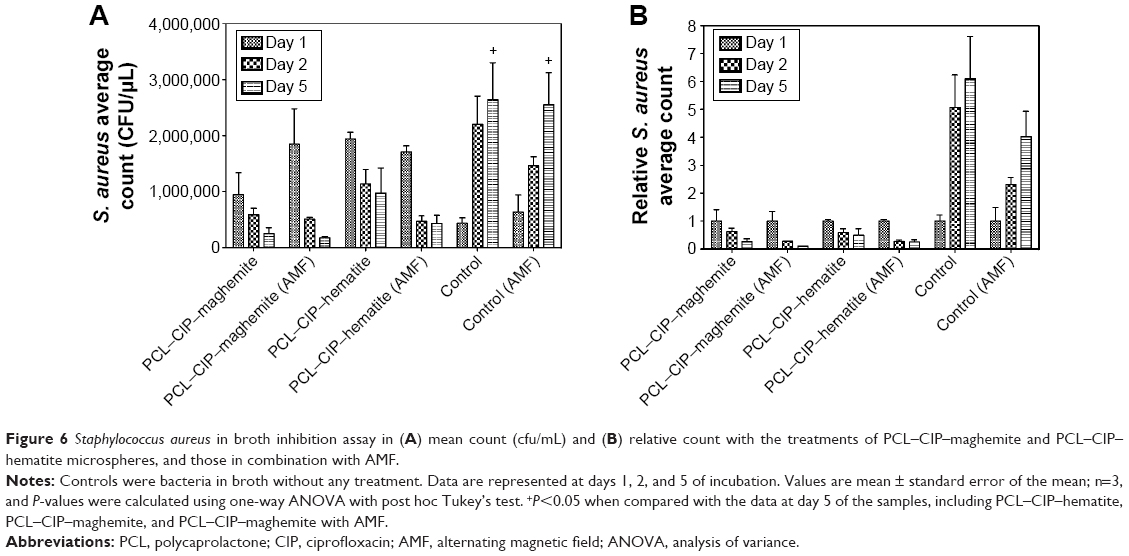 | Figure 6 Staphylococcus aureus in broth inhibition assay in (A) mean count (cfu/mL) and (B) relative count with the treatments of PCL–CIP–maghemite and PCL–CIP–hematite microspheres, and those in combination with AMF. |
The bioavailability of CIP is reduced by 50% when coadministered with iron compound.36 Masadeh et al reported that the use of iron oxide NPs (Fe2O3) in combination with CIP almost abolished its inhibitory effects on bacterial planktonic growth and biofilm formation.37 However, S. aureus was not included in that study. Another study demonstrated that Escherichia coli growth increased after γ-Fe2O3 treatment.38 Azam et al reported that Fe2O3 NPs exhibited the least bactericidal activity when compared with zinc oxide NPs, copper (II) oxide NPs, and tetracycline (standard antibiotics) against S. aureus.39 We also observed higher bacterial growth after treatment with maghemite (γ-Fe2O3), hematite (α-Fe2O3), PCL-maghemite, and PCL-hematite after 2 days of culture when compared to PCL–CIP–maghemite and PCL–CIP–hematite (data not shown). Importantly, the results of the present study suggested that treatment with PCL–CIP–maghemite and PCL–CIP–hematite containing MIC50 of CIP inhibited S. aureus growth after 2 days and 5 days of culture, and the inhibitory effects were stronger when applied with AMF.
Rakshit et al reported an adsorption mechanism of CIP on Fe3O4 NPs via inner-sphere complexation and suggested that the adsorption was a pH-dependent binding of the carboxyl functional group of CIP on Fe3O4 NPs.40 Tang et al suggested that CIP adsorption on reduced graphene oxide (Fe3O4) was relatively high at pH 5.0–7.0, and the dominant mechanisms in the sorption were π–π electron interaction and electrostatic repulsions.41 Although the present study did not investigate the adsorption mechanism of CIP on maghemite or hematite, we hypothesize that CIP could interact with iron oxide NPs during the polymer encapsulation process and/or partial PCL degradation and may alter the antibacterial property of CIP toward S. aureus.
