")
Back to Journals » International Journal of Nanomedicine » Volume 13
Magnetic nanoparticle hyperthermia potentiates paclitaxel activity in sensitive and resistant breast cancer cells
Authors Rivera-Rodriguez A , Chiu-Lam A , Morozov VM, Ishov AM , Rinaldi C
Received 14 April 2018
Accepted for publication 25 May 2018
Published 23 August 2018 Volume 2018:13 Pages 4771—4779
DOI https://doi.org/10.2147/IJN.S171130
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Angelie Rivera-Rodriguez,1 Andreina Chiu-Lam,2 Viacheslav M Morozov,3,4 Alexander M Ishov,3,4 Carlos Rinaldi1,2,4
1J. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Gainesville, FL, USA; 2Department of Chemical Engineering, University of Florida, Gainesville, FL, USA; 3Department of Anatomy and Cell Biology, University of Florida College of Medicine, Gainesville, FL, USA; 4UF Health Cancer Center Gainesville, FL, USA
Introduction: Overcoming resistance to antimitotic drugs, such as paclitaxel (PTX), would represent a major advance in breast cancer treatment. PTX induces mitotic block and sensitive cells exit mitosis dying by mitotic catastrophe. Resistant cells remain in block and continue proliferation after drug decay, denoting one of the PTX resistance mechanisms. Mild hyperthermia (HT) triggers mitotic exit of PTX-pretreated cells, overcoming PTX resistance and suggesting HT-forced mitotic exit as a promising strategy to potentiate PTX.
Methods and results: Superparamagnetic iron oxide nanoparticles (SPIONs) were used to deliver mild HT at 42°C in PTX-pretreated breast adenocarcinoma MCF-7 cells sensitive and resistant to PTX. To evaluate mechanism of cell death, cells were classified based on nuclear morphology into interphase, mitotic, micronucleated, and apoptotic. The combined PTX→SPION treatment resulted in an increase in the percentage of micronucleated cells, an indication of forced mitotic exit. Importantly, in PTX-resistant cells, the combination therapy using SPION HT helps to overcome resistance by reducing the number of cells relative to the control.
Conclusion: SPION HT potentiates PTX by significantly reducing cell survival, suggesting potential of combined treatment for future clinical translation.
Keywords: iron oxide nanoparticles, chemotherapy, drug resistance, hyperthermia, taxanes
Introduction
Paclitaxel (PTX, taxol) is an antimitotic drug that was originally isolated from the Pacific yew tree. This drug was approved by the US Food and Drug Administration and is commonly used for the treatment of ovarian, breast, lung, head, and neck cancer, and Kaposi sarcoma.1,2 More than 50% of breast cancer patients are resistant to taxanes at first line of treatment, and around 80% become resistant during second line of treatment.3,4 As a result, in the USA alone, >60,000 women treated with taxanes will not benefit from the therapy.
The mechanism of action of taxanes is to block cancer cells during cell division (mitosis) through reversible binding to tubulin, which results in microtubule hyper-stabilization.2 Such inhibition of microtubule dynamics activates the spindle assembly checkpoint (SAC), which prompts a persistent mitotic arrest. PTX-sensitive cells die by a mechanism known as mitotic catastrophe, a biochemical event characterized by slow and steady degradation of cyclin B (anaphase-promoting complex/cyclosome substrate).5–8 When cyclin B levels drop below a threshold, cells exit mitosis by micronuclei formation, failing the next round of cell division by undergoing apoptosis, necrosis, or senescence.8 Mutations in these pathways associated with cell death are responsible for the most common causes of PTX resistance. Resistant cancer cells remain in mitosis until drugs clear and then continue proliferation, resulting in PTX resistance in both preclinical breast cancer models and breast cancer patients.8,9
In previous work, Giovinazzi et al hypothesized that mitotic exit should be targeted in order to overcome PTX resistance mechanisms.6 They identified that PTX-induced mitotic block is sensitive to physiological hyperthermia (HT, also referred to as heat shock), suggesting a sequential strategy of treatment (Figure 1). Thus, manipulation of PTX-induced mitotic block and forced mitotic exit through the application of HT could enhance the efficacy of taxane therapy in breast cancer and may help overcome PTX resistance.
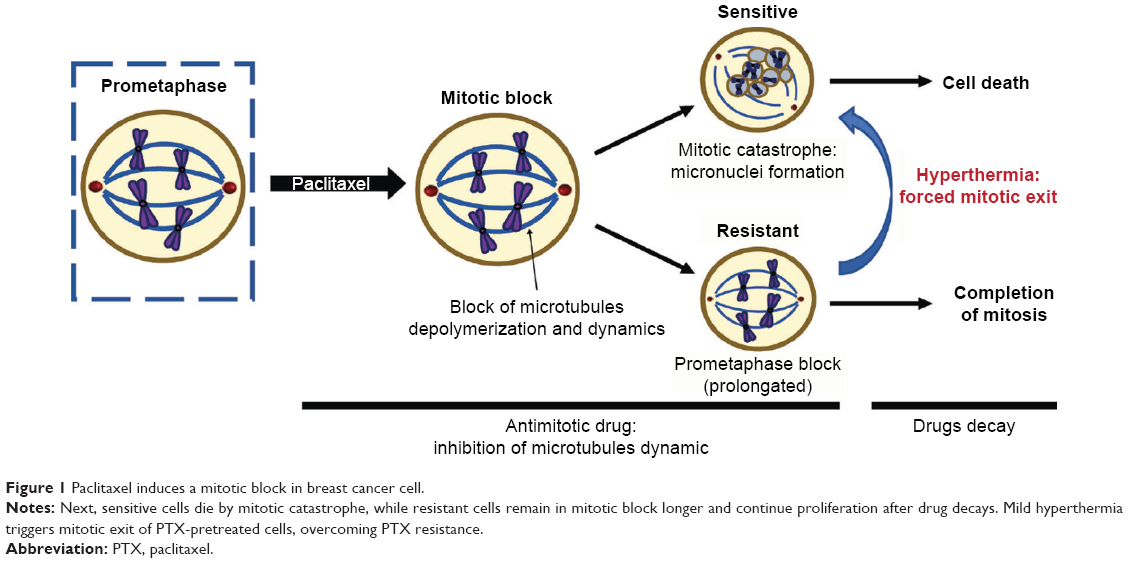 | Figure 1 Paclitaxel induces a mitotic block in breast cancer cell. |
HT has been known to be an effective potentiator of chemotherapy, but clinical application has been limited due to the difficulty in achieving controlled temperature delivery while sparing healthy tissue.10–13 Nanoscale heat generation represents an attractive alternative to conventional methods of HT because heat can be generated and constrained within the area of interest through a combination of nanoparticle localization and spatial control of the means of actuating heat release.14,15 Superparamagnetic iron oxide nanoparticles (SPIONs) in combination with alternating magnetic fields (AMFs) have been studied as a means to apply HT in cancer treatment, in what is variably called magnetic fluid HT, magnetic nanoparticle HT, or magnetic HT.16 The use of SPION HT was successfully translated for the treatment of glioblastoma multiforme in Europe, suggesting potential application in other types of cancer.17–19 SPIONs are regarded as biocompatible and biodegradable, and can be engineered to achieve high heating rates and maintain colloidal stability in biological environments through the use of engineered surface coatings.20–25 Another potential advantage of SPION HT over other HT treatments is that nanoscale heat could activate lysosomal death pathways, killing cancer cells selectively without the need of temperature rise.26,27 Furthermore, SPION heating can be achieved deep in the body, and instrumentation to generate AMFs suitable for SPION-based thermal therapy in patients are feasible and commercially available.28
Nanoscale heat generation using SPIONs has shown great potential in combination with chemotherapeutics. Various studies have demonstrated that under identical heat doses, nanoscale heating is more effective at potentiating cancer drugs, possibly due to additional damage to cell membranes, lysosomes, microtubules, and the proteolytic apparatus of cancer cells.29–32
In this contribution, we show that nanoscale heat generation by SPIONs potentiates PTX activity and overcomes PTX resistance. The combination treatment of PTX and SPION HT enhanced cell killing compared with PTX alone or combination of PTX with external (ext) mild HT on breast cancer cell lines. These results suggest the potential benefits of SPION HT in the development of new therapies or in enhancing outcomes of current therapies.
Materials and methods
Nanoparticle synthesis
Iron oxide nanoparticles were synthesized through the co-precipitation method as described by Mérida et al23 Briefly, deionized water was degassed with nitrogen for 30 minutes and an aqueous solution of iron (II) and iron (III) salts was prepared using a molar ratio Fe3+:Fe2+ of 2:1. The iron solution was sonicated and degassed, and the reaction was heated to 85°C, followed by the addition of ammonium hydroxide (Thermo Fisher Scientific, Waltham, MA, USA). The reaction was kept at 85°C for 1 hour, and the pH was maintained between 8 and 9 by adding small aliquots of NH4OH as needed. The resultant iron oxide solution was cooled at room temperature (RT) and peptized using tetramethylammonium hydroxide (Alfa Aesar, Tewksbury, MA, USA).
To coat the particle with poly(ethylene) glycol (PEG), oleic acid (OA, 90%, Sigma-Aldrich, St. Louis, MO, USA) was adsorbed onto the particles by adding 15 g OA/g SPIONs, followed by ultrasonication (Q700, Qsonica Sonicators, Newtown, CT, USA) for 15 minutes. The mixture was heated to 50°C and allowed to react for 2 hours. SPIONs were precipitated using twice the volume of ethanol (200 proof, Decon Labs, King of Prussia, PA, USA) and magnetically decanted to recover the particles, followed by suspension in toluene (>98%, Sigma-Aldrich).
PEG ligand exchange
For monomethoxy PEG (mPEG) oxidation, mPEG-5000 Da (Sigma-Aldrich) was dissolved in acetone (12.5 w/v %) followed by a 12-hour reaction with Jones Reagent (4 v/v %). Then, the reaction was stopped with isopropanol and activated charcoal. The oxidized mPEG solution was vacuum filtered and placed in a rotary evaporator (Rotovap, Buchi Corporation, New Castle, DE, USA). The reduced solution was mixed with 1 M hydrochloric acid (40 w/v %) and poured into a separatory funnel to be washed with ethyl ether. The aqueous layer was collected and poured into the separatory funnel to extract the oxidized mPEG using methylene chloride. The organic solution was then dried in the rotovap, washed with cold diethyl ether and dried in a vacuum oven at RT. The resulting oxidized mPEG was melted in the rotovap at 120°C and conjugated to 3-aminopropyltriethoxisilane (TCI America, Portland, OR, USA) at 3.8 w/w % for 2 hours at 500 mbar.
For PEG ligand exchange of OA-coated NPs, a polymer/particle solution of 0.4 mg/mL in dry toluene was prepared at a 5:1 molar ratio of PEG silane:OA. The solution was shaken vigorously and acetic acid was added at a molar ratio of 1:2 of PEG silane:acetic acid. The particle/polymer solution was shaken at 150 rpm for 72 hours. After the reaction, particles were washed and precipitated in cold diethyl ether, and dried in the vacuum oven at RT. Then, the particles were suspended in deionized water, dialyzed, and sterilized by filtration.