Fibroblast and macrophage proliferation
The percentages of fibroblast (24 hours) and macrophage (56 hours) viability are shown in Figure 7A and B, respectively. Fibroblast viability after treatment with the samples was more than 100% compared with the control (polystyrene). When the concentration of sample treatments was decreased from 4 μg/mL to 0.5 μg/mL, the fibroblast viability increased in all sample groups. Specifically, fibroblast viability was significantly increased in the presence of PCL–CIP–hematite compared with that in the presence of maghemite, hematite, and PCL–CIP–maghemite with particle concentrations of 1 μg/mL and 0.5 μg/mL. Macrophage viability was also more than 100% compared with the control (Figure 7B). The results revealed no significant differences in the macrophage cultures among the groups of samples after 56 hours. The results of both cell cultures confirmed the biocompatibility of the chemically synthesized iron oxide NPs and PCL–CIP–NPs at concentrations of 0.5–4 μg/mL with fibroblasts in vitro. These findings also suggest that iron oxide NPs and PCL–CIP–NPs do not cause macrophages (inflammatory cells) to grow aggressively during the early stage of cell culture (56 hours), and cell viability was similar to that of the control (polystyrene).
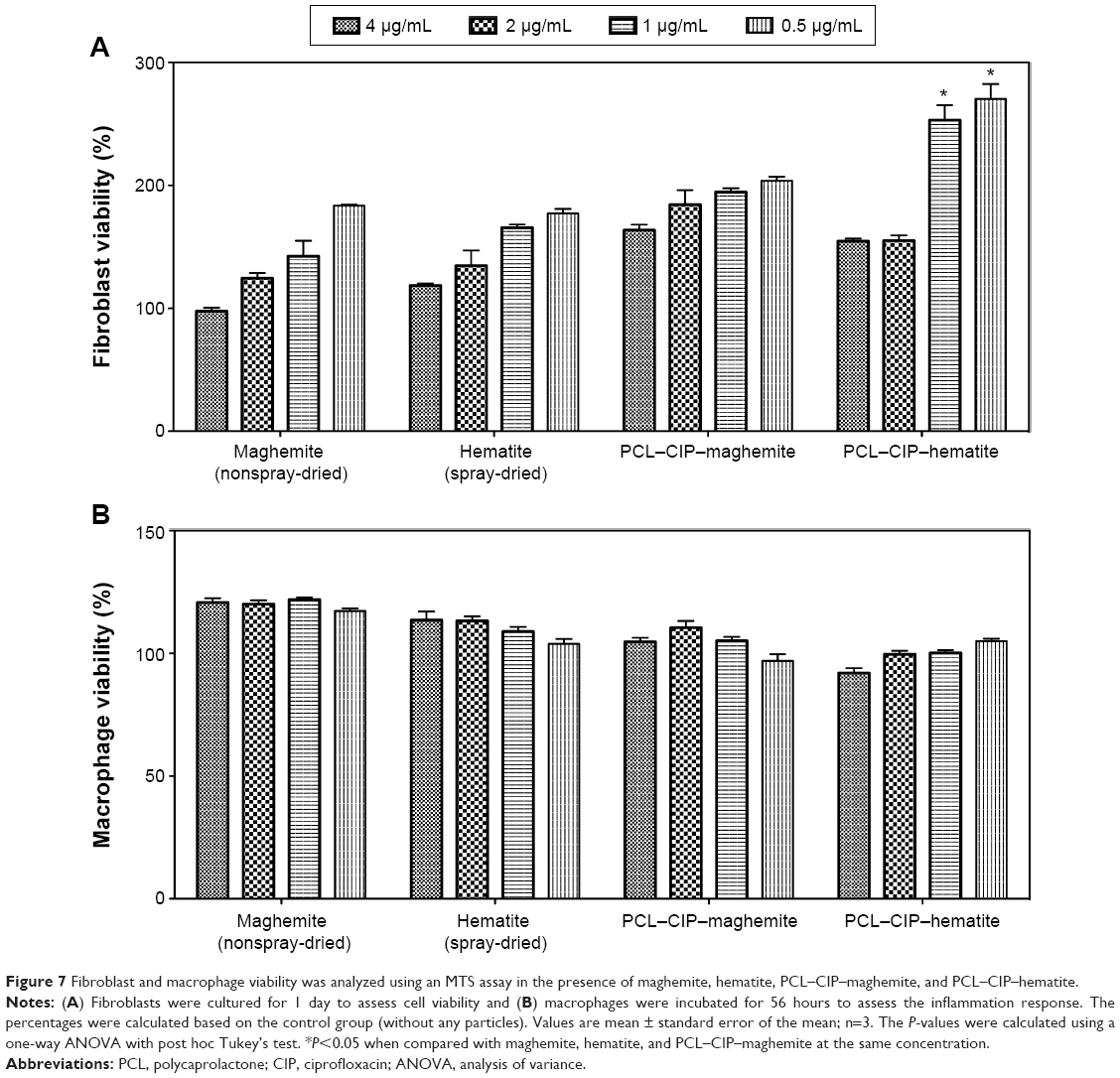 | Figure 7 Fibroblast and macrophage viability was analyzed using an MTS assay in the presence of maghemite, hematite, PCL–CIP–maghemite, and PCL–CIP–hematite. |
In vitro drug release study
A schematic representation of the PCL–CIP–NPs is shown in Figure 8A. The CIP release profiles of the PCL–CIP–NPs were determined after incubation under an AMF. The magnetic flux density (magnetic field) in Gauss was plotted vs time in seconds during rotation, as shown in Figure 8B. This figure shows the maximum field range of 10 G and the minimum field range of -5 G. The release profiles of the PCL–CIP–NPs in PBS (1 M, pH 7.4) with or without the application of an AMF are shown in Figure 8C and D. All samples exhibited a considerable amount of drug release immediately after incubation with a rotation rate of 225 rpm after 1 day. The percentage of drug release at day 1 from the PCL–CIP–maghemite was ~51%, that from the PCL–CIP–maghemite with an AMF was ~78%, and that from the PCL–CIP–hematite both with and without an AMF was almost the same, ~36% and ~39%, respectively. The results indicated that the immediate release of CIP from PCL–CIP–NP microspheres was increased by ~27% in the presence of an AMF for PCL–CIP–maghemite and ~3% for PCL–CIP–hematite.
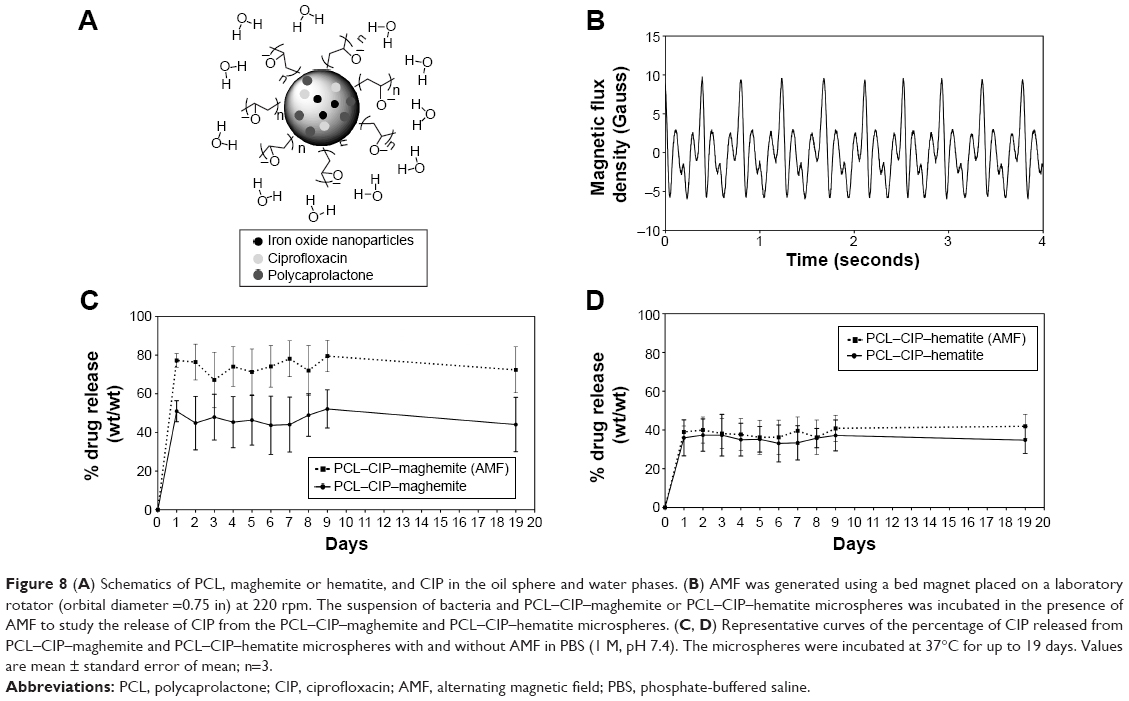 | Figure 8 (A) Schematics of PCL, maghemite or hematite, and CIP in the oil sphere and water phases. (B) AMF was generated using a bed magnet placed on a laboratory rotator (orbital diameter =0.75 in) at 220 rpm. The suspension of bacteria and PCL–CIP–maghemite or PCL–CIP–hematite microspheres was incubated in the presence of AMF to study the release of CIP from the PCL–CIP–maghemite and PCL–CIP–hematite microspheres. (C, D) Representative curves of the percentage of CIP released from PCL–CIP–maghemite and PCL–CIP–hematite microspheres with and without AMF in PBS (1 M, pH 7.4). The microspheres were incubated at 37°C for up to 19 days. Values are mean ± standard error of mean; n=3. |
PCL degradation occurs very slowly over more than a year.42,43 The degradation process of PCL first proceeds via surface or bulk degradation, which involved hydrolytic cleavage of the polymer backbone only at the surface.42 Such initial hydration occurs in the first 6 months. No weight loss of PCL (molecular weight ~20–50 kD) is observed during the initial (or partial) phase of the biodegradation process.43 Bosworth and Downes studied in vitro degradation behavior of the PCL fibers in PBS at 37°C at pH 7.4.44 Their findings from high-performance liquid chromatography analysis revealed that PCL became more hydrophobic and, hence, is more likely to comprise longer length chains. A slight increase in the degradation by-products (no significant difference over 90 days) was detected, however, and may have been accelerated by autocatalysis occurring secondary to the accumulation of acidic breakdown products. Moreover, the results in that study from electrospray-mass spectrometry confirmed that PCL fibers underwent partial bulk degradation over 90 days. Because PCL has a very long degradation time (months), the absorbance values measured at 270 nm in the present study should only be due to the CIP at the observed time scale.