Nanoparticle characterization
All nanoparticle characterizations were performed following protocols described by Mérida et al23 Most characterizations were carried out with suspensions of 10 mg/mL, except for transmission electron microscopy (TEM, 200CX, JEOL, Peabody, MA, USA) and dynamic light scattering (DLS), which used concentrations of 60 and 0.1 mg/mL, respectively. DLS (Zeta PALS, Brookhaven Instruments, Holtsville, NY, USA) was used to determine the hydrodynamic diameter of the particles at RT. Dynamic magnetic susceptibility (DMS) measurements were used to evaluate the mechanism of magnetic relaxation of the nanoparticles in a frequency range of 1 Hz to 160 kHz and applied field amplitude of 0.5 mT, using a susceptometer (DynoMag, Imego, Goteborg, Sweden). Equilibrium magnetic measurements were made using a superconducting quantum interference device magnetometer (MPMS3, Quantum Design, San Diego, CA, USA), to obtain magnetization curves at 300 K and magnetic field between −7 and 7 T. The iron content in liquid suspensions was determined using a colorimetric assay based on complexation of Fe2+ with 1,10-phenanthroline. Absorbance values were obtained using an ultraviolet-visible spectrophotometer (UV-2600, Shimadzu Scientific Instruments, Inc., Portland, OR, USA) at 508 nm and used to determine iron concentration in each sample. Samples and calibration standards were run in triplicate. The specific absorption rate (SAR) of the nanoparticles in liquid supension was obtained by exposing samples to an AMF produced by the coil of an induction heater (EasyHeat 8310 LI, Ambrell, Scottsville, NY, USA). All measurements were run in triplicate, at 51 kA/m (36 kA/m rms) and 343 kHz. The size and shape of the nanoparticle inorganic cores were determined via TEM and analyzed by counting at least 200 particles per image (n=3, ImageJ 1.48 v, NIH). A log-normal size distribution was used to obtain the number weighted mean core diameter and geometric deviation.
Cell culture
MCF-7 parental breast adenocarcinoma and a PTX-resistant subline were a kind gift from Dr Parissenti.33 The cells were cultured in DMEM (Thermo Fisher Scientific), high glucose, supplemented with 10% fetal bovine serum, 1% glutamine, and 1% penicillin, and grown in a humidified incubator at 37°C and 5% CO2.
Drug and HT treatment
Approximately 3,500 viable cells were seeded in the wells of an 8-well strip plate (Corning Incorporated, Tewksbury, MA, USA) and grown for 24 hours. Seven conditions were established for the experiments: control, PTX, SPIONs at 37°C, ext HT, SPIONs HT, PTX → ext HT (“→”: followed by), and PTX → SPION HT. PTX (LC Labs., Woburn, MA, USA) solution in media was added to a final concentration of 1, 2.5, and 25 nM, and incubated for a total of 18 hours. After 16 hours of incubation with PTX, iron oxide magnetic nanoparticles were added to the cells at a concentration of 1.5 mg Fe/mL in media. The 8-well strip was positioned inside a 14-loop coil (1.5″ diameter × 5.5″ length) and a sterilized fiber optic temperature probe was used to record the temperature rise of the samples. The magnetic field (0–37 kA/m at 270 kHz) was turned on and mild HT was applied at 42°C for 2 hours using the induction heater (EasyHeat 8310 LI, Ambrell). For ext HT, the cells were exposed for 2 hours at 42°C in a humidified incubator. After PTX and HT treatments, the cells were washed twice with PBS and media, and 100 μL of fresh media was added to each well. The cells were incubated at 37°C for 4 days post treatment.
Image analysis
After treatment, cell nuclei were stained with Hoechst 33342 (Thermo Fisher Scientific) and fixed with 2% paraformaldehyde. A series of 4 fluorescent images, at 10× magnification, were taken, following an established pattern in 4 wells of the 8-well strip for each of the 7 conditions (16 images in total per condition), using a fluorescence microscope (Zeiss Axio Observer, Thornwood, NY, USA). The fluorescent images were analyzed by counting the total number of cells per image in each well using the Cell Counter plugin in ImageJ (ImageJ 1.48 v, NIH). The number of cells per well was normalized relative to control cells. Also, cells were classified based on nuclear morphology (interphase, mitotic, micronuclei, and apoptotic) in selected conditions. At least 1,000 cells were evaluated for each experimental condition.
Statistical analysis
To determine if differences in treatments were statistically significant, a one-way analysis of variance (ANOVA) was performed with Sidak’s post hoc test. Cell numbers for each condition were normalized against the control and error propagation was performed from at least 3 replicates.