In a previous study, CIP was conjugated to the hydroxyl ends of polyesters, which were synthesized using caprolactone or polyethylene glycol (PEG) as initiators.45 Degradation of the polymer then occurred at 37°C in an aqueous buffered solution at pH 7.4, and the CIP release was quantified by UV–Vis spectrophotometry at 277 nm. The conjugated drug release rate with PEG was more than that with caprolactone for monomers and initiators. The addition of PEG, which is a hydrophilic initiator, accelerates the drug release rate (29% CIP after 35 days at pH 7.4) than the addition of caprolactone. Another study showed that CIP release from the PCL matrix soaking in PBS at pH 7.4 at 80°C and 150°C was remarkably slow, with <5% release after 56 days.46 The findings suggested that the slow release was due to the slow degradation of PCL. Furthermore, in that study, PCL was activated with 5 M NaOH for 72 hours at 20°C to resemble in vivo behavior before the PCL degradation study; yet, no change was observed during the first 7 days and <10% weight loss was detected after 82 days. Another previous study soaked PCL scaffolds under simulated physiologic conditions (PBS at 37°C), in which PCL scaffolds treated with NaOH for 24 hours showed only a 20% weight loss after about 4 years of incubation.47 Evidence from many previous studies suggests that PCL did not likely interfere with the absorbance measurement of CIP at 270 nm in the present study, because PCL does not readily degrade for up to 19 days in PBS at pH 7.4. CIP and PCL are both hydrophobic in nature and could interact among PCL chains chemically, as previously described.44 Therefore, this could be the reason why the percentages of drug release observed in the present study did not reach 100%, as CIP (hydrophobic drug) was trapped in a PCL (hydrophobic polymer) matrix that did not degrade over 19 days in the present study.
The immediate release of CIP entrapped within PCL–CIP–NPs was observed 1 day after application of the AMF. The percentages of drug release (wt/wt) of PCL–CIP–NPs were significantly increased (P<0.05, two tailed, unpaired Student’s t-test) at all time points under AMF by ~27% in the PCL–CIP–maghemite group, except days 3, 5, 8, and 19, whereas there was no statistically significant difference in the PCL–CIP–hematite group. The overall percentage of drug release from PCL–CIP–maghemite (AMF) was significantly greater than that from the PCL–CIP–hematite (AMF) (P<0.05, two tailed, unpaired Student’s t-test) at all time points by ~35%. These findings suggest that antibiotics carried by PCL–NP microspheres can be released with the application of an AMF. Therefore, this method of delivery can reduce the dose and frequency of antibiotic administration to inhibit infection. Moreover, antibiotics can be guided for delivery to the site of the orthopedic implant using a magnetic field.
Conclusion
The findings of the present study demonstrated that the release of CIP entrapped within the PCL–iron oxide NPs increased after application of an AMF due to the inhibition of S. aureus for 1 day, 2 days, and 5 days in vitro. CIP was immediately released from the PCL–CIP–NPs by applying an AMF, and the release remained constant for up to 19 days. Drug release from the PCL–CIP–maghemite and the PCL–CIP–hematite was enhanced by AMF compared with the same sample without applying an AMF by ~27% (P<0.05) and ~3% (P not significant), respectively. The chemically synthesized hematite (α-Fe2O3), maghemite (γ-Fe2O3), and PCL–CIP–NPs used in the present study were biocompatible with fibroblasts and macrophages. The bacterial density decreased when the culture was incubated with the PCL–CIP–NPs, regardless of the presence of the AMF, from days 1 to 5 of incubation. Moreover, drug release was increased under incubation with the application of AMF due to the enhanced suppression of bacterial growth in the presence of the PCL–CIP–NPs from days 1 to 5, compared with the same group without the application of AMF. Additional studies that focus on optimizing the effects of magnetic field strength are warranted. The use of iron oxide NPs for targeted delivery and stimulated release of drugs represents a promising alternative external control technique for effective antibiotic treatment.
Acknowledgments
The authors would like to thank Ms Mary Ellenburg for her technical assistance in bacteria inhibition study. This work was supported by the US Army Medical Research and Materiel Command under contract W81XWH-08-1-0333.
Disclosure
The authors report no conflicts of interest in this work.
References
Pankhurst QA, Connolly J, Jones SK, Dobson J. Applications of magnetic nanoparticles in biomedicine. J Phys D Appl Phys. 2003;36:R167–R181. | ||
Berry CC, Curtis ASG. Functionalisation of magnetic nanoparticles for applications in biomedicine. J Phys D Appl Phys. 2003;36:R198–R206. | ||
Moroz P, Jones SK, Gray BN. Magnetically mediated hyperthermia: current status and future directions. Int J Hyperthermia. 2002;18(4):267–284. | ||
Mahmoudi M, Simchi A, Milani AS, Stroeve P. Cell toxicity of superparamagnetic iron oxide nanoparticles. J Colloid Interface Sci. 2009;336(2):510–518. | ||
Coey JMD. Magnetism and Magnetic Materials. Cambridge: Cambridge University Press; 2010. | ||
Hoare T, Santamaria J, Goya GF, et al. A magnetically triggered composite membrane for on-demand drug delivery. Nano Lett. 2009;9(10):3651–3657. | ||