Results
Nanoparticle characterization
SPIONs were synthesized by the co-precipitation method, coated with PEG (5,000 Da), and their physical, magnetic, and hydrodynamic sizes and magnetic properties characterized. Figure 2A, shows the TEM micrograph of the PEG-coated SPION. TEM images were analyzed using the particle analysis feature of ImageJ to obtain the physical diameter distribution of the SPIONs. Figure 2B shows the nanoparticles’ magnetic, physical, and hydrodynamic diameter distributions, which have lognormal medians of 13, 15, and 40 nm, respectively. The PEG brush layer is ~12.5 nm thick providing the nanoparticles a biocompatible coating that makes them colloidally stable in biological environments.25
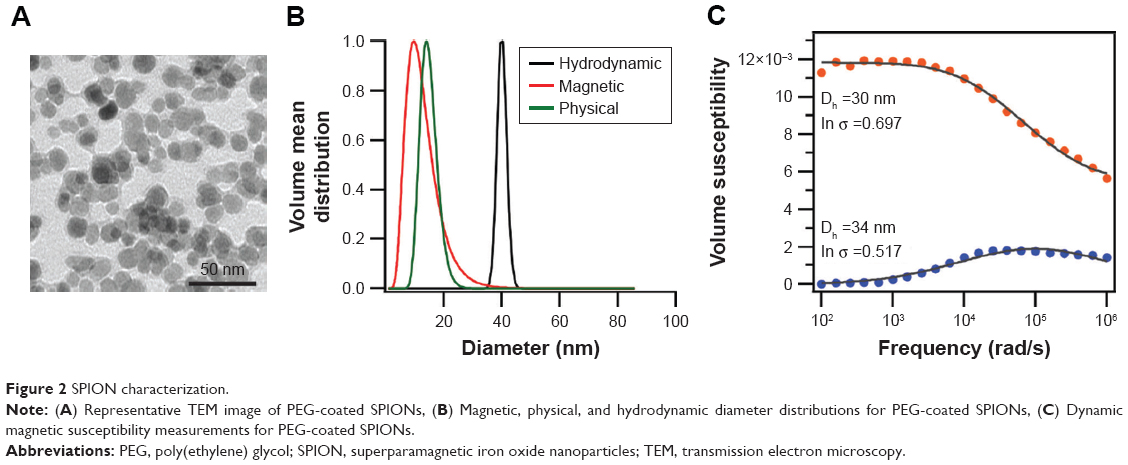 | Figure 2 SPION characterization. |
The SAR of the particles at a field of 51 kA/m and 343 kHz was 533 W/gFe, which is sufficient to achieve rapid heating rates under the AMF conditions used in our experiments. DMS measurements (Figure 2C) suggest that the nanoparticles have both Néel and Brownian relaxation components at a fraction of 0.48 and 0.52, respectively.34 At the moment, it is still being debated as to what the contributions of Néel and Brownian mechanisms are to cell death in experiments, such as this. Néel particles are expected to be better heaters in biological environments, whereas the heating of Brownian particles will depend on their coupling with extracellular matrix.35 For in vitro work, the concentration of iron in the SPIONs was determined as previously described, and proper dilutions of SPION in media were performed in order to obtain a final concentration of 1.5 mgFe/mL.
PTX and HT treatment
Experiments were performed to determine the effect of SPION HT alone or in combination with PTX in the MCF-7 cells that are sensitive (wild type [WT]) and resistant (taxol resistant [TR]) to PTX. The results are shown in Figure 3. SPION suspensions were prepared in media at a concentration of 1.5 mgFe/mL, at which there was no evidence of cytotoxic effect on cells at 37°C for 2-hour exposure (Figure 3). Ext HT at 42°C for 2 hours had a mild cytotoxic effect on WT MCF-7 cells and negligible effect on TR cells. In contrast, SPION HT at 42°C resulted in greater cytotoxicity in both WT and TR cells, with a reduction of 60% of the total cell number. Combination treatments of PTX with ext HT or SPION HT were performed at increasing doses of PTX (1, 2.5, and 25 nM). PTX and ext HT combination treatments showed a negligible effect when compared with PTX alone. On the other hand, PTX with SPION HT combination treatment showed a greater effect for both MCF-7 WT and TR cells, with cell survival below 10% for WT. In the TR cells, the combination therapy of PTX and SPION HT reduced the number of cells by >65% in the treatments with low PTX concentration, and there was around 80% cell reduction at a PTX concentration of 25 nM.
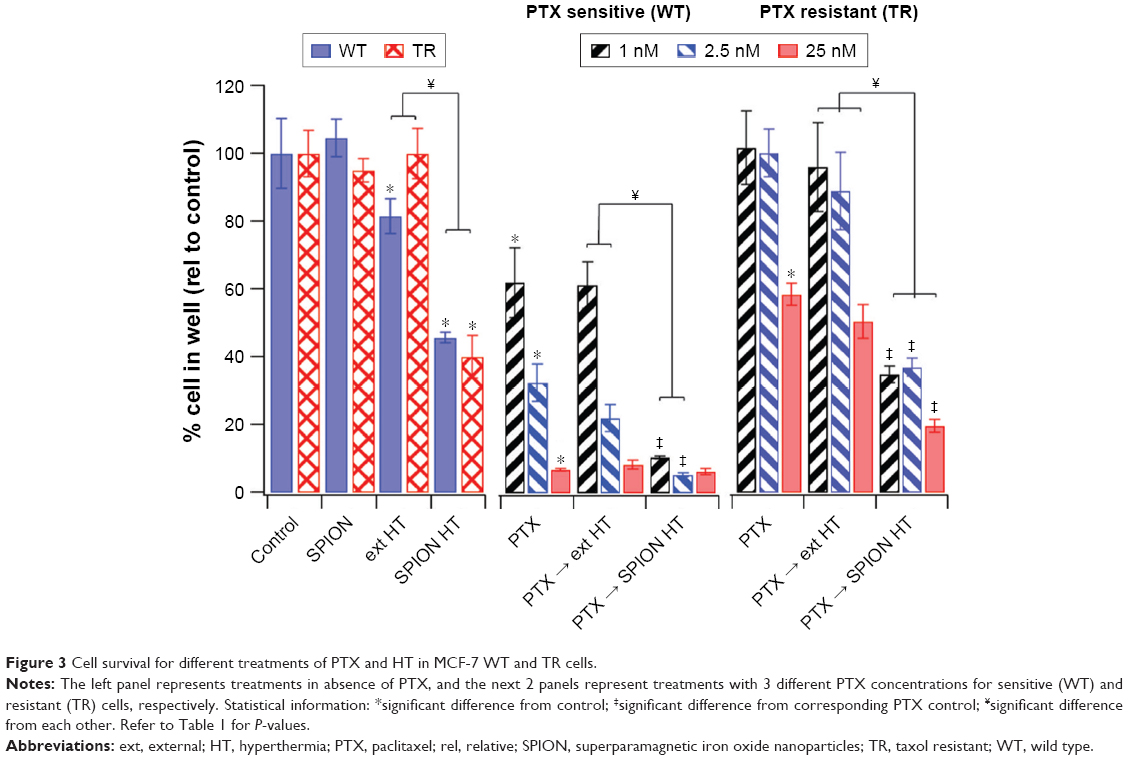 | Figure 3 Cell survival for different treatments of PTX and HT in MCF-7 WT and TR cells. |
Statistical significance was determined by one-way ANOVA with Sidak’s post hoc test and the results of comparisons are summarized in Table 1. We found that TR cells are less susceptible to heat when exposed to ext HT, compared with WT cells. However, both cell sublines are equally affected by SPION HT treatment (in the absence of PTX). There is also a statistically significant difference between HT and SPION HT treatments in both cell lines, suggesting that local heating produced by SPIONs is more effective in cell killing. Comparisons between PTX alone and PTX→HT show no statistically significant difference for both cell lines, in contrast with PTX→SPION HT, which shows a statistically significant improvement in outcome over cells treated with PTX alone.
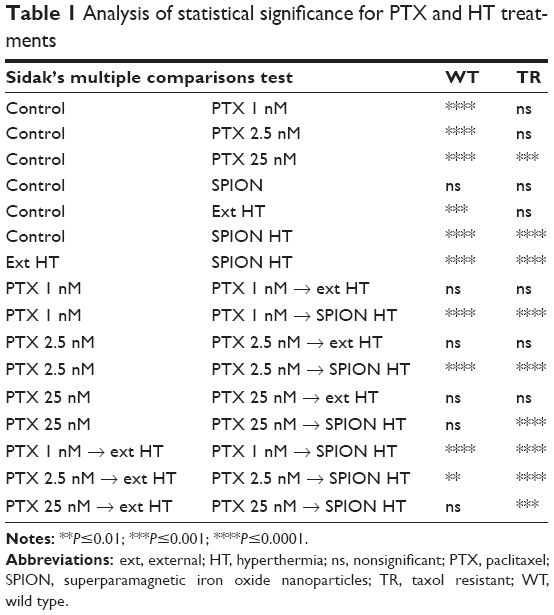 | Table 1 Analysis of statistical significance for PTX and HT treatments |
Analysis of treatment outcome by nuclei morphology
In order to better understand the effect of each treatment, the cell nuclei were visualized and classified as interphase, mitotic, micronucleated (MN), and apoptotic (Figure 4). Cells in interphase or mitotic stages were regarded as normal cells. In contrast, MN morphology is an indication of forced mitotic exit, and apoptotic morphology indicates cells that had undergone significant damage and are entering stages of programmed cell death. Figure 4A shows representative images for nuclei morphologies and Figure 4B summarizes the corresponding percentages of cells classified by nuclei morphology in several treatment conditions for MCF-7 WT and TR cells. For both cell types, PTX→SPION HT resulted in a greater increase in the percentage of MN cells. Also, there was an increase in the percentage of apoptotic cells for the PTX→SPION HT treatment groups in the TR cells. SPION HT by itself did not cause an increase in micronuclei formation, but it did increase the percentage of apoptotic cells in the TR cell line. These results suggest that SPION HT potentiates the PTX effect by significantly reducing cell survival, and, in contrast to the ext HT, triggers programmed cell death.
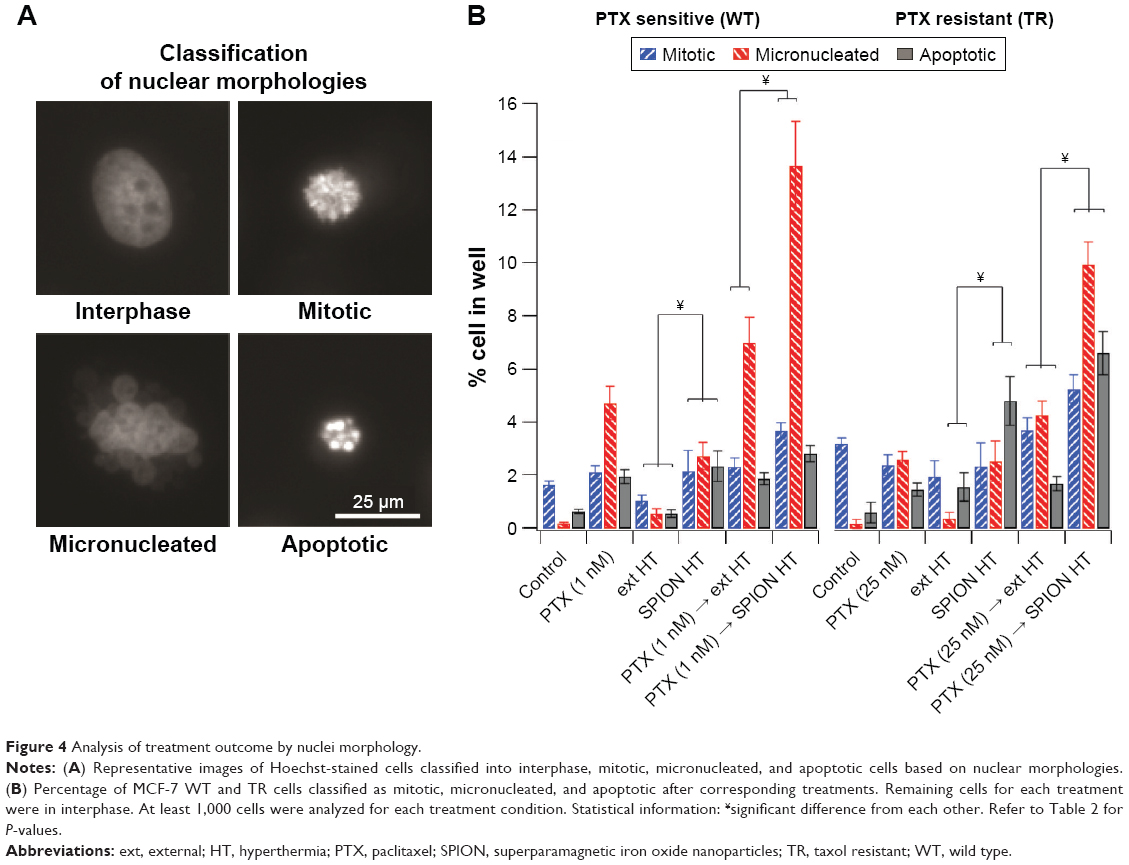 | Figure 4 Analysis of treatment outcome by nuclei morphology. |
Analysis for statistical significance (Table 2) shows that ext HT alone did not cause any difference compared with the control in the percentage of classified cells, whereas SPION HT alone shows a significant difference in MN and apoptotic cell percentages in both sensitive and resistant sublines. When compared with PTX alone, both combination treatments were more effective at increasing micronuclei formation in both sublines, but not necessarily were efficient at increasing the percentage of apoptotic cells. However, both ext and SPION HT treatments alone were significantly different at producing MN and apoptotic cells, with higher percentage of both in cells treated with SPION HT alone. Moreover, both combination treatments show a significant difference with a P-value<0.0001 in resistant cells for the 3 cell classifications. For the sensitive cells, a significant difference was seen in mitotic and MN cell formation.