Tai LA, Tsai PJ, Wang YC, Wang YJ, Lo LW, Yang CS. Thermosensitive liposomes entrapping iron oxide nanoparticles for controllable drug release. Nanotechnology. 2009;20(13):135101. | ||
Muller-Schulte D, Schmitz-Rode T. Thermosensitive magnetic polymer particles as contactless controllable drug carriers. J Magn Magn Mater. 2006;302(1):267–271. | ||
Zhang J, Misra RD. Magnetic drug-targeting carrier encapsulated with thermosensitive smart polymer: core-shell nanoparticle carrier and drug release response. Acta Biomater. 2007;3(6):838–850. | ||
Hu S-H, Tsai C-H, Liao C-F, Liu D-M, Chen S-Y. Controlled rupture of magnetic polyelectrolyte microcapsules for drug delivery. Langmuir. 2008;24(20):11811–11818. | ||
Hu S-H, Chen S-Y, Liu D-M, Hsiao C-S. Core/single-crystal-shell nanospheres for controlled drug release via a magnetically triggered rupturing mechanism. Adv Mater. 2008;20(14):2690–2695. | ||
Hu S-H, Liu T-Y, Huang H-Y, Liu D-M, Chen S-Y. Magnetic-sensitive silica nanospheres for controlled drug release. Langmuir. 2007;24(1):239–244. | ||
Dilnawaz F, Singh A, Mohanty C, Sahoo SK. Dual drug loaded superparamagnetic iron oxide nanoparticles for targeted cancer therapy. Biomaterials. 2010;31(13):3694–3706. | ||
Li R, Wu R, Zhao L, et al. Folate and iron difunctionalized multiwall carbon nanotubes as dual-targeted drug nanocarrier to cancer cells. Carbon N Y. 2011;49(5):1797–1805. | ||
Subbiahdoss G, Sharifi S, Grijpma DW, et al. Magnetic targeting of surface-modified superparamagnetic iron oxide nanoparticles yields antibacterial efficacy against biofilms of gentamicin-resistant staphylococci. Acta Biomater. 2012;8(6):2047–2055. | ||
Sirivisoot S, Pareta RA, Webster TJ. A conductive nanostructured polymer electrodeposited on titanium as a controllable, local drug delivery platform. J Biomed Mater Res A. 2011;99(4):586–597. | ||
Sirivisoot S, Pareta R, Webster TJ. Electrically controlled drug release from nanostructured polypyrrole coated on titanium. Nanotechnology. 2011;22(8):085101. | ||
Sirivisoot S, Harrison BS. Skeletal myotube formation enhanced by electrospun polyurethane carbon nanotube scaffolds. Int J Nanomedicine. 2011;6:2483–2497. | ||
Xu H, Cheng L, Wang C, Ma X, Li Y, Liu Z. Polymer encapsulated upconversion nanoparticle/iron oxide nanocomposites for multimodal imaging and magnetic targeted drug delivery. Biomaterials. 2011;32(35):9364–9373. | ||
Wang S, Guo S, Cheng L. Disodium norcantharidate loaded poly([var epsilon]-caprolactone) microspheres: I. Preparation and evaluation. Int J Pharm. 2008;350(1–2):130–137. | ||
Pitt CG. Poly(ε-caprolactone) and its copolymers. In: Chasin M, Langer R, editors. Biodegradable Polymer as Drug Delivery Systems. New York, NY: Marcel Decker Inc; 1990:71–120. | ||
Kim SY, Lee YM. Taxol-loaded block copolymer nanospheres composed of methoxy poly(ethylene glycol) and poly(ε-caprolactone) as novel anticancer drug carriers. Biomaterials. 2001;22(13):1697–1704. | ||
Lamprecht A, Torres HR, Schäfer U, Lehr C-M. Biodegradable microparticles as a two-drug controlled release formulation: a potential treatment of inflammatory bowel disease. J Control Release. 2000;69(3):445–454. | ||
Lamprecht A, Ubrich N, Hombreiro Pérez M, Lehr C, Hoffman M, Maincent P. Influences of process parameters on nanoparticle preparation performed by a double emulsion pressure homogenization technique. Int J Pharm. 2000;196(2):177–182. | ||
Gristina AG. Biomaterial-centered infection: microbial adhesion versus tissue integration. Science. 1987;237(4822):1588–1595. | ||
Khalil H, Williams RJ, Stenbeck G, Henderson B, Meghji S, Nair SP. Invasion of bone cells by Staphylococcus epidermidis. Microbes Infect. 2007;9(4):460–465. | ||