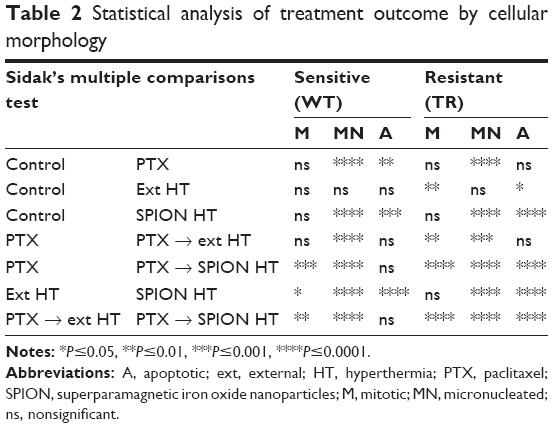 | Table 2 Statistical analysis of treatment outcome by cellular morphology |
Discussion
Currently, there are several ways to treat breast cancer, depending on the type and stage. Among chemotherapeutic agents, antimitotic drugs, such as taxanes are commonly used to treat breast cancer. This microtubule poison is a cytotoxic drug that has proven its efficacy in a large number of malignancies. However, its clinical success has been limited by toxicity and drug resistance.6
Despite extensive research on PTX, the mechanisms underlying clinical resistance remain poorly understood. PTX-induced hyper-polymerization of microtubules can affect cellular machinery at multiple levels, suggesting several mechanisms of PTX resistance. In interphase cells, microtubule hyper-polymerization blocks cytoplasmic transport, resulting in abnormal dynamics of signaling proteins.36 The major effect of PTX on microtubule hyper-polymerization is the activation of the SAC, compromising the proper attachment of microtubules, which prompts a SAC-dependent mitotic block.37 PTX-induced hyper-polymerization of microtubules is reversible, by resuming normal microtubule dynamics after PTX washout in cell culture or PTX decay in patients. While cells resistant to PTX remain in mitotic block longer, cells sensitive to PTX exit mitosis by a mechanism known as mitotic catastrophe.5 The biochemical mechanism of mitotic catastrophe in the presence of an active SAC is not understood, however, it is well established that, as soon as levels of cyclin B drop below a threshold, cells exit mitosis in an aberrant MN G1 stage, a morphological marker of mitotic catastrophe.38 MN cells often fail the next round of cell division by undergoing apoptosis, necrosis, or senescence.39–41
To overcome taxane resistance and more efficiently kill tumor cells, two approaches have been previously investigated: prevent mitotic exit or force it. Unfortunately, the first one was not effective due to severe side effects in clinical applications.42 In order to force mitotic exit of PTX-induced mitotic block, mild HT (41°C–42°C) can be used to kill resistant cells in PTX-pretreated cells.6 Mild HT at the end of PTX treatment, when cells are accumulated in mitosis, significantly increases cell death compared with PTX or mild HT alone, suggesting efficacy of combined treatment.6
HT has been shown to be an effective potentiator of chemotherapy, but clinical applications have been limited due to poor control of temperature rise in the target tissue, while sparing nearby healthy tissue.43 Nanoscale heat generation represents an attractive alternative to conventional methods of HT, as heat can be generated and constrained within the area of interest through a combination of nanoparticle localization and spatial control of the means of actuating heat release.
Nanoscale thermal treatment using SPIONs has not shown any cytotoxicity effects in MCF-7 cells in vitro at a concentration of 1.5 mgFe/mL. SPIONs can be engineered to achieve high heating rates and maintain colloidal stability in biological environments.25 As previously shown20,21 and demonstrated here in Figure 2, SPIONs were coated with PEG for colloidal stability and biocompatibility, and its magnetic properties and energy dissipation rates make them suitable for Magnetic Fluid HT in both in vitro and in vivo models.44,45 Also, SPION HT is more effective at potentiating cancer drugs due to additional damage caused by nanoscale heating to cell membranes, lysosomes, microtubules, and the proteolytic apparatus of cancer cells.29–31
Our results indicate that SPION HT is more effective than ext HT at potentiating response to PTX in sensitive and resistant cell lines. Most importantly, combination of SPION HT with PTX in the resistant cell line leads to significant reduction in cell survival at a low PTX concentration that, otherwise, had no effect. Despite this, we still observe about 20%–40% survival in TR cell subline. We need to consider that PTX only affects cells during mitosis, and cells were only exposed to the drug for 18 hours. In addition, MCF-7 cells have a doubling time of ~38 hours and cell-cycle synchronization was not performed because of its poor relevance in clinical translation. Furthermore, three different concentrations of PTX (including half inhibitory concentration) were selected for this study because the clinically relevant concentration in cell culture studies has been unclear.46 However, combination treatment using PTX and SPIONs kills cells more efficiently than PTX or HT treatments alone, and combination treatment of PTX with ext HT (Figure 3). These results suggest treatment advantages provided by nanoscale heating that help to potentiate PTX and overcome resistance. This suggests the importance of developing new therapies that enhance current therapy outcomes. In that regard, SPION HT in combination with PTX shows promising results.
To help address the mechanism of cell death and differential survival on MCF-7 WT and TR cells upon combination treatments, we evaluated nuclei morphology to discriminate between interphase, mitotic, MN, and apoptosis at the end of treatment (Figure 4). For both cell lines, the percentage of MN cells (indicative for mitotic catastrophe, the required outcome of PTX treatment) increases from PTX treatment alone, to combination of PTX with ext HT, and is further elevating in combination of PTX with SPION HT, showing better treatment outcomes. The level of apoptosis increases from a 0.5% (control) to a 5% and 8% in SPION HT and PTX→SPION HT, respectively in resistant cell line. On the other hand, we did not observe a significant increase of apoptosis in sensitive cells. Indeed, the process of apoptosis may occur as a secondary event to follow PTX-induced mitotic block, or can be elevated by higher PTX concentrations,7 that can be a subject of the follow-up study.
Conclusion
We show that PTX and SPION HT treatment enhance the outcome in cell killing compared with PTX alone or combination of PTX with ext mild HT on breast adenocarcinoma cell line. Importantly, in PTX-resistant cells, PTX and SPION HT potentiate PTX effects overcoming PTX resistance. Further studies are needed to better understand the mechanism behind the treatment, in order to identify HT targets for future clinical translation. Our results suggest the potential benefits of SPION HT in developing new therapies that help enhance breast cancer therapy outcomes.
Acknowledgments
This work was supported by the National Science Foundation Graduate Research Fellowship (Grant number DGE-1315138); and the National Cancer Institute (Grant number R21CA198820). We thank Dr Parissenti from Northern Ontario School of Medicine for providing parental and PTX-resistant MCF-7 cells.
Disclosure
The authors report no conflicts of interest in this work
References
Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9(10):790–803. | ||
Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. | ||
Bonneterre J, Spielman M, Guastalla JP, et al. Efficacy and safety of docetaxel (Taxotere) in heavily pretreated advanced breast cancer patients: the French compassionate use programme experience. Eur J Cancer. 1999;35(10):1431–1439. | ||
Crown J, O’Leary M, Ooi WS. Docetaxel and paclitaxel in the treatment of breast cancer: a review of clinical experience. Oncologist. 2004;9(Suppl 2):24–32. | ||
Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16(12):1194–1200. | ||
Giovinazzi S, Bellapu D, Morozov VM, Ishov AM. Targeting mitotic exit with hyperthermia or APC/C inhibition to increase paclitaxel efficacy. Cell Cycle. 2013;12(16):2598–2607. | ||
Lindsay CR, Scholz A, Morozov VM, Ishov AM. Daxx shortens mitotic arrest caused by paclitaxel. Cell Cycle. 2007;6(10):1200–1204. | ||
Schnerch D, Follo M, Krohs J, Felthaus J, Engelhardt M, Wäsch R. Monitoring APC/C activity in the presence of chromosomal misalignment in unperturbed cell populations. Cell Cycle. 2012;11(2):310–321. | ||
McGrogan BT, Gilmartin B, Carney DN, McCann A. Taxanes, Microtubules and Chemoresistant Breast Cancer. Biochim Biophys Acta. 2008; 1785(2):96–132. | ||
Eskander RN, Cripe J, Bristow RE. Intraperitoneal chemotherapy from Armstrong to HIPEC: challenges and promise. Curr Treat Options Oncol. 2014;15(1):27–40. | ||
Helm CW. CurrentStatus and Future Directions of Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy in the Treatment of Ovarian Cancer. Surg Oncol Clin N Am. 2012:21(4);645–663. | ||
Mulier S, Claes JP, Dierieck V, et al. Survival benefit of adding Hyperthermic IntraPEritoneal Chemotherapy (HIPEC) at the different time-points of treatment of ovarian cancer: review of evidence. Curr Pharm Des. 2012;18(25):3793–3803. | ||
Oyeniyi J, Wu J, Liu D, et al. Treatment of carcinomatosis using cytoreductive surgery and hyperthermic intraperitoneal chemotherapy in adolescents and young adults. Am J Surg. 2015;209(4):610–615. | ||
Dhavalikar R, Rinaldi C. Theoretical Predictions for Spatially-Focused Heating of Magnetic Nanoparticles Guided by Magnetic Particle Imaging Field Gradients. J Magn Magn Mater. 2016;419:267–273. | ||
Hensley D, Tay ZW, Dhavalikar R, et al. Combining magnetic particle imaging and magnetic fluid hyperthermia in a theranostic platform. Phys Med Biol. 2017;62(9):3483–3500. | ||
Laurent S, Dutz S, Häfeli UO, Mahmoudi M. Magnetic fluid hyperthermia: focus on superparamagnetic iron oxide nanoparticles. Adv Colloid Interface Sci. 2011;166(1–2):8–23. | ||
Jordan A, Scholz R, Wust P, Fähling H, Roland Felix, Roland F. Magnetic fluid hyperthermia (MFH): Cancer treatment with AC magnetic field induced excitation of biocompatible superparamagnetic nanoparticles. J Magn Magn Mater. 1999;201(1–3):413–419. | ||