Stratton C. Fluoroquinolone antibiotics: properties of the class and individual agents. Clin Ther. 1992;14(3):348–375. | ||
Jeong YI, Na HS, Seo DH, et al. Ciprofloxacin-encapsulated poly(dl-lactide-co-glycolide) nanoparticles and its antibacterial activity. Int J Pharm. 2008;352(1–2):317–323. | ||
Armstrong EP, Friedman AD. Bone and joint infections. In: DiPiro JT, Talbert RL, Yee GC, et al, editors. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw-Hill; 2011:2029–2039. | ||
Lew DP, Waldvogel FA. Use of quinolones in osteomyelitis and infected orthopaedic prosthesis. Drugs. 1999;58(Suppl 2):85–91. | ||
Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; Sixteenth Informational Supplement. Wayne, PA: Clinical and Laboratory Standards Institute; 2006. CLSI document M100-S16 [ISBN 1-56238-625-5]. | ||
Wiegand I, Hilpert K, Hancock RE. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc. 2008;3(2):163–175. | ||
Pascual-Reguera MI, Parras GP, Diamp AM. Solid-phase UV spectrophotometric method for determination of ciprofloxacin. Microchem J. 2004;77(1):79–84. | ||
Kang YS, Risbud S, Rabolt JF, Stroeve P. Synthesis and characterization of nanometer-size Fe3O4 and γ-Fe2O3 particles. Chem Mater. 1996;8:2209–2211. | ||
Bee A, Massart R, Neveu S. Synthesis of very fine maghemite particles. J Magn Magn Mater. 1995;149:6–9. | ||
Lode H. Evidence of different profiles of side effects and drug-drug interactions among the quinolones – the pharmacokinetic standpoint. Chemotherapy. 2001;47(Suppl 3):24–31. [discussion 44–48]. | ||
Masadeh MM, Karasneh GA, Al-Akhras MA, et al. Cerium oxide and iron oxide nanoparticles abolish the antibacterial activity of ciprofloxacin against gram positive and gram negative biofilm bacteria. Cytotechnology. 2015;67(3):427–435. | ||
He S, Feng Y, Gu N, Zhang Y, Lin X. The effect of γ-Fe2O3 nanoparticles on Escherichia coli genome. Environ Pollut. 2011;159(2):3468–3473. | ||
Azam A, Ahmed AS, Oves M, Khan MS, Habib SS, Memic A. Antimicrobial activity of metal oxide nanoparticles against Gram-positive and Gram-negative bacteria: a comparative study. Int J Nanomedicine. 2012;7:6003–6009. | ||
Rakshit S, Sarkar D, Elzinga EJ, Punamiya P, Datta R. Mechanisms of ciprofloxacin removal by nano-sized magnetite. J Hazard Mater. 2013;24(6–247):221–226. | ||
Tang Y, Guo H, Xiao L, Yu S, Gao N, Wang Y. Synthesis of reduced graphene oxide/magnetite composites and investigation of their adsorption performance of fluoroquinolone antibiotics. Colloids Surf A Physicochem Eng Asp. 2013;424:74–80. | ||
Woodruff MA, Hutmacher DW. The return of a forgotten polymer – polycaprolactone in the 21st century. Prog Polym Sci. 2010;35:1217–1256. | ||
Sinha VR, Bansal K, Kaushik R, Kumria R, Trehan A. Poly-ε-caprolactone microspheres and nanospheres: an overview. Int J Pharm. 2004;1(18):1–23. | ||
Bosworth LA, Downes S. Physicochemical characterization of degrading polycaprolactone scaffolds. Polym Degrad Stab. 2010;95:2269–2276. | ||
Sobczak M. Synthesis and characterization of polyester conjugates of ciprofloxacin. Eur J Med Chem. 2010;45:3844–3849. | ||
Puga AM, Rey-rico A, Magarinos B, Alvarez-Lorenzo C, Concheiro A. Hot melt poly-e-caprolactone/poloxamine implantable matrices for sustained delivery of ciprofloxacin. Acta Biomater. 2012;8(4):1507–1518. | ||
Lam CXF, Teoh SH, Hutmacher DW. Comparison of the degradation of polycaprolactone and polycaprolactone–(beta-tricalcium phosphate) scaffolds in alkaline medium. Polym Int. 2007;56:718–728. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.