Maier-Hauff K, Ulrich F, Nestler D, et al. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J Neurooncol. 2011;103(2):317–324. | ||
Thiesen B, Jordan A. Clinical applications of magnetic nanoparticles for hyperthermia. Int J Hyperthermia. 2008;24(6):467–474. | ||
Barrera C, Herrera AP, Bezares N, et al. Effect of poly(ethylene oxide)-silane graft molecular weight on the colloidal properties of iron oxide nanoparticles for biomedical applications. J Colloid Interface Sci. 2012;377(1):40–50. | ||
Barrera C, Herrera AP, Rinaldi C. Colloidal dispersions of monodisperse magnetite nanoparticles modified with poly(ethylene glycol). J Colloid Interface Sci. 2009;329(1):107–113. | ||
Creixell M, Herrera AP, Latorre-Esteves M, Ayala V, Torres-Lugo M, Rinaldi C. The effect of grafting method on the colloidal stability and in vitro cytotoxicity of carboxymethyl dextran coated magnetic nanoparticles. J Mater Chem. 2010;20(39):8539–8547. | ||
Mérida F, Chiu-Lam A, Bohórquez AC, et al. Optimization of synthesis and peptization steps to obtain iron oxide nanoparticles with high energy dissipation rates. J Magn Magn Mater. 2015;394:361–371. | ||
Unni M, Uhl AM, Savliwala S, et al. Thermal Decomposition Synthesis of Iron Oxide Nanoparticles with Diminished Magnetic Dead Layer by Controlled Addition of Oxygen. ACS Nano. 2017;11(2):2284–2303. | ||
Wahajuddin, Arora S. Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers. Int J Nanomedicine. 2012;7:3445–3471. | ||
Creixell M, Bohórquez AC, Torres-Lugo M, Rinaldi C. EGFR-targeted magnetic nanoparticle heaters kill cancer cells without a perceptible temperature rise. ACS Nano. 2011;5(9):7124–7129. | ||
Domenech M, Marrero-Berrios I, Torres-Lugo M, Rinaldi C. Lysosomal membrane permeabilization by targeted magnetic nanoparticles in alternating magnetic fields. ACS Nano. 2013;7(6):5091–5101. | ||
Jordan A, Scholz R, Maier-Hauff K, et al. Presentation of a new magnetic field therapy system for the treatment of human solid tumors with magnetic fluid hyperthermia. J Magn Magn Mater. 2001;225(1–2):118–126. | ||
Alvarez-Berrios MP, Castillo A, Mérida F, Mendez J, Rinaldi C, Torres-Lugo M. Enhanced proteotoxic stress: one of the contributors for hyperthermic potentiation of the proteasome inhibitor bortezomib using magnetic nanoparticles. Biomater Sci. 2015;3(2):391–400. | ||
Alvarez-Berríos MP, Castillo A, Rinaldi C, Torres-Lugo M. Magnetic fluid hyperthermia enhances cytotoxicity of bortezomib in sensitive and resistant cancer cell lines. Int J Nanomedicine. 2014;9:145–153. | ||
Lee JS, Rodríguez-Luccioni HL, Méndez J, et al. Hyperthermia induced by magnetic nanoparticles improves the effectiveness of the anticancer drug cis-diamminedichloroplatinum. J Nanosci Nanotechnol. 2011;11(5):4153–4157. | ||
Torres-Lugo M, Rinaldi C. Thermal potentiation of chemotherapy by magnetic nanoparticles. Nanomedicine (Lond). 2013;8(10):1689–1707. | ||
Hembruff SL, Laberge ML, Villeneuve DJ, et al. Role of drug transporters and drug accumulation in the temporal acquisition of drug resistance. BMC Cancer. 2008;8:318. | ||
Maldonado-Camargo L, Torres-Díaz I, Chiu-Lam A, Hernández M, Rinaldi C. Estimating the contribution of Brownian and Néel relaxation in a magnetic fluid through dynamic magnetic susceptibility measurements. J Magn Magn Mater. 2016;412:223–233. | ||
Fortin JP, Gazeau F, Wilhelm C. Intracellular heating of living cells through Néel relaxation of magnetic nanoparticles. Eur Biophys J. 2008;37(2):223–228. | ||
Thadani-Mulero M, Nanus DM, Giannakakou P. Androgen receptor on the move: boarding the microtubule expressway to the nucleus. Cancer Res. 2012;72(18):4611–4615. | ||
Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8(5):379–393. | ||
Mantel C, Guo Y, Lee MR, et al. Cells enter a unique intermediate 4N stage, not 4N-G1, after aborted mitosis. Cell Cycle. 2008;7(4):484–492. | ||
Erenpreisa JE, Ivanov A, Dekena G, et al. Arrest in metaphase and anatomy of mitotic catastrophe: mild heat shock in two human osteosarcoma cell lines. Cell Biol Int. 2000;24(2):61–70. | ||
Roninson IB, Broude EV, Chang B-D. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resistance Updates. 2001;4(5):303–313. | ||
Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15(7):1153–1162. | ||
Janssen A, Medema RH. Mitosis as an anti-cancer target. Oncogene. 2011;30(25):2799–2809. | ||
van der Zee J. Heating the patient: a promising approach? Ann Oncol. 2002;13(8):1173–1184. | ||
Kozissnik B, Bohorquez AC, Dobson J, Rinaldi C. Magnetic fluid hyperthermia: advances, challenges, and opportunity. Int J Hyperthermia. 2013;29(8):706–714. | ||
Tasci TO, Vargel I, Arat A, Guzel E, Korkusuz P, Atalar E. Focused RF hyperthermia using magnetic fluids. Med Phys. 2009;36(5):1906–1912. | ||
Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–2681. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